Introduction
Chronic obstructive pulmonary disease (COPD) is a
common and frequently occurring respiratory disease with high
morbidity and mortality. COPD has become an important public health
problem due to the economic burden it imposes on society (1). Numerous studies have shown that
cigarette smoke exposure is the predominant risk factor for the
development of COPD (2). COPD is
characterized by an abnormal inflammatory response, airway
obstruction, alveolar destruction and apoptosis (3). Pulmonary vascular endothelial cells
are one of the main components of pulmonary vessels. Apoptosis and
the pulmonary vascular endothelial cell inflammatory response
increase in some patients with COPD, which may aggravate
morphologic alterations of lung tissues and lead to significant
abnormalities in lung function indices and a corresponding decline
in lung function (4). However, the
mechanism of cigarette-induced COPD causing apoptosis and
inflammation of pulmonary vascular endothelial cells has not been
established.
Carnitine palmitoyltransferase 1A (Cpt1a) is a key
enzyme located to the inner mitochondrial membrane and involved in
the regulation of fatty acid oxidation. Cpt1a transports fatty
acids from the cytoplasm to the mitochondria for subsequent fatty
acid oxidation (5). Fatty acid
oxidation protects against endothelial cell apoptosis and lung
injury induced hyperoxia in neonatal mice (6). Metabolism of fatty acid in
endothelial cells were found to be dysregulated in patients with
COPD. Oxidation of fatty acids reduction increases apoptosis in
lung endothelial cells treated with cigarette smoke extract
(7). In addition, Cpt1a deficiency
can inhibit endothelial cell proliferation and angiogenesis
(8). Our previous study showed
that L-carnitine upregulates Cpt1a, promotes an increase in
substrate fatty acids and inhibits cigarette-induced endothelial
cell apoptosis (9). However, the
role of Cpt1a in pulmonary vascular endothelial cells in COPD
remains to be elucidated.
Therefore, the hypothesis of the current study is
that Cpt1a alleviates cigarette smoke-induced chronic obstructive
pulmonary disease by promoting fatty acid oxidation to inhibit the
inflammatory response and cell apoptosis. The main contents of the
present study were as follows: i) clarifying the correlation
between Cpt1a and lung apoptosis, inflammation and lung function in
patients with COPD at the clinical level; ii) confirming whether
Cpt1a can treat cigarette-induced COPD in an animal model; and iii)
clarifying the mechanism underlying Cpt1a protection of COPD
lungs.
Materials and methods
Subject recruitment and sample
preparation
All animals were housed in accordance with the Guide
for the Care and Use of Laboratory Animals. All procedures of the
present study were approved by the Ethics Committee of the Second
Hospital of Shanxi Medical University (CMTT number 2013012) and in
accordance with international standards. Prior to the clinical
study, the subjects (n=20) were informed of the nature, purpose,
possible benefits and risks of the trial and the subjects
voluntarily confirmed their consent to participate. The inclusion
criteria were as follows: i) Patients who met the Global Initiative
for Chronic Obstructive Lung Disease (GOLD) in 2015 with severe
COPD, ii) no bronchiectasis, iii) no bronchial asthma, iv) no heart
failure and v) patients who underwent pulmonary resection. The
exclusion criteria were as follows: patients i) with severe hepatic
and renal dysfunction, ii) hematologic diseases, iii) usage of
immunosuppressants in recent 3 months, or iv) severe immune system
diseases. All patients were detected for lung function which was
diagnosed as different degrees of dyspnoea, shortness of breath,
cough, chronic cough and other symptoms. In addition, patients
(n=10) without COPD who underwent pulmonary resection in The Second
Hospital of Shanxi Medical University during the same period were
selected as the control group. Postoperatively, the lung tissues of
all selected patients were collected and frozen in a -80˚C
refrigerator.
Animal model
The tails of healthy adult C57BL/6 mice (5 groups,
10 mice/group, 50% male and 50% female, Age: 8~10 weeks, weight
18~22 g, 12 h light/12 h dark, Temperature is 18~22˚C, humidity,
50-60%, all mice purchased from Beijing Vital River Laboratory
Animal Technology Co., Ltd., China)were injected with
pGLVU6/GFP-short hairpin (sh)RNA control, pGLVU6/GFP-shRNA Cpt1a,
pGLVU6/GFP control and pGLVU6/GFP-Cpt1a lentivirus to establish
mouse models with knockdown of Cpt1a or overexpression of Cpt1a. At
two weeks after the injection, the above four groups of mice were
placed in a self-made tobacco smoke inhalation exposure device
(30x40x90 cm plexiglass cuboid with nine evenly distributed
circular exhaust holes, 2 cm in diameter, on the cover) and a
wooden rectangular box containing cigarettes (Furong brand, China
Tobacco Hubei Industrial LLC) was placed in it. The principal
combustion products of cigarette were as follows: nicotine content
in smoke, 1.2 mg/cigarette; carbon monoxide content in smoke, 14
mg/cigarette; and tar content, 15 mg/cigarette. The control group
were exposed to smoke-free air and raised normally. To establish
the model of COPD in mice, the mice in the model group were exposed
to cigarette smoke four times a day in the device. A total of six
cigarettes were lit each time and left burning for 1 h, with the
smoke concentration in the closed box reaching 100-120
mg/m3. The mice were allowed to breathe smoke-free air
for 30 min between two times of smoke exposure. The procedures were
conducted six days per week and lasted for four weeks. The
experiment included five groups (n=10): control group;
pGLVU6/GFP-shRNA control + COPD group; pGLVU6/GFP-shRNA Cpt1a +
COPD group; pGLVU6/GFP control + COPD group; and pGLVU6/GFP-Cpt1a +
COPD group.
Murine lung function testing
The lung function of mice was tested by using Buxco
Fine Pointe Series Whole Body Plethysmography (Buxco Research
Systems). The test indexes were as follows: Tidal volume (TV), peak
expiratory flow (PEF), 50% expiratory flow (EP50), forced
expiratory volume in 0.3 seconds (FEV0.3) and forced vital capacity
(FVC).
Detection of differences in primary
pulmonary microvascular endothelial cells (PMVECs) fatty acid
oxidation
PMVECs from different experimental groups were
serum-starved in 12-well plates, then washed with warm PBS. The
cells were incubated with 14C-labeled FAO (Fatty acid
oxidation (FAO) medium consisting of DMEM-low glucose (Invitrogen;
Thermo Fisher Scientific, Inc.), 0.25 µCi/ml (1-14C) palmitate,
0.25 µCi/ml (1-14C) oleate, 50 µM palmitate, 50 µM oleate, 0.5%
BSA, 1 mM carnitine and 12.5 mM HEPES (pH-7.4) at 37˚C for 3 h. The
procedures were thrice repeated for each group. After 3 h, the
culture medium was collected from each well and an equal portion of
the culture medium was distributed to a sealed trapping device.
14CO2 was removed from the medium fraction by
adding perchloric acid and captured in NaOH, which was collected
and analyzed by liquid scintillation counting to determine the
complete FAO ratio of CO2. The acidified medium was
collected, refrigerated and centrifuged 5 min at 16,000 x g and
4˚C. The acid soluble metabolite (ASM) of FAO was determined by
liquid scintillation counting analysis of the equal samples. Cells
were thrice rinsed with cold Hank's balanced salt solution (HBSS)
and lysed with SDS lysis buffer. The protein concentration of the
lysate was determined by the BCA assay. The results of FAO were
expressed as a percentage of CO2.
Determination of ceramide
The lung tissue homogenate sample from mice was
placed into the centrifuge tube, then the internal standard mix was
added. After vortex oscillation blending, the sample was
centrifuged (1,000 x g, 4-8˚C, 5 min) and the supernatant was
collected for later detection. Ceramide was analysed by LC-MS.
