Introduction
Concomitant presence of two simultaneous genomic
losses is a rare event and in most such cases it is difficult to
attribute the symptoms to one of the two affected genomic regions.
3q deletion syndrome is a genomic disorder characterized by a great
variability of phenotypes associated to the extension of deletion.
The clinical features of 3q deletion syndrome range from
intellectual disability, motor developmental delay, congenital
heart disease, renal and gastrointestinal malformations, autism,
congenital hypothyroidism, epilepsy and brain anomalies (1,2). The
corpus callosum is the primary commissural region of the brain
consisting of white matter tracts connecting cerebral hemispheres.
Its primary function is to integrate and transfer information from
cerebral hemispheres to process sensory, motor, and high-level
cognitive signals (3).
Agenesis or dysplasia of the corpus callosum is a
brain malformation with variable clinical expression reported in
many syndromes with predominantly genetic etiologies. Dysplasia and
dysgenesis of the corpus callosum are nonspecific descriptions that
imply defective development of the corpus callosum. The term
dysplasia is applied when the morphology of the corpus callosum is
altered as a congenital trait. For instance, the corpus callosum
may be hump-shaped, kinked, or a striped corpus callosum that lacks
an anatomically distinct genu and splenium (4,5).
Agenesis of corpus callosum is characterized by a complete absence
of corpus callosum. Agenesis and dysplasia of the corpus callosum
have an incidence of 0.5 to 70 in 10,000 individuals, and their
prevalence in children with developmental disabilities is about 230
in 10,000 (2.3%) (6). Corpus
callosum defects are frequently associated with other fetal
malformations, as ventriculomegaly often reported in fetuses with
agenesis or dysplasia of the corpus callosum (7,8).
Fetal ventriculomegaly, defined as dilation of the cerebral
ventricles, is a common cerebral anomaly often detected with
prenatal ultrasound scan with a prevalence of 0.3 to 1.5 per 1,000
live births (9). This condition is
typically categorized as mild (10-12 mm), moderate (13-15 mm) or
severe (>15) ventriculomegaly (10,11).
Fetus with severe ventriculomegaly is known to have a poor
prognosis in accordance with survival and neurodevelopment outcome.
However, the prognosis for infants with mild-to-moderate
ventriculomegaly is widely variable, which makes genetic counseling
challenging in clinical practice (12). Terminal 12p deletion, instead,
represents one of the rarest subtelomeric imbalances (13). Refereed clinical features of
constitutional deletions involving the terminal band of the short
arm of chromosome 12 (12p13.3) depends on variation in deletion
size, and can involves growth retardation, schizophrenia,
microcephaly, dysmorphic facial features, muscular hypotonia, and
other congenital abnormalities (13). In this case report we present a
prenatal diagnosis of a fetus with novel interstitial deletion of
12.87 Mb at chromosome region 3q13q21.2 and paternal inherited
microdeletion of 1.2 Mb at chromosome region 12p13.3 presenting a
corpus callosum dysplasia and a mild ventriculomegaly at fetal
ultrasound scan. This case report is expected to provide a further
reference for clinicians to identify complex syndromes in prenatal
period.
