Introduction
Myoclonic epilepsy with ragged red fibers (MERRF)
syndrome is a type of mitochondrial encephalomyopathy characterized
by progressive myoclonic epilepsy, ataxia and RRFs, mostly caused
by the 8344A>G mitochondrial DNA mutation. Myoclonic epilepsy is
only present in 20% of patients and can be either continuous or
intermittent, which is usually evoked and aggravated by light and
other activities. It was reported that most of the patients with
generalized tonic-clonic seizures, and some with atonic and absence
seizures and focal seizures (1).
Generalized epilepsy might represent a putative negative prognostic
factor as it was very common to all deceased patients (2). In addition, ataxia, eyelid ptosis,
hearing loss, myopathic signs and symptoms, cognitive impairment,
neuropathy, exercise intolerance and multiple lipomatosis are also
common clinical features (2).
Most patients with MERRF syndrome (80%) have a
positive family history but phenotypes among family members vary
significantly, which might partially be explained by the difference
in the heteroplasmy of mutated mtDNA (2). MERRF syndrome in adolescents may
progress very rapidly, with a fatal outcome, but in early adulthood
or childhood, it often progresses slowly. The mean age of onset was
reported to be 35 years old approximately (2), and the eldest age of death was
reported to be 79 years old and the youngest age of death was 7
years old (3).
The 8344A>G mutation in the mitochondrially
encoded tRNA lysine gene has been found in ~80% of patients with
MERRF syndrome (4), and the
prevalence of this point mutation in the adult population varies
from 0.39:100,000 to 1.5:100,000 (5,6). The
3243A>G mutation in the mitochondrially encoded tRNA-Leu (UUA/G)
1 (MT-TL1) gene is even more prevalent (1). However, to the best of our knowledge,
there is no report of an association between mitochondrial
3302A>G mutation in the MT-TL1 gene and MERRF syndrome so far.
The present report describes a case of MERRF syndrome with the
3302A>G mutation.
Case report
A 37-year-old woman presented to the Jiangxi
Provincial People's Hospital (Nanchang, China) in December 2018
complaining of tiredness after doing relatively little housework
and weight loss after the birth of a second child at the age of 25
years, since then the symptoms had slowly worsened. Starting at the
age of 32 years, the patient had felt proximal limb weakness, noted
an unstable gait and occasionally fell. In the 2 weeks before
admission to the hospital, paroxysmal jitters had repeatedly (1-5
times/day; ~1 min each time) occurred in the limbs and trunk, with
occasional loss of consciousness.
The patient's mother had a history of fatigue and
weight loss and died of a lung infection. The patient's son (15
years old) and daughter (13 years old) reported no symptoms, but
whole-exome sequencing with next-generation sequencing (NGS) method
on the Illumina HiSeq 3000 Sequencing Systems in the Laboratory of
the Wuhan Kindstar Diagnostics Co., Ltd. with the use of Clinvar
(accession number: VCV000689871.2) and the Revised Cambridge
Reference Sequence (GenBank accession number: NC_012920.1) in the
Mitomap databases for the validated NGS assay (7-9)
showed the presence of a mitochondrial 3302A>G mutation. HiSeq
3000 Sequencing Systems in the Laboratory of the Wuhan Kindstar
Diagnostics Co., Ltd. with the use of Clinvar (accession number:
VCV000689871.2) and the Revised Cambridge Reference Sequence
(GenBank accession number: NC_012920.1) in the Mitomap databases
for the validated NGS assay (7-9)
showed the presence of a 3302A>G mutation.
The patient was short (height, 150cm) and slender
(body weight, 28.5kg) with normal limb muscle tone. Neurological
examination showed decreased deep tendon reflexes, a positive
Romberg test and positive finger-to-nose and heel-knee-shin tests.