Agilent 6530 Q-TOF was used to identify and analyse levels of
ceramide, which was confirmed by comparing retention times and
tandem mass spectrometry data with standard compounds. The results
were corrected for naturally occurring 13C impurity of the tracers.
MassHunter Quantitative Analysis software (Agilent Technologies,
Inc.) was used to quantify the ceramide content.
Western blotting
Protein was extracted from RIPA lysate (Huaxingbio)
containing protease inhibitor (Thermo Fisher Scientific, Inc.). The
tissues or cells were ultrasonically disrupted, then centrifuged at
14,000 x g and 4˚C for 15 min. The supernatant was collected and
the protein concentration was determined using the BCA assay.
Proteins (30 µg) were separated by SDS-PAGE (10%) together with a
pre-stained protein ladder (Thermo Fisher Scientific, Inc.), then
transferred to nitrocellulose membranes (MilliporeSigma, blocked
with 5% non-fat milk in Tris-Buffered saline and Tween-20 (TBST; 20
mmol/l Tris-Cl, 150 mmol/l NaCl, 0.05% Tween 20, pH 7.4) at 4~8˚C
for 2 h and incubated overnight with primary antibody (cat. no.
#97361; 1:1,000, CST) at 4˚C. After being washed with buffer, the
membranes were incubated with secondary antibodies(Anti-rabbit IgG,
HRP-linked Antibody, cat. no. #7074, 1:1,000, CST, USA) at 4˚C for
40 min. The relative protein content was determined by Bio-Rad
laser imaging system (Bio-Rad Laboratories, Inc.) and Image Lab
software v6.1 (Bio-Rad Laboratories, Inc.) after the DyLight
800-labeled secondary antibody (1:10,000 dilution; KPL, Inc. ) was
incubated at room temperature for 1 h the next day(Visualization
reagent kit, Cat. No. P0020, Beyotime Institute of Biotechnology).
Primary antibody Cpt1a (cat. no. 97361, 1:1,000) secondary
Antibodies (Cat. No. #7074,1:1,000) and GAPDH (cat. no. 5174,
1:1,000) were purchased from Cell Signaling Technology, Inc.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA in tissues(cells number: 2x106)
was extracted using TRIzol® reagent (Thermo Fisher
Scientific, Inc.). The first-stand cDNA was synthesized using First
Strand cDNA Synthesis kit with gDNA Eraser according to the
manufacturer's protocol. PCR was performed using cycling
conditions: Denaturation 95˚C for 30 sec, annealing 60˚C for 40 sec
and extension at 72˚C for 60 sec; 35 cycles) (Takara, Osaka,
Japan). RT-qPCR was performed with SYBR Green Master Mix to examine
the relative mRNA levels of indicated genes with an AJ qTOWER 2.2
Real-Time PCR system (Analytik Jena AG) by using a quantitative
real-time PCR kit (Takara Bio, Inc.). Sequences for RT-qPCR primers
were: Mouse Cpt1a, 5'-CTCCGCCTGAGCCATGAAG-3', mouse GAPDH:
5'-AGGTCGGTGTGAACGGATTTG3'. GAPDH was used as an internal control.
Relative gene expression level was calculated by 2-ΔΔCq
method (10).
Haematoxylin and eosin staining
Lung samples were dissected and fixed at 18~25˚C in
4% paraformaldehyde solution (Sangon Biotech Co., Ltd.) for 72 h.
Tissues were embedded in paraffin (Sangon Biotech Co., Ltd.).
Sections of lung were cut at 3-4 µm and prepared for haematoxylin
and eosin staining by standard procedures. Samples were immersed in
xylene and alcohol, stained with hematoxylin for 5 min, stained
with eosin for 3 min and re-immersed in alcohol and xylene. The
sections were counterstained with Harris haematoxylin (Sangon
Biotech Co., Ltd.) and normal IgG (Merck Millipore Sigma Aldrich)
as a negative control. Images were captured with a brightfield DM4B
microscope (Leica Microsystems GmbH).
Enzyme-linked immunosorbent assay
(ELISA)
Lung samples were collected at enrolment and
immediately stored at -80˚C in a single biologic resource centre.
Lung tissues inflammatory factors (TGFβ, IL-6, IL-β and TNF-α) were
determined by commercial ELISA kits (cat. nos. 70-EK981-96,
70-EK206/3-96, 70-EK201B/3-96 and 70-EK282/4-96) purchased from
Multisciences (Lianke, Hangzhou, China) Biotech Co., Ltd.,
according to the manufacturer's instructions. The detection
threshold was 0.156 and 1.56 ng/ml. Samples, reagents and buffers
were prepared strictly in accordance with the manufacturer's
guideline.
Mouse PMVEC isolation
Primary PMVECs were isolated by following these
steps. C57BL/6J mice (8-10 weeks old, both male and female) were
anesthetized by abdominal injection with pentobarbitone sodium (100
mg/kg body weight). and lungs were removed. The lung samples were
enzymatically digested by a mouse lung dissociation kit (Miltenyi
Biotec GmbH). Following removal of CD45+ cells,
CD45-cells were collected, washed and incubated with
CD31-conjugated beads (Invitrogen; Thermo Fisher Scientific, Inc.).
CD31+ cells were enriched using a MACS column and
magnetic field by protocol of reagent kits. For magnetic
separation, MACS ART MS Columns were placed into a MiniMACS
Separator, rinsed once with 1 ml of MACS ART Binding Buffer
(discarded after flow-through), and the CD45- cells
suspension was then placed in the column. The PMVECs, which were
bound to CD31-conjugated magnetic microbeads, were then retained in
the column. That was because the column was placed in MiniMACS
Separator, which is basically a magnet forming magnetic field,
which causes the retention of magnetically labeled cells. This
CD31+ cells was then washed by adding 4 ml of medium and
centrifuged 10 min at 1,000 rpm in 4~8˚C. The freshly isolated
cells were considered as passage 0, which was cultured in dish
coated with human fibronectin (30 µg/ml). Cells within 5 passages
were used for experiments.
Haematoxylin and eosin and TUNEL
staining
Haematoxylin-eosin staining is a basic method of
histology and pathological examination as the haematoxylin produces
crisp, intense blue nuclei providing optimal contrast to the
eosin-stained cytoplasm. TUNEL staining detects the DNA breaks
formed when DNA fragmentation occurs in the last phase of
apoptosis. Lung samples were dissected and fixed at 18~25˚C in 4%
paraformaldehyde solution (Sangon Biotech Co., Ltd.) for 72 h.
Tissue was embedded in paraffin (Sangon Biotech Co., Ltd.).
Sections of lung were cut at ~3-4 µm and prepared for haematoxylin
and eosin staining by standard procedures. Then the tissue wax was
cut into serial 5 µm sections. After being reconstructed by
protease K, the sections were stained by haematoxylin and eosin.
The cell samples were collected, washed, fixed and then stained by
TUNEL and DAPI. Histological sections were observed in six randomly
selected fields for analysis) under a Flirorescent microscope
(Nikon Eclipse 80i; Nikon Corporation).
Flow cytometric assay for Annexin
v-positive cells
Flow cytometry was used to measure Annexin
V-positive cells that are considered as apoptotic cells by
Flowjo7.6.1 (Treestar Inc., Ashland, OR). Briefly, cells were
collected using the TrypLE (Thermo Fisher Scientific, Inc.). After
washed with cold phosphate buffered saline, Annexin V binding
buffer (300 µl) was added to resuspend cells. A total
1x105 cells were collected and incubated with 100 µl
Annexin V binding buffer along with 5 µl of fluorescein
isothiocyanate conjugated Annexin V (Thermo Fisher Scientific,
Inc.) and 5 µl of propidium iodide (Life Technology) for 20 min at
room temperature. Finally, Annexin V binding buffer (500 µl) was
added and mixed gently. FC-500 (Beckman Coulter, Inc.) was used to
detect the Annexin V-positive cells with a total of 20,000 events
analyzed. The apoptotic rate was calculated by percentage of early
and late apoptotic cells.