Case report
Patient
A 35-years-old pregnant woman secundigravida,
without a remarkable family history, come in our medical center
with a suspicion of a corpus callosum defect after a first
ultrasound scan at 22 weeks. Then, we performed a second ultrasound
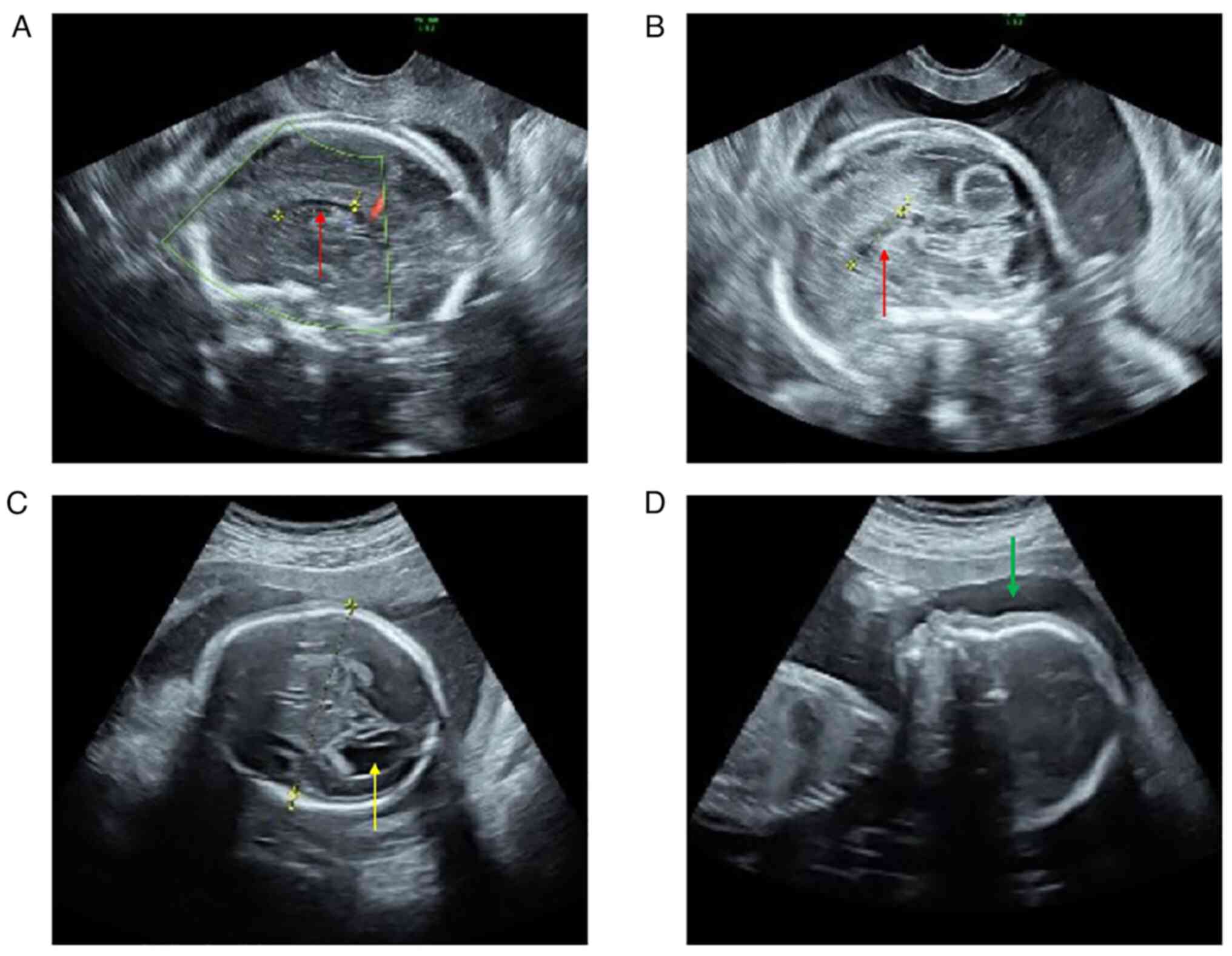
scan confirming the presence of dysplasia of corpus callosum
(Fig. 1A and B) and a mild unilateral ventriculomegaly
(Fig. 1C). Afterwards the woman
underwent amniocentesis at 23 weeks of gestation to investigate the
presence of chromosomal abnormalities. Corpus callosum dysplasia
was diagnosed visualizing the morphology and dimension of corpus
callosum by a trans-abdominal ultrasound scan. The corpus callosum
appears subtle and thin, in particular the body and the splenium
were not present, whereas the genu was present but appears thin.
The prenatal ultrasound showed a normal femur length of 39 mm and
normal length (290 mm). Measurements of the biparietal diameter and
head circumference were 62 and 239 mm, respectively. Moreover, the
sagittal ultrasound view shows also a flat forehead with a
flattened facial profile (Fig.
1D). Parents were both healthy and non-consanguineous, despite
the father referred a mild muscle hypotonia, several episodes of
shoulder and hip dislocations, and generalized joint laxity. After
genetic counselling, considering the relevant ultrasound clinical
features and the chromosomal aberrations, the couple decided to
terminate the pregnancy without performing magnetic resonance
imaging or neurosonography and it was not possible to perform the
autopsy.
Methods
Amniotic fluid was collected at 23 weeks of
gestation. Measurements of the biparietal diameter and head
circumference were obtained from a transverse axial plane of the
fetal head. The femur length was measured in a longitudinal scan.
