Introduction
Hereditary spherocytosis (HS), an inherited
condition characterized by the presence of spherocytes in
peripheral blood smears, is the most prevalent cause of haemolytic
anaemia (1,2). Although HS is a global disease, its
incidence has increased in Northern Europe to ~1 in 2,000 cases
(3). In East Asia, research from
Korea and Japan has revealed that HS is the most common congenital
haemolytic anaemia (4,5). The predicted prevalence in China is
1.27 per 100,000 for men and 1.49 per 100,000 for women (6). Currently, known mutant genes include
ankyrin 1 (ANK1), spectrin α erythrocytic 1 (SPTA1), spectrin β
erythrocytic (SPTB), solute carrier family 4 member 1 (SLC4A1) and
erythrocyte membrane protein band 4.2 (EPB42), which encode the
erythrocyte membrane proteins ankyrin, spectrin, band 3 and band
4.2, respectively. Mutations in these genes lead to abnormalities
or malfunctions of their respective proteins (1,7-9).
Approximately 90% of the cases of HS are inherited as autosomal
dominant due to mutations in ANK1, SPTB and SLC4A1. A further 10%
of the cases of HS are inherited as autosomal recessive due to
mutations in SPTA1 and EPB42(10).
HS has a heterogeneous clinical manifestation,
ranging from asymptomatic compensated haemolysis to transfusion
dependence (1,11-13).
The diagnostic foundation for this condition is a negative Coombs
test, the presence of spherocytes and anaemia with a positive
family history. Therapy is directed at preventing or minimizing the
effects of chronic haemolysis and anaemia. Although there is no
treatment for erythrocyte membrane abnormalities, available options
include folic acid and erythropoietin supplements, as well as blood
transfusions, splenectomy and cholecystectomy, depending on
individual disease severity (7,14,15).
As a diagnostic tool, whole-exome sequencing based
on next-generation sequencing technology has considerably
contributed to the discovery of novel mutations that cause
Mendelian diseases (16-18).
A previous study reported a novel ANK1 c.4276C>T (p.R1426*)
nonsense mutation identified by NGS in a patient with HS. However,
Sanger sequencing confirmed that neither the parents or younger
brother carried this mutation (19). Thus, to the best of our knowledge,
reports of such mutations, particularly mutations within the ANK1
gene, and their clinical characteristics in families with HS, are
rare. The present case report describes the identification of an HS
pedigree with a novel ANK1 mutation using next-generation
sequencing technology, thereby expanding the spectrum of ANK1
mutations. Moreover, the related literature is reviewed.
Case report
A 24-year-old male patient (proband) was
hospitalized in July 2021 at the Affiliated Traditional Chinese
Medicine Hospital of Southwest Medical University (Luzhou, China),
with a 12-year history of unexplained anaemia and recurrent
jaundice. A physical examination showed pale conjunctiva and an
enlarged spleen. The patient had no history of any other diseases.
Moreover, no history of smoking, drinking or drug use was reported,
and the parents were not consanguineous. However, the patient's
father, paternal aunt and paternal grandmother reported a similar
medical history of anaemia and jaundice.
For investigating the cause of anaemia, tests for
whole blood cell count, liver function, serum iron, serum ferritin,
transferrin saturation, red blood cell osmotic fragility and
eosin-5'-maleimide (EMA) binding, as well as the Coombs test, were
performed on the patient, the patient's father and the paternal
grandmother, while tests for whole blood cell count, liver function
and EMA binding were performed on the paternal aunt (Table I). The patient, father and paternal
aunt had compensated anaemia with an elevated reticulocyte ratio.
The test results of the patient and family members showed a
negative Coombs test, increased red blood cell osmotic fragility
and decreased mean channel fluorescence levels in the EMA binding
test.
 | Table IBlood test results of the patient and
the family members. |
Table I
Blood test results of the patient and
the family members.
| Characteristic | Reference range | Patient | Father | Aunt | Grandmother |
|---|
| Red blood cells, n
(x1012/l) | 4.30-5.80 | 4.03 | 3.56 | 3.67 | 1.90 |
| Hemoglobin, g/l | 130-175 | 139 | 124 | 117 | 64 |
| Mean corpuscular
volume, fl | 82-100 | 101.20 | 103.90 | 91.90 | 106.70 |
| MHC, pg | 27-34 | 34.40 | 34.70 | 31.90 | 33.60 |
| MHC concentration,
g/l | 316-354 | 340 | 334 | 347 | 315 |
| Reticulocytes, % | 0.90-2.20 | 8.94 | 9.92 | 8.87 | 11.01 |
| Osmotic
fragility | (-) | ↑ | ↑ | NA | ↑ |
| Spherocytosis | (-) | (+) | (+) | NA | (+) |
| Eosin-5'-maleimide,
% | 100 | 87.91 | 85.94 | 87.59 | 96.30 |
| Coombs test | (-) | (-) | (-) | NA | (-) |
| Iron, µmol/l | 10.60-36.70 | 19 | 47.50 | NA | 33.60 |
| Ferritin,
ng/ml | 25-280 | 322.43 | >1,000 | NA | 891.42 |
| Serum total
bilirubin, µmol/l | 0-23 | 96.10 | 69.90 | 26.80 | 29.40 |
| Direct bilirubin,
µmol/l | 0-7 | 11.50 | 21 | 8.40 | 11.10 |
| Indirect bilirubin,
µmol/l | 0-20 | 84.60 | 48.90 | 18.40 | 18.30 |
| Unbound
iron-binding capacity, µmol/l | 34-48 | 29.80 | 6.70 | NA | 7.10 |
| Total iron-binding
capacity, µmol/l | 50-77 | 48.80 | 54.20 | NA | 40.70 |
The patient underwent a bone marrow aspiration and
bone marrow smear. The patient and family members underwent a
peripheral blood smear test. The bone marrow and peripheral blood
smears were performed using Wright's staining. Buffered Wright
stain was added for 5 min to fix and stain the specimen, and then
the slides were left to stand for 5 min after adding an equal
amount of ultrapure water, all at room temperature. The slides were
then washed, blotted and assessed via light microscopy. The results
of the bone marrow smear and peripheral blood smear were analysed
by experienced physicians in the Department of Pathology of The
Affiliated Traditional Chinese Medicine Hospital of Southwest
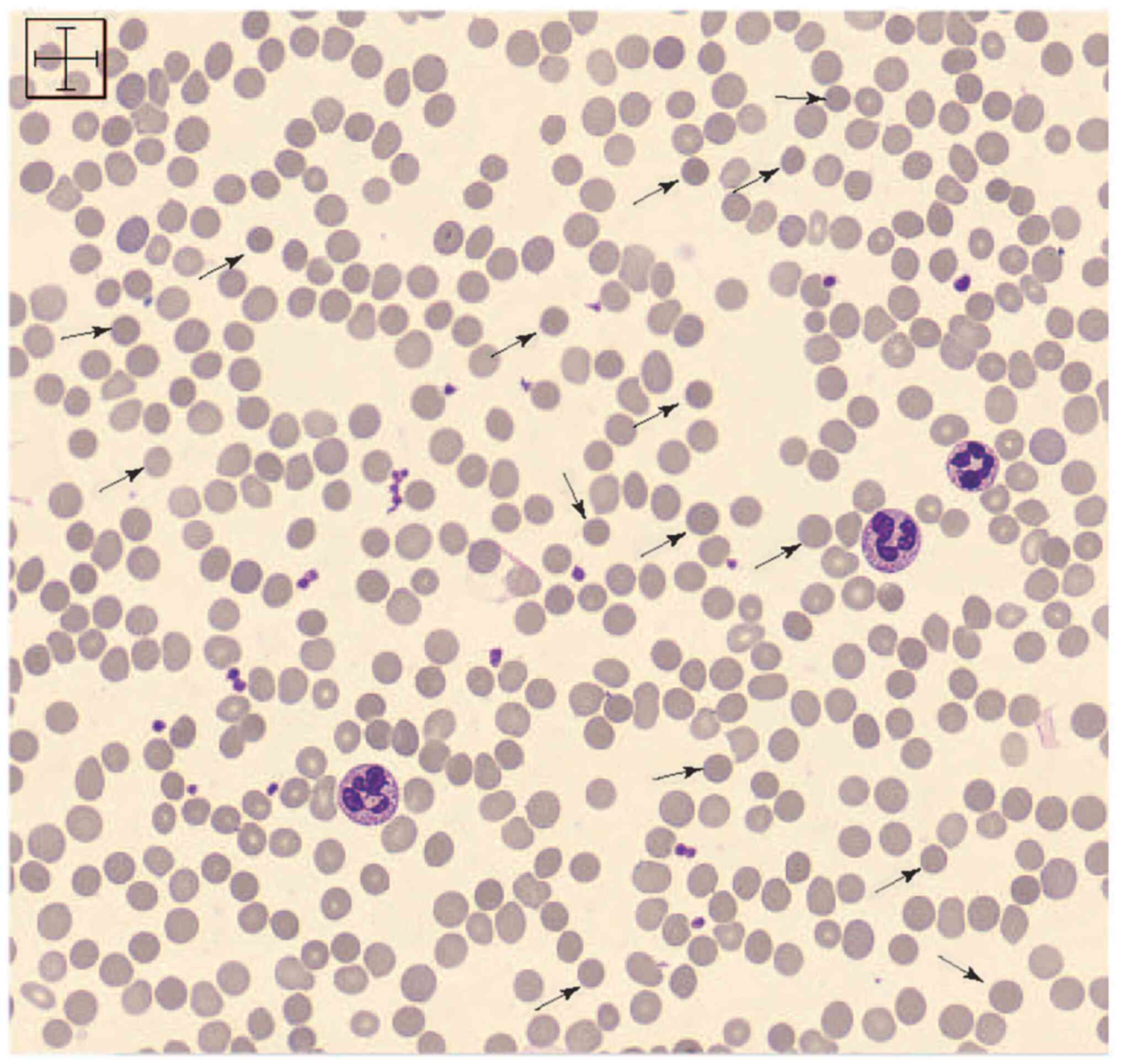
Medical University. Spherocytes were found in the peripheral blood
smears (Fig. 1). All other causes
of anaemia were excluded via bone marrow aspiration.
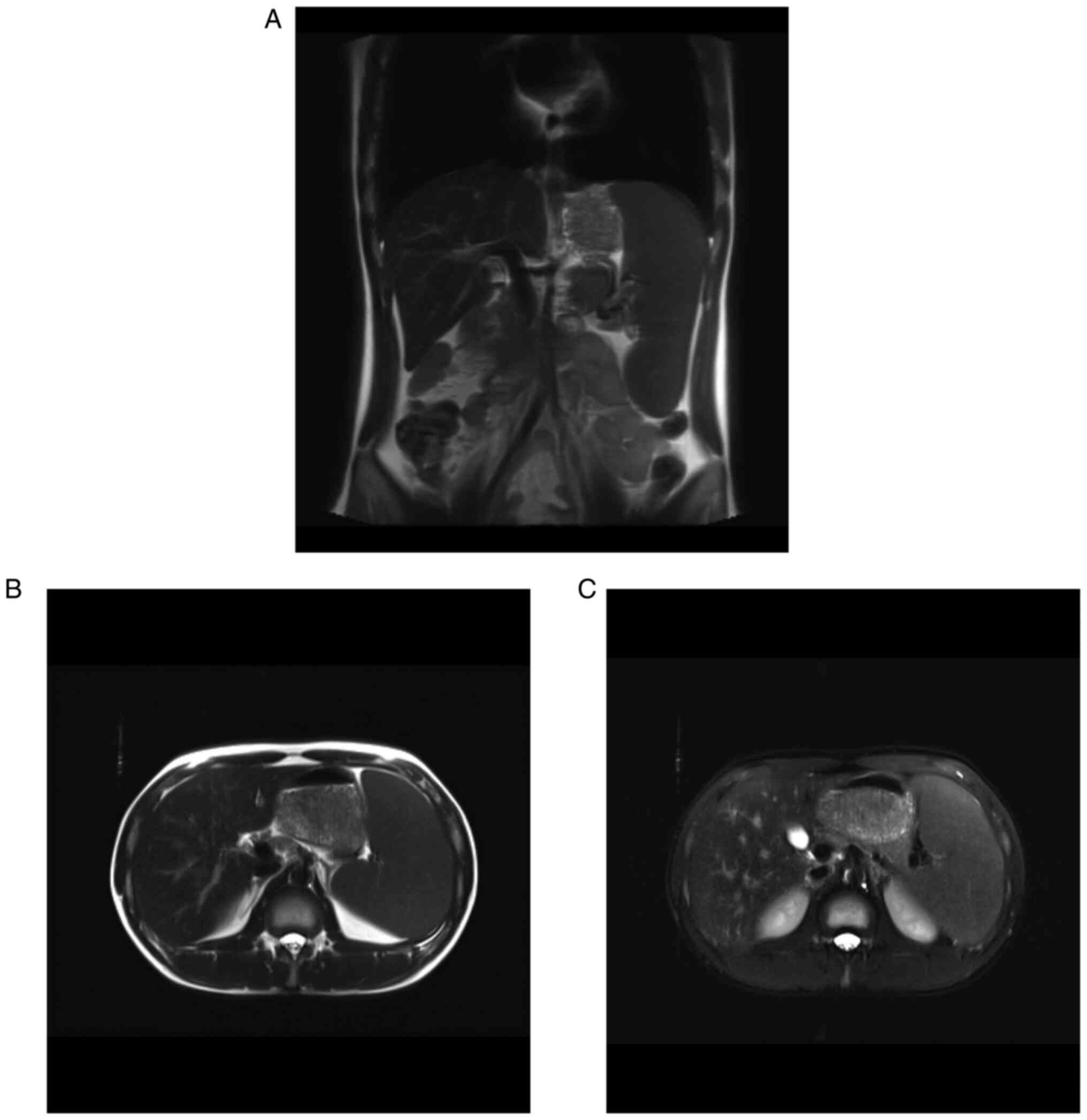
Abdominal magnetic resonance imaging (MRI) of the
patient (Fig. 2) and father
revealed splenomegaly and iron deposition in the spleen. A blood
sample from the patient was sent to Guangzhou KingMed Diagnostics
Group Co., Ltd., for whole-exome sequencing to identify possible
mutations. Genomic DNA collection and whole-exome sequencing were
performed according to the manufacturer's instructions as
previously described (20). A
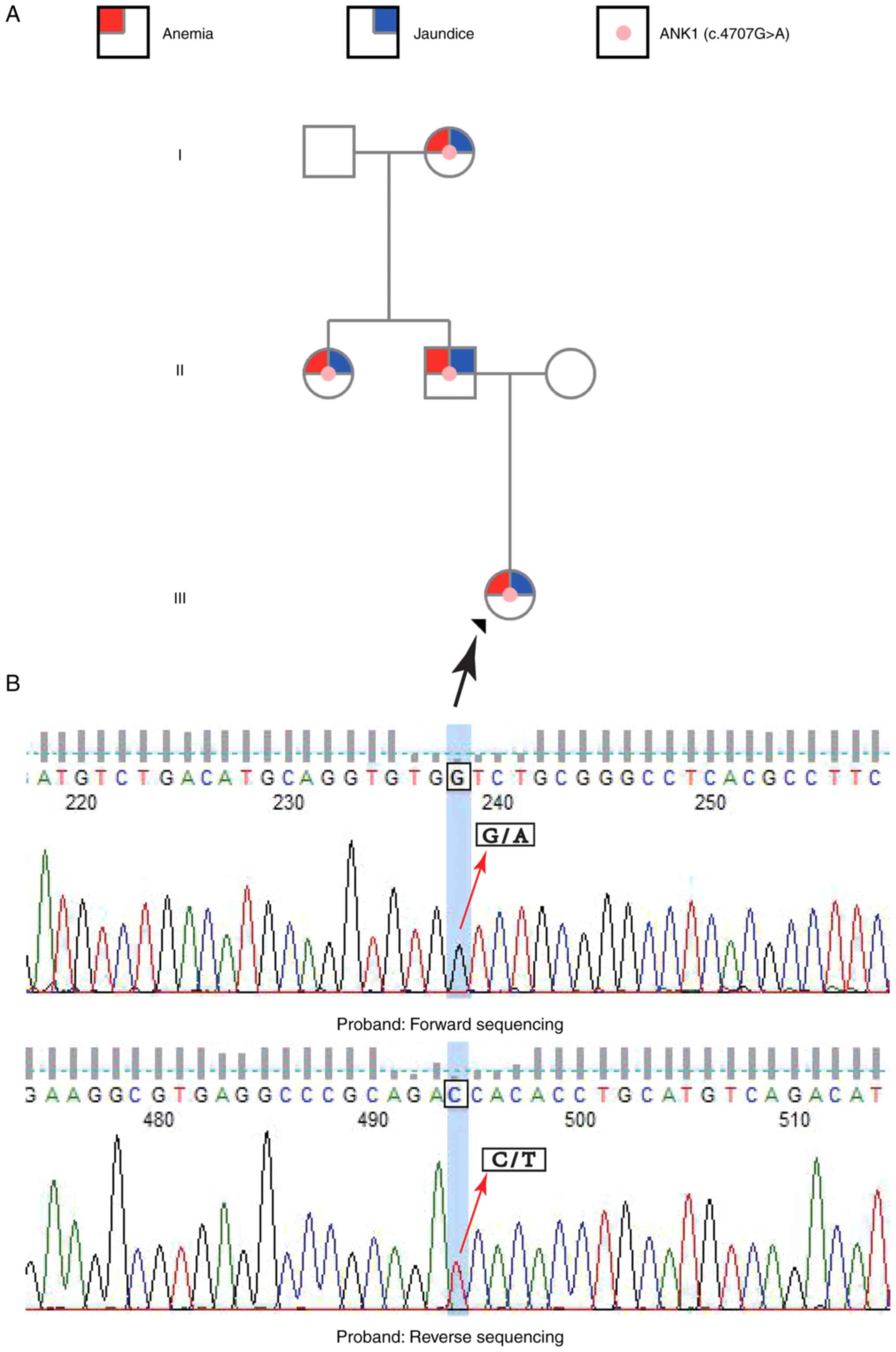
novel mutation in ANK1, c.4707G>A (p.Trp1569*), was identified
in the patient. The genetic pedigree map and sequencing results for
the analysed family members are shown in Fig. 3. The heterozygous mutation was
verified by Sanger sequencing at Guangzhou KingMed Diagnostics
Group Co., Ltd. (Fig. 3), which
showed an overlap of signals for G/A and C/T at position 4707 of
the ANK1 gene in the chromatograms of the forward and reverse
sequencing results, respectively. Furthermore, Sanger sequencing
also confirmed that the mutation was present in the patient's
father, aunt and grandmother (data not shown). The c.4707G>A
(p.Trp1569*) mutation produced a truncated ANK1 protein, thereby
impairing ANK1 function. This variation was not found in four major
databases [Human Gene Mutation Database (https://digitalinsights.qiagen.com/products-overview/clinical-insights-portfolio/human-gene-mutation-database/),
ESP6500siv2 (https://esp.gs.washington.edu/drupal/), 1000 Genomes
(https://www.internationalgenome.org/)
and dbSNP147 (https://www.ncbi.nlm.nih.gov/snp/)] and was not
identified in the patient's mother or grandfather upon Sanger
sequencing (data not shown).
All affected family members are currently undergoing
regular follow-ups every 3-6 months. Folic acid supplementation and
blood transfusion are administered when necessary. Neither the
patient nor the affected family members have received a
splenectomy.
Discussion
The present study describes an HS pedigree with four
family members involved. According to related guidelines (21,22),
whole-exome sequencing was performed in the patient and a novel
ANK1 mutation, c.4707G>A (p.Trp1569*), was identified. In recent
years, an increasing number of novel mutations related to HS have
been identified with second-generation sequencing (23). Moreover, second-generation
sequencing has become an affordable and accessible test for
diagnosing HS, as genetic sequencing costs have decreased
significantly.
The genotypes and protein phenotypes of HS vary
widely, with ANK1 mutations accounting for 35-65% of cases in
Northern Europe, which is similar to the prevalence of ANK1
mutations in the Korean population (ANK1 mutations result in 66.66%
of cases) (24,25). By contrast, most cases of HS in the
Japanese population are due to mutations in EPB42 (45%), whereas in
China, ANK1 and SPTB have been reported as the most commonly
mutated genes, each being present in 45% of patients (5,26).
Thus, deficiencies in ankyrin alone or ankyrin and spectrin
combined, caused by mutations in ANK1 or ANK1 and SPTB,
respectively, are the major causes of HS in China. The present
report identified a novel HS-related mutation in ANK1.
Ankyrin is the main protein responsible for coupling
the erythrocyte plasma membrane and the underlying cytoskeleton; it
interacts with spectrin to maintain the biconcave disc shape of
these cells, allowing them to undergo reversible deformation while
traversing the microvasculature without changing the surface area
(27). ANK1 mutations are
responsible for the change from the biconcave disc shape to the
spherical shape. This limits erythrocyte deformability and triggers
mechanical rupture haemolysis in capillary network-rich organs such
as the liver and spleen. The ANK1 gene is localized to 8P11.21 and
contains 49 exons that encode three major structural domains,
including the N-terminal binding, central spectral binding and
C-terminal regulatory domains, of which the C-terminal regulatory
domain is the most mutable (28).
Recent studies have found that ANK1 mutations in Chinese patients
with HS are mainly observed in the N-terminal membrane
protein-binding domain, the two ZU5 domains, the UPA domain and the
death domain, with clinical manifestations of different types of
ANK1 mutations appearing in other structural domains being random
(29,30). The mutation in ANK1 identified in
the current study was a rare nonsense mutation located in the gene
sequence encoding the C-terminal regulatory domain, resulting in
autosomal dominant inheritance, which led to the development of
clinical symptoms in three generations of the family.
Studies have shown that clinical manifestations of
HS might be related to the ANK1 mutation site. Mutations in the
central spectral binding domain of ANK1 reportedly elicit more
severe anaemia and higher rates of erythrocyte deformation.
Variants affecting the death domain are associated with a low mean
corpuscular volume (MCV) and a low mean corpuscular haemoglobin
(MCH) level (25,29). However, even the same ANK1
mutations may result in heterogeneous disease severity in different
pedigrees (7,10). As a novel truncated mutation
discovered at the end of the C-terminal regulatory domain, to the
best of our knowledge, there is almost no information about its
related clinical manifestations. As for truncated mutations at the
end of the C-terminal regulatory domain, Wang et al
(19) reported an ANK1
c.4276C>T (p.R1426*) nonsense mutation at the C-terminal
regulatory domain in a patient with HS. However, Sanger sequencing
confirmed that none of the other family members carried this
mutation. To date, the present study is the first pedigree report
of an ANK1 truncated mutation at the end of the C-terminal
regulatory domain with multiple patients involved. Most individuals
in the present patient's family had compensated anaemia, indicating
mild HS and potentially explaining their normal MCV, MCH, and MCH
concentration (MCHC) levels, which are typically reduced in
patients with HS. Additionally, several studies have shown normal
or increased MCHC level in patients with HS (29,31,32).
Multiple episodes of haemolysis may cause excessive
iron deposition in the spleen of adult patients with HS. Therefore,
these events must be differentiated from other iron overload
diseases, such as hereditary hemochromatosis (33). In the present case, abdominal MRI
showed spleen iron deposition in the patient and the father, which
is seldom reported in patients with HS. As bone marrow aspiration
and whole exosome sequencing excluded other iron overload causes,
it was considered that the iron deposition in the spleen of the
patient and the father was caused by chronic haemolytic
anaemia.
Osmotic gradient ektacytometry is a robust
measurement for diagnosing HS (34). However, it is not widely available
for the clinical diagnosis of HS in mainland China. Therefore, the
combination of EMA testing and second-generation sequencing is
recommended in the Chinese and South Korean guidelines for HS
diagnosis (21,22). In the present case, EMA testing and
second-generation sequencing helped to diagnose HS.
In conclusion, the present study reported an HS
pedigree with a novel ANK1 mutation and unique clinical
manifestation. Affordable and accessible second-generation
sequencing helped to diagnose HS and discover a novel HS-related
mutation. Individuals in this pedigree showed a mild clinical
manifestation, which might be related to the novel truncated
mutation at the end of the C-terminal regulatory domain of ANK1.
This novel mutation pedigree expands the mutation spectrum of
ANK1-related HS. Further studies are warranted to show the
pathogenesis of this novel c.4707G>A (p.Trp1569*) mutation for
HS.
Acknowledgements
Not applicable.
Funding
Funding: The whole-exome and Sanger sequencing analysis were
supported by funding from the National Traditional Chinese Medicine
Clinical Research Base Construction Unit of The Affiliated
Traditional Chinese Medicine Hospital of Southwest Medical
University [grant no. (2020) 33], the Luzhou ‘Jiucheng
Talents-Scientific and Technological Innovation Team’ [grant no.
(2021) 162] and the Scientific Research Team Cultivation Project of
the Affiliated Traditional Chinese Medicine Hospital of Southwest
Medical University [grant no. (2022) 68, 2022-CXTD-04].
Availability of data and materials
The datasets generated and/or analysed during the
current study are available in the NCBI Sequence Read Archive
repository (https://dataview.ncbi.nlm.nih.gov/object/PRJNA884085?reviewer=5isvljlpqtfr33fnpmdl3bjcq0)
under the accession no. SRR21700253.
Authors' contributions
JW, SY, JC and XZ designed the study. XZ and MP
collected the data. XZ drafted the manuscript. YY, YZ and DZ
analyzed and interpreted the genetic data. MP and ZP followed up
with the patient and analyzed the clinical data. JW, SY and JC
revised the manuscript. JW and XZ confirm the authenticity of all
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of The Affiliated Traditional Chinese Medicine Hospital
of Southwest Medical University (approval no. KY2022010). The study
was conducted in accordance with the Declaration of Helsinki.
Written informed consent was obtained from all individuals involved
in the study.
Patient consent for publication
Written informed consent was obtained for the
publication of clinical data and genetic analysis data, including
the addition of genetic information to the NCBI repository.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Perrotta S, Gallagher PG and Mohandas N:
Hereditary spherocytosis. Lancet. 372:1411–1426. 2008.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Zamora EA and Schaefer CA: Hereditary
spherocytosis. In: StatPearls [Internet]. StatPearls Publishing,
Treasure Island, FL, 2022.
|
|
3
|
King MJ, Garçon L, Hoyer JD, Iolascon A,
Picard V, Stewart G, Bianchi P, Lee SH and Zanella A: International
Council for Standardization in Haematology. ICSH guidelines for the
laboratory diagnosis of nonimmune hereditary red cell membrane
disorders. Int J Lab Hematol. 37:304–325. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Shim YJ, Jung HL, Shin HY, Kang HJ, Choi
JY, Hah JO, Lee JM, Lim YT, Yang EJ, Baek HJ, et al:
Epidemiological study of hereditary hemolytic anemia in the Korean
pediatric population during 1997-2016: A nationwide retrospective
cohort study. J Korean Med Sci. 35(e279)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Yawata Y, Kanzaki A, Yawata A, Doerfler W,
Ozcan R and Eber SW: Characteristic features of the genotype and
phenotype of hereditary spherocytosis in the Japanese population.
Int J Hematol. 71:118–135. 2000.PubMed/NCBI
|
|
6
|
Wang C, Cui Y, Li Y, Liu X and Han J: A
systematic review of hereditary spherocytosis reported in Chinese
biomedical journals from 1978 to 2013 and estimation of the
prevalence of the disease using a disease model. Intractable Rare
Dis Res. 4:76–81. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bolton-Maggs PH, Langer JC, Iolascon A,
Tittensor P and King MJ: General Haematology Task Force of the
British Committee for Standards in Haematology. Guidelines for the
diagnosis and management of hereditary spherocytosis-2011 update.
Br J Haematol. 156:37–49. 2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Delaunay J: The molecular basis of
hereditary red cell membrane disorders. Blood Rev. 21:1–20.
2007.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Narla J and Mohandas N: Red cell membrane
disorders. Int J Lab Hematol. 39 (Suppl 1):S47–S52. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Tole S, Dhir P, Pugi J, Drury LJ, Butchart
S, Fantauzzi M, Langer JC, Baker JM, Blanchette VS, Kirby-Allen M
and Carcao MD: Genotype-phenotype correlation in children with
hereditary spherocytosis. Br J Haematol. 191:486–496.
2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Eber SW, Armbrust R and Schröter W:
Variable clinical severity of hereditary spherocytosis: Relation to
erythrocytic spectrin concentration, osmotic fragility, and
autohemolysis. J Pediatr. 117:409–416. 1990.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Friedman EW, Williams JC and Van Hook L:
Hereditary spherocytosis in the elderly. Am J Med. 84 (3 Pt
1):513–516. 1988.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Whitfield CF, Follweiler JB,
Lopresti-Morrow L and Miller BA: Deficiency of alpha-spectrin
synthesis in burst-forming units-erythroid in lethal hereditary
spherocytosis. Blood. 78:3043–3051. 1991.PubMed/NCBI
|
|
14
|
Abdullah F, Zhang Y, Camp M, Rossberg MI,
Bathurst MA, Colombani PM, Casella JF, Nabaweesi R and Chang DC:
Splenectomy in hereditary spherocytosis: Review of 1,657 patients
and application of the pediatric quality indicators. Pediatr Blood
Cancer. 52:834–837. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Alizai NK, Richards EM and Stringer MD: Is
cholecystectomy really an indication for concomitant splenectomy in
mild hereditary spherocytosis? Arch Dis Child. 95:596–599.
2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Biesecker LG and Green RC: Diagnostic
clinical genome and exome sequencing. N Engl J Med. 370:2418–2425.
2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Rabbani B, Mahdieh N, Hosomichi K, Nakaoka
H and Inoue I: Next-generation sequencing: Impact of exome
sequencing in characterizing Mendelian disorders. J Hum Genet.
57:621–632. 2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yang Y, Muzny DM, Xia F, Niu Z, Person R,
Ding Y, Ward P, Braxton A, Wang M, Buhay C, et al: Molecular
findings among patients referred for clinical whole-exome
sequencing. JAMA. 312:1870–1879. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wang X, Yi B, Mu K, Shen N, Zhu Y, Hu Q
and Lu Y: Identification of a novel de novo ANK1 R1426* nonsense
mutation in a Chinese family with hereditary spherocytosis by NGS.
Oncotarget. 8:96791–96797. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wei Y, He Y and Guo X: Clinical phenotype
and genetic analysis of twins with congenital coagulation factor V
deficiency. J Pediatr Hematol Oncol. 44:e482–e486. 2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kim Y, Park J and Kim M: Diagnostic
approaches for inherited hemolytic anemia in the genetic era. Blood
Res. 52:84–94. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Xue J, He Q, Xie XJ, Su AL and Cao SB: A
clinical and experimental study of adult hereditary spherocytosis
in the Chinese population. Kaohsiung J Med Sci. 36:552–560.
2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang R, Yang S, Xu M, Huang J, Liu H, Gu W
and Zhang X: Exome sequencing confirms molecular diagnoses in 38
Chinese families with hereditary spherocytosis. Sci China Life Sci.
61:947–953. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Eber SW, Gonzalez JM, Lux ML, Scarpa AL,
Tse WT, Dornwell M, Herbers J, Kugler W, Ozcan R, Pekrun A, et al:
Ankyrin-1 mutations are a major cause of dominant and recessive
hereditary spherocytosis. Nat Genet. 13:214–218. 1996.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Park J, Jeong DC, Yoo J, Jang W, Chae H,
Kim J, Kwon A, Choi H, Lee JW, Chung NG, et al: Mutational
characteristics of ANK1 and SPTB genes in hereditary spherocytosis.
Clin Genet. 90:69–78. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Qin L, Nie Y, Zhang H, Chen L, Zhang D,
Lin Y and Ru K: Identification of new mutations in patients with
hereditary spherocytosis by next-generation sequencing. J Hum
Genet. 65:427–434. 2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Salomao M, Zhang X, Yang Y, Lee S, Hartwig
JH, Chasis JA, Mohandas N and An X: Protein 4.1R-dependent
multiprotein complex: New insights into the structural organization
of the red blood cell membrane. Proc Natl Acad Sci USA.
105:8026–8031. 2008.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Lux SE, John KM and Bennett V: Analysis of
cDNA for human erythrocyte ankyrin indicates a repeated structure
with homology to tissue-differentiation and cell-cycle control
proteins. Nature. 344:36–42. 1990.PubMed/NCBI View
Article : Google Scholar
|
|
29
|
Wang X, Zhang A, Huang M, Chen L, Hu Q, Lu
Y and Cheng L: Genetic and clinical characteristics of patients
with hereditary spherocytosis in Hubei Province of China. Front
Genet. 11(953)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang D, Song L, Shen L, Zhang K, Lv Y, Gao
M, Ma J, Wan Y, Gai Z and Liu Y: Mutational characteristics of
causative genes in Chinese hereditary spherocytosis patients: A
report on fourteen cases and a review of the literature. Front
Pharmacol. 12(644352)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Aggarwal A, Jamwal M, Sharma P, Sachdeva
MUS, Bansal D, Malhotra P and Das R: Deciphering molecular
heterogeneity of Indian families with hereditary spherocytosis
using targeted next-generation sequencing: First South Asian study.
Br J Haematol. 188:784–795. 2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Michaels LA, Cohen AR, Zhao H, Raphael RI
and Manno CS: Screening for hereditary spherocytosis by use of
automated erythrocyte indexes. J Pediatr. 130:957–960.
1997.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Kowdley KV, Brown KE, Ahn J and Sundaram
V: ACG clinical guideline: Hereditary hemochromatosis. Am J
Gastroenterol. 114:1202–1218. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Llaudet-Planas E, Vives-Corrons JL,
Rizzuto V, Gómez-Ramírez P, Sevilla Navarro J, Coll Sibina MT,
García-Bernal M, Ruiz Llobet A, Badell I, Velasco-Puyó P, et al:
Osmotic gradient ektacytometry: A valuable screening test for
hereditary spherocytosis and other red blood cell membrane
disorders. Int J Lab Hematol. 40:94–102. 2018.PubMed/NCBI View Article : Google Scholar
|