Introduction
Atherosclerosis (AS) is an inflammatory disease of
the large and middle arteries characterized by abnormal aggregation
of subcutaneous lipids, especially oxidized low-density lipoprotein
(ox-LDL), and macrophage-derived foam cells and is a major cause of
death from cardiovascular disease (1). Ox-LDL can act as the main trigger of
aseptic inflammation by recruiting and attaching to a variety of
inflammatory cells, mainly monocytes and neutrophils and
aggravating the inflammatory response that damages blood vessels
(2). Increased levels of
intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion
molecule-1 (VCAM-1) and E-selectin are observed in atherosclerotic
vascular endothelial cells (3,4).
Ox-LDL induced increased adhesion activity of endothelial cells,
which is mainly manifested by significantly increased cell adhesion
molecules expressed by endothelial cells (5), to contribute to endothelia cell
damage.
Currently, statins are the first-line drugs in
clinical treatment of AS. Statins play an anti-AS role mainly by
improving vascular endothelial function (6), inhibiting proliferation and migration
of vascular smooth muscle cells (7) and inhibiting formation of foam cells
(8). Statin can significantly
induce the expression of Krüppel like transcription factor 2
(KLF2), a member of the zinc finger subfamily of transcription
factors and a member of the SPL/Krüppel like transcription factor
family (9,10). KLF2 is evolutionarily conserved
between rodents and humans and maintains endothelial homeostasis
through anti-inflammatory, anti-thrombosis, anti-oxidation and
anti-proliferation effects in endothelial cells (11,12).
However, the protective role of KLF2 in the process of endothelial
cell injury has not been fully elucidated. Further exploration of
the regulatory mechanism of KLF2 on endothelial injury may provide
a new target for the clinical treatment of AS.
KLF2 has also been shown to be involved in the
differentiation of monocytes. KLF2 expression is reduced during the
differentiation of monocytes to macrophages (13), which negatively regulates the
pro-inflammatory activation of monocytes/macrophages and is also an
important regulator of innate immune response (13,14).
Myeloid cell-specific knockout of KLF2 enhances the adhesion of
macrophages to endothelial cells and induces the uptake of ox-LDL
by macrophages (10,15), thereby increasing the formation of
macrophage-derived foam cells. The appearance of foam cells is
considered to be one of the early manifestations of AS and the
potential extracellular and intracellular lipid deposition is also
a key trigger factor for the progression of AS lesions (16,17).
Therefore, it is important to enhance the outflow of cholesterol
from macrophages in the treatment process of AS, which can
effectively reduce the formation of macrophage-derived foam cells
and contribute to plaque stability.
Autophagy may play an active role in cholesterol
effluent from macrophages. A recent study showed that interfering
with KLF2 inhibited Beclin and LC3 levels in abdominal aortic
aneurysms, suggesting that KLF2 activates autophagy-related genes
in smooth muscle cells (18).
Simvastatin maintains microvascular function by inhibiting RAC1,
thereby releasing RAB7, activating autophagy and increasing the
expression of KLF2, which in turn further promotes the activation
of autophagy (19). However, it
remains to be elucidated whether KLF2 plays a role in the process
of macrophage foaming, and whether KLF2 can participate in the
process of cholesterol effusion in macrophages by regulating
autophagy is worth exploring.
Therefore, the present study aimed to investigate
whether KLF2 could protect against endothelial cell injury and
promote cholesterol excretion from foam cells through
autophagy.
Materials and methods
Cell culture
Human umbilical vein endothelial cells (HUVECs) and
human acute monocytic leukemia cells (THP-1 monocytes) were
purchased from CoBioer Biosciences Co., Ltd. HUVECs were cultured
in DMEM medium (Gibco; Thermo Fisher Scientific Inc.) supplemented
with 10% FBS in a humidified atmosphere containing 5%
CO2 at 37˚C. An in vitro AS model was established
by the induction of 100 µg/ml ox-LDL (MilliporeSigma) for HUVECs
for 24 h. For ox-LDL treatment, the cells were exposed to 100 µg/ml
of ox-LDL for 24 h after transfection.
THP-1 monocytes were cultured in RPMI-1640 medium
(Gibco; Thermo Fisher Scientific Inc.) supplemented with 10% FBS
(Thermo Fisher Scientific, Inc.) in a humidified atmosphere
containing 5% CO2 at 37˚C. THP-1 monocytes were treated
with 50 nM phorbol-12-myristate-13-acetate (PMA) for 48 h to induce
their differentiation into macrophages. Then, THP-1 derived
macrophages were continuously treated with 50 µg/ml ox-LDL for 48 h
to form THP-1 macrophage-derived foam cells.
Cell transfection
HUVECs or THP-1 derived macrophages were seeded into
6-well plates at the density of 1x106 cells/well. The
sequence of KLF2 (5'-CAACAGCGTGCTGGACTTCA-3') was cloned into a
pcDNA3.1 vector to generate overexpressing plasmid (pcDNA3.1-KLF2)
by GenePharma Co., Ltd. and the empty pcDNA3.1 vector (pcDNA3.1-NC)
was used as a negative control. The accession no. of this KLF2 was
NC_000019. Transfection of 20 µg pcDNA3.1 plasmids into HUVECs or
THP-1 derived macrophages was performed at 37˚C for 6 h by using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the kit instructions. At 48 h
post-transfection, reverse transcription-quantitative (RT-q) PCR
and western blot analysis were used to examine gene expression
levels.
RT-qPCR
HUVECs or THP-1 monocytes in each group (24-well
plates at a density of 2x105 cells per well) were
treated with TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) to obtain the total RNA according to the
manufacturer's protocols. RNA purity was assessed by
spectrophotometric analysis (CWBIO) wherein the A260/280 ratios
were between 1.8 and 2.2. Equal RNA was used for cDNA generation by
using a PrimeScript RT reagent kit with a gDNA eraser (Takara Bio,
Inc.) according to the manufacturer's protocols. Subsequently, SYBR
Premix Ex Taq II (Takara Bio, Inc.) was then used to amplify cDNA
through qPCR according to the manufacturer's protocols using an ABI
Prism 7500 Fast Real-time PCR instrument (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The amplification efficiencies of
the qPCRs were 90-110%. The thermocycling conditions were: Initial
denaturation at 95˚C for 10 min; followed by 40 cycles of 95˚C for
1 min and 60˚C for 1 min. The sequences of oligonucleotide primers
used for qPCR were: KLF2 forward, 5'-AGACCTACACCAAGAGTTCGCATC-3'
and reverse, 5'-ATCGCACAGATGGCACTGGAATG-3'; ICAM-1 forward,
5'-GCCCGAGCTCAAGTGTCTAA-3' and reverse, 5'-GGAGAGCACATTCACGGCA-3';
VCAM-1 forward, 5'-AATTTATGTGTGTGAAGGAG-3' and reverse,
5'-GCATGTCATATTCACAGAA-3'; E-selectin forward,
5'-TGTGGGTCTGGGTAGGAACC-3' and reverse,
5'-AGCTGTGTAGCATAGGGCAAG-3'; ATP-binding cassette transporters A1
(ABCA1) forward, 5'-GCTGGTGTGGACCCTTACTC-3' and reverse,
5'-GCAGCTTCATATGGCAGCAC-3'; ATP-binding cassette transporter G1
(ABCG1) forward, 5'-TGTCTGATGGCCGCTTTCTC-3' and reverse,
5'-GGACCCATAATGGCCACCAA-3'; scavenger receptor BI (SR-BI) forward,
5'-ATGACTCCTGAGTCCTCGCT-3' and reverse, 5'-GGTCAGCGTTGAGGAAGTGA-3';
peroxisome proliferator-activated receptor-γ (PPARγ) forward,
5'-CCAGAAGCCTGCATTTCTGC-3' and reverse, 5'-CACGGAGCTGATCCCAAAGT-3';
liver X receptor (LXR) α forward, 5'-TCTGGACAGGAAACTGCACC-3' and
reverse, 5'-AAGAATCCCTTGCAGCCCTC-3'; GAPDH forward,
5'-CCTCAAGATCATCAGCAATG-3' and reverse, 5'-CCATCC
ACAGTCTTCTGGGT-3'. The 2-ΔΔCq
method was used to analyze the relative expression of KLF2, ICAM-1,
VCAM-1 and E-selectin (20), which
were normalized to GAPDH. The experiment was repeated three
times.