Cytogenetic analysis was performed on cultured amniocytes by
G-banding according to standard procedures. At last, 16 metaphases
were analyzed. Chromosome analysis of parental blood samples was
performed using GTG-banding techniques on PHA-stimulated blood
lymphocytes. Array comparative genomic hybridization (aCGH)
analysis was performed on DNA from cultured amniocytes and DNA from
parental blood to characterize the extent of deletion, using 44K
platform (Agilent Technologies, Santa Clara, CA) as previously
reported (8). Briefly 500 ng of
the proband first and parents later with the relative sex-matched
reference DNA were processed according to the manufacturer's
protocol. Fluorescence was scanned in a dual-laser scanner
(Innoscan 710, Agilent Technologies, Santa Clara, CA) and images
were extracted and analyzed through Agilent Feature Extraction
Software. The position of oligomers refers to the Human genome
February 2009 (version GRCh37, hg19) assembly. For genes pLI
(probability of loss intolerance) and %HI (haploinsufficiency rank)
scores were retrieved from Decipher (https://www.deciphergenomics.org/). pLI score
indicates the probability that a gene is intolerant to a Loss of
Function (LoF) mutation. The higher the score, the more likely the
gene is involved in a dominant disease, and the lower the pLI
score, the more likely it is to indicate a recessive disease gene.
Genes with high pLI scores (pLI≥0.9) are extremely LoF intolerant,
whereby genes with low pLI scores (pLI≤0.1) are LoF tolerant. HI
stays for Haploinsufficiency, wherein a single functional copy of a
gene is insufficient to maintain normal function and is a major
cause of dominant disease.
High ranks of %HI (e.g., 0-10%) indicate that a gene
is more likely to exhibit haploinsufficiency, low ranks of %HI
(e.g., 90-100%) suggest a gene is less likely to exhibit
haploinsufficiency (14).
Findings
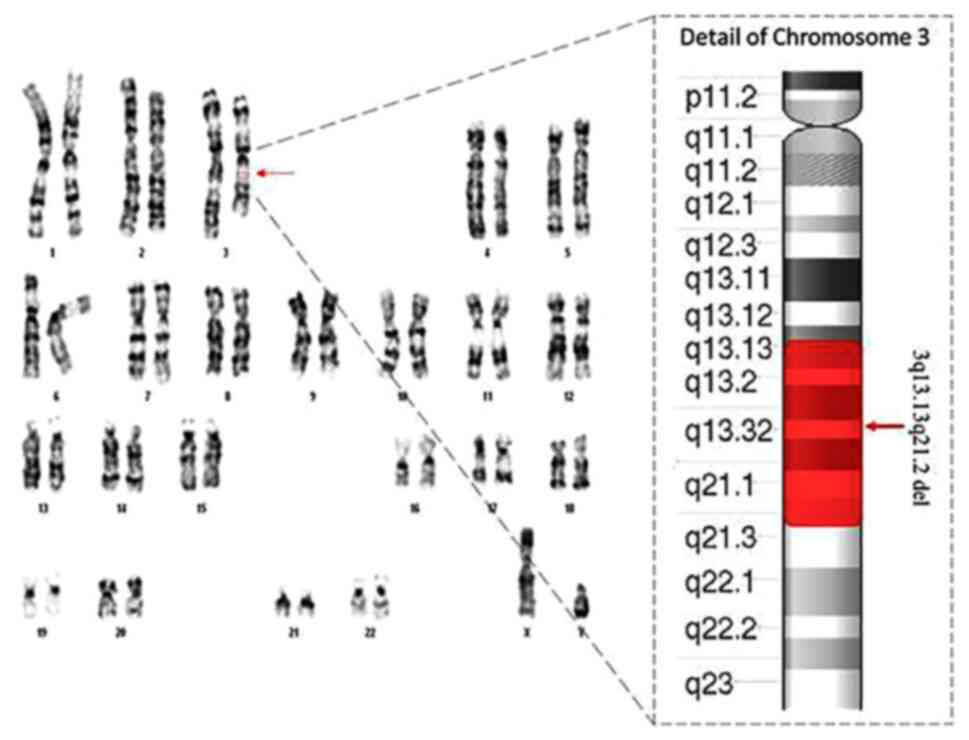
The result of fetal karyotype indicated a
chromosomal structural anomaly. Specifically, a reduction in length
of long arm of one chromosome 3 with an anomalous banding pattern
involving bands q13.1 and q21 (Fig.