Assessment of the cranial nerves was unremarkable. Biochemical
tests showed increased serum lactate dehydrogenase (305 IU/l;
normal range: 114-240 IU/l), normal level of creatine kinase and
increased serum lactate (3.34 mmol/l; normal range: 0.5 to 2.2
mmol/l) and cerebrospinal fluid (CSF) lactate (2.55 mmol/l; normal
range: 0-2.0 mmol/l). Myopathy-related electromyographic activities
in bilateral quadriceps, right deltoid and sternocleidomastoid
muscles, and right thoracic paraspinal muscles at T10 and T11
levels were found.
For comparison purposes, corresponding magnetic
resonance imaging (MRI)/magnetic resonance spectroscopy (MRS) and
histopathological pictures from a ‘healthy’ control subject are
also shown (Fig. 1). The control
subject was a 47-year-old woman who was admitted to the Jiangxi
Provincial People's Hospital owing to repeated convulsions of all
four limbs for 3 months with aggravation and limb weakness for 20
days. The control subject started to have limb convulsions without
any stimulation 3 months before admission, which happened 3
times/week for 10 min each time and with no symptoms between
episodes. In the 20 days before admission, the convulsion attack
frequency increased to 1-2 times/day, with an attack duration of
10-20 min. Following admission, a series of tests were performed on
the control subject and normal electroencephalographic
examinations, hypocalcemia (blood calcium, 1.71 mmol/l; normal
range: 2.0-2.6 mmol/l), hyperphosphatemia (blood phosphate, 2.16
mmol/l; normal range: 0.73-1.35 mmol/l) and a low blood parathyroid
hormone level (12.34 pg/l; normal range: 15-65 pg/l), were
recorded. A diagnosis of hypoparathyroidism was considered. Head
MRI examinations were performed to determine if the
hypoparathyroidism was caused by pituitary lesions, but normal
findings were shown. In addition, the blood creatine kinase level
was found to be elevated (creatine kinase, 860 IU/l; normal range,
40-200 IU/l) and an electromyographic examination of the limbs
suggested suspicious myogenic myopathy. However, no abnormality was
found in the histopathological examination of the right quadriceps
femoris muscle. Therefore, hypoparathyroidism myopathy was excluded
from the diagnosis and the increased blood creatine kinase in the
patient was considered as a result of limb convulsions.
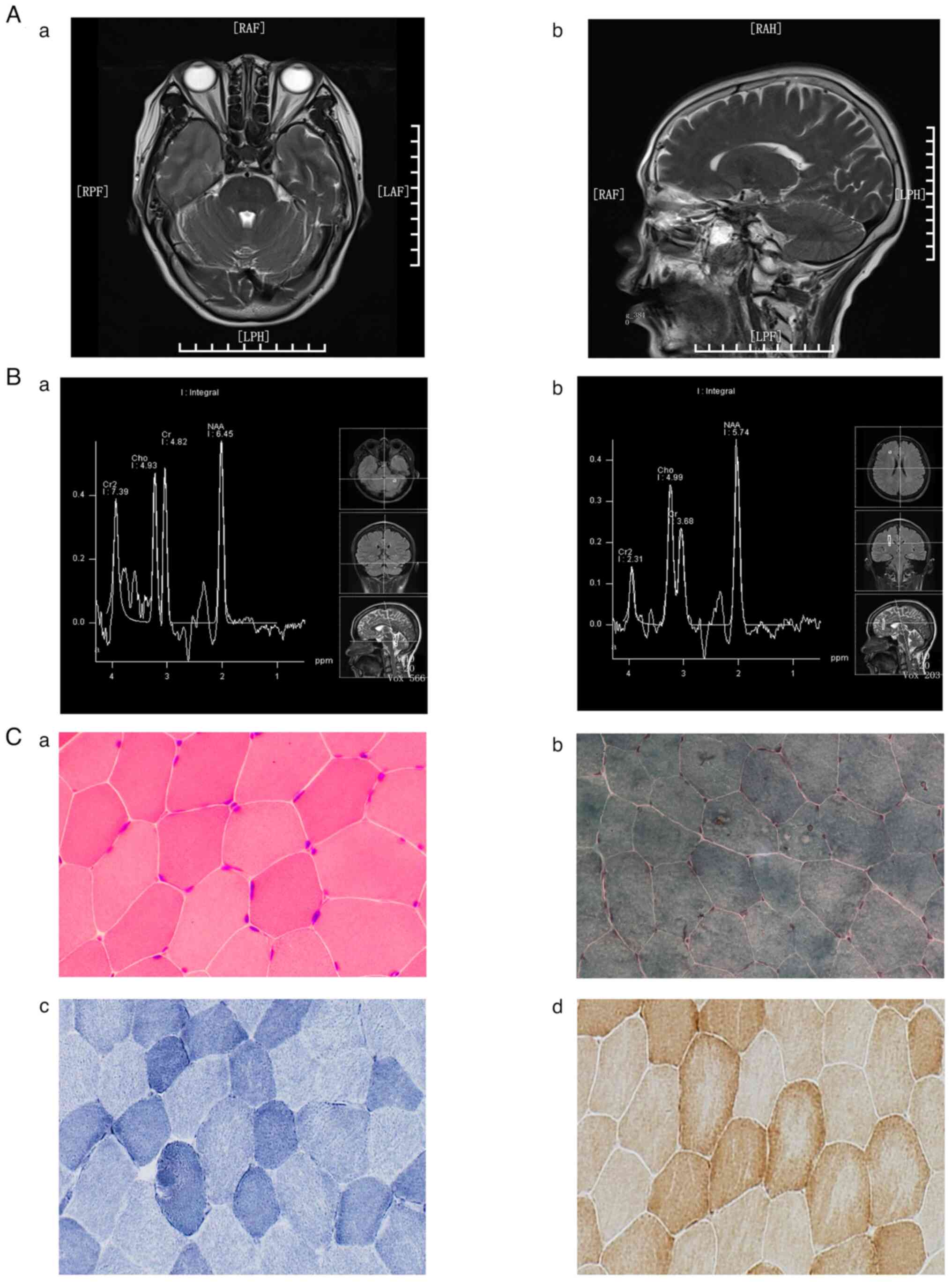 | Figure 1MRI, MRS and histopathological
examinations in the control subject. (Aa) Transverse and (Ab)
sagittal views of the head MRI T2-weighted image. MRS of (Ba) the
left cerebellum and (Bb) the anterior horn of the right lateral