Western blotting
For the detection of nuclear factor erythroid
2-related factor 2 (Nrf2), nucleoprotein and cytoplasmic protein
extraction kit obtained from Nanjing KeyGen Biotech Co., Ltd. was
used to extract the nucleoprotein and cytoplasmic proteins from
HUVECs or THP-1 monocytes. For the detection of other proteins,
HUVECs or THP-1 monocytes in each group were homogenized in RIPA
lysis buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1% NP-40, 0.5%
sodium deoxycholate, 0.1% SDS; Beyotime institute of
Biotechnology], which was centrifuged at 4˚C at 850 x g for 15 min
to collect the total protein. The protein concentration was
determined by bicinchoninic acid (BCA) method. The equivalent
amount of 50 µg protein was separated by 12% SDS-PAGE and then
transferred to a PVDF membranes (MilliporeSigma) which were then
blocked with 5% non-fat milk for 2 h at room temperature.
Subsequently, the membranes were incubated with primary antibodies
as KLF2 (cat. no. ab236507; 1:1,000; Abcam), ICAM-1 (cat. no.
ab282575; 1:1,000; Abcam), VCAM-1 (cat. no. ab174279; 1:1,000;
Abcam), E-selectin (cat. no. 20894-1-AP; 1:800; ProteinTech Group,
Inc.), ABCA1 (cat. no. 96292S; 1:1,000; Cell Signaling Technology,
Inc.), ABCG1 (cat. no. ab52617; 1:1,000; Abcam), SR-BI (cat. no.
ab217318; 1:1,000; Abcam), PPARγ (cat. no. 16643-1-AP; 1:1,000;
ProteinTech Group, Inc.), LXRα (cat. no. 14351-1-AP; 1:1,000;
ProteinTech Group, Inc.), Nrf2 (cat. no. ab62352; 1:1,000; Abcam),
Lamin B (cat. no. 12987-1-AP; 1:1,000; ProteinTech Group, Inc.),
LC3II (cat. no. 4108S; 1:1,000; Cell Signaling Technology, Inc.),
Beclin1 (cat. no. 3495T; 1:1,000; Cell Signaling Technology, Inc.),
p62 (cat. no. 18420-1-AP; 1:1,000; ProteinTech Group, Inc.) and
GAPDH (cat. no. 5174T; 1:1,000; Cell Signaling Technology, Inc.) at
4˚C overnight. Following incubation with the horseradish
peroxidase-conjugated anti-rabbit secondary antibody (cat. no.
7074P2, 1:3,000; Cell Signaling Technology, Inc.) for 1 h at room
temperature, protein bands were visualized by a chemiluminescent
detection system (ECL; Cytiva) using an ECL reagent (Thermo Fisher
Scientific, Inc.). Bands grey values were semi-quantified analyzed
using ImageJ software (v1.46; National Institutes of Health).
Protein expression was normalized to the internal reference gene
GAPDH or Lamin B.
CCK-8 assay
HUVECs (5x103 cells per well) were seeded
into 96-well plates and transfection performed for 48 h. Then, the
transfected HUVECs were treated with 100 µg/ml of ox-LDL for 24 h.
After PBS washing, each well was added with 10 µl CCK-8 solution
and the 96-well plates were incubated at 37˚C for 2 h. The
absorbance of each well was detected by a microplate reader
(Bio-Rad Laboratories, Inc.) at 450 nm.
ELISA
HUVECs were cultured in 96-well plates at a density
of 1x104 cells/ml. HUVECs were transfected for 48 h and
then exposed to ox-LDL (100 µg/ml) for 24 h, then the medium was
obtained and centrifuged at 4˚C at 850 x g for 10 min to collect
the culture supernatant. The levels of TNF-α, IL-6 and IL-1β in the
supernatant was assessed using a TNF-α ELISA Kit (cat. no. PT518),
IL-1β ELISA Kit (cat. no. PI305) and IL-6 ELISA Kit (cat. no.
PI330) all from Beyotime Institute of Biotechnology.
Oil Red O staining
After THP-1 monocytes were induced with 50 nM PMA
and/or 50 µg/ml ox-LDL, Oil Red O staining was used to evaluate the
formation of THP-1 macrophage-derived foam cells. Following the
treatment, cells were fixed with 4% paraformaldehyde
(MilliporeSigma) for 30 min and then incubated with filtered 0.5%
Oil Red O solution (MilliporeSigma) for 15 min at room temperature.
Images were captured under a confocal microscope (Olympus
Corporation) and positive Oil Red O areas (staining areas) was used
to reflect the total lipid content. The % positive Oil Red O areas
was calculated by ImageJ (v.1.8.0; National Institutes of
Health).
Liquid scintillation counting
Cholesterol efflux was analyzed as previously
described (21). The macrophages
and foam cells were cultured and labeled with 0.4 µCi/ml
[3H]-cholesterol for 24 h and incubated overnight in
RPMI-1640 medium with 0.1% BSA to allow equilibration of
[3H]-cholesterol in all cellular pools. Following PBS
washing, equilibrated [3H] cholesterol-labeled cells
were incubated in RPMI-1640 medium containing 0.1% BSA with or
without 15 µg/ml apoA-I for 12 h. A 150 µl sample of efflux medium
was obtained at and passed through a 0.45 µm filter to remove any
floating cells. Monolayers were washed twice in PBS and cellular
lipids were extracted with isopropanol. Medium and cell-associated
[3H] cholesterol was then determined by liquid
scintillation counting through detecting the α-rays derived from
[3H] cholesterol. Percent efflux was evaluated according
to the equation: [total media counts/(total cellular counts + total
media counts)] x100%.
Statistical analysis
All of the experiments were repeated three times and
the data were conformed to normal distribution using Shapiro-Wilk
test. Data are presented as means ± standard deviation and analyzed
by GraphPad 8.0 (GraphPad Software, Inc.). Two-group comparisons
were analyzed with Student's t-test. Comparisons between three or
more groups were analyzed by One way analysis of variance, followed
by Tukey's test. P<0.05 was considered to indicate a
statistically significant difference.