2). To investigate the breaking points of chromosomal deletion
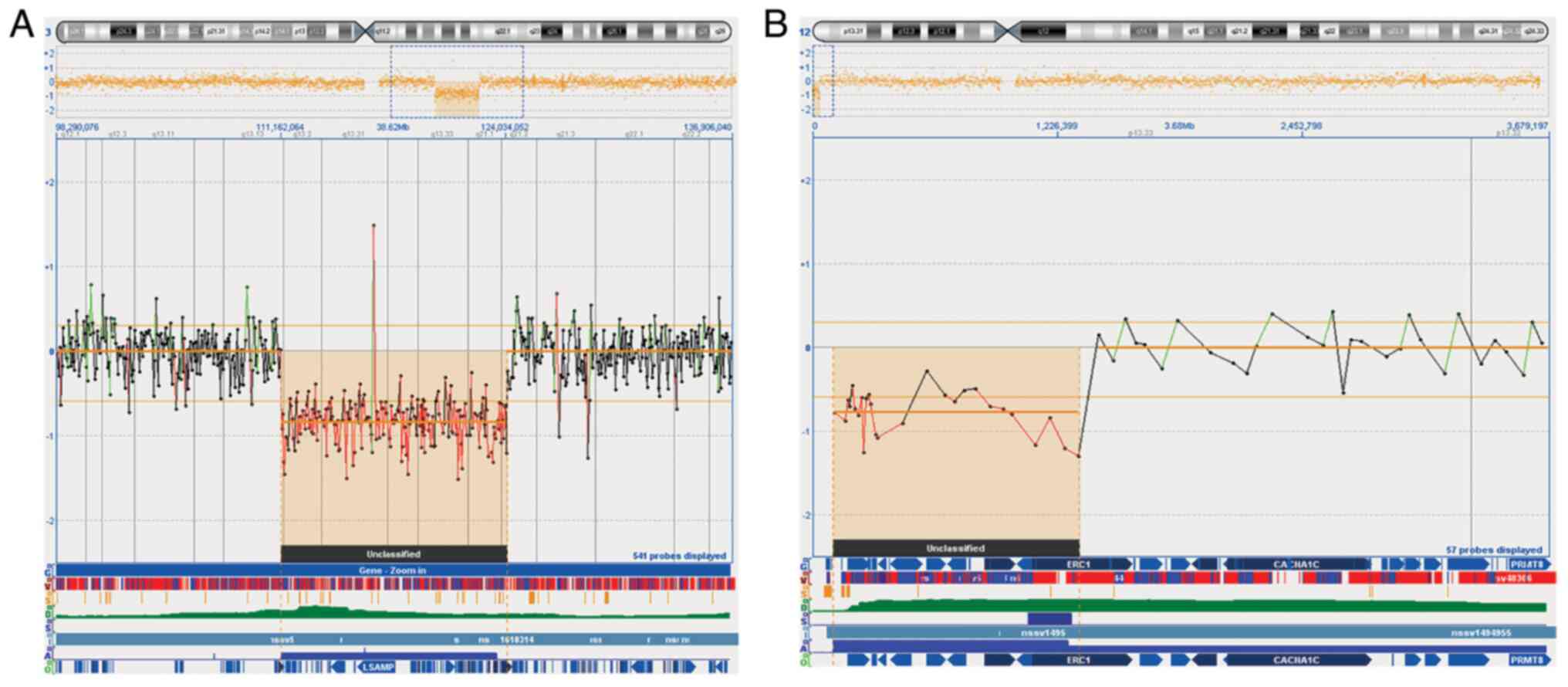
aCGH analysis was performed using a 44K array platform. The result
confirmed a fetal 12.87 Mb deletion in chromosomal band 3q13q21 arr
[hg19] 3q13.13q21.2 (111162064_124034052) (Fig. 3A), detecting a further
microdeletion of 1.2 Mb in chromosomal band 12p13.33, arr [hg19]
12p13.33 (100698_1327097) x1 not visible with standard karyotype
(Fig. 3B). To investigate the
origin of deletions, aCGH was performed on both parents. The
results showed a paternal inherited origin of 12p13.33
microdeletion. Investigation for aforementioned 12.87 Mb deletion
of 3q chromosome by DECIPHER database reveals 81 OMIM genes
(Table SI), among those genes the
highest rank of %HI (0-10%) scores were reported for 6 genes:
LSAMP, ZBTB20, GSK3B, NAA50, CASR, GTF2E1. Whereas, genes with
highest pLI scores (pLI≥0.9) are reported for 12 genes: USF3,
KALRN, ARHGAP31, STXBP5L, ADCY5, KPNA1, LSAMP, ZBTB20, CD86, GSK3B,
CD200, NECTIN3. About 1.23 Mb deletion of 12p chromosome by
DECIPHER database reveals 9 OMIM genes (Table SII), among those genes the high
%HI (0-10%) score was reported for only ERC1 gene, whereas, genes
with rank pLI>0.9 were reported for 2 genes: KDM5A and WNK1.
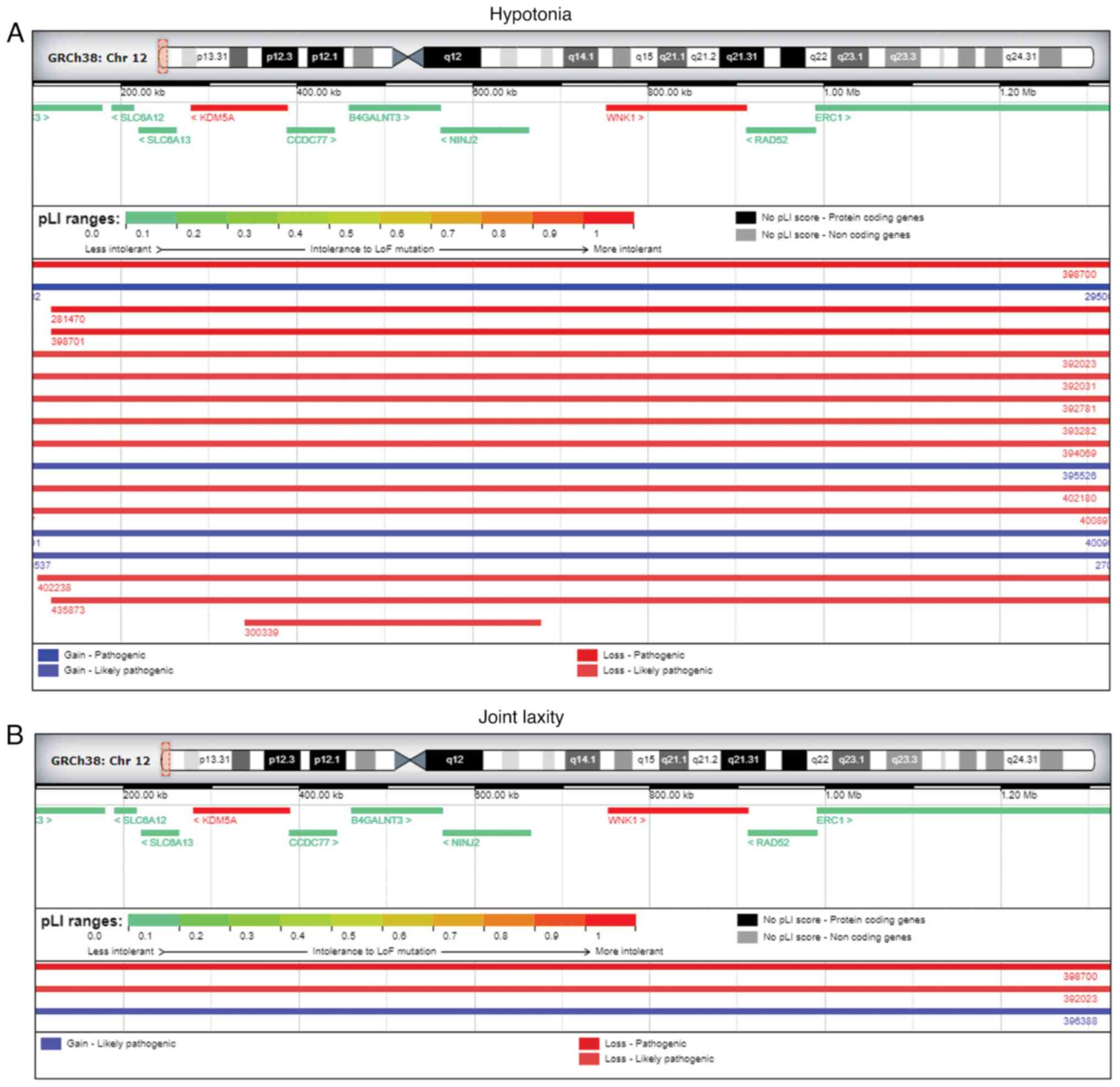
Additional DECIPHER analysis for 12p13.3 microdeletion revealed a
total of 78 patients showing a complete overlapping for several
pathogenic or likely pathogenic deletions. Among the 78 patients,
12 of them reported muscular hypotonia (Fig. 4A), with 2 of them reporting joint
laxity as well (Fig. 4B),
interestingly similar clinical features were referred by the father
of the fetus during the genetic counseling. Furthermore, among the
78 patients showing a complete overlapping deletion with our case,
we also found a patient (ID 395656) with ventriculomegaly and none
with dysplasia of the corpus callosum. Additional insights about
patient ID 395656 showed that it carried a 12p deletion of 10 Mb,
ten-fold bigger if compared with the deletion of our case covering
225 gene, some of them like CCND2, PHC1 and NTF3 gene, has been
previously reported to be involved in brain formation (15-17).
Since the involvement of a huge number of genes, many of them could
most likely contribute to the ventriculomegaly phenotype of patient
ID 395656. Moreover, we didn't find genes suggestive of any
epigenetic effects in this region, nor imprinted genes has been
reported on chromosome 12.
Discussion
The presented case is unique in harboring two
specific deletions in 3q13.13q21.2 and 12p13.33. According to the
literature search, there were no reports describing any case with
interstitial deletion of the long arm of chromosome 3 and terminal
microdeletion of the short arm of chromosome 12. Concomitant
presences of two simultaneous genomic losses are rare and in most
such cases it is difficult to attribute the symptoms to one of the
two affected genomic regions, making genotype-phenotype correlation
extremely difficult. Here, we report a prenatal diagnosis of a male
fetus presenting ultrasounds evidence of corpus callosum dysplasia
and ventriculomegaly showing a 3q13q21 deletion and 12p13.33
microdeletion. Litterature search for 3q13.13q21.2 deletion
revealed previously described post natal cases showing several
clinical phenotypes including skeletal malformations included
scoliosis, lordosis, thoracic kyphosis, joint contractures, and
peripheral malformations affecting the hands and feet, corpus
callosum malformations, ventriculomegaly, alobar
holoprosen-cephaly, skull malformations, autism, attention deficits
and epilepsy (2). Molin et
al (2), described an
overlapping deletion of about 580 kb at 3q13.31 defined as shortest
region of overlapping (SRO). Comparative phenotypic evaluation has
showed that 5 patients among the 24 with SRO had agenesis of the
corpus callosum and 3 patients presented ventriculomegaly, evoking
the clinical features of the case presented in this study. This 580
kb segment includes four OMIM genes: DRD3, ZNF80, TIGIT, and
ZBTB20. Among them, two genes, ZBTB20 and DRD3 were previsoly
associated to brain development delay (2). ZBTB20 encodes a transcriptional
repressor expressed in the cerebellum and corpus callosum (18) and it is a cell fate determinant for
hippocampal neurons (19,20), whereas DRD3 encodes a dopamine
receptor presents in the limbic system (21). Given their roles in neural
development, haploinsufficiency of ZBTB20 and DRD3 genes may
contribute to the corpus callosum and cerebellar malformations.
However, as mentioned, proximal long arm of chromosome 3 is a gene
dense region, hence, the involvement of a huge number of genes
could most likely contribute to the complex phenotypic features of
fetus development.
In this case we detected also a terminal 12p
deletion reported as one of the rarest subtelomeric imbalance
(13). Previous cases with
terminal 12p deletion presented a phenotypic spectrum ranging from
a normal development to development delay, facial dysmorphism and
microcephaly (22,23). Comparative phenotypic evaluations
of literature and databases has showed that most commonly reported
clinical features are intellectual disability or development delay,
microcephaly, muscular hypotonia, scoliosis and small for
gestational age (24,25). Several genomic structural
variations were detected in 12p13.3 region, not association with
healthy individuals was found in literature for this 12p
microdeletion (23). Searching for
12p13.3 microdeletion on DECIPHER database highlighted the presence
of 3 genes wiht highest score for %HI (0-10%) and pLI (pLI≥0.9):
KDM5A, WNK1, and ERC1 genes. KDM5A family have been strongly linked
to a wide range of neurodevelopment disorders (26). ERC1 can potentially be accounted
for the etiology of autism spectrum disorders (22,27,28).
WNK kinases have a function in the nervous system, since whole
genome exome sequencing identified variants in WNK1 in patients
affected by Charcot-Marie-Tooth a form of autosomic recesive
peripheral neuropathy (29). In
12p13.3 microdeletion of our case we detected a partial deletion
(exons 1-11) of ERC1 gene sufficient to cause a non-production of
ERC1 protein. Moreover, