Introduction
Cardiovascular disease is one of the deadliest
diseases worldwide and acute myocardial infarction has become one
of the leading causes of death (1-4).
Timely blood reperfusion to the ischemic heart tissue can limit the
size of myocardial infarction and reduce mortality. Nevertheless,
this treatment can further induce serious secondary injury to the
myocardium known as myocardial ischemia/reperfusion injury (MIRI)
(5-7).
MIRI has complex pathological mechanisms, such as mitochondrial
disorders, calcium overload and the production of reactive oxygen
(8-10).
Inflammation plays an important role in the pathophysiology of MIRI
(11,12). MIRI activates the
immunity-inflammation responses and increases the production of
inflammasomes, leading to inflammatory cell infiltration and
increased cytokine release in the heart (11,13,14).
Likewise, hypoxia/reoxygenation (H/R), a model of MIRI, leads to
the excessive release of inflammatory factors TNF-α and IL-6 from
cardiac myoblast H9c2 cells and causes excessive death of H9c2
cells (15,16). Therefore, the pharmacological
inhibition of cardiomyocyte inflammation can effectively protect
the myocardium against MIRI.
Emodin, as an anthraquinone derivative extracted
from traditional Chinese medicine rhubarb, possesses
anti-inflammation, immune regulation and antioxidant properties
(17,18). Emodin attenuates MIRI in rodents
via various cellular functions and signaling pathways, but the
precise mechanism underlying the attenuation of MIRI by emodin by
modulating the inflammation response remains unknown (19,20).
A type of small non-coding RNA known as microRNA
(miRNA/miR) negatively regulates gene expression during
post-transcriptional processes by binding to the target mRNAs
(21). MiRNAs are instrumental in
the development of cardiac diseases, including MIRI (22). For instance, miR-384-5p alleviates
MIRI in rats via the inhibition of the expression of Beclin-1 to
suppress autophagy (23). miR-322
prevents MIRI by attenuating FBXW7-caused oxidative stress
(24). However, whether emodin can
attenuate inflammation in MIRI by regulating miRNAs and its
upstream transcription factors remains unknown.
In the present study, it was further confirmed that
emodin pretreatment could relieve ischemia/reperfusion (I/R) injury
and inflammatory responses of mice myocardium. By using RNA
sequencing combined with reverse transcription-quantitative PCR
(RT-qPCR) and western blotting, emodin was shown to inhibit the
expression of transcription factor RUNX1 and thus downregulated the
transcriptional level of miR-142-3p in MIRI. The expression of
RUNX1/miR-142-3p was confirmed both in vivo or in
vitro. The present study's findings support an undiscovered
model, whereby emodin inhibited RUNX1/miR-142-3p pathway and thus
upregulated DRD2, which acts as an anti-inflammatory mediator that
suppresses the NF-κB-dependent inflammation and prevents MIRI.
Materials and methods
Animals
A total of 80 male ICR mice (6-8-weeks old, weighing
25±3 g) were obtained from Beijing Vital River Laboratory Animal
Technology Co., Ltd. and housed in a temperature-controlled animal
room (23±1˚C), with relative humidity between 50-60% and daylight
between 8:00-19:00. Food and water were freely accessible for the
mice. Each group contained 5-6 mice in every independent experiment
and a total of 80 mice were used in the project. All animal
experiments were approved by the Animal Care and Use Committee of
The Affiliated Hospital of Qingdao University (approval no.
QYFYWZLL26907; approval date, March 20, 2021). Mice used in
experiments were euthanized 24 h after establishing an MIRI model
by intraperitoneal administration of an overdose of pentobarbital
sodium (200 mg/kg), and the heart samples were harvested. After
establishing the MIRI model, mouse health was monitored every other
hour, including the ability to breathe normally, eat normally and
move freely. Some of the mice were euthanized prior to the
experimental endpoint if they went into dyspnea, lethargy, shock or
decreased activity.
Experimental protocol for construction
of MIRI model
ICR mice were randomly divided into three groups,
namely: Sham group (control), MIRI group and emodin
(MedChemExpress) treatment group (MIRI + emodin). Mice were
intragastrically administrated with 10 mg/kg emodin or saline
(vehicle) once a day for 7 consecutive days (day 1-7) before
inducing the MIRI model. The MIRI model was performed 1 h after the
last emodin treatment on day seven. To establish the MIRI model,
the mice were anesthetized by intraperitoneal administration of 2%
pentobarbital sodium (50 mg/kg) and were ventilated by using a
rodent respirator. The thoracic cavity was opened, and the
pericardium was cut open to expose the heart. The left anterior
descending branch (LAD) of the coronary artery was ligated 2-mm
away from its origin with surgical sutures for 60 min. A successful
ligation was confirmed by the change of myocardial color from red
to grayish white. The mice in the sham group were also
anesthetized, and a suture was just passed under the LAD without
obstruction. After a 60 min ligation of the LAD, myocardial blood
flow was restored for 24 h to induce I/R injury before the heart
was harvested for subsequent analysis.
Determination of area at risk (AAR)
and infarct area
For the measurement of AAR and infarct area, 24 h
after MIRI, the heart samples of the mice were perfused with Evans
blue (1%) and subsequently incubated with 2.0% triphenyltetrazolium
chloride (TTC) at 37˚C for 20 min. The non-ischemic myocardium was
stained blue with Evans Blue. AAR was stained red by TTC, and the
infarct area appeared pale after staining. The left ventricle (LV),
AAR and infarct areas were determined using Image J 1.42q software
(National Institutes of Health). The percentages of infarct
area/AAR and AAR/LV were defined as the dead and ischemia level of
myocardium, respectively.
Western blotting
The mice cardiac tissues (60-80 mg) or rat cardiac
myoblast H9c2 cells were lysed in RIPA buffer (Thermo Fisher
Scientific, Inc.). The supernatant was quantified with Pierce BCA
protein assay reagent kit (Thermo Fisher Scientific, Inc.). Total
protein (20 µg per sample) was loaded and separated in 10% or 12%
SDS-PAGE, and transferred to a PVDF membrane (MilliporeSigma).
After blocking in TBST (0.1% Tween-20) containing 5% fat-free milk
at room temperature for 1 h, the membrane was incubated with
primary anti-RUNX1 (1:1,000; cat. no. ab229482; Abcam), anti-DRD2
(1:500; cat. no. bs-20730R; BIOSS), anti-phosphorylated (p-)P65
subunit of NF-κB (1:1,000; cat. no. ab76302; Abcam), anti-NF-κB P65
subunit (1:1,000; cat. no. bs-20355R; BIOSS), anti-TNF-α (1:500;
cat. no. bs-2081R; BIOSS), anti-IL-6 (1:500; cat. no. ab259341;
Abcam) or anti-β-tubulin (1:1,000; cat. no. ab18207; Abcam)
antibodies respectively, overnight at 4˚C. After washing three
times in TBST, the membranes were incubated with HRP-conjugated
goat anti-rabbit secondary antibody (1:10,000; cat. no. ab191866;
Abcam) for 1.5 h at room temperature. Protein band signals were
detected using ECL program (Thermo Fisher Scientific, Inc.) under
chemiluminescence detector (Bio-Rad Laboratories). The expression
of target protein was normalized to β-tubulin using Image Lab
Software (Bio-Rad Laboratories).
Hematoxylin-eosin (H&E) and Masson
staining
Paraformaldehyde (4%) was used to immobilize
myocardial tissue samples at 4˚C for 24 h. Subsequently, samples
were dehydrated with gradient ethanol (70-100%) for 30 min before
treating with 50 and 100% xylene, respectively, at room temperature
for 1.5 h for transparency. After embedding in paraffin, the
processed tissues were cut into 5-µm thick slices. After
deparaffinization in an incubator (60˚C; 2 h), followed by
incubation twice with 100% xylene for 20 min and rehydration in
descending alcohol (100-70%) at room temperature, the slices were
stained with Masson's Trichrome Stain kit (Beijing Solarbio Science
& Technology Co., Ltd.) or H&E (Beijing Solarbio Science
& Technology Co., Ltd.) according to the products'
instructions. The results were observed under a light microscope
(Eclipse TS100; Nikon Corporation) and images were captured.
Immunohistochemistry
The 5-µm thick paraffin slices of myocardium were
deparaffinized in an incubator (60˚C; 2 h) and rehydrated in a
descending alcohol series before washing with distilled water.
After antigen recovery, 3% H2O2 was used to
eliminate the activity of endogenous peroxidase. The processed
slices were blocked with 5% BSA for 30 min at room temperature
before incubation with anti-TNF-α (1:200; cat. no. ab205587; Abcam)
in a sealed wet box at 4˚C overnight, and then incubated with
HRP-conjugated anti-rabbit secondary antibody (1:2,000; ab191866;
Abcam) at room temperature for 1 h. After incubation with
diaminobenzidine reagents (cat. no. DA1016; Beijing Solarbio
Science & Technology Co., Ltd.) for color detection, the slices
were counterstained with hematoxylin at room temperature for 3 min
and images were captured under a light microscope (Eclipse TS100;
Nikon Corporation).
Creatine kinase (CK), CK-MB and
lactate dehydrogenase (LDH) assay
The expression levels of creatine kinase, CK-MB and
lactate dehydrogenase in the serum of the mice were measured using
Creatine Kinase Assay kit (cat. no. BC1145, Beijing Solarbio
Science & Technology Co., Ltd.), CK-MB Elisa kit (cat. no.
SEKM-0152; Beijing Solarbio Science & Technology Co., Ltd.) and
LDH Activity Detection kit (cat. no. BC0685, Beijing Solarbio
Science & Technology Co., Ltd.), respectively.
Cell culture and establishment of H/R
model
Rat cardiac myoblast H9c2 cells were obtained from
the Cell Bank of the Chinese Academy of Sciences and cultured with
Dulbecco's modified Eagle medium (Gibco; Thermo Fisher Scientific,
Inc.) with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc.) and 100 U/ml streptomycin and penicillin (Gibco; Thermo
Fisher Scientific, Inc.) in normal gas mixture (95% air and 5%
CO2) at 37˚C. The H9c2 cells were incubated in gas
mixture containing 94% N2, 5% CO2 and 1%
O2 for 3 h to induce hypoxia injury before removing to a
normoxic incubator for another 3 h to maintain reoxygenation
(19,25).
Cell transfection
Lipofectamine® 3000 (Invitrogen) was used
in transfection. Negative control (NC) mimic (cat. no.
miR1N0000001-1-5) and miR-142-3p mimic (cat. no. miR10000155-1-5)
were obtained from Guangzhou RiboBio Co., Ltd. NC mimic or miR
mimic at a final concentration of 50 nM were transfected to a H9c2
cell-seeded 6-well plate at 37˚C for 4 h. The complementary DNAs
(cDNAs) of RUNX1 and DRD2 were subcloned to pcDNA3.1 vector
(Invitrogen: Thermo Fisher Scientific, Inc.), and 2 µg/well of
these cDNAs were transfected into a H9c2 cell-seeded 6-well plate
at 37˚C for 4 h. The control (without H/R injury) and H/R groups
were transfected with pcDNA3.1 vector as the negative control.
Subsequent experiments were performed 48 h after transfection.
Inhibition of miR-142-3p and
overexpression of DRD2 in vivo
The mmu-miR-142-3p antagomir (miR-142-3p anti-ago;
cat. no. miR-311620131514-4-5) and negative control antagomir (NC
anti-ago; cat. no. miR3N0000001-4-5) were purchased from Guangzhou
RiboBio Co., Ltd., and administered by tail vein injection with 5
nmol three times (once every two days). The MIRI models were
established after 20 days from the first injection. To determine
the effect of DRD2 in MIRI, recombinant adeno-associated virus
(AAV) expressing DRD2 (AAV-DRD2) was purchased from Hanbio
Biotechnology Co., Ltd. and administered once via a 50-µl tail vein
injection, while the control and MIRI groups were injected with AAV
control (AAV-Ctrl; Hanbio Biotechnology Co., Ltd.). The MIRI models
were established 20 days after the injection.
RT-qPCR analysis
Total RNAs were extracted from myocardium or H9c2
cells using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). A total of 1 µg of extracted total RNA was
reverse-transcribed to cDNAs by using a Reverse Transcription kit
(Takara Bio, Inc.) according to the manufacturer's instructions.
Primers were obtained from Sangon Biotech Co., Ltd. RT-qPCR was
performed using SYBR® Green Master Mix (Takara Bio.
Inc.) in a 20 µl reaction system containing specific primers, and
primer sequences are listed in Table
SI. The mRNA levels of target genes were detected using the
CFX96 real time PCR detection system (Bio-Rad Laboratories). For
miR-142-3p (sequence: 5'-UGUAGUGUUUCCUACUUUAUGGA-3') analysis, the
stem-loop reverse transcription primer sequence was
5'-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCCATA-3', and the
qPCR forward primer was 5'-TGTAGTGTTTCCTACTT-3' and the reverse
primer was 5'-GTGCAGGGTCCGAGGT-3'. The thermocycling conditions
used for RT-qPCR were as follows: Cycle at 94˚C for 5 min, followed
by 40 cycles (10 sec at 94˚C for denaturation, 15 sec at 60˚C for
annealing and 20 sec at 72˚C for extension) and a final elongation
at 72˚C for 5 min. The relative expression levels of miRNA and
genes were normalized to that of internal control U6 and GAPDH,
respectively, and analyzed by using 2-∆∆Cq method
(26).
RNA purification
Total RNAs were extracted from the mice hearts of
MIRI and MIRI + emodin groups by using TRIzol® reagent
(cat. no. 15596026; Invitrogen; Thermo Fisher Scientific, Inc.) at
24 h after performing MIRI. RNA samples were stored in dry ice and
submitted to Shanghai Personal Biotechnology Co., Ltd. for
high-throughput sequencing. Quality control for the RNA samples was
carried out by using an Agilent 2100 Bioanalyzer with Agilent RNA
6000 Nano Kit (Agilent Technologies, Inc.). RNA samples with the
standard of total RNAs >8 µg, concentration >250 ng/µl, the
ratio of 28S/18S >1.5 and the RNA integrity number >8 were
selected for establishment of mRNA and miRNA libraries.
Library preparation and RNA
sequencing
The mRNAs were separated from the total RNAs using
the olig-dT method, and cut into 200 bp fragments by
Mg2+ randomly. Fragments were reverse transcribed to
cDNAs, which were purified and end modified before being connected
to adapters. The cDNA fragments of 200-300 bp were chosen for PCR
amplification and library construction. RNAs (18-30 nt) were
separated from total RNAs, and selected for construction for
microRNA sequencing library. The final library was amplified with
phi29 DNA polymerases (Thermo Fisher Scientific, Inc.) to make a
DNA nanoball (DNB) which had >300 copies of one molecule. DNBs
were loaded into the patterned nanoarray and single end 150 base
reads were generated on the DNBSEQ-T7 sequencing platform (MGI; BGI
Group). Quality control of raw reads was performed using the
SOAPnuke software (BGI Group). The following reads were filtered:
i) The reads containing the adaptor, ii) the reads whose N content
was >5%, and iii) low-quality reads (reads with bases having a
quality score <10 as the proportion of total bases in the reads
that were >20% as low-quality reads). The filtered clean reads
obtained from each sample were packed in the form of FASTQ, with an
average size of 6.62 Gb. Hierarchical Indexing for Spliced
Alignment of Transcripts (HISTAT) was used to map RNA-sequencing
reads to the reference genome. Firstly, the HISTAT global FM index
was used to anchor the position of partial sequences in each read
on the genome, and then the partial genome indexes of these
alignment positions was used to align the remaining sequences of
each read to extend the alignment area. Differentially expressed
genes were analyzed with Bioconductor DESeq2 version 1.12.3
(https://www.rdocumentation.org/packages/DESeq2). The
criteria for determining differentially expressed miRNAs and
transcription factors are the log2 fold-change
(log2FC) >2 or <-2 with a q value <0.05 and the
log2FC >1 or <-1 with q value <0.05,
respectively.
Prediction of potential transcription
factors (TFs) and target genes of miRNAs
Based on the differentially expressed miRNAs, the
upstream TFs were predicted using TransmiR v2.0 database
(http://www.cuilab.cn/transmir), an
easy-accessible public tool, which integrates experimentally
verified TF-miRNA regulatory relationships from the publications
(27). In addition, the downstream
target genes of miRNAs were also predicted using miRWalk database
(http://mirwalk.umm.uni-heidelberg.de/), which is
freely available and presents validated and predicted information
on miRNA-target interaction (28).
ELISA
The cytokines released from the myocardium were
measured by using TNF-α (cat. no. ab208348; Abcam) and IL-6 (cat.
no. ab222503; Abcam) ELISA kits. Assays were performed according to
the manufacturer's instructions. The absorbance at 450 nm was
measured using a Sunrise microplate reader (Tecan Group, Ltd.).
Statistical analysis
The data were collected from at least three
independent experiments and statistical analysis was performed
using one-way analysis of variance (ANOVA) followed by Tukey's
multiple comparisons test in GraphPad Prism 7.0 (GraphPad Software,
Inc.). All data were expressed as the mean ± standard deviation.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Emodin protects against MIRI in
mice
To investigate the underlying molecular mechanism
for natural emodin attenuation of MIRI, emodin was intragastrically
administrated to the mice for 7 consecutive days before inducing
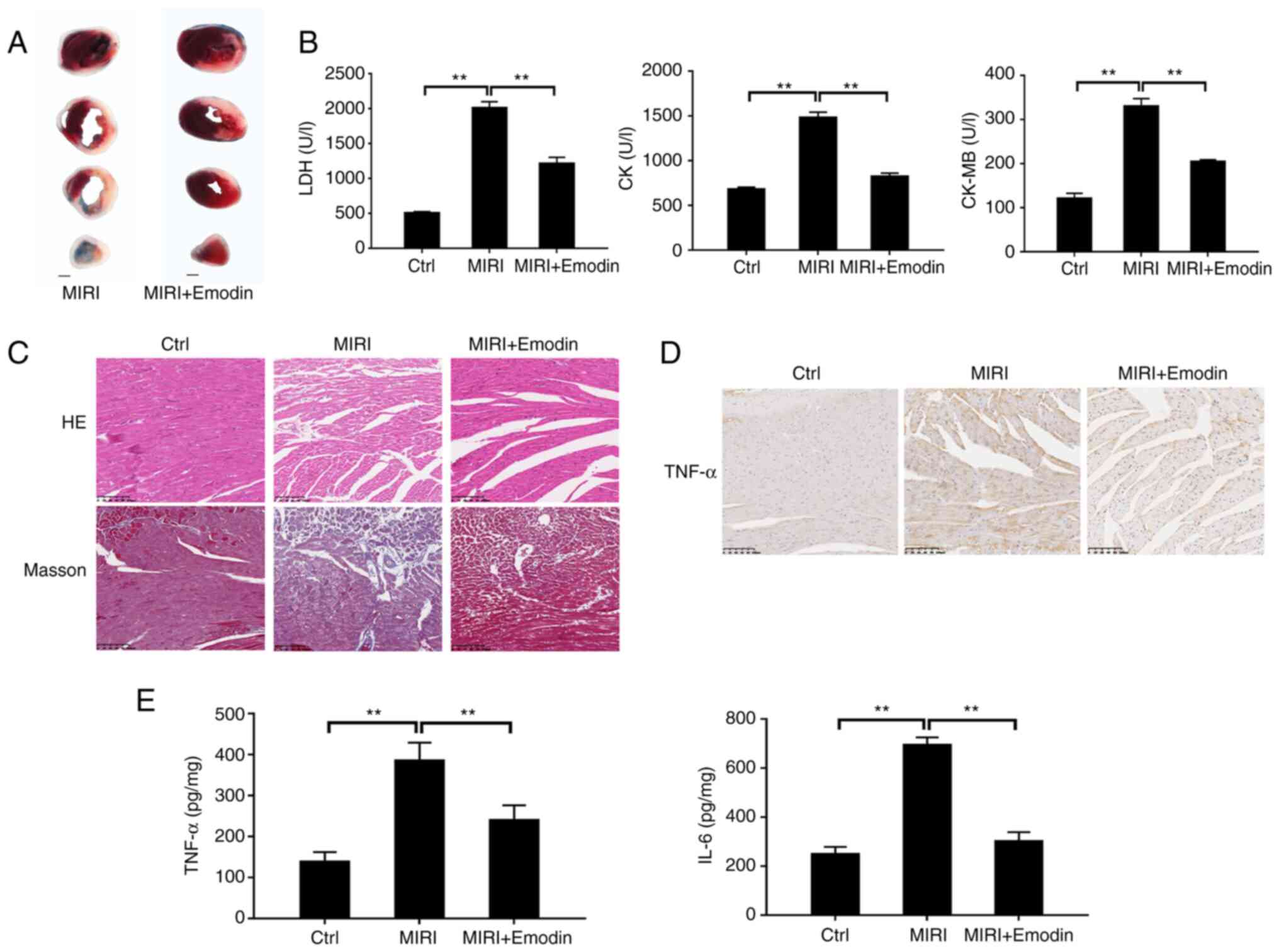
the MIRI model. As shown in Fig.
1A, Evans Blue/TTC staining showed that I/R resulted in
myocardial infarction. By contrast, pretreatment with emodin
markedly attenuated the size of myocardial infarction induced by
I/R, and the productions of LDH, CK and CK-MB were subsequently
significantly suppressed compared with the MIRI group (Fig. 1B). Moreover, the alleviative effect
was confirmed using histopathological staining. H&E staining
images displayed that the cardiomyocytes in the MIRI group were
disordered and swollen, and the sarcolemma was disrupted compared
with the control (sham) group (Fig.
1C). Pretreatment with emodin markedly reversed these
pathological changes. The Masson staining results showed that the
myocardial collagen fiber deposition (stained blue) in the MIRI
group increased, and emodin pretreatment markedly reduced the
collagen deposition (Fig. 1C).
Next, the regulative action of emodin on the process
of inflammatory responses after MIRI was tested.
Immunohistochemical staining displayed an upregulated expression of
TNF-α in the myocardial sections after I/R injury compared with the
control group (Fig. 1D), and
emodin pretreatment suppressed the expression of TNF-α. The TNF-α
and IL-6 secretions from myocardium evoked by I/R injury were also
significantly reduced in the emodin pretreatment group (Fig. 1E). Therefore, the inhibition of
inflammation response by emodin prevented MIRI.
Emodin suppresses the RUNX1/miR-142-3p
pathway in either MIRI or hypoxia/reoxygenation injury
To determine the molecular mechanism underlying
inhibition of inflammation during MIRI by emodin, myocardium from
MIRI and MIRI + emodin groups were collected and RNA transcripts
and microRNA sequencing were performed using the DNBSEQ-T7
sequencing platform. The sequencing data were normalized to the
MIRI group. As shown in Table
SII, 34 differentially expressed miRNAs were screened out. To
determine the key transcription factors (TFs) that regulated the
expression of these differentially expressed miRNAs, the TransmiR
v2.0 database was used to match the regulatory networks between the
differentially expressed TFs and miRNAs. A total of four pairs of
differentially expressed regulatory networks between TFs and miRNAs
were matched (Table SIII), among
which only two pairs displayed consistent trends of downregulation.
For an unprejudiced analysis, the RUNX1/miR-142-3p pathway with the
most obvious change was uncovered as the candidate for the
modulation of MIRI-induced inflammation by emodin (Table SIII).
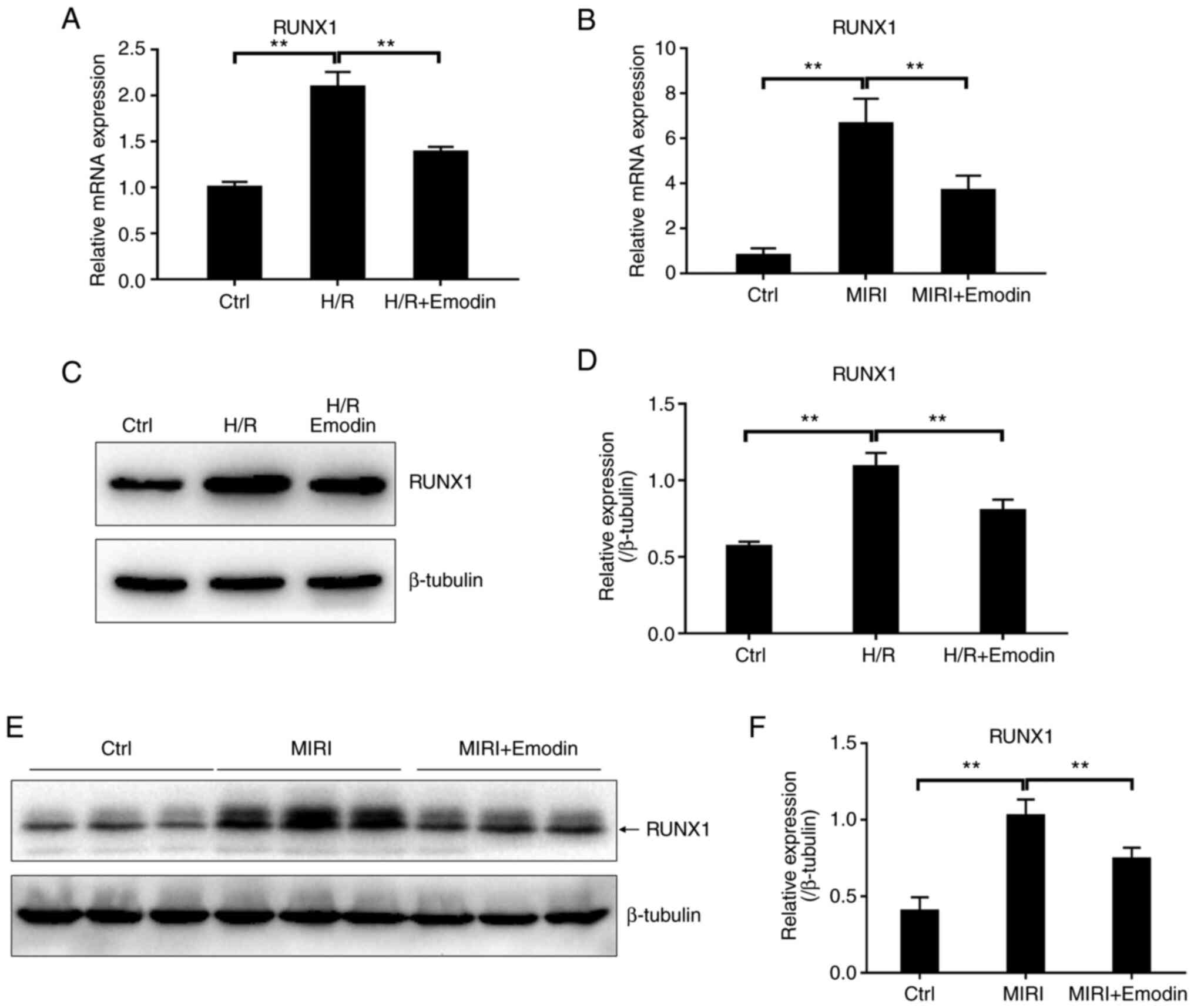
The present study examined whether the level of
miR-142-3p could be regulated by the transcription factor RUNX1 in
the pathological process of MIRI. The mRNA expression levels of
transcription factor RUNX1 were significantly increased after H/R
injury in H9c2 cells or MIRI (Fig.
2A and B). RUNX1 protein
levels were also detected using western blotting and showed a
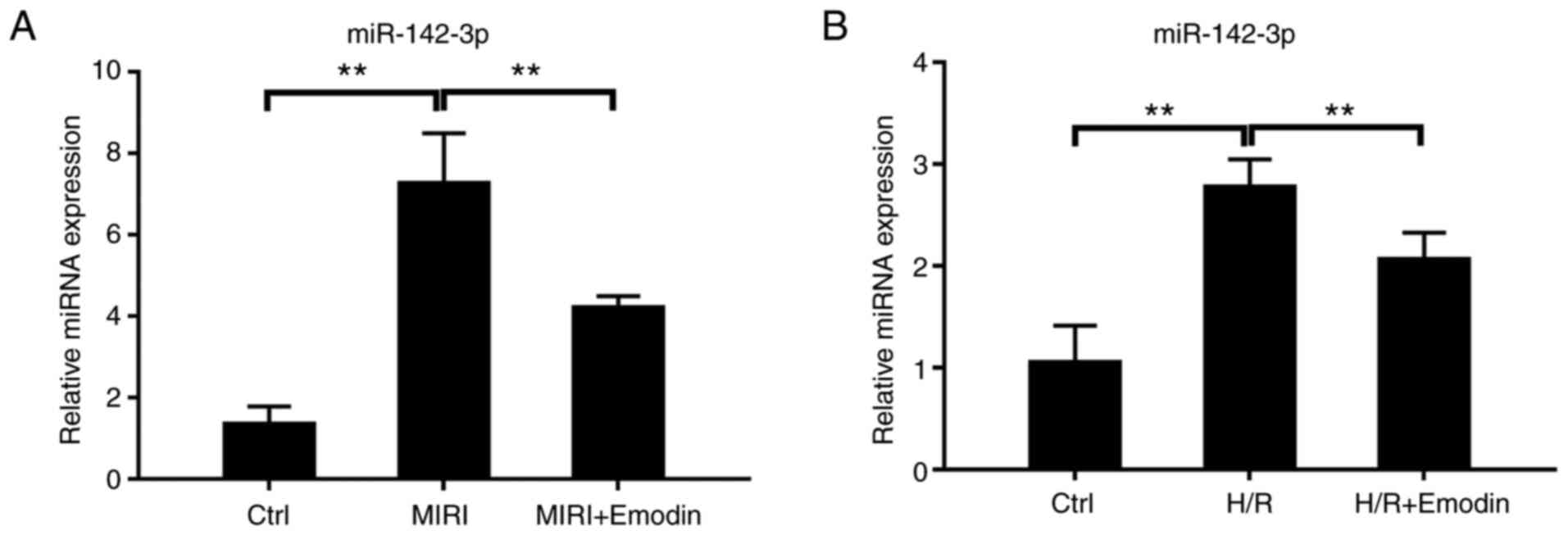
consistent trend with the mRNA level (Fig. 2C-F). As expected, the expression of
miR-142-3p was also significantly increased after H/R injury in
H9c2 cells or MIRI (Fig. 3A and
B). These results further
identified that RUNX1 acted as a transcription factor, and that it
also promoted the expression of miR-142-3p in cardiomyocytes.
Next, the inhibitory effect of emodin on the
RUNX1/miR-142-3p pathway was verified. Pretreatment with emodin
attenuated the expression levels of either RUNX1 or miR-142-3p in
both the myocardium with I/R injury and in H9c2 cells with H/R
compared with the model groups (Figs.
2 and 3). These results
supported that the RUNX1/miR-142-3p pathway was downregulated in
emodin-pretreated groups based on bioinformatical analysis
(Table SIII). Therefore, the
overactive RUNX1/miR-142-3p pathway was involved in the
inflammatory process of MIRI, and emodin could suppress the
RUNX1/miR-142-3p pathway.
MiR-142-3p negatively regulates the
expression of DRD2
With few exceptions, miRNAs theoretically negatively
regulate the expression of their targeted mRNAs (29). To identify the downstream mRNAs
targeted by miR-142-3p, two subsets of genes were defined, among
which subset one represents predicted targets for miR-142-3p by
using the miRWalk database and subset two represents upregulated
differentially expressed genes (log2FC >2 with q
value <0.05) in the emodin-treated group based on transcriptome
sequencing. The 12 overlapped genes between subset one and two are
defined as the potential targets for miR-142-3p (Table SIV).
To further validate the key gene targeted by
miR-142-3p, the present study used RT-qPCR to detect the expression
of these 12 potential genes in H9c2 cells overexpressing miR-142-3p
mimic. As shown in Fig. S1,
transfection with miR-142-3p mimic in H9c2 cells for 48 h caused a
significant increase in the expression of miR-142-3p compared with
the negative control mimic group. The mRNA levels of KCNA2, TNIP3
and DRD2 in the miR-142-3p mimic-overexpressed group were
significantly downregulated compared with the NC mimic group, among
which DRD2 showed the most obvious downregulation (Fig. 4A). Accordingly, DRD2 was chosen as
a candidate. To further confirm this, a miR-142-3p antagomir (miR
anti-ago) was used to reduce miR-142-3p levels in mice myocardium
(Fig. S1B), and the protein
expression was detected using western blotting. As expected, the
protein level of DRD2 was significantly increased in the miR-142-3p
antagomir (anti-ago) pretreated myocardium after I/R injury
compared with the MIRI group (Fig.
4B and C). Besides, the
protein level of DRD2 also significantly increased in the
emodin-treated H9c2 cells with H/R injury compared with the
untreated group (Fig. 4D and
E). These data further confirmed
that emodin treatment attenuated the level of miR-142-3p, which
negatively regulated the expression of DRD2. The protein level of
DRD2 was significantly increased in the emodin-pretreated
myocardium after I/R injury compared with the MIRI group (Fig. 4F and G). Therefore, emodin upregulated DRD2
after MIRI by downregulating miR-142-3p.
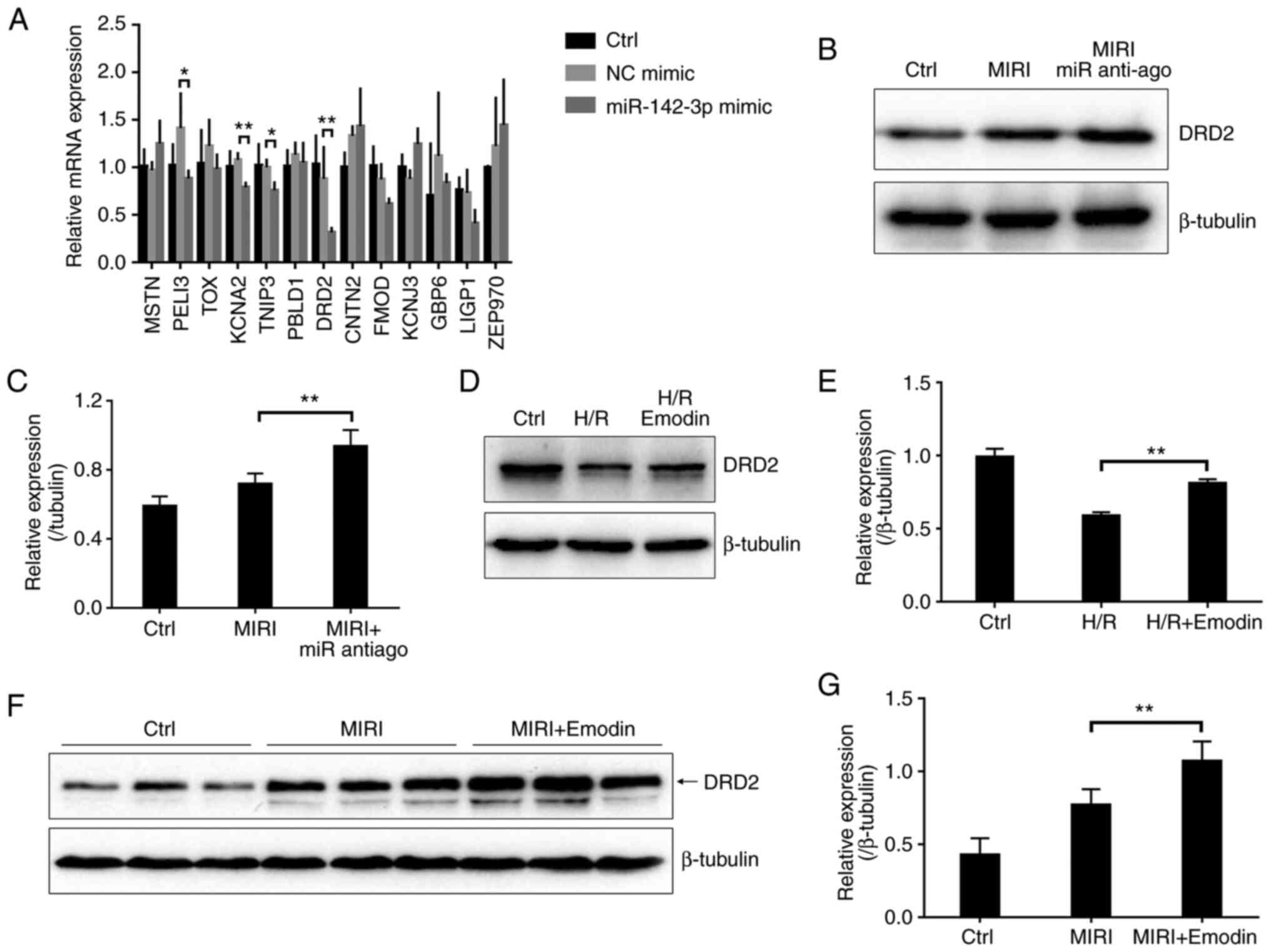 | Figure 4miR-142-3p negatively regulates the
expression of DRD2. (A) Reverse transcription-quantitative PCR for
the identification of the downstream target gene of miR-142-3p by
overexpressing miR-142-3p mimic in H9c2 cells. n=4. (B) The DRD2
protein levels were detected by western blotting in cardiac tissues
with MIRI or miR-142-3p antagomir (anti-ago) treatment. (C) Summary
for the protein expression of DRD2 based on (B), n=3. (D) DRD2
protein levels were detected by western blotting in H9c2 cells with
H/R injury or emodin treatment. (E) Summary for the protein
expression of DRD2 based on (D), n=3. (F) DRD2 protein levels were
measured using western blotting in cardiac tissues with MIRI or
emodin treatment. Three bands represent three independent samples.
(G) Summary for the protein expression of DRD2 based on (F), n=3.
*P<0.05, **P<0.01. NC, negative
control; MIRI, myocardial ischemia/reperfusion injury; anti-ago,
antagomir; DRD2, dopamine receptor D2; Ctrl, control; miR,
microRNA; H/R, hypoxia/reoxygenation. |
RUNX1/miR-142-3p/DRD2 pathway can
regulate the NF-κB-mediated inflammatory response in H9c2 cells
with H/R injury
Next, RUNX1, miR-142-3p mimic and DRD2 were
overexpressed in H9c2 cells and their roles were evaluated in the
modulation of inflammation (Fig.
S2). As shown in Fig. 5A and
B, the production of cytokine
TNF-α in the RUNX1 overexpression group and IL-6 in the RUNX1 and
miR-142-3p mimic overexpression groups from H9c2 cells after H/R
injury were significantly increased compared with the H/R injury
group. In addition, the p-P65 subunit of NF-κB was upregulated in
these two groups compared with the H/R injury group (Fig. 5C). Conversely, the DRD2
overexpression group showed a significantly reduced production of
TNF-α and IL-6 compared with the H/R group (Fig. 5A and B), and the p-P65 subunit of NF-κB was
also downregulated (Fig. 5C).
Therefore, RUNX1 and miR-142-3p acted as pro-inflammatory factors,
and DRD2 acted as an anti-inflammatory factor to regulate the
NF-κB-mediated inflammatory responses.
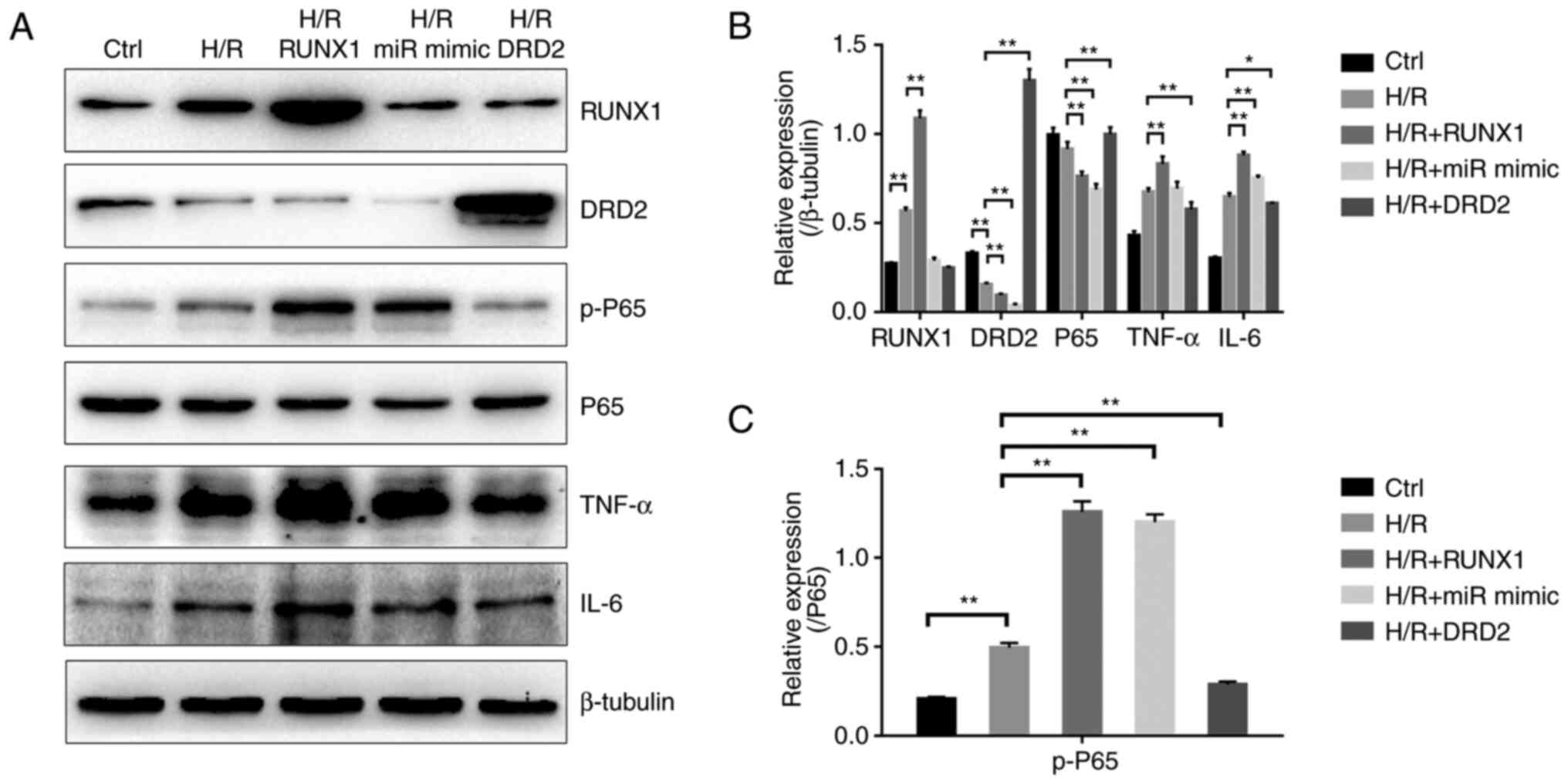 | Figure 5RUNX1/miR-142-3p/DRD2 pathway
regulates the NF-κB-dependent inflammatory responses in H9c2 cells
with H/R injury. (A) The NF-κB dependent inflammatory response was
regulated by overexpression of RUNX1, miR-142-3p mimic and DRD2 in
H9c2 cells with H/R injury and detected using western blotting. (B)
Bar diagram showing the protein expression levels of RUNX1, DRD2,
P65, TNF-α and IL-6 based on (A). n=3. (C) Bar diagram showing the
protein expression of functional p-P65 subunit of NF-κB based on
(A). n=3. *P<0.05, **P<0.01. H/R,
hypoxia/reoxygenation; DRD2, dopamine receptor D2; p-,
phosphorylated-; miR, microRNA; Ctrl, control. |
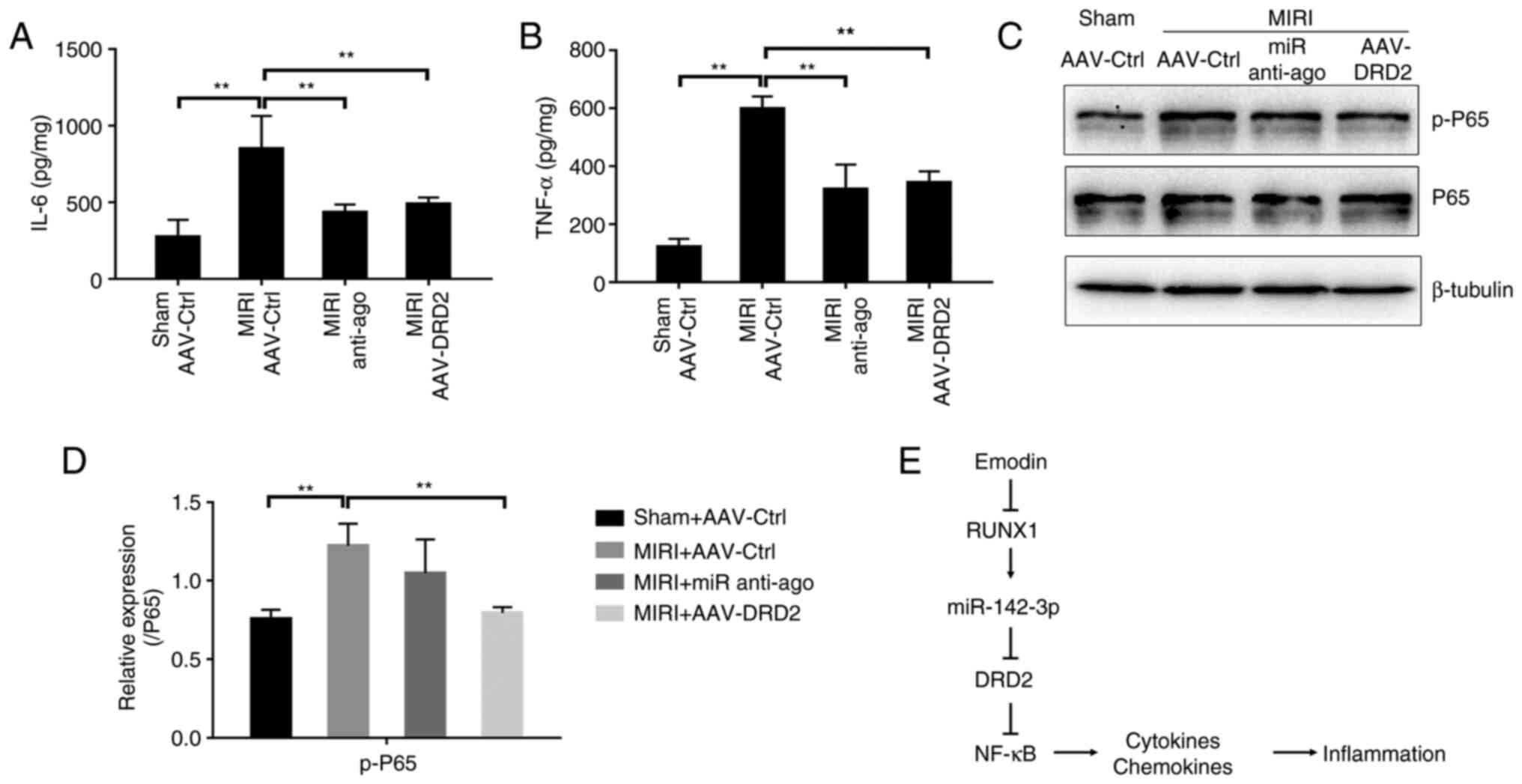
Either the inhibition of miR-142-3p or
overexpression of DRD2 attenuates NF-κB-mediated inflammatory
response in MIRI model of mice
Next, miR-142-3p anti-ago or adeno-associated virus
(AAV) carrying DRD2 (AAV-DRD2) were used to further validate the
roles of miR-142-3p and DRD2 in modulating the inflammation in
vivo after MIRI. As shown in Fig.
S3, tail vein injection with AAV-DRD2 caused a significant
increase of DRD2 in mice cardiac tissues 20 days after the first
injection compared with the AAV control (AAV-Ctrl) injection group.
The secretions of TNF-α and IL-6 from the myocardium were
significantly inhibited in the anti-ago-pretreated and
AAV-DRD2-overexpressed groups after MIRI compared with the MIRI
group (Fig. 6A and B). AAV-DRD2-overexpression also resulted
in a significant decrease in p-P65 protein, and the anti-ago
treatment showed a downregulated trend (Fig. 6C and D). Therefore, the inhibition of
miR-142-3p or activation of DRD2 suppresses the NF-κB-mediated
inflammatory process. Taken all together, the present study
presented a novel model, in which the inhibition of the expression
of RUNX1 and miR-142-3p by emodin lead to the upregulation of DRD2
which acted as an anti-inflammatory factor to suppress
NF-κB-dependent inflammation and prevent MIRI (Fig. 6E).
Discussion
The present study aimed to investigate the link
between emodin treatment and inflammation responses during MIRI.
Results showed that emodin suppressed the expression of
RUNX1/miR-142-3p in cardiomyocytes and contributed to an increased
level of DRD2. Then, DRD2 inhibited the phosphorylation of NF-κB,
thus reducing the production of inflammatory cytokines TNF-α and
IL-6 (Fig. 6E). These findings not
only described a novel molecular basis underlying the action of
emodin attenuation of MIRI, but also suggested that the
pharmacological inhibition of RUNX1/miR-142-3p or activation of
DRD2 may lead to a therapeutic potential for MIRI.
A number of cytokines and chemokines are released
from the myocardium after I/R injury, leading to either apoptosis
or cell death (30,31). NF-κB regulates the transcription of
various pro-inflammatory genes implicated in disease processes
(32). Emodin exerts
anti-inflammatory action by inhibiting the transcription factor
NF-κB (33,34). For example, emodin displays its
anti-arthritic action by inhibiting the NF-κB pathway and
pro-inflammatory factors in an arthritic model of mice induced by
collagen (35,36). Besides, emodin can relieve corneal
injury and improve the corneal structure by inhibiting the
activation of NF-κB and c-JunN-terminal kinase (37,38).
Although emodin inhibits the NF-κB-dependent inflammation in MIRI,
the link between emodin and NF-κB is poorly reported (39). By using an unprejudiced analysis
through RNA sequencing combination with qPCR and western blotting,
the present study identified the downregulated transcription factor
RUNX1 as the downstream target regulated by emodin. The present
study further verified that RUNX1 controlled the expression of
miR-142-3p and may serve as the initial effector to modulate
emodin-mediated anti-inflammatory action in cardiomyocytes.
RUNX1-overexpression can directly enhance the inflammatory
responses in H9c2 cells. This finding is consistent with the
observation that the downregulation of RUNX1 prevents cardiac
remodeling (40,41). Therefore RUNX1, as a
proinflammatory factor, participates in MIRI and the inhibition of
RUNX1 may present a novel therapeutic strategy for MIRI. In the
present study, the RUNX1-triggered miR-142-3p/DRD2 pathway was
further confirmed in MIRI models and H9c2 cells with the H/R model.
These findings complement the gap of signaling pathways between
emodin and NF-κB, and suggest that the RUNX1/miR-142-3p/DRD2
pathway acts as a novel therapeutic target for MIRI and emodin may
provide a therapeutic potential for MIRI.
The toxicity of emodin has been previously assessed
(42). According to the United
States National Toxicology Program (CAS no. 518-82-1), emodin at
daily oral doses of 15, 35 or 70 mg/kg for 12 months does not
affect the survival, body weight and food consumption of mice
(43). Emodin at an oral dose of
100 mg/day has been investigated in a clinical trial (trial no.
NCT00801268) for autosomal dominant polycystic kidney disease in
the USA. A type of Chinese herb tablet, Sanhuangpian (Chinese FDA
approval no. Zhunzi Z37021116) containing main active ingredient,
emodin 12 mg, has been approved in China for inflammatory diseases.
Therefore, the application of emodin in clinical practice may
improve the treatment of MIRI.
MiRNAs, as small non-coding RNAs, modulate the
transcription level of various genes that are implicated in the
determination of apoptosis, necrosis, inflammation and fibrosis
(44-46).
The role of miRNAs in MIRI has been extensively studied throughout
the last decade. Early changes (increase or decrease) of miRNAs
occur in the myocardium in response to MIRI (47-49).
The pharmacological or genetic regulation of these miRNAs can
govern the pathological process of MIRI; therefore, miRNAs are
regarded as therapeutic targets for heart diseases (47,50-52).
In the present study, the miR-142-3p level decreased in response to
emodin and further confirmed it as a pro-inflammatory factor in
MIRI. Therefore, the specific inhibition of miR-142-3p could
provide an alternative therapeutic strategy for MIRI. The mRNA of
DRD2 as a target of miR-142-3p was also verified in the present
study.
DRD2 is an anti-inflammatory critical component that
controls innate immunity in the central nervous system (53). This is consistent with the present
study's findings that either inhibition of miR-142-3p by specific
antagomir or reduction of the transcription of miR-142-3p by the
emodin/RUNX1 pathway could increase DRD2 expression levels in mice
myocardium. In addition, DRD2-overexpression in H9c2 cells or
myocardium of mice significantly reduced the levels of TNF-α, IL-6
and phosphorylated NF-κB. Although it cannot be excluded that
emodin and miR-142-3p may directly regulate the NF-κB cytoplasmic
binding or activity, these results at least partially show that
upregulation of DRD2 by the emodin/RUNX1/miR-142-3p axis could
suppress the NF-κB-mediated inflammatory responses.
In summary, the present study not only presented a
novel molecular basis for protection against MIRI by emodin, but
also suggested that the pharmacological modulation of the
RUNX1/miR-142-3p/DRD2 pathway to suppress inflammatory responses of
cardiomyocytes may be beneficial for the treatment of MIRI or other
related heart diseases. Moreover, emodin may provide a potential
therapeutic approach for MIRI.
Supplementary Material
RT-qPCR determination of miR-142-3p
levels in H9c2 cells or cardiac tissues of mice. (A) Transfection
with miR-142-3p (miR) mimic in H9c2 cells for 48 h caused a
significant increase of miR-142-3p level compared with the NC mimic
group. n=4. (B) Tail vein injection with miR anti-ago caused a
significant decrease of miR-142-3p levels in mice cardiac tissues 7
days after the first injection compared with the NC antagomir group
detected by RT-qPCR. n=4. **P<0.01. RT-qPCR, reverse
transcription-quantitative PCR; miR, microRNA; Ctrl, control; NC,
negative control; miR anti-ago, miR-142-3p antagomir.
RUNX1 and DRD2 proteins are
successfully overexpressed in H9c2 cells. (A) RUNX1 protein levels
in H9c2 cells transfected with Runx1 plasmid for 48 h were detected
using western blotting. (B) Summary for the protein expression of
RUNX1 based on (A). n=3. (C) DRD2 protein levels in H9c2 cells
transfected with DRD2 plasmid for 48 h were detected using western
blotting. (D) Summary for the protein expression of DRD2 based on
(C). n=3. **P<0.01. RUNX1, runt-related transcription
factor 1; DRD2, dopamine receptor D2; Ctrl, control.
Expression of DRD2 protein in mice
cardiac tissues detected using western blotting. (A) Representative
western blotting images showed that tail vein injection with
AAV-DRD2 caused a significant increase of DRD2 in mice cardiac
tissues 20 days after the first injection compared with the
AAV-Ctrl injection group. (B) Bar diagram showed the protein
expression of DRD2 based on (A). n=3. **P<0.01. DRD2,
dopamine receptor D2; AAV, adeno-associated virus; Ctrl,
control.
Primer sequences for reverse
transcription-quantitative PCR.
Differentially expressed miRNAs
(log2FC > 2 or < -2 with a q value of no more than
0.05). MIRI + emodin mice versus MIRI mice.
A total of four pairs of matched
differentially expressed regulatory networks between TFs and
miRNAs.
Overlapped 12 potential genes between
subset one and subset two.
Acknowledgements
Not applicable.
Funding
Funding: This project was financially supported by grants from
the Qingdao 2020 Scientific Research Plan of Traditional Chinese
Medicine (grant no. 2020-zyy058) and Youth Scientific Research Fund
Project of the Affiliated Hospital of Qingdao University (grant no.
3095).
Availability of data and materials
The raw RNA sequencing data reported in this paper
have been deposited in the Genome Sequence Archive (GSA) in
National Genomics Data Center, China National Center for
Bioinformation, Chinese Academy of Sciences (GSA accession number:
CRA007195), and that are publicly accessible at https://ngdc.cncb.ac.cn/gsa/browse/CRA007195. The
datasets used and/or analyzed during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
XZ, XL and LX designed this project. XZ, QQ, XL, YW
and FL performed experiments and analyzed data. XZ and QQ wrote the
manuscript. All authors have read and approved the final
manuscript. XZ and QQ confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
All animal experiments were approved by the Animal
Care and Use Committee of The Affiliated Hospital of Qingdao
University (approval no. QYFYWZLL26907).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Edmondson D and von Känel R:
Post-traumatic stress disorder and cardiovascular disease. Lancet
Psychiatry. 4:320–329. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Andersson C and Vasan RS: Epidemiology of
cardiovascular disease in young individuals. Nat Rev Cardiol.
15:230–240. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Rout A, Tantry US, Novakovic M, Sukhi A
and Gurbel PA: Targeted pharmacotherapy for ischemia reperfusion
injury in acute myocardial infarction. Expert Opin Pharmacother.
21:1851–1865. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Xiong YY, Gong ZT, Tang RJ and Yang YJ:
The pivotal roles of exosomes derived from endogenous immune cells
and exogenous stem cells in myocardial repair after acute
myocardial infarction. Theranostics. 11:1046–1058. 2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Chen M, Li X, Yang H, Tang J and Zhou S:
Hype or hope: Vagus nerve stimulation against acute myocardial
ischemia-reperfusion injury. Trends Cardiovasc Med. 30:481–488.
2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Keeley EC, Boura JA and Grines CL: Primary
angioplasty versus intravenous thrombolytic therapy for acute
myocardial infarction: A quantitative review of 23 randomised
trials. Lancet. 361:13–20. 2003.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Davidson SM, Ferdinandy P, Andreadou I,
Bøtker HE, Heusch G, Ibáñez B, Ovize M, Schulz R, Yellon DM,
Hausenloy DJ, et al: Multitarget strategies to reduce myocardial
ischemia/reperfusion injury: JACC review topic of the week. J Am
Coll Cardiol. 73:89–99. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chang JC, Lien CF, Lee WS, Chang HR, Hsu
YC, Luo YP, Jeng JR, Hsieh JC and Yang KT: Intermittent hypoxia
prevents myocardial mitochondrial Ca2+ overload and cell
death during ischemia/reperfusion: The role of reactive oxygen
species. Cells. 8(564)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kawaguchi M, Takahashi M, Hata T, Kashima
Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J,
et al: Inflammasome activation of cardiac fibroblasts is essential
for myocardial ischemia/reperfusion injury. Circulation.
123:594–604. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wallert M, Ziegler M, Wang X, Maluenda A,
Xu X, Yap ML, Witt R, Giles C, Kluge S, Hortmann M, et al:
α-Tocopherol preserves cardiac function by reducing oxidative
stress and inflammation in ischemia/reperfusion injury. Redox Biol.
26(101292)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sun X, Wei Z, Li Y, Wang J, Hu J, Yin Y,
Xie J and Xu B: Renal denervation restrains the inflammatory
response in myocardial ischemia-reperfusion injury. Basic Res
Cardiol. 115(15)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ong SB, Hernández-Reséndiz S,
Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA and
Hausenloy DJ: Inflammation following acute myocardial infarction:
Multiple players, dynamic roles, and novel therapeutic
opportunities. Pharmacol Ther. 186:73–87. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Chen X, Li X, Zhang W, He J, Xu B, Lei B,
Wang Z, Cates C, Rousselle T and Li J: Activation of AMPK inhibits
inflammatory response during hypoxia and reoxygenation through
modulating JNK-mediated NF-κB pathway. Metabolism. 83:256–270.
2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Chen J, Jiang Z, Zhou X, Sun X, Cao J, Liu
Y and Wang X: Dexmedetomidine preconditioning protects
cardiomyocytes against hypoxia/reoxygenation-induced necroptosis by
inhibiting HMGB1-mediated inflammation. Cardiovasc Drugs Ther.
33:45–54. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Dong X, Fu J, Yin X, Cao S, Li X, Lin L
and Huyiligeqi and Ni J: Emodin: A review of its pharmacology,
toxicity and pharmacokinetics. Phytother Res. 30:1207–1218.
2016.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Dong X, Zeng Y, Liu Y, You L, Yin X, Fu J
and Ni J: Aloe-emodin: A review of its pharmacology, toxicity, and
pharmacokinetics. Phytother Res. 34:270–281. 2020.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
Ye B, Chen X, Dai S, Han J, Liang X, Lin
S, Cai X, Huang Z and Huang W: Emodin alleviates myocardial
ischemia/reperfusion injury by inhibiting gasdermin D-mediated
pyroptosis in cardiomyocytes. Drug Des Devel Ther. 13:975–990.
2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li Q, Gao J, Pang X, Chen A and Wang Y:
Molecular mechanisms of action of emodin: As an anti-cardiovascular
disease drug. Front Pharmacol. 11(559607)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297.
2004.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Hu F, Zhang S, Chen X, Fu X, Guo S, Jiang
Z and Chen K: MiR-219a-2 relieves myocardial ischemia-reperfusion
injury by reducing calcium overload and cell apoptosis through
HIF1α/NMDAR pathway. Exp Cell Res. 395(112172)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhang C, Liang R, Gan X, Yang X, Chen L
and Jian J: MicroRNA-384-5p/beclin-1 as potential indicators for
epigallocatechin gallate against cardiomyocytes ischemia
reperfusion injury by inhibiting autophagy via PI3K/Akt pathway.
Drug Des Devel Ther. 13:3607–3623. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chen Z, Su X, Shen Y, Jin Y, Luo T, Kim
IM, Weintraub NL and Tang Y: MiR322 mediates cardioprotection
against ischemia/reperfusion injury via FBXW7/notch pathway. J Mol
Cell Cardiol. 133:67–74. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yu SY, Dong B, Fang ZF, Hu XQ, Tang L and
Zhou SH: Knockdown of lncRNA AK139328 alleviates myocardial
ischaemia/reperfusion injury in diabetic mice via modulating
miR-204-3p and inhibiting autophagy. J Cell Mol Med. 22:4886–4898.
2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Tong Z, Cui Q, Wang J and Zhou Y: TransmiR
v2.0: An updated transcription factor-microRNA regulation database.
Nucleic Acids Res. 47 (D1):D253–D258. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wang LJ, Qiu BQ, Yuan MM, Zou HX, Gong CW,
Huang H, Lai SQ and Liu JC: Identification and validation of
dilated cardiomyopathy-related genes via bioinformatics analysis.
Int J Gen Med. 15:3663–3676. 2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ogier-Denis E, Fasseu M, Vandewalle A and
Laburthe M: MicroRNAs and intestinal pathophysiology. Med Sci
(Paris). 23:509–514. 2007.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yu LM, Dong X, Xue XD, Xu S, Zhang X, Xu
YL, Wang ZS, Wang Y, Gao H, Liang YX, et al: Melatonin attenuates
diabetic cardiomyopathy and reduces myocardial vulnerability to
ischemia-reperfusion injury by improving mitochondrial quality
control: Role of SIRT6. J Pineal Res. 70(e12698)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ekshyyan O and Aw TY: Apoptosis in acute
and chronic neurological disorders. Front Biosci. 9:1567–1576.
2004.PubMed/NCBI View
Article : Google Scholar
|
|
32
|
Atreya I, Atreya R and Neurath MF:
NF-kappaB in inflammatory bowel disease. J Intern Med. 263:591–596.
2008.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Huang Q, Lu G, Shen HM, Chung MC and Ong
CN: Anti-cancer properties of anthraquinones from rhubarb. Med Res
Rev. 27:609–630. 2007.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhu T, Zhang W, Feng SJ and Yu HP: Emodin
suppresses LPS-induced inflammation in RAW264.7 cells through a
PPARγ-dependent pathway. Int Immunopharmacol. 34:16–24.
2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhu X, Zeng K, Qiu Y, Yan F and Lin C:
Therapeutic effect of emodin on collagen-induced arthritis in mice.
Inflammation. 36:1253–1259. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ding QH, Ye CY, Chen EM, Zhang W and Wang
XH: Emodin ameliorates cartilage degradation in osteoarthritis by
inhibiting NF-κB and Wnt/β-catenin signaling in-vitro and in-vivo.
Int Immunopharmacol. 61:222–230. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Chen GL, Zhang JJ, Kao X, Wei LW and Liu
ZY: Emodin ameliorates lipopolysaccharides-induced corneal
inflammation in rats. Int J Ophthalmol. 8:665–669. 2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kitano A, Saika S, Yamanaka O, Ikeda K,
Okada Y, Shirai K and Reinach PS: Emodin suppression of ocular
surface inflammatory reaction. Invest Ophthalmol Vis Sci.
48:5013–5022. 2007.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Wu Y, Tu X, Lin G, Xia H, Huang H, Wan J,
Cheng Z, Liu M, Chen G, Zhang H, et al: Emodin-mediated protection
from acute myocardial infarction via inhibition of inflammation and
apoptosis in local ischemic myocardium. Life Sci. 81:1332–1338.
2007.PubMed/NCBI View Article : Google Scholar
|
|
40
|
McCarroll CS, He W, Foote K, Bradley A,
McGlynn K, Vidler F, Nixon C, Nather K, Fattah C, Riddell A, et al:
Runx1 deficiency protects against adverse cardiac remodeling after
myocardial infarction. Circulation. 137:57–70. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Li X, Zhang S, Wa M, Liu Z and Hu S:
MicroRNA-101 protects against cardiac remodeling following
myocardial infarction via downregulation of runt-related
transcription factor 1. J Am Heart Assoc. 8(e013112)2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Cui Y, Chen LJ, Huang T, Ying JQ and Li J:
The pharmacology, toxicology and therapeutic potential of
anthraquinone derivative emodin. Chin J Nat Med. 18:425–435.
2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Oshida K, Hirakata M, Maeda A, Miyoshi T
and Miyamoto Y: Toxicological effect of emodin in mouse testicular
gene expression profile. J Appl Toxicol. 31:790–800.
2011.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Ghafouri-Fard S, Shoorei H and Taheri M:
Non-coding RNAs participate in the ischemia-reperfusion injury.
Biomed Pharmacother. 129(110419)2020.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Suzuki HI and Miyazono K: Emerging
complexity of microRNA generation cascades. J Biochem. 149:15–25.
2011.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Fan ZX and Yang J: The role of microRNAs
in regulating myocardial ischemia reperfusion injury. Saudi Med J.
36:787–793. 2015.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ong SB, Katwadi K, Kwek XY, Ismail NI,
Chinda K, Ong SG and Hausenloy DJ: Non-coding RNAs as therapeutic
targets for preventing myocardial ischemia-reperfusion injury.
Expert Opin Ther Targets. 22:247–261. 2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Xing X, Guo S, Zhang G, Liu Y, Bi S, Wang
X and Lu Q: miR-26a-5p protects against myocardial
ischemia/reperfusion injury by regulating the PTEN/PI3K/AKT
signaling pathway. Braz J Med Biol Res. 53(e9106)2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Wang JX, Zhang XJ, Li Q, Wang K, Wang Y,
Jiao JQ, Feng C, Teng S, Zhou LY, Gong Y, et al: MicroRNA-103/107
regulate programmed necrosis and myocardial ischemia/reperfusion
injury through targeting FADD. Circ Res. 117:352–363.
2015.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Zhao J, Li X, Hu J, Chen F, Qiao S, Sun X,
Gao L, Xie J and Xu B: Mesenchymal stromal cell-derived exosomes
attenuate myocardial ischaemia-reperfusion injury through
miR-182-regulated macrophage polarization. Cardiovasc Res.
115:1205–1216. 2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Hullinger TG, Montgomery RL, Seto AG,
Dickinson BA, Semus HM, Lynch JM, Dalby CM, Robinson K, Stack C,
Latimer PA, et al: Inhibition of miR-15 protects against cardiac
ischemic injury. Circ Res. 110:71–81. 2012.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Chaudhuri AD, Choi DC, Kabaria S, Tran A
and Junn E: MicroRNA-7 regulates the function of mitochondrial
permeability transition pore by targeting VDAC1 expression. J Biol
Chem. 291:6483–6493. 2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Shao W, Zhang SZ, Tang M, Zhang XH, Zhou
Z, Yin YQ, Zhou QB, Huang YY, Liu YJ, Wawrousek E, et al:
Suppression of neuroinflammation by astrocytic dopamine D2
receptors via αB-crystallin. Nature. 494:90–94. 2013.PubMed/NCBI View Article : Google Scholar
|