Results
KLF2 overexpression attenuates
ox-LDL-induced endothelial cell injury
The expression of KLF2 in HUVECs was downregulated
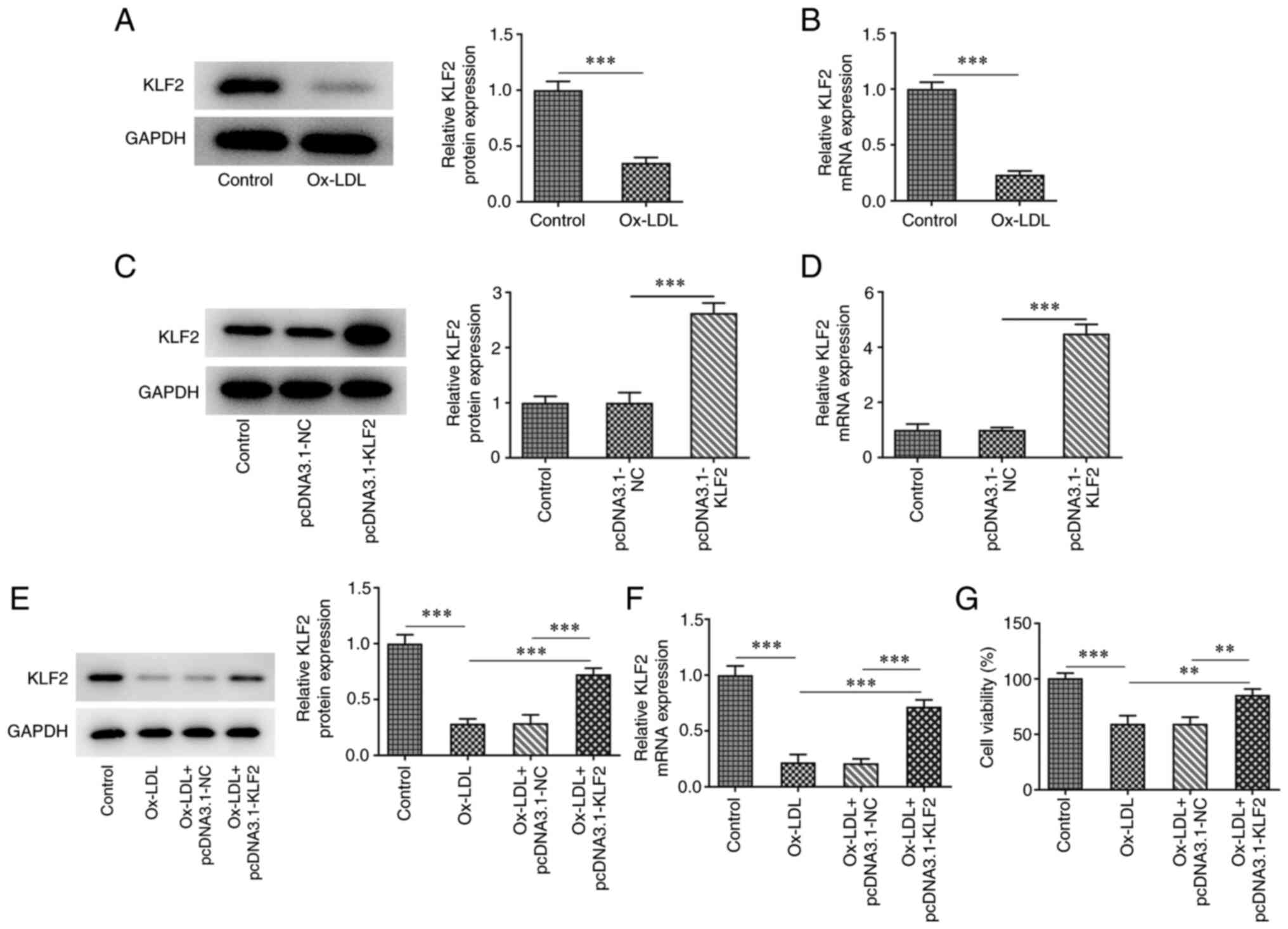
after ox-LDL induction compared to the control group (Fig. 1A and B). The expression of KLF2 in HUVECs
transfected with pcDNA3.1-KLF2 was upregulated (Fig. 1C and D). When ox-LDL-induced HUVECs were
transfected with pcDNA3.1-KLF2, KLF2 protein and mRNA expression
was significantly upregulated (Fig.
1E and F). The result of
Fig. 1E indicated that ox-LDL
suppressed the HUVECs viability while the further KLF2
overexpression improved the decreased viability of ox-LDL-induced
HUVECs (Fig. 1G). The levels of
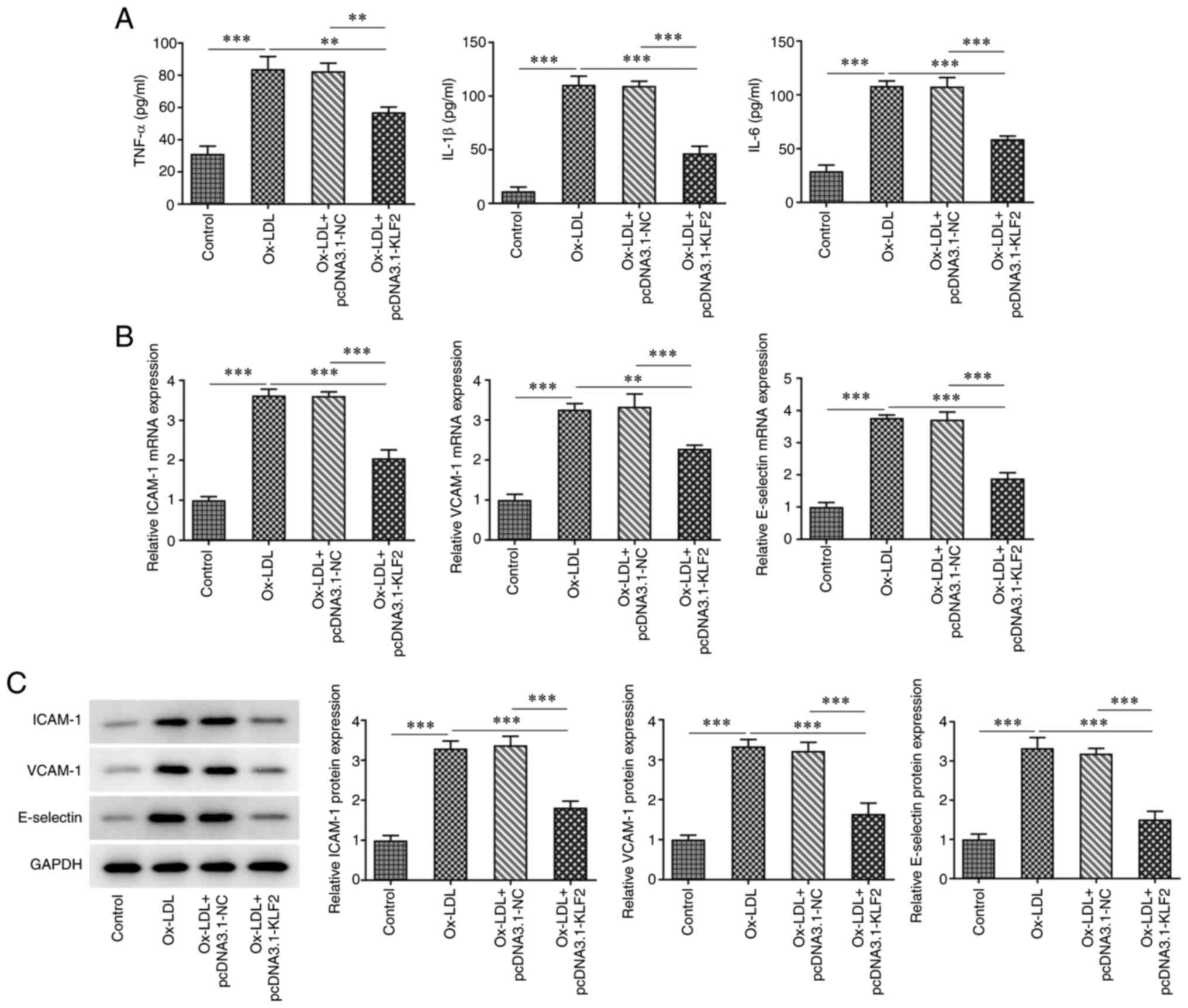
TNF-α, IL-1β and IL-6 were increased in ox-LDL-induced HUVECs,
which was suppressed by KLF2 overexpression (Fig. 2A). The expression of adhesion
factors (ICAM-1, VCAM-1 and E-selectin) was increased in HUVECs
treated with ox-LDL and decreased in ox-LDL-induced HUVECs
transfected with pcDNA3.1-KLF2 (Fig.
2B and C). These data
suggested that KLF2 overexpression attenuated ox-LDL-induced
endothelial cell injury.
KLF2 overexpression inhibits the
formation of THP-1 macrophage-derived foam cells
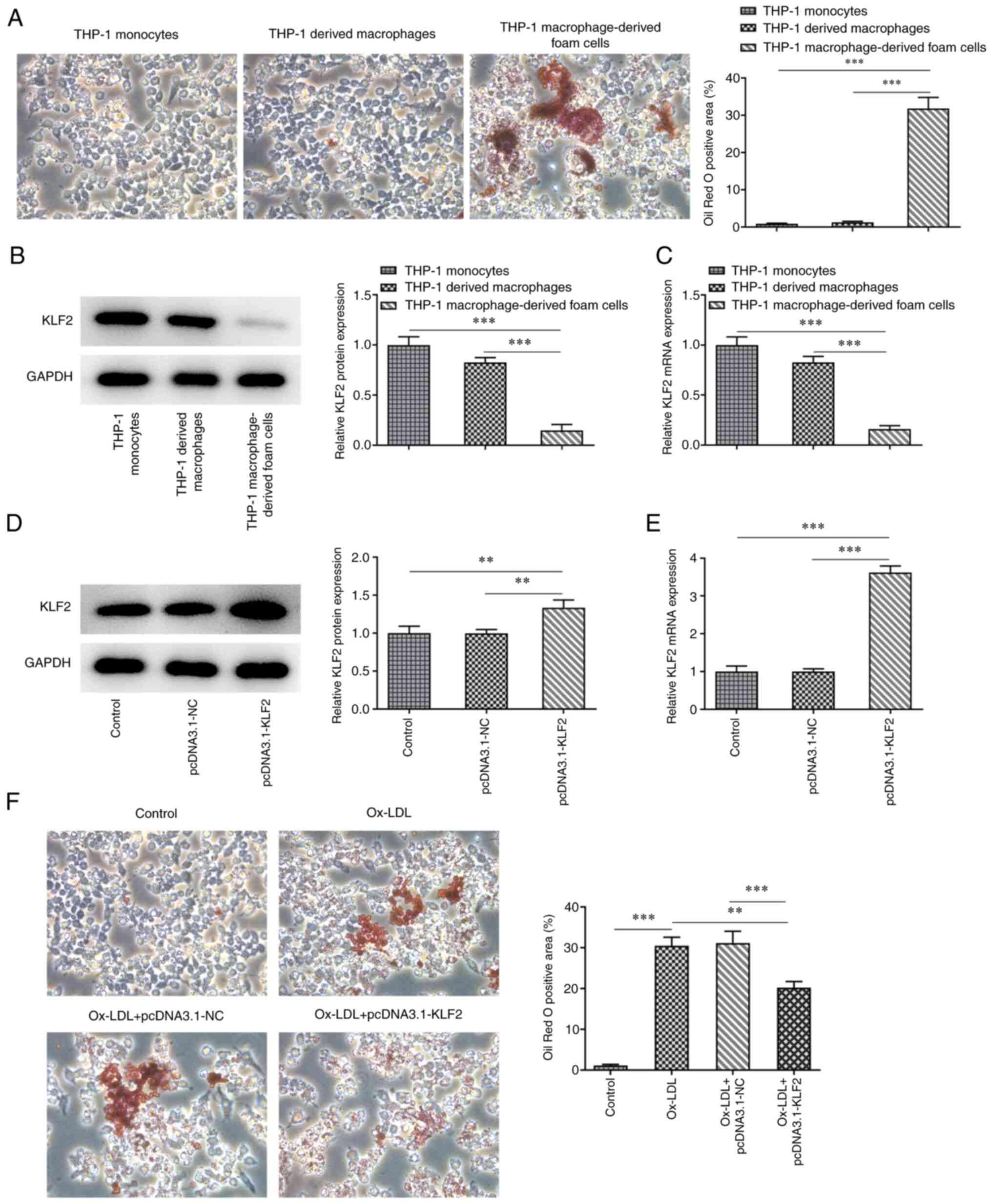
After PMA induced THP-1 monocytes for 48 h, the
cells were fusiform in shape with extended pseudopods to
differentiate into macrophages. The mature macrophages were
differentiated. The mature differentiated macrophages were
incubated with ox-LDL for 48 h and the cells were stained with oil
red O. Under the light microscope, there were a large number of red
lipid particles in the cells and the cell volume increased
(Fig. 3A). The expression of KLF2
in THP-1 macrophage-derived foam cells was decreased compared with
that in THP-1 monocytes and THP-1 derived macrophages (Fig. 3B and C). After THP-1 derived macrophages
(control) were transfected with pcDNA3.1-KLF2, KLF2 expression was
upregulated (Fig. 3D and E). A large number of red lipid particles
were observed in the THP-1 derived macrophages induced by ox-LDL,
which were decreased by KLF2 overexpression (Fig. 3F). On the whole, KLF2
overexpression inhibited the formation of THP-1 macrophage-derived
foam cells.
KLF2 overexpression promotes lipid
efflux in THP-1 macrophage-derived foam cells
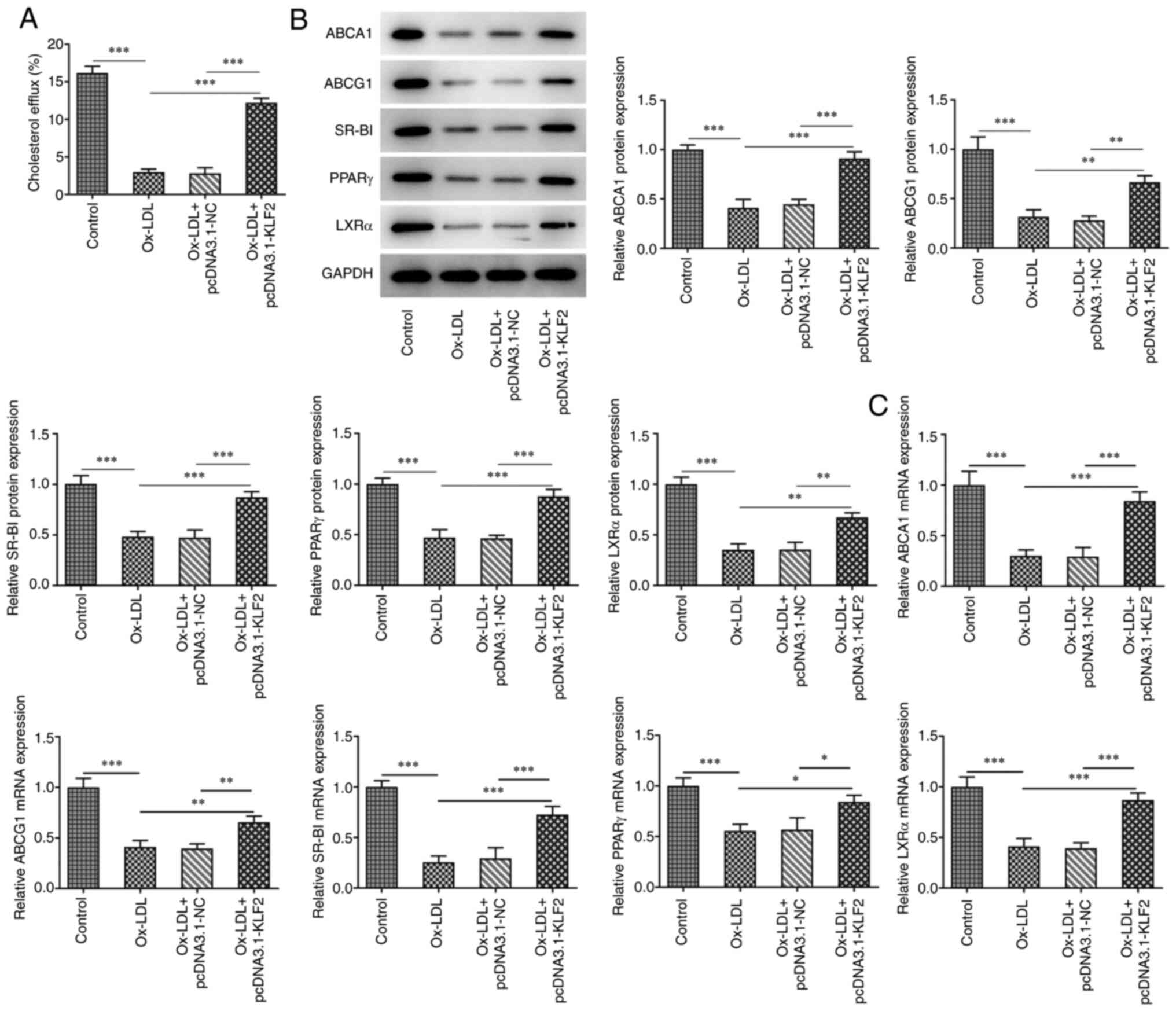
As shown in Fig.
4A, Ox-LDL reduced the cholesterol efflux in THP-1 derived
macrophages, which was promoted by KLF2 overexpression. Ox-LDL
suppressed the expression of cholesterol efflux regulatory proteins
(ABCA1, ABCG1, SR-BI, PPARγ and LXRα) in THP-1 derived macrophages
and KLF2 overexpression could reverse the above changes (Fig. 4B). Consistently, the mRNA
expression levels of ABCA1, ABCG1, SR-BI, PPARγ and LXRα showed the
same changing tendency as their protein expression levels (Fig. 4C). In brief, KLF2 overexpression
promoted lipid efflux in THP-1 macrophage-derived foam cells.
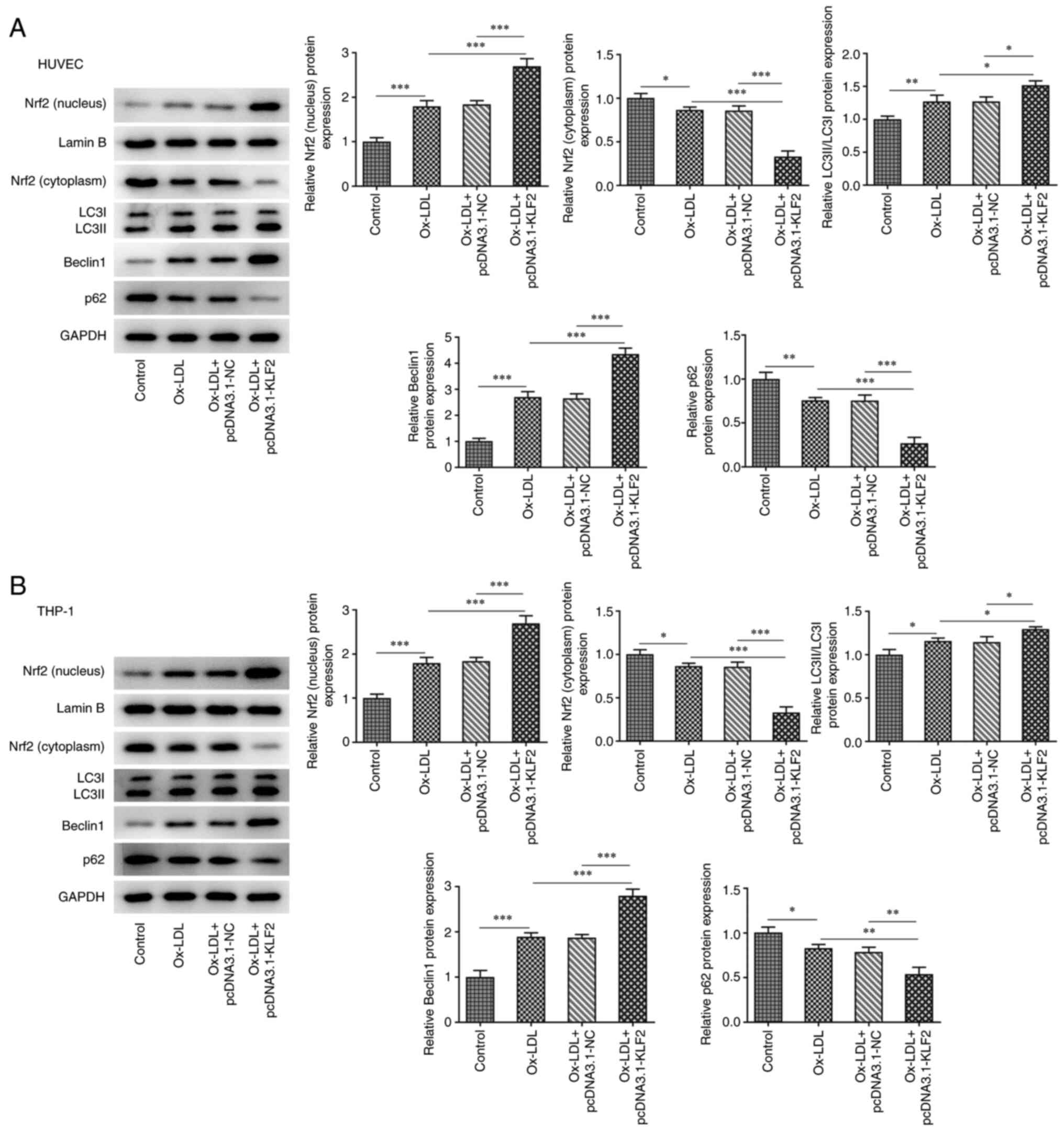
KLF2 overexpression activities Nrf2
expression and enhances autophagy in ox-LDL-induced HUVECs and
THP-1 macrophage derived foam cells
In HUVECs, ox-LDL promoted the expression of Nrf2
(nucleus), LC3II/LC3I and Beclin1 while suppressing the expression
of Nrf2 (cytoplasm) and p62. KLF2 overexpression further enhanced
the expression of Nrf2 (nucleus), LC3II/LC3I and Beclin1 while
downregulating the expression of Nrf2 (cytoplasm) and p62 in ox-LDL
induced HUVECs (Fig. 5A). As shown
in Fig. 5B, the expression of Nrf2
(nucleus), LC3II/LC3I and Beclin1 was increased while the
expression of Nrf2 (cytoplasm) and p62 was decreased in THP-1
derived macrophages induced by ox-LDL and the expression of Nrf2
(nucleus), LC3II/LC3I and Beclin1 was further increased while the
expression of Nrf2 (cytoplasm) and p62 was further decreased by
KLF2 overexpression in THP-1 derived macrophages induced by ox-LDL.
These observations revealed that KLF2 overexpression activates Nrf2
expression and enhances autophagy in ox-LDL-induced HUVECs and
THP-1 macrophage-derived foam cells.
Discussion
The expression of KLF2 can be significantly induced
by statins in the clinical treatment of AS. The present study
indicated that KLF2 expression was decreased in ox-LDL induced
HUVECs. In vivo animal experiments have shown that the
specific loss of KLF2 in endothelial cells is prone to the
occurrence of AS (22). In
addition, KLF2 can also reduce endothelial permeability and enhance
endothelial barrier function by activating endothelial RAP guanine
nucleotide exchange factor 3(23).
Fork head transcription factor 1, regulated by KLF2, directly
inhibits the activation of endothelial inflammasome and delays the
formation of AS lesions (24). In
the present study, KLF2 overexpression improved viability and
alleviated inflammation of ox-LDL-induced HUVECs, which indicated
that KLF2 could protect against injury in HUVECs induced by
ox-LDL.
Aggregation of macrophage-derived foam cells in the
vascular wall is the main pathological feature of AS (25,26).
Macrophages phagocytize excess ox-LDL, resulting in intracellular
lipid metabolism disorder and excessive lipid accumulation in
macrophages formed foam cells. The formation of foam cells is
closely associated with excessive lipid uptake by cells and
intracellular bile sterol outflow disorder (27). It has been shown that inhibiting
cholesterol outflow can lead to cholesterol accumulation in
macrophages and accelerate the formation of foam cells (25). Therefore, the key to inhibiting the
formation of foam cells depends on the cholesterol outflow pathway.
There are three vectors for cholesterol outflow: ABCA1, ABCG1 and
SR-BI (28). PPARγ is expressed in
a variety of vascular cells, including monocytes/macrophages
(29), endothelial cells (30) and smooth muscle cells (31), which are involved in the AS
process. As members of the nuclear receptor superfamily, PPARα and
LXRα bind corresponding ligands and then form active heterodimer
with retinoid X receptor, which is mainly involved in lipid
metabolism and promotes cholesterol excretion (32). The present study demonstrated that
KLF2 overexpression suppressed the formation of macrophage-derived
foam cells by increasing cholesterol outflow through the promotion
of the expression of ABCA1, ABCG1, SR-BI, PPARγ and LXRα.
Under basal conditions, autophagy may help maintain
cell balance. As a stress response to hunger or oxidative,
autophagy can be activated as an adaptive process (33). Evidence suggests that dysregulation
of autophagy could result in AS and that autophagy is implicated in
endothelial cells response to pathophysiological stimuli during AS
(34). Autophagy participates in
the defense mechanism against oxidative stress and inflammation,
thereby preventing vascular cell death (35). Ox-LDL is reported to be an inducer
of autophagy in HUVECs (36,37),
which is in agreement with the present study, as evidenced by
increased LC3II/LC3I, Beclin1 expression and decreased p62
expression in HUVECs. Additionally, a previous study shows that
enhanced autophagy can improve age-related phenotypes, reduce
age-related heart and kidney disease and improve the health status
of mice (38). It can also protect
the body from stress injury and delay the development of AS
(39). Macrophage autophagy serves
an important role in the occurrence and development of AS by
promoting the efflux of intracellular cholesterol (40). Defective autophagy of macrophages
in AS impairs cholesterol metabolism and inflammatory body
activation (41,42). Ox-LDL exposure has been shown to
promote the expression of autophagosome marker proteins in
cultures, possibly by increasing oxidative stress and inflammation
(43,44). However, there is currently
disagreement regarding the reported autophagic activity in
ox-LDL-treated macrophages, as some reports suggest that autophagic
flux is inhibited in ox-LDL-stimulated macrophages (45,46)
and others insisted that that ox-LDL could lead to activation of
autophagy (47,48). The present study also suggested
that autophagy was induced after ox-LDL exposure in THP-1 derived
macrophages. It has been reported that metformin could inhibit foam
cell formation by activating KLF2-mediated autophagy (49). KLF2 possibly contributes to
regulation of autophagy in a model of acute liver injury (19). KLF-autophagy pathway modulates the
life span of nematodes and regulates mammalian age-associated
vascular dysfunction (50). A
recent study showed that interfering with KLF2 inhibits Beclin and
LC3 levels in abdominal aortic aneurysms (18). The present study demonstrated that
overexpression of KLF2 increased the autophagy in ox-LDL-induced
HUVECs and THP-1 macrophage derived foam cells.
Nrf2 is a transcription factor involved in cellular
redox homeostasis. Under oxidative stress, Nrf2 separates from its
inhibitor KEAP-1 and translocates into the nucleus, leading to
transcriptional activation of cellular defense genes (51,52).
Nrf2 activation can protect human coronary endothelial cells from
oxidative stress and knockdown of Nrf2 expression promotes hydrogen
peroxide-induced apoptosis (53).
Hu et al (54) found that
activation of Nrf2 signaling pathway reduced the level of reactive
oxygen species in vascular endothelial cells and inhibited NLRP3
dependent endothelial cell pyrolysis. Induction of Nrf2/HO-1
activation can upregulate the expression of ABCA1 and ABCG1 and
enhance cholesterol excretion (55,56).
Wang et al (57) found that
Nrf2 induces the formation of cytolysosome and enhances autophagy
activity, thereby inhibiting tumor cell apoptosis. It is noteworthy
that Nrf2 inducer upregulates the gene and protein expression of
autophagy-related molecules and also enhances autophagic flux in
diabetic mouse aorta, suggesting that Nrf2 could activate autophagy
in AS (58). Notably, KLF2 can
stimulate Nrf2 to enter into the nucleus and initiate activation of
the Nrf2 pathway (59) and
artesunate enhances nuclear translocation of Nrf2 in vascular
smooth muscle cells by upregulating KLF2 expression (60). The present study indicated that
KLF2 overexpression stimulated Nrf2 to transport into the nucleus
and enhanced autophagy by increasing the expression of LC3II/LC3I
and Beclin1 and decreasing the p62 expression in ox-LDL induced
HUVECs and THP-1 macrophage-derived foam cells. The addition of
some siRNA-Nrf2 or some inhibitor of autophagy might provide
stronger evidence for the present findings, which is a limitation
of the present study and will be conducted in the next
experiments.
In conclusion, KLF2 expression was decreased in
ox-LDL induced HUVECs and THP-1 macrophage-derived foam cells. In
addition, KLF2 alleviated endothelial cell injury and promotes
lipid outflow by inhibiting the formation of THP-1
macrophage-derived foam cells through enhancing autophagy mediated
by Nrf2. The findings provided a promising target for the treatment
of AS. Some limitations that need to be addressed later. The
addition of some siRNA-Nrf2 or some inhibitor of autophagy might
provide stronger evidence for the present findings. Whether the
changes in steady-state nuclear and cytoplasmic levels of Nrf2 are
transcriptional, due to changes in the stability of Nrf2, or
release of Nrf2 from KEAP1 in the cytoplasm needs to be explored.
The use of commercial fluorescent sensors (LC3B-RFP and LC3B-GFP)
and electron microscopy to measure the process of autophagy will be
considered in future study.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZT and XD conceived and designed the experiments.
HR, YL, HY and QL performed the experiments. ZT, HR and XD analyzed
the data and wrote the manuscript. ZT and YL confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang Y and Kou J: Research progress of
oxLDL in atherosclerotic thrombosis. Chin J Cardiovasc Rehabil Med.
30:344–347. 2021.
|
|
2
|
Trpkovic A, Resanovic I, Stanimirovic J,
Radak D, Mousa SA, Cenic-Milosevic D, Jevremovic D and Isenovic ER:
Oxidized low-density lipoprotein as a biomarker of cardiovascular
diseases. Crit Rev Clin Lab Sci. 52:70–85. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wojakowski W and Gminski J: Soluble
ICAM-1, VCAM-1 and E-selectin in children from families with high
risk of atherosclerosis. Int J Mol Med. 7:181–185. 2001.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Davies MJ, Gordon JL, Gearing AJ, Pigott
R, Woolf N, Katz D and Kyriakopoulos A: The expression of the
adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human
atherosclerosis. J Pathol. 171:223–229. 1993.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhong XJ, Chen TW, Chen YH, Chen FH,
Zhou-Xue LI, Liu LL, Liu MQ and Huang QR: Effects of ox-LDL on the
proaggregation and proadhesion-related molecules expression of
vascular endothelial cells. Chin J Arterioscler. 9(e89877)2014.
|
|
6
|
Sen-Banerjee S, Mir S, Lin Z, Hamik A,
Atkins GB, Das H, Banerjee P, Kumar A and Jain MK: Kruppel-like
factor 2 as a novel mediator of statin effects in endothelial
cells. Circulation. 112:720–726. 2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chandrasekar B, Mummidi S, Mahimainathan
L, Patel DN, Bailey SR, Imam SZ, Greene WC and Valente AJ:
Interleukin-18-induced human coronary artery smooth muscle cell
migration is dependent on NF-kappaB- and AP-1-mediated matrix
metalloproteinase-9 expression and is inhibited by atorvastatin. J
Biol Chem. 281:15099–15109. 2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Takemoto M and Liao JK: Pleiotropic
effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase
inhibitors. Arterioscler Thromb Vasc Biol. 21:1712–1719.
2001.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wu Z and Wang S: Role of kruppel-like
transcription factors in adipogenesis. Dev Biol. 373:235–243.
2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Atkins GB, Wang Y, Mahabeleshwar GH, Shi
H, Gao H, Kawanami D, Natesan V, Lin Z, Simon DI and Jain MK:
Hemizygous deficiency of Krüppel-like factor 2 augments
experimental atherosclerosis. Circ Res. 103:690–693.
2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Novodvorsky P and Chico TJ: The role of
the transcription factor KLF2 in vascular development and disease.
Prog Mol Biol Transl Sci. 124:155–188. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Parmar KM, Larman HB, Dai G, Zhang Y, Wang
ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA Jr and
García-Cardeña G: Integration of flow-dependent endothelial
phenotypes by Kruppel-like factor 2. J Clin Invest. 116:49–58.
2006.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Das H, Kumar A, Lin Z, Patino WD, Hwang
PM, Feinberg MW, Majumder PK and Jain MK: Kruppel-like factor 2
(KLF2) regulates proinflammatory activation of monocytes. Proc Natl
Acad Sci USA. 103:6653–6658. 2006.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mahabeleshwar GH, Kawanami D, Sharma N,
Takami Y, Zhou G, Shi H, Nayak L, Jeyaraj D, Grealy R, White M, et
al: The myeloid transcription factor KLF2 regulates the host
response to polymicrobial infection and endotoxic shock. Immunity.
34:715–728. 2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lingrel JB, Pilcher-Roberts R, Basford JE,
Manoharan P, Neumann J, Konaniah ES, Srinivasan R, Bogdanov VY and
Hui DY: Myeloid-specific Krüppel-like factor 2 inactivation
increases macrophage and neutrophil adhesion and promotes
atherosclerosis. Circ Res. 110:1294–1302. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Frostegård J: Immunity, atherosclerosis
and cardiovascular disease. BMC Med. 11(117)2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Summerhill VI, Grechko AV, Yet SF, Sobenin
IA and Orekhov AN: The atherogenic role of circulating modified
lipids in atherosclerosis. Int J Mol Sci. 20(3561)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Salmon M, Spinosa M, Zehner ZE, Upchurch
GR and Ailawadi G: Klf4, Klf2, and Zfp148 activate
autophagy-related genes in smooth muscle cells during aortic
aneurysm formation. Physiol Rep. 7(e14058)2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Guixé-Muntet S, de Mesquita FC, Vila S,
Hernández-Gea V, Peralta C, García-Pagán JC, Bosch J and
Gracia-Sancho J: Cross-talk between autophagy and KLF2 determines
endothelial cell phenotype and microvascular function in acute
liver injury. J Hepatol. 66:86–94. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hu YW, Wang Q, Ma X, Li XX, Liu XH, Xiao
J, Liao DF, Xiang J and Tang CK: TGF-beta1 up-regulates expression
of ABCA1, ABCG1 and SR-BI through liver X receptor alpha signaling
pathway in THP-1 macrophage-derived foam cells. J Atheroscler
Thromb. 17:493–502. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Jain MK, Sangwung P and Hamik A:
Regulation of an inflammatory disease: Krüppel-like factors and
atherosclerosis. Arterioscler Thromb Vasc Biol. 34:499–508.
2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Huang RT, Wu D, Meliton A, Oh MJ, Krause
M, Lloyd JA, Nigdelioglu R, Hamanaka RB, Jain MK, Birukova A, et
al: Experimental lung injury reduces Krüppel-like factor 2 to
increase endothelial permeability via regulation of RAPGEF3-Rac1
signaling. Am J Respir Crit Care Med. 195:639–651. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhuang T, Liu J, Chen X, Zhang L, Pi J,
Sun H, Li L, Bauer R, Wang H, Yu Z, et al: Endothelial Foxp1
suppresses atherosclerosis via modulation of Nlrp3 inflammasome
activation. Circ Res. 125:590–605. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhang M, Li L, Xie W, Wu JF, Yao F, Tan
YL, Xia XD, Liu XY, Liu D, Lan G, et al: Apolipoprotein A-1 binding
protein promotes macrophage cholesterol efflux by facilitating
apolipoprotein A-1 binding to ABCA1 and preventing ABCA1
degradation. Atherosclerosis. 248:149–159. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Chistiakov DA, Bobryshev YV and Orekhov
AN: Macrophage-mediated cholesterol handling in atherosclerosis. J
Cell Mol Med. 20:17–28. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Fisher EA: Regression of atherosclerosis:
The journey from the liver to the plaque and back. Arterioscler
Thromb Vasc Biol. 36:226–235. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Yu XH, Fu YC, Zhang DW, Yin K and Tang CK:
Foam cells in atherosclerosis. Clin Chim Acta. 424:245–252.
2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Castrillo A and Tontonoz P: PPARs in
atherosclerosis: The clot thickens. J Clin Invest. 114:1538–1540.
2004.PubMed/NCBI View
Article : Google Scholar
|
|
30
|
Vattulainen-Collanus S, Akinrinade O, Li
M, Koskenvuo M, Li CG, Rao SP, de Jesus Perez V, Yuan K, Sawada H,
Koskenvuo JW, et al: Loss of PPARγ in endothelial cells leads to
impaired angiogenesis. J Cell Sci. 129:693–705. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Bruemmer D, Blaschke F and Law RE: New
targets for PPARgamma in the vessel wall: Implications for
restenosis. Int J Obes (Lond). 29 (Suppl 1):S26–S30.
2005.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ge CX, Yu R, Xu MX, Li PQ, Fan CY, Li JM
and Kong LD: Betaine prevented fructose-induced NAFLD by regulating
LXRα/PPARα pathway and alleviating ER stress in rats. Eur J
Pharmacol. 770:154–164. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang S, Guo C, Chen Z, Zhang P, Li J and
Li Y: Vitexin alleviates ox-LDL-mediated endothelial injury by
inducing autophagy via AMPK signaling activation. Mol Immunol.
85:214–221. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Li X, Zhou J, Dou Y, Shi Y, Wang Y, Hong
J, Zhao J, Zhang J, Yuan Y, Zhou M and Wei X: The protective
effects of angelica organic acid against ox-LDL-induced autophagy
dysfunction of HUVECs. BMC Complement Med Ther.
20(164)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ding Z, Liu S, Wang X, Khaidakov M, Dai Y
and Mehta JL: Oxidant stress in mitochondrial DNA damage, autophagy
and inflammation in atherosclerosis. Sci Rep.
3(1077)2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lin HH: In vitro and in vivo
atheroprotective effects of gossypetin against endothelial cell
injury by induction of autophagy. Chem Res Toxicol. 28:202–215.
2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Che J, Liang B, Zhang Y, Wang Y, Tang J
and Shi G: Kaempferol alleviates ox-LDL-induced apoptosis by
up-regulation of autophagy via inhibiting PI3K/Akt/mTOR pathway in
human endothelial cells. Cardiovasc Pathol. 31:57–62.
2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Fernández ÁF, Sebti S, Wei Y, Zou Z, Shi
M, McMillan KL, He C, Ting T, Liu Y, Chiang WC, et al: Disruption
of the beclin 1-BCL2 autophagy regulatory complex promotes
longevity in mice. Nature. 558:136–140. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kitada M, Ogura Y and Koya D: The
protective role of Sirt1 in vascular tissue: Its relationship to
vascular aging and atherosclerosis. Aging (Albany NY). 8:2290–2307.
2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Ouimet M, Ediriweera H, Afonso MS,
Ramkhelawon B, Singaravelu R, Liao X, Bandler RC, Rahman K, Fisher
EA, Rayner KJ, et al: microRNA-33 regulates macrophage autophagy in
atherosclerosis. Arterioscler Thromb Vasc Biol. 37:1058–1067.
2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Grootaert MO, da Costa Martins PA, Bitsch
N, Pintelon I, De Meyer GR, Martinet W and Schrijvers DM: Defective
autophagy in vascular smooth muscle cells accelerates senescence
and promotes neointima formation and atherogenesis. Autophagy.
11:2014–2032. 2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Cao H, Jia Q, Yan L, Chen C, Xing S and
Shen D: Quercetin suppresses the progression of atherosclerosis by
regulating MST1-mediated autophagy in ox-LDL-induced RAW264.7
macrophage foam cells. Int J Mol Sci. 20(6093)2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Mollace V, Gliozzi M, Musolino V, Carresi
C, Muscoli S, Mollace R, Tavernese A, Gratteri S, Palma E, Morabito
C, et al: Oxidized LDL attenuates protective autophagy and induces
apoptotic cell death of endothelial cells: Role of oxidative stress
and LOX-1 receptor expression. Int J Cardiol. 184:152–158.
2015.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Fan X, Wang J, Hou J, Lin C, Bensoussan A,
Chang D, Liu J and Wang B: Berberine alleviates ox-LDL induced
inflammatory factors by up-regulation of autophagy via AMPK/mTOR
signaling pathway. J Transl Med. 13(92)2015.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Gu HF, Li HZ, Tang YL, Tang XQ, Zheng XL
and Liao DF: Nicotinate-curcumin impedes foam cell formation from
THP-1 cells through restoring autophagy flux. PLoS One.
11(e0154820)2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Li G, Peng J, Liu Y, Li X, Yang Q, Li Y,
Tang Z, Wang Z, Jiang Z and Wei D: Oxidized low-density lipoprotein
inhibits THP-1-derived macrophage autophagy via TET2
down-regulation. Lipids. 50:177–183. 2015.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Huang B, Jin M, Yan H, Cheng Y, Huang D,
Ying S and Zhang L: Simvastatin enhances oxidized-low density
lipoprotein-induced macrophage autophagy and attenuates lipid
aggregation. Mol Med Rep. 11:1093–1098. 2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Zhang BC, Zhang CW, Wang C, Pan DF, Xu TD
and Li DY: Luteolin attenuates foam cell formation and apoptosis in
Ox-LDL-stimulated macrophages by enhancing autophagy. Cell Physiol
Biochem. 39:2065–2076. 2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Wu H, Feng K, Zhang C, Zhang H, Zhang J,
Hua Y, Dong Z, Zhu Y, Yang S and Ma C: Metformin attenuates
atherosclerosis and plaque vulnerability by upregulating
KLF2-mediated autophagy in apoE-/-
mice. Biochem Biophys Res Commun. 557:334–341. 2021.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Hsieh PN, Zhou G, Yuan Y, Zhang R,
Prosdocimo DA, Sangwung P, Borton AH, Boriushkin E, Hamik A,
Fujioka H, et al: A conserved KLF-autophagy pathway modulates
nematode lifespan and mammalian age-associated vascular
dysfunction. Nat Commun. 8(914)2017.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Kobayashi A, Kang MI, Okawa H, Ohtsuji M,
Zenke Y, Chiba T, Igarashi K and Yamamoto M: Oxidative stress
sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to
regulate proteasomal degradation of Nrf2. Mol Cell Biol.
24:7130–7139. 2004.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Zhang DD, Lo SC, Cross JV, Templeton DJ
and Hannink M: Keap1 is a redox-regulated substrate adaptor protein
for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol.
24:10941–10953. 2004.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Donovan EL, McCord JM, Reuland DJ, Miller
BF and Hamilton KL: Phytochemical activation of Nrf2 protects human
coronary artery endothelial cells against an oxidative challenge.
Oxid Med Cell Longev. 2012(132931)2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Hu Q, Zhang T, Yi L, Zhou X and Mi M:
Dihydromyricetin inhibits NLRP3 inflammasome-dependent pyroptosis
by activating the Nrf2 signaling pathway in vascular endothelial
cells. Biofactors. 44:123–136. 2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Liu Z, Wang J, Huang E, Gao S, Li H, Lu J,
Tian K, Little PJ, Shen X, Xu S and Liu P: Tanshinone IIA
suppresses cholesterol accumulation in human macrophages: Role of
heme oxygenase-1. J Lipid Res. 55:201–213. 2014.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Lu Q, Tang SL, Liu XY, Zhao GJ, Ouyang XP,
Lv YC, He PP, Yao F, Chen WJ, Tang YY, et al:
Tertiary-butylhydroquinone upregulates expression of ATP-binding
cassette transporter A1 via nuclear factor E2-related factor 2/heme
oxygenase-1 signaling in THP-1 macrophage-derived foam cells. Circ
J. 77:2399–2408. 2013.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Wang J, Liu Z, Hu T, Han L, Yu S, Yao Y,
Ruan Z, Tian T, Huang T, Wang M, et al: Nrf2 promotes progression
of non-small cell lung cancer through activating autophagy. Cell
Cycle. 16:1053–1062. 2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Lazaro I, Lopez-Sanz L, Bernal S, Oguiza
A, Recio C, Melgar A, Jimenez-Castilla L, Egido J, Madrigal-Matute
J and Gomez-Guerrero C: Nrf2 activation provides atheroprotection
in diabetic mice through concerted upregulation of antioxidant,
anti-inflammatory, and autophagy mechanisms. Front Pharmacol.
9(819)2018.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Fledderus JO, Boon RA, Volger OL, Hurttila
H, Ylä-Herttuala S, Pannekoek H, Levonen AL and Horrevoets AJ: KLF2
primes the antioxidant transcription factor Nrf2 for activation in
endothelial cells. Arterioscler Thromb Vasc Biol. 28:1339–1346.
2008.PubMed/NCBI View Article : Google Scholar
|
|
60
|
He LH, Gao JH, Yu XH, Wen FJ, Luo JJ, Qin
YS, Chen MX, Zhang DW, Wang ZB and Tang CK: Artesunate inhibits
atherosclerosis by upregulating vascular smooth muscle
cells-derived LPL expression via the KLF2/NRF2/TCF7L2 pathway. Eur
J Pharmacol. 884(173408)2020.PubMed/NCBI View Article : Google Scholar
|