ventricle. Histopathological examinations through (Ca) H&E,
(Cb) MGT, (Cc) SDH and (Cd) COX staining. Normal MRI and MRS
results were recorded. In addition, no ragged red fibers were found
on H&E and MGT staining, and no muscle fibers with excessive
SDH or absent COX staining were found. (Ca and Cb, x400
magnification; Cc and Cd, x200 magnification). MRI, magnetic
resonance imaging; MRS, magnetic resonance spectroscopy; MGT,
modified Gomori trichrome; SDH, succinate dehydrogenase; COX,
cytochrome c oxidase; RAF, Right anterior foot; LAF, Left
anterior foot; RPF, Right posterior foot; LPH, Left posterior head;
Cr2, creatine 2; Cho, Choline; Cr, Creatine; NAA,
N-acetylaspartate. |
For the aforementioned histopathological
examination, a 0.5x0.5x1-cm piece of muscle was taken from the
right quadriceps femoris (15 cm above the knee) of the patient and
the control subject with informed consent; the muscle tissue was
frozen with isopentane pre-chilled in liquid nitrogen and attached
on the cryostat chuck. The tissue block was allowed to equilibrate
to the cryostat temperature (-25˚C) before cutting 7-µm sections
from it and placing them onto slides. The sections of skeletal
muscle tissue were processed and stained according to standard
protocols with hematoxylin and eosin (H&E), modified Gomori
trichrome (MGT), succinate dehydrogenase (SDH) and cytochrome
c oxidase (COX), respectively, as previously published
(10). Briefly, the sections were
stained at room temperature with Harris' hematoxylin for 3 min and
with 1% eosin for ≥20 sec after rinsing in running distilled water
for H&E staining, or with Harris' hematoxylin for 5 min and
with Gomori trichrome mixture for 10 min (until green) after
rinsing in distilled water for MGT staining. In addition, the
sections were incubated flat in a damp atmosphere at 37˚C for 90
min for SDH staining or in a cytochrome oxidase incubating medium
at 37˚C for 3 h for COX staining. All chemical reagents were
purchased from Sinopharm Chemical Regent Co., Ltd. Compared with
the histopathological findings in the control subject (Fig. 1), the histopathological examination
of the skeletal muscle from the patient demonstrated RRFs
containing aggregates of abnormal mitochondria that appeared
subsarcolemmally and scattered through some muscle fibers as
inhomogeneous purple-red granular areas in H&E staining, and
abnormal aggregates of mitochondria appeared as reddish blotches,
mostly at the periphery of muscle fibers in MGT staining (Fig. 2). In addition, muscle fibers with
excessive SDH staining and negative COX staining were found in the
patient (Fig. 2), but not in the
control subject (Fig. 1).
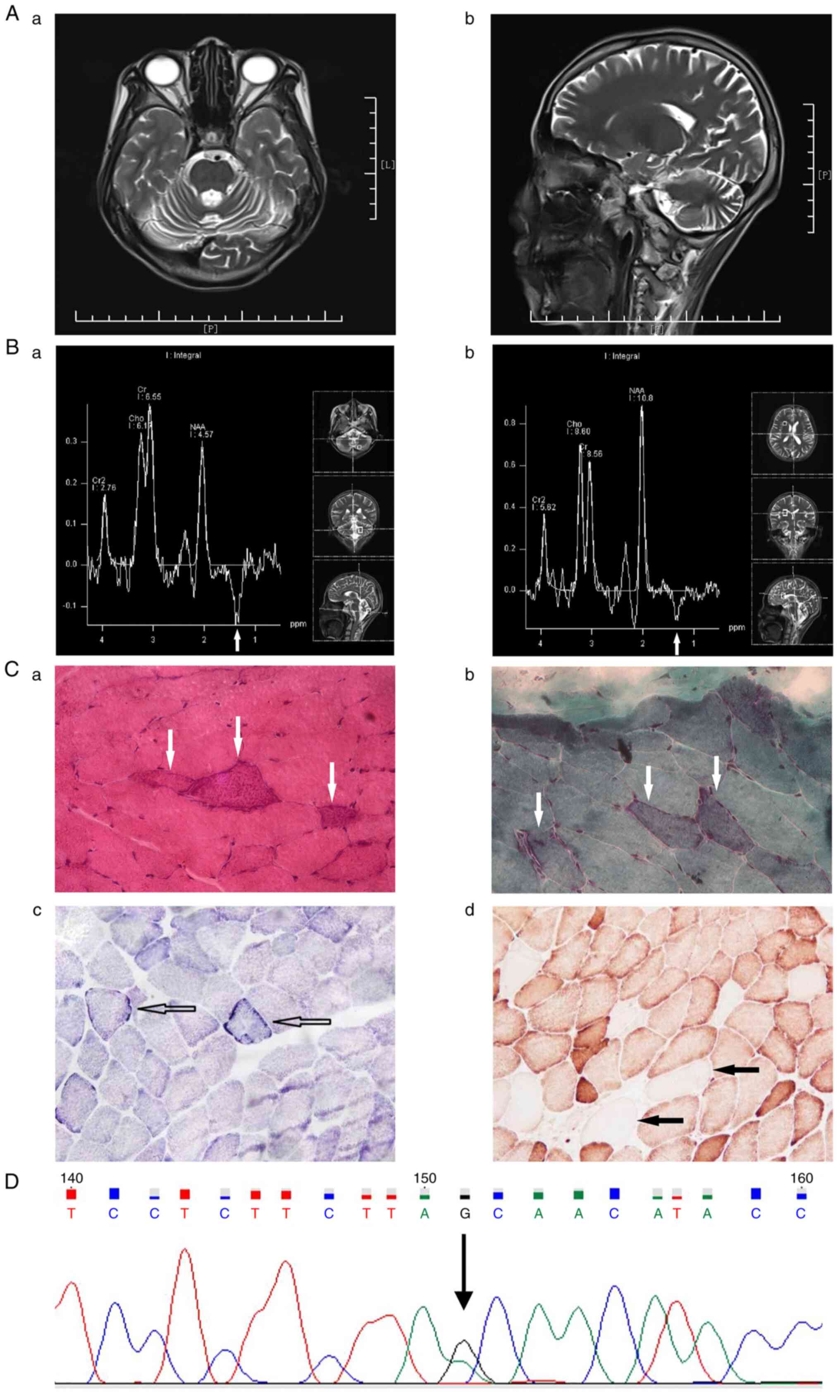 | Figure 2MRI imaging, MRS and histopathological
studies in the patient. (Aa) Transverse and (Ab) sagittal views of
the head MRI T2-weighted image. MRS of (Ba) the left cerebellum and
(Bb) the anterior horn of the right lateral ventricle showed
inversed lactate peak. (Ca) H&E staining showing ‘ragged red
fibers’, as indicated by white arrows. (Cb) MGT staining showing
abnormal aggregates of mitochondria appearing as a reddish blotch,
as indicated by white arrows. (Cc) SDH staining showing muscle
fibers with excessive SDH, as indicated by empty arrows. (Cd). COX
staining showing muscle fibers with absent COX staining, as
indicated by solid arrows. (D) Mitochondrial 3302A>G mutation in
the MT-TL1 gene. (Ca and Cb, x400 magnification; Cc and Cd, x200
magnification). MRI, magnetic resonance imaging; MRS, magnetic
resonance spectroscopy; MGT, modified Gomori trichrome; SDH,
succinate dehydrogenase; COX, cytochrome c oxidase; RAF,
Right anterior foot; LAF, Left anterior foot; RPF, Right posterior
foot; LPH, left posterior head; Cr2, creatine 2; Cho, Choline; Cr,
Creatine; NAA, N-acetylaspartate. |
Compared with the results of the head MRI in the
control, the cerebellar sulci of the patient were significantly
widened and the fourth ventricle was significantly enlarged
(Fig. 2). At the same time, the
inverted lactate peaks could be seen at 1.33 ppm in the MRS of the
patient (Fig. 2), but not in the
MRS of the control (Fig. 1); the
inverted lactate peak in the left cerebellum was larger and deeper
than that at the anterior horn of the right lateral ventricle in
the patient (Fig. 2). In addition,
compared with that in the control subject, the left cerebellum, but
not the anterior horn of the right lateral ventricle, was atrophied
in the patient, and the anterior horn of the right lateral
ventricle had no lesions in the patient (Fig. 2). Moreover, a mitochondrial
3302A>G mutation in the MT-TL1 gene was identified in the blood
sample from the patient (Fig. 2)
and multiple electroencephalographic spike waves were found. Chest
computed tomography showed bilateral bronchiectasis and pulmonary
infection, as well as atelectasis in the right middle lobe.
Therefore, the patient was diagnosed with MERRF syndrome with the
3302A>G mutation.
The patient was treated with antiepileptic drug
levetiracetam (500 mg, bid), idebenone (30 mg, tid), coenzyme Q10
(10 mg, tid), vitamin B2 (20 mg, tid), and levocarnitine (1 g,
tid), and the patient's myoclonic seizure were controlled, and
fatigue after activity was improved at discharge. The patient
follow-up was performed every 6 months. After discharge, the
patient got respiratory infection several times, and recently the
patient passed away at home due to severe respiratory infection.
The patient's son and daughter are currently asymptomatic.
Discussion
MERRF syndrome is a multisystem disorder
characterized by myoclonus, which often starts as the first symptom
in childhood after normal early development (11). The classic features of MERRF
syndrome include myoclonic epilepsy, ataxia and RRFs. In addition,
hearing loss, short stature, optic atrophy and cardiomyopathy, as
well as occasionally pigmented retinopathy and lipoma, may also be
present (2). The main symptoms in
the present patient were consistent with the clinical
manifestations of MERRF syndrome.
MRI often reveals cortical atrophy, basal ganglia
calcification, white matter abnormalities and brain stem atrophy in
patients with MERRF syndrome (11). In the present patient, cerebellar
atrophy was shown. Moreover, MRS showed inverted lactic acid peaks
in the cerebellum and basal ganglia, which was consistent with the
increase in lactic acid level in the serum and CSF. Increased
lactic acid levels at rest and after moderate activity indicate
mitochondrial dysfunction, and elevated levels of lactic acid in
the serum and CSF are important indicators for MERRF syndrome
(12).
Although RRF is a marker of MERRF syndrome, it may
be missing in 5-10% of cases (11). The presence of RRFs and
COX-negative fibers in muscle biopsy, as observed in the present
patient, often confirms the diagnosis of MERRF syndrome (12). The manifestations of mitochondrial
myopathy in the patient were prominent, but myoclonic epilepsy was
not the first symptom, as in patients with the 8344A>G or
3243A>G mutation.
The 3302A>G mutation is related to the lack of
respiratory chain complex I, which impairs the processing of RNA19
and leads to the accumulation of RNA precursor RNA19 in the muscles
(13). The inefficient cleavage of
the precursor RNA19 and defects in aminoacylation due to tRNA
structural changes cause a decrease in mitochondria (14). These changes may explain the main
symptoms, such as chronically progressive adult myopathy and
exercise intolerance, in patients with the 3302A> G mutation.
Patients with the 3302A>G mutation may have symptoms of
mitochondrial myopathy, encephalopathy, lactic acidosis and stroke
(MELAS syndrome) (15). However,
it is considered that the 3302A>G mutation can also cause
abnormalities in other systems (14). It has been reported a high mutation
load of the 3302A>G mutation can lead to fatal cardiorespiratory
failure (16). In addition, some
patients may develop type II diabetes mellitus and polycystic ovary
syndrome (17). Nevertheless,
symptoms of the muscular system appear earlier and are more
prominent (18), as shown in the
present case.
As the phenotype of this gene locus (mitochondrial
3302A>G) is highly heterogeneous, symptoms may vary and the age
of onset may not be limited to adulthood. The phenotypic
heterogeneity in the patient with MERRF syndrome reported in the
present study was demonstrated by chronic progressive adult
myopathy, stroke-like episodes, lactic acidosis, ataxia and
myoclonic epilepsy.
Acknowledgements
The authors would like to thank Ms. Zhen Li of the
Neurological Institute of Jiangxi Province for tissue sectioning
and histological staining.
Funding
Funding: This case report was supported by Jiangxi Provincial
People's Hospital (grant no. 2019-009).
Availability of data and materials
All data generated and/ or analyzed during this
study are included in this published article.
Authors' contributions
GH, YW and DY collected, analyzed and interpreted
the patient data, and advised on treatment. GH performed the
pathological examination. GH, YW and DY confirmed the diagnosis and
confirm the authenticity of all the raw data. GH and DY wrote the
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from the
patient and control subject.
Patient consent for publication
The patient and control subject gave their consent
for the publication of this case report and accompanying
images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bindoff LA and Engelsen BA: Mitochondrial
diseases and epilepsy. Epilepsia. 53 (Suppl 4):S92–S97.
2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mancuso M, Orsucci D, Angelini C, Bertini
E, Carelli V, Comi GP, Minetti C, Moggio M, Mongini T, Servidei S,
et al: Phenotypic heterogeneity of the 8344A>G mtDNA ‘MERRF’
mutation. Neurology. 80:2049–2054. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lopriore P, Gomes F, Montano V, Siciliano
G and Mancuso M: Mitochondrial epilepsy, a challenge for
neurologists. Int J Mol Sci. 23(13216)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Finsterer J, Zarrouk-Mahjoub S and
Shoffner JM: MERRF classification: Implications for diagnosis and
clinical trials. Pediatr Neurol. 80:8–23. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bornstein R, Gonzalez B and Johnson SC:
Mitochondrial pathways in human health and aging. Mitochondrion.
54:72–84. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Rahman S: Mitochondrial diseases and
status epilepticus. Epilepsia. 59 (Suppl 2):S70–S77.
2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Andrews RM, Kubacka I, Chinnery PF,
Lightowlers RN, Turnbull DM and Howell N: Reanalysis and revision
of the Cambridge reference sequence for human mitochondrial DNA.
Nat Genet. 23(147)1999.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Veenin K, Wattanasirichaigoon D,
Suktitipat B, Noojarern S, Lertrit P, Tim-Aroon T, Kaewsutthi S and
Treepongkaruna S: Association of mitochondrial DNA polymorphisms
with pediatric-onset cyclic vomiting syndrome. Front Pediatr.
10(876436)2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wong LC, Chen T, Wang J, Tang S, Schmitt
ES, Landsverk M, Li F, Wang Y, Zhang S, Zhang VW and Craigen WJ:
Interpretation of mitochondrial tRNA variants. Genet Med.
22:917–926. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Dubowitz V, Sewry C, Oldfors A and Lane R:
Muscle Biopsy: A Practical Approach. 4th ed. Elsevier, London,
pp21-24, 2013.
|
|
11
|
Hahn A, Schanzer A, Neubauer BA, Gizewski
E, Ahting U and Rolinski B: MERRF-like phenotype associated with a
rare mitochondrial trnaile mutation (m.4284 G>A).
Neuropediatrics. 42:148–151. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Lorenzoni PJ, Scola RH, Kay CS, Arndt RC,
Silvado CE and Werneck LC: MERRF: Clinical features, muscle biopsy
and molecular genetics in Brazilian patients. Mitochondrion.
11:528–532. 2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Siira SJ, Shearwood AJ, Bracken CP,
Rackham O and Filipovska A: Defects in RNA metabolism in
mitochondrial disease. Int J Biochem Cell Biol. 85:106–113.
2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Maniura-Weber K, Helm M, Engemann K,
Eckertz S, Mollers M, Schauen M, Hayrapetyan A, von Kleist-Retzow
JC, Lightowlers RN, Bindoff LA and Wiesner RJ: Molecular
dysfunction associated with the human mitochondrial 3302A>G
mutation in the MTTL1 (mt-tRNALeu(UUR)) gene. Nucleic Acids Res.
34:6404–6415. 2006.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Goto M, Komaki H, Saito T, Saito Y,
Nakagawa E, Sugai K, Sasaki M, Nishino I and Goto Y: MELAS
phenotype associated with m.3302A>G mutation in mitochondrial
tRNA(Leu(UUR)) gene. Brain Dev. 36:180–182. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
van den Bosch BJ, de Coo IF, Hendrickx AT,
Busch HF, de Jong G, Scholte HR and Smeets HJ: Increased risk for
cardiorespiratory failure associated with the A3302G mutation in
the mitochondrial DNA encoded tRNALeu(UUR) gene. Neuromuscul
Disord. 14:683–688. 2004.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ding Y, Zhuo G and Zhang C: The
mitochondrial tRNALeu(UUR) A3302G mutation may be associated with
insulin resistance in woman with polycystic ovary syndrome. Reprod
Sci. 23:228–233. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hutchison WM, Thyagarajan D, Poulton J,
Marchington DR, Kirby DM, Manji SS and Dahl MH: Clinical and
molecular features of encephalomyopathy due to the A3302G mutation
in the mitochondrial tRNA (Leu(UUR)) gene. Arch Neurol.
62:1920–1923. 2005.PubMed/NCBI View Article : Google Scholar
|