Statistical analysis
Experiments of cell cultivation were performed with
six biological replicates with a total of five times of technical
repetitions for measurements. Data are expressed as mean ± standard
error of the mean. One-way analysis of variance with the post
Tukey's test was used to determine whether there is any statistical
significance between the means of groups. The Student-Newman-Keuls
test and Pearson's correlation analysis were used to examine which
specific groups of means were statistically different. P<0.05
was considered to indicate a statistically significant
difference.
Results
Correlation between Cpt1a levels of
lung tissues and lung function in patients with COPD
To determine the correlation between Cpt1a
expression and lung function in patients with COPD, clinical data
were analysed and lung tissues from patients with COPD were
collected for the following tests. In this study, 30 samples were
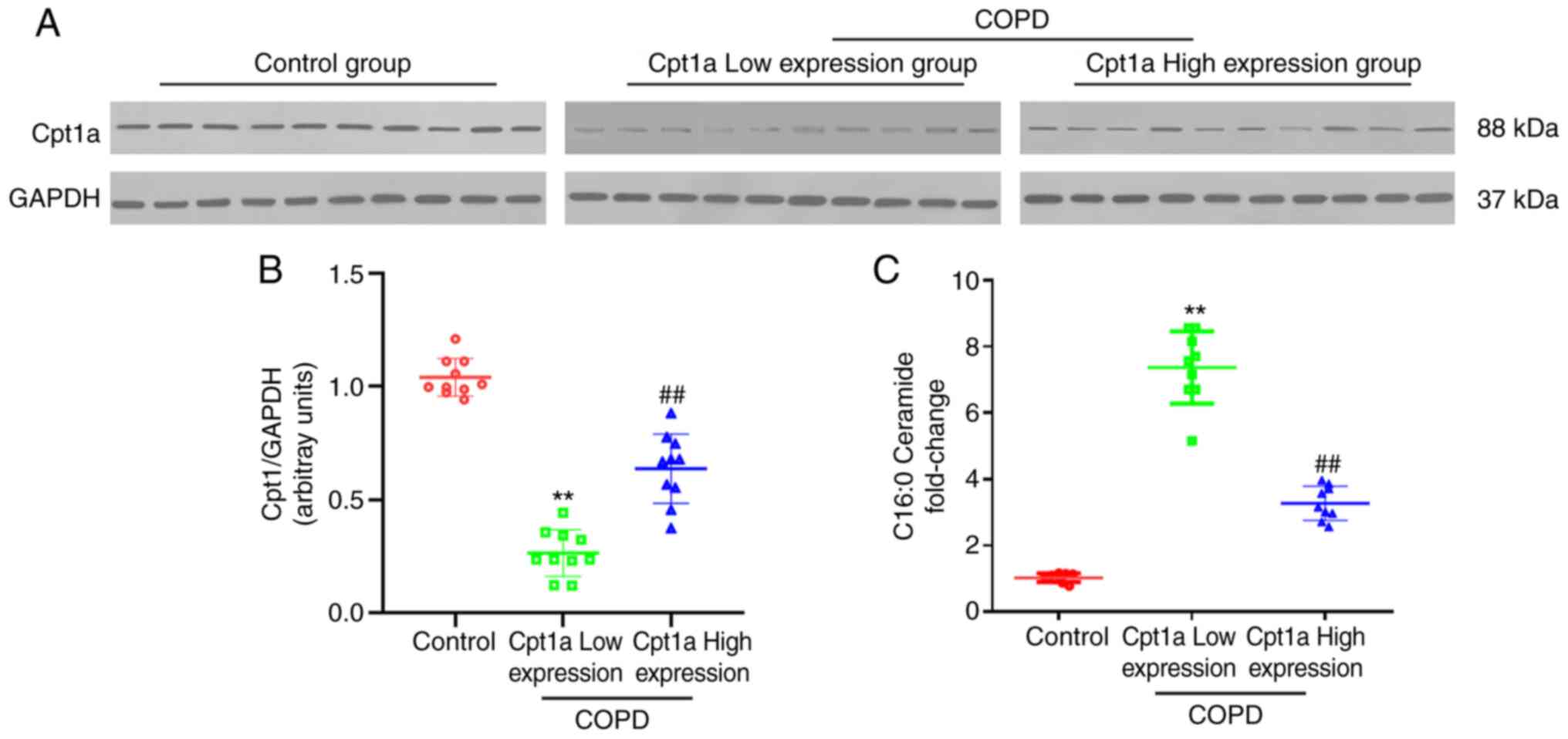
collected. Clinical baselines and data are presented in Table SI. The results of western blotting
showed that compared with the control group, the expression of
Cpt1a was decreased significantly in lung tissues from patients
with COPD. Significant differences in Cpt1a expression were
detected between patients with COPD which were then divided into
high and low Cpt1a expression groups (Fig. 1A and B). A comparison of lung function data in
patients with high and low expression of Cpt1a suggested
significant differences in lung function. The lung function indices
of patients with high expression of Cpt1a were clearly improved
compared with patients with low expression of Cpt1a (Table I). It was also shown that ceramide
in the high expression of Cpt1a COPD group was significantly lower
than the low expression of Cpt1a COPD group (Fig. 1C). The above findings suggested
that Cpt1a may be involved in protecting lung function in patients
with COPD.
 | Table IComparison of lung function Indices
between patients with COPD with differential expression of Cpt1a
and the control group. |
Table I
Comparison of lung function Indices
between patients with COPD with differential expression of Cpt1a
and the control group.
| Indicator | Control (n=10) | COPD (n=10) Cpt1a
low expression | COPD (n=10) Cpt1a
high expression | P-value |
|---|
| Tidal volume
(ml) | 2.73±0.32 |
1.47±0.25a |
1.87±0.31b | 0.001 |
| Peak expiratory
flow (ml/s) | 37.15±0.18 |
16.38±2.13a |
20.48±1.56b | 0.003 |
| 50% expiratory flow
(ml/s) | 1.79±0.20 |
1.35±0.22a |
1.56±1.19b | 0.021 |
| Forced expiratory
volume in 0.3 seconds (ml) | 4.42±0.33 |
2.33±0.32a |
3.43±0.12b | 0.013 |
| Forced expiratory
volume in 0.3 sec/forced vital capacity (%) | 87.61±4.41 |
64.49±4.30a |
74.73±5.30b | 0.001 |
Cpt1a is critically involved in
apoptotic and inflammatory responses in the lung tissues of
patients with COPD
Lung inflammation and apoptosis are the most basic
pathologic features of patients with COPD. Previous studies have
shown that Cpt1a is involved in apoptosis of pulmonary
microvascular endothelial cells (6,7,9), but
the correlation between Cpt1a expression and apoptosis and
inflammation of lung tissues of patients with COPD has not been
established. Based on the different levels of Cpt1a expression in
lung tissues of patients with COPD, patients were divided into high
and low Cpt1a expression groups and the following test results were
obtained: Haematoxylin and eosin staining showed that compared with
the low Cpt1a expression COPD, the high Cpt1a expression COPD group
had less pathologic damage under the microscope, with a smaller
amount of inflammatory cell infiltration and more complete lung
tissue morphology (Fig. 2A). TUNEL
staining showed significantly decreased apoptosis in the high Cpt1a
expression COPD group (Fig. 2B).
Additionally, compared with the low Cpt1a expression group, the
expression of inflammatory factors (TGF-β, TNF-α, IL-6 and IL-1β)
in lung tissues of the high Cpt1a expression COPD group was
decreased (Figs. 2C and S1). Thus, Cpt1a appeared to be involved
in the maintenance of lung morphology and exhibit anti-apoptosis
and anti-inflammation properties in patients with COPD.
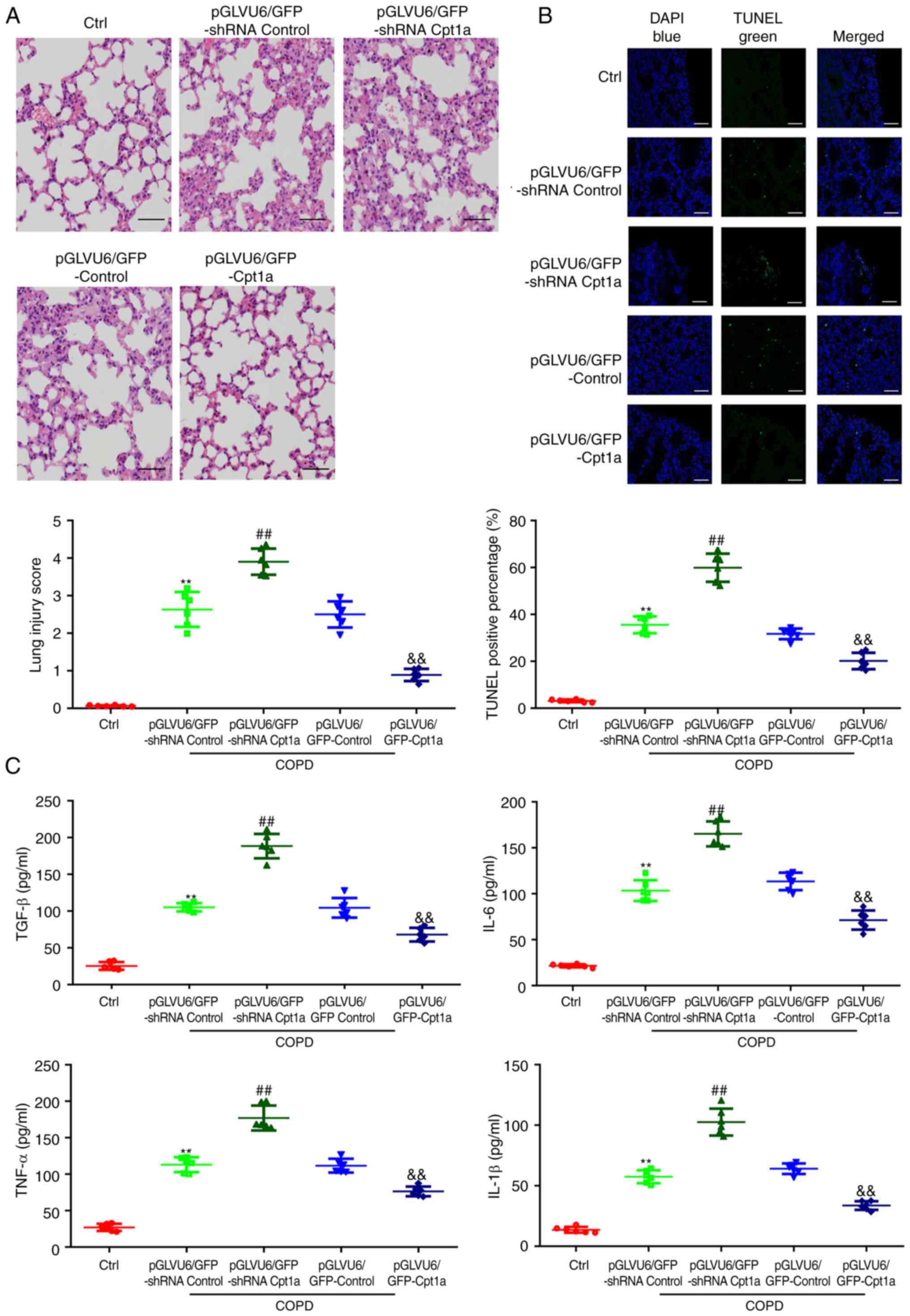
Cpt1a augmentation ameliorates the
COPD-induced apoptosis and inflammation of the lung tissues in COPD
mice
To identify the role of Cpt1a in patients with COPD,
lentivirus mediated Cpt1a knockout and overexpression models were
established and the following tests were carried out. Transfection
of pGLVU6/GFP shRNA Cpt1a and pGLVU6/GFP Cpt1a significantly
reduced or elevated the levels of Cpt1a protein and mRNA in lung
tissues, respectively (Fig. S2A
and B). Haematoxylin and eosin
staining showed that compared with COPD model mice, knockdown of
Cpt1a significantly aggravated the COPD-induced morphologic
disorder of lung tissues, resulted in inflammatory cell
infiltration, smooth muscle hyperplasia and partial rupture and
fusion of alveolar wall. However, COPD mice with overexpression of
Cpt1a had less pathologic damage and inflammatory cell infiltration
(Figs. 3A and S2A-D). TUNEL staining showed that Cpt1a
knockout aggravated lung cell apoptosis in COPD mice, while Cpt1a
overexpression showed the opposite result (Fig. 3B). In addition, in vivo
results also showed that overexpression of Cpt1a significantly
alleviated the lung inflammatory response (Fig. 3C) in COPD mice.
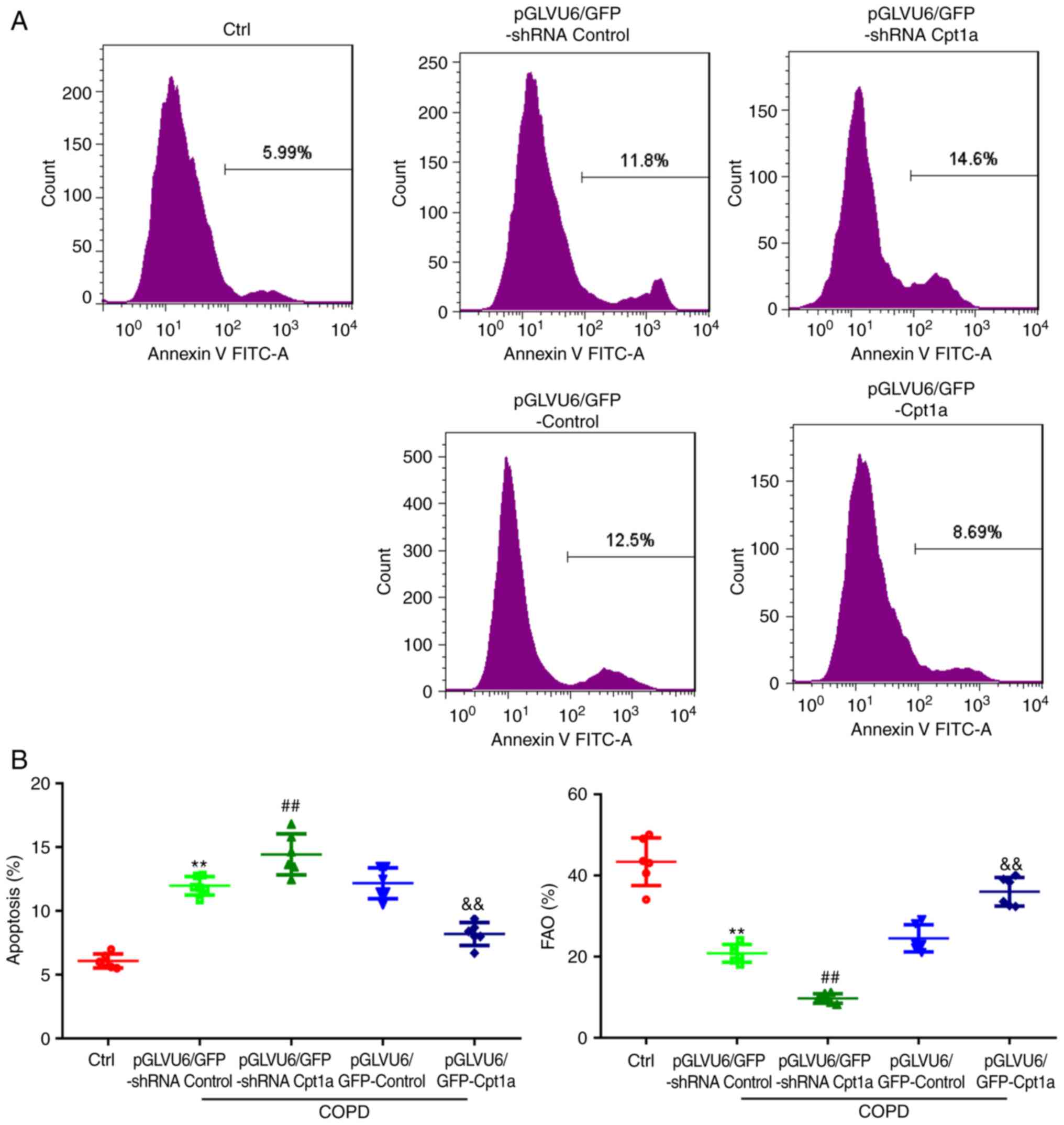
The results of previous studies in our laboratory
showed that Cpt1a is mainly expressed in pulmonary microvascular
endothelial cells and participates in anti-apoptosis. However,
whether overexpression of Cpt1a reduces apoptosis of PMVECs in COPD
mice is unknown. Therefore, lung microvascular endothelial cells
were isolated from COPD mice. Annexin V-PI flow analysis showed
that compared with COPD model mice, knockdown of Cpt1a
significantly aggravated the apoptosis of PMVECs induced by COPD,
while overexpression of Cpt1a alleviated the apoptosis induced by
COPD (Fig. 4A). It was also found
that Cpt1a significantly promoted fatty acid oxidation of primary
PMVECs in COPD mice (Fig. 4B).
Therefore, Cpt1a alleviated COPD-induced lung tissue disorders in
COPD mice.
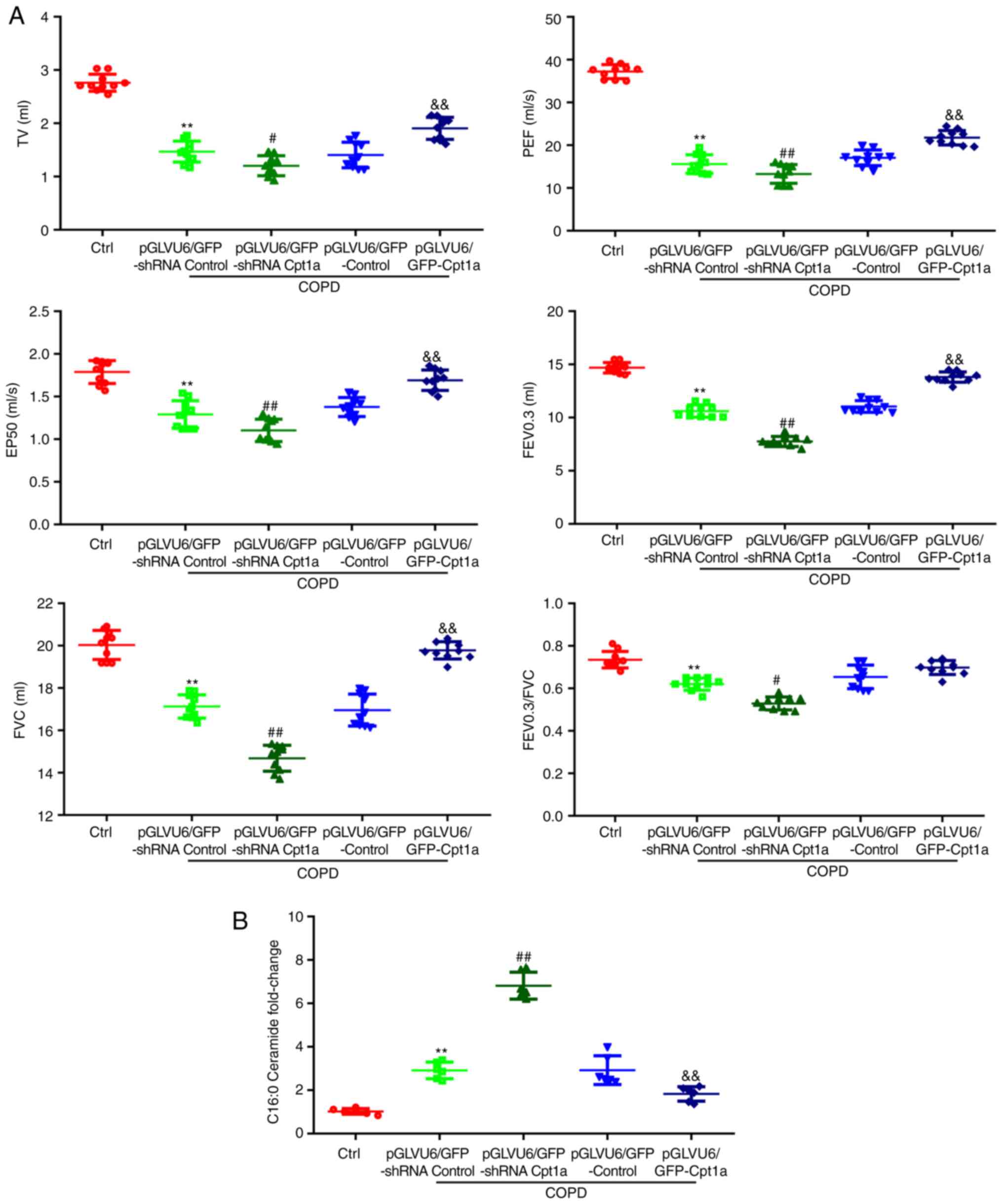
Cpt1a overexpression significantly
improves the lung function in COPD animals
The results of our previous experiments showed that
there is a correlation between the difference in Cpt1a expression
in clinical patients and the lung function. To identify whether
Cpt1a served a role in the treatment of patients with COPD, the
present study examined the lung function of the two murine models
in vivo. Knockdown of Cpt1a significantly decreased lung
function indices compared with COPD mice and overexpression of
Cpt1a improved the lung function indices of COPD mice (Fig. 5 and Table II). Expression of ceramide in lung
tissues of COPD mice with overexpression of Cpt1a was decreased,
which is consistent with the clinical results. These results
suggest that Cpt1a could induced lung function disorders in
patients with COPD.
| Table IIDetection of lung function indices of
model mice. |
Table II
Detection of lung function indices of
model mice.
| Group | Tidal volume
(ml) | Peak expiratory
flow (ml/s) | 50% expiratory flow
(ml/s) | Forced expiratory
volume in 0.3 sec (ml) | Forced vital
capacity (ml) | Forced expiratory
volume in 0.3 sec/forced vital capacity (%) |
|---|
| Ctrl | 2.76±0.16 | 37.21±1.66 | 1.79±0.13 | 14.69±0.49 | 20.03±0.69 | 0.73±0.04 |
| pGLVU6/GFP-shRNA
Control | 1.47±0.20 | 15.60±2.17 | 1.29±0.16 | 10.62±0.58 | 17.13±0.55 | 0.62±0.03 |
| pGLVU6/GFP-shRNA
Cpt1a | 1.20±0.19 | 13.28±2.19 | 1.10±0.13 | 7.75±0.48 | 14.68±0.61 | 0.53±0.03 |
|
pGLVU6/GFP-Control | 1.41±0.24 | 17.07±1.85 | 1.38±0.11 | 11.04±0.55 | 16.96±0.75 | 0.65±0.06 |
|
pGLVU6/GFP-Cpt1a | 1.91±0.21 | 21.76±1.68 | 1.69±0.12 | 13.80±0.48 | 19.78±0.41 | 0.70±0.03 |
| Pa | 0.001 | 0.0123 | 0.024 | 0.001 | 0.0342 | 0.0453 |
| Pb | 0.001 | 0.001 | 0.001 | 0.002 | 0.001 | 0.0234 |
Discussion
The prevalence of COPD has increased in recent
years, especially among young individuals. COPD has become one of
the commonest diseases affecting the health and quality of life of
individuals worldwide (2,11). COPD is characterized by apoptosis
and inflammation of pulmonary vascular endothelial cells. Increased
apoptosis and inflammation of lung tissues result in abnormal lung
function and aggravate the course of COPD (12). It has been reported that the
expression of Cpt1a is decreased in pulmonary microvascular
endothelial cell lines from patients with COPD (5), but whether Cpt1a participates in the
protection of lung function in patients with COPD is unclear. The
present study showed for the first time that the difference in
Cpt1a expression in lung tissues of patients with COPD is related
to lung function. The higher the expression of Cpt1a, the more
improved the lung function indices, the lower the apoptosis rate
and the lower the inflammatory response. Overexpression of Cpt1a
was shown to alleviate lung damage, inhibit apoptosis and the
inflammatory response of pulmonary microvascular endothelial cells
in patients with COPD and serve a role in the treatment of COPD by
promoting the rate of substrate fatty acid oxidation and inhibiting
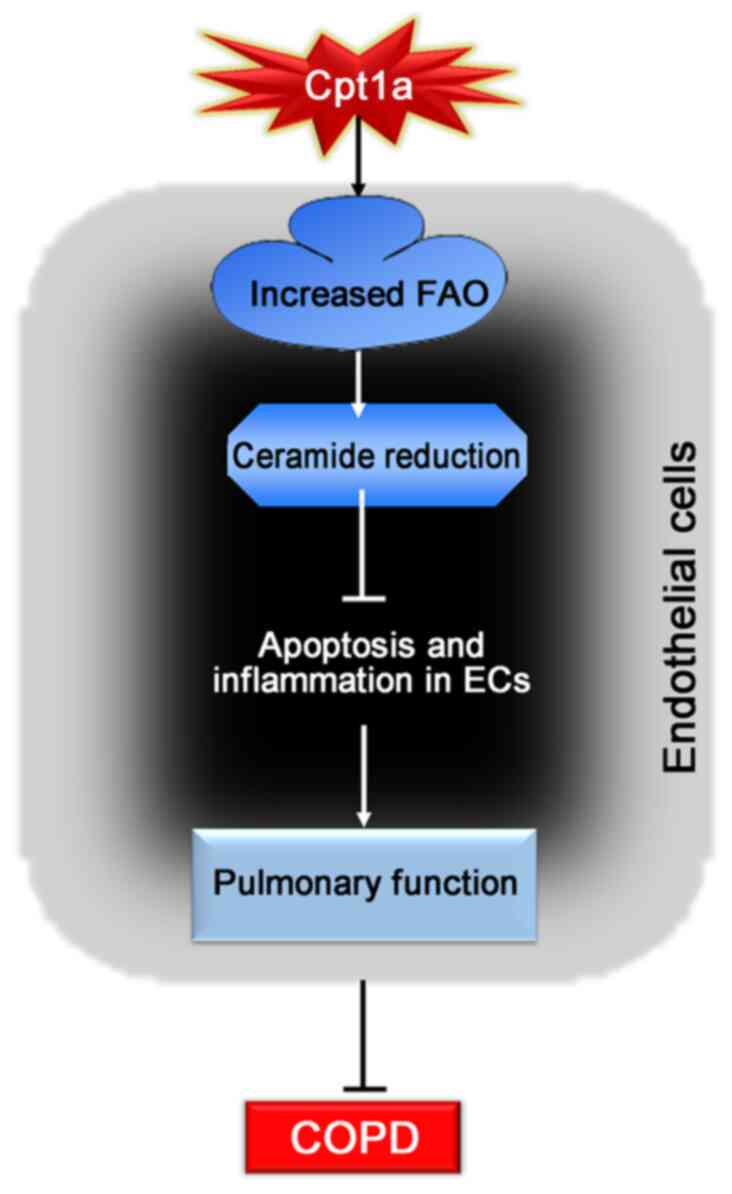
the production of ceramide (Fig.
6).
The CPT system consists of two separate proteins
located in the outer (Cpt1) and inner (Cpt2) mitochondrial
membranes (13). Clinical data
have shown that Cpt2 deficiency leads to sudden foetal death
(14). Cpt1 is the key molecule of
mitochondrial fatty acid oxidation, composing of Cpt1a, Cpt1b and
Cpt1c (15), which are expressed
primarily in the liver and lungs, skeletal muscles and brain,
respectively (14,16,17).
Our previous studies showed that Cpt1a is expressed in lung
endothelial cells, where Cpt1b and Cpt1c are minimally expressed,
which is consistent with previous reports (9,18).
Therefore, the present study examined the role of Cpt1a in lung
tissues of patients with COPD in detail. Basic studies have shown
that Cpt1a-mediated fatty acid oxidation is related to the invasion
and metastasis of colon cancer and promotes the occurrence and
development of cancer (19-21).
Cpt1a also inhibits the proliferation and migration of lung cancer
cells (20). In view of these
findings, available information on the importance of Cpt1a in some
tissues is limited and controversial. The role of Cpt1a in
pulmonary vascular endothelial cells is not fully understood and it
is unclear whether Cpt1a is involved in maintenance of pulmonary
homeostasis. The present study showed for the first time that Cpt1a
plays an irreplaceable role in regulating lung function. It found
differences in the expression of Cpt1a in the lung tissues of
clinical patients with COPD and found that lung function indices in
the group with high Cpt1a expression were improved compared with
the group with low Cpt1a expression. Consistent with the clinical
results, knockout and knockdown of Cpt1a in vivo showed that
Cpt1a reversed pulmonary dysfunction in COPD mice. Cpt1a could be
used as a clinical molecular target to improve lung dysfunction in
patients with COPD.
Tobacco smoke is deadly and has more than 7,000
chemicals, 69 of which are verified as carcinogens (21). Smoking is one important risk
factors for COPD and ~80% of patients with COPD were induced by
smoking (22). Toxic particles of
inhaled smoke induce airway inflammation that is exacerbated in
patients with COPD (23). Smoking
does great harm to human beings and is a social problem. Smoking
cessation has been confirmed by a large number of studies to be an
effective way to prevent COPD, delay airflow restriction and slow
down deterioration of lung function (24,25).
The current study created a cigarette smoke-induced COPD murine
model. Consistent with the previous research results, the present
study confirmed the harmful effects of cigarette smoke on lung
function through lung function testing. Cigarette smoke increased
inflammation and apoptosis in lung tissues, caused morphologic
changes of lung tissues and decreased the expression of Cpt1a, the
protective factor of lung endothelial cells.
Pulmonary vascular endothelial injury (apoptosis and
an inflammatory response) is a common pathologic manifestation in
the progression of COPD (7-9).
The balance between endothelial injury and anti-injury affects the
occurrence and development of COPD (26). Several studies have been conducted
to determine how to delay endothelial apoptosis and inhibit the
inflammatory response (7-9,26).
Our previous study suggests that Cpt1a is the key downstream
molecule of L-carnitine, which could inhibit apoptosis of pulmonary
vascular endothelial cells in COPD (9), but whether Cpt1a is involved in
anti-apoptosis and anti-inflammation of pulmonary vascular
endothelial cells in COPD remains to be elucidated. The results of
haematoxylin and eosin staining, TUNEL staining and ELISA revealed
that the level of Cpt1a expression in patients with COPD was
directly associated with infiltration of inflammatory cells,
apoptosis and the expression of lung tissue inflammatory factors.
Consistent with the clinical results, inflammatory cell
infiltration, apoptosis and inflammatory factor expression in lung
tissues of the COPD model with Cpt1a knockdown in vivo were
increased and overexpression of Cpt1a reversed this trend. In
addition, primary pulmonary microvascular endothelial cells were
isolated from model mice and the results showed that Cpt1a
inhibited apoptosis of pulmonary microvascular endothelial cells.
These results indicate that Cpt1a plays an important role in
pulmonary endothelial cells and subsequent function regulation.
Ceramide is an important lipid molecule that
regulates cell differentiation, proliferation, apoptosis, aging and
other life activities (27).
Studies have shown that ceramide could promote airway inflammation
and airway hyperresponsiveness and play an important role in the
pathogenesis of asthma (28), COPD
(29) and acute lung injury
(30). It has been reported that
ceramide promotes the expression of MMP-9 in lung epithelial cells
by activating the JAK2-STAT3 pathway, thus promoting the
development of airway remodelling in lung tissues (31). Upregulation of ceramide is involved
in apoptosis of lung epithelial cells (32). Intratracheal instillation of
ceramide in mice leads to apoptosis of alveolar epithelium and
endothelial cells, resulting in enlargement of air cavity (33). Inhibition of ceramide synthesis by
Fumonisin B1, an inhibitor of ceramide synthase, reduces apoptosis
of epithelial cells and airway inflammation (34). Nevertheless, it is unclear whether
Cpt1a is involved in the production of ceramide in lung tissues of
patients with COPD. Ceramide in lung tissues of patients with COPD
was detected by LC-MS, which revealed that ceramide production was
decreased in the lung tissues of patients with COPD with high
expression of Cpt1a, while the opposite trend was shown in the low
Cpt1a expression group. Moreover, the results of in vivo
animal model are consistent with the clinical results. These
results suggest that Cpt1a is involved in mediating ceramide
production in lung tissues and serves a crucial role in the
progression of COPD.
Fatty acid is an important component of blood lipids
and the main product of lipid digestion. Fatty acid oxidation plays
an essential role in regulating triglyceride metabolism (35). As the main site for fatty acid
oxidation, mitochondria provide the ester acyl CoA synthase needed
for fatty acid activation, then perform transmembrane transfer,
fatty acid β oxidation and acetyl CoA thorough oxidation, thus
providing energy (36). The
functional state of fatty acid oxidation is closely related to the
occurrence and development of a number of pathologic processes,
such as lipid accumulation, lipid peroxidation damage and insulin
resistance (36). Normally, fatty
acids are transported into mitochondria by Cpt1 on the
mitochondrial membrane, then undergo the process of fatty acid
β-oxidation. The remaining fatty acids can be oxidized under the
action of peroxisomes and microsomes to produce reactive oxygen
species (37). Abundant clinical
data indicate abnormal mitochondrial fatty acid oxidation of lung
endothelial cells in patients with COPD. Abnormal expression of
Cpt1 leads to abnormal fatty acid oxidation, thus disrupting liver
function (38). Our previous
results showed that L-carnitine treatment promotes the expression
of Cpt1a and oxidation of fatty acids in endothelial cells and
alleviates the apoptosis of endothelial cells induced by cigarette
smoke (9). In the current study,
primary pulmonary microvascular endothelial cells from a murine
model were isolated to measure the fatty acid acidification rate.
It was found that overexpression of Cpt1a promoted fatty acid
oxidation in pulmonary microvascular endothelial cells of the COPD
murine model, while the knockdown of Cpt1a led to the opposite
results.
In conclusion, the present study showed for the
first time that overexpression of Cpt1a could alleviate lung
dysfunction and reduce inflammatory response and apoptosis of lung
tissues in COPD mice and protect cigarette-induced COPD by
promoting the oxidation rate of substrate fatty acids, thus
inhibiting the production of ceramide to suppress apoptosis of
endothelial cells and inflammatory responses. The data suggested
that Cpt1a may be a potential new target for the treatment of
patients with COPD.
Supplementary Material
Identification of mice model. (A) The
expression of Cpt1a in lung tissues of mice was detected by western
blotting. (B) The mRNA level of Cpt1a in lung tissues of mice was
detected by reverse transcription-quantitative PCR. n=6 per group.
**P<0.01 vs. Control; ##P<0.01 vs.
pGLVU/GFP-shRNA control; &&P<0.01 vs.
pGLVU/GFP control. Cpt1a, carnitine palmitoyltransferase 1A; COPD,
chronic obstructive pulmonary disease.
Typical standard curve for
inflammatory factors, mouse ELISA. Typical standard curve for (A)
TGF-β, (B) IL-6, (C) TNF-α and (D) IL-1β. OD, optical density.
COPD Clinical baseline characteristics
of patients and controls.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Shanxi Province Key
R&D Program (International Science and Technology Cooperation)
Project (approval no. 201903D421058); Special Fund Project for the
Central Government to Guide Local Science and Technology
Development (approval no. YDZX20191400004736); Fund program for the
Scientific Activities of Selected Returned Overseas Professionals
in Shanxi Province (approval no. R02201); Fund program for the
Scientific Activities of Selected Returned Overseas Professionals
in Shanxi Province (approval no. 20210026), The Education
Department of Shanxi Province Foundation for Graduate Education
Innovation (approval no. 2018BY068) and Health Commission of Shanxi
Province Foundation for Higher Education Technology Innovation
(approval no. 2017050).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HZ and LL performed the conception and design of the
study, and drafted and revised the manuscript. LL, YZ and JG
analyzed data and prepared figures. LL, GY, SZ and DR performed the
experiments and manuscript review. HZ, GY and LL confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
the Second Hospital of Shanxi Medical University (CMTT number
2013012). All patients were informed in detail about the objective
and methods of this study and signed a consent form. All procedures
of the present study, including animals and patients, were approved
by the Ethics Committee of the Second Hospital of Shanxi Medical
University (CMTT number: 2013012) and in accordance with
international standards.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lareau SC, Fahy B, Meek P and Wang A:
Chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care
Med. 199:P1–P2. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mannino DM and Buist AS: Global burden of
COPD: Risk factors, prevalence, and future trends. Lancet.
370:765–773. 2007.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Rao W, Wang S, Duleba M, Niroula S, Goller
K, Xie J, Mahalingam R, Neupane R, Liew AA, Vincent M, et al:
Regenerative metaplastic clones in COPD lung drive inflammation and
fibrosis. Cell. 181:848–864. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hisata S, Racanelli AC, Kermani P,
Schreiner R, Houghton S, Palikuqi B, Kunar B, Zhou A, McConn K,
Capili A, et al: Reversal of emphysema by restoration of pulmonary
endothelial cells. J Exp Med. 218(e20200938)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Raud B, Roy DG, Divakaruni AS, Tarasenko
TN, Franke R, Ma EH, Samborska B, Hsieh WY, Wong AH, Stüve P, et
al: Etomoxir actions on regulatory and memory T cells are
independent of Cpt1a-mediated fatty acid oxidation. Cell Metab.
28:504–515. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yao H, Gong J, Peterson AL, Lu X, Zhang P
and Dennery PA: Fatty acid oxidation protects against
hyperoxia-induced endothelial cell apoptosis and lung injury in
neonatal mice. Am J Respir Cell Mol Biol. 60:667–677.
2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Gong J, Zhao H, Liu T, Li L, Cheng E, Zhi
S, Kong L, Yao W and Li J: Cigarette smoke reduces fatty acid
catabolism, leading to apoptosis in lung endothelial cells:
Implication for pathogenesis of COPD. Front Pharmacol.
10(941)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Schoors S, Bruning U, Missiaen R, Queiroz
KC, Borgers G, Elia I, Zecchin A, Cantelmo AR, Christen S, Goveia
J, et al: Fatty acid carbon is essential for dNTP synthesis in
endothelial cells. Nature. 520:192–197. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yao RQ, Ren C, Xia ZF and Yao YM:
Organelle-specific autophagy in inflammatory diseases: A potential
therapeutic target underlying the quality control of multiple
organelles. Autophagy. 17:385–401. 2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Çolak Y, Afzal S, Nordestgaard BG, Lange P
and Vestbo J: Importance of early COPD in young adults for
development of clinical COPD: Findings from the copenhagen general
population study. Am J Respir Crit Care Med. 203:1245–1256.
2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wong J, Magun BE and Wood LJ: Lung
inflammation caused by inhaled toxicants: A review. Int J Chron
Obstruct Pulmon Dis. 11:1391–401. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Li F, Jiang T, Li Q and Ling X:
Camptothecin (CPT) and its derivatives are known to target
topoisomerase I (Top1) as their mechanism of action: Did we miss
something in CPT analogue molecular targets for treating human
disease such as cancer? Am J Cancer Res. 7:2350–2394.
2017.PubMed/NCBI
|
|
14
|
Bonnefont JP, Djouadi F, Prip-Buus C,
Gobin S, Munnich A and Bastin J: Carnitine palmitoyltransferases 1
and 2: Biochemical, molecular and medical aspects. Mol Aspects Med.
25:495–520. 2004.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Casals N, Zammit V, Herrero L, Fadó R,
Rodríguez-Rodríguez R and Serra D: Carnitine palmitoyltransferase
1C: From cognition to cancer. Prog Lipid Res. 61:134–148.
2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wolfgang MJ, Kurama T, Dai Y, Suwa A,
Asaumi M, Matsumoto SI, Cha SH, Shimokawa T and Lane MD: The
brain-specific carnitine palmitoyltransferase-1c regulates energy
homeostasis. Proc Natl Acad Sci USA. 103:7282–7287. 2006.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Gao XF, Chen W, Kong XP, Xu AM, Wang ZG,
Sweeney G and Wu D: Enhanced susceptibility of Cpt1c knockout mice
to glucose intolerance induced by a high-fat diet involves elevated
hepatic gluconeogenesis and decreased skeletal muscle glucose
uptake. Diabetologia. 52:912–920. 2009.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Jiang Z, Knudsen NH, Wang G, Qiu W, Naing
ZZC, Bai Y, Ai X, Lee CH and Zhou X: Genetic control of fatty acid
β-oxidation in chronic obstructive pulmonary disease. Am J Respir
Cell Mol Biol. 56:738–748. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Xiong X, Wen YA, Fairchild R, Zaytseva YY,
Weiss HL, Evers BM and Gao T: Upregulation of CPT1A is essential
for the tumor-promoting effect of adipocytes in colon cancer. Cell
Death Dis. 11(736)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tan Z, Xiao L, Tang M, Bai F, Li J, Li L,
Shi F, Li N, Li Y, Du Q, et al: Targeting CPT1A-mediated fatty acid
oxidation sensitizes nasopharyngeal carcinoma to radiation therapy.
Theranostics. 8:2329–2347. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hecht SS: Tobacco carcinogens, their
biomarkers and tobacco-induced cancer. Nat Rev Cancer. 3:733–744.
2003.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Rabe KF and Watz H: Chronic obstructive
pulmonary disease. Lancet. 389:1931–1940. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hogg JC and Timens W: The pathology of
chronic obstructive pulmonary disease. Ann Rev Pathol. 4:435–459.
2009.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Brandsma CA, Van den Berge M, Hackett TL,
Brusselle G and Timens W: Recent advances in chronic obstructive
pulmonary disease pathogenesis: From disease mechanisms to
precision medicine. J Pathol. 250:624–635. 2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Taylor JD: COPD and the response of the
lung to tobacco smoke exposure. Pulm Pharmacol Ther. 23:376–383.
2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Schupp JC, Adams TS, Cosme C Jr, Raredon
MSB, Yuan Y, Omote N, Poli S, Chioccioli M, Rose KA, Manning EP, et
al: Integrated single-cell atlas of endothelial cells of the human
lung. Circulation. 144:286–302. 2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Castro BM, Prieto M and Silva LC:
Ceramide: A simple sphingolipid with unique biophysical properties.
Prog Lipid Res. 54:53–67. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Choi Y, Kim M, Kim SJ, Yoo HJ, Kim SH and
Park HS: Metabolic shift favoring C18:0 ceramide accumulation in
obese asthma. Allergy. 75:2858–2866. 2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ekroos K, Lavrynenko O, Titz B, Pater C,
Hoeng J and Ivanov NV: Lipid-based biomarkers for CVD, COPD, and
aging-A translational perspective. Prog Lipid Res.
78(101030)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
McVey MJ, Weidenfeld S, Maishan M, Spring
C, Kim M, Tabuchi A, Srbely V, Takabe-French A, Simmons S, Arenz C,
et al: Platelet extracellular vesicles mediate transfusion-related
acute lung injury by imbalancing the sphingolipid rheostat. Blood.
137:690–701. 2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tsurumaki H, Katano H, Sato K, Imai R,
Niino S, Hirabayashi Y and Ichikawa S: WP1066, a small molecule
inhibitor of the JAK/STAT3 pathway, inhibits ceramide
glucosyltransferase activity. Biochem Biophys Res Commun.
491:265–270. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kurinna SM, Tsao CC, Nica AF, Jiffar T and
Ruvolo PP: Ceramide promotes apoptosis in lung cancer-derived A549
cells by a mechanism involving c-Jun NH2-terminal kinase. Cancer
Res. 64:7852–7856. 2004.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yeganeh B, Lee J, Bilodeau C, Lok I,
Ermini L, Ackerley C, Caniggia I, Tibboel J, Kroon A and Post M:
Acid sphingomyelinase inhibition attenuates cell death in
mechanically ventilated newborn rat lung. Am J Respir Crit Care
Med. 199:760–772. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Kim SH, Singh MP, Sharma C and Kang SC:
Fumonisin B1 actuates oxidative stress-associated colonic damage
via apoptosis and autophagy activation in murine model. J Biochem
Mol Toxicol. 22(e22161)2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Rosenthal MD: Fatty acid metabolism of
isolated mammalian cells. Prog Lipid Res. 26:87–124.
1987.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Houten SM, Violante S, Ventura FV and
Wanders RJA: The biochemistry and physiology of mitochondrial fatty
acid β-oxidation and its genetic disorders. Ann Rev Physiol.
78:23–44. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Han J, Qu H, Han M, Ding Y, Xie M, Hu J,
Chen Y and Dong H: MSC-induced lncRNA AGAP2-AS1 promotes stemness
and trastuzumab resistance through regulating CPT1 expression and
fatty acid oxidation in breast cancer. Oncogene. 40:833–847.
2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Dai J, Liang K, Zhao S, Jia W, Liu Y, Wu
H, Lv J, Cao C, Chen T, Zhuang S, et al: Chemoproteomics reveals
baicalin activates hepatic CPT1 to ameliorate diet-induced obesity
and hepatic steatosis. Proc Natl Acad Sci USA. 115:E5896–E5905.
2018.PubMed/NCBI View Article : Google Scholar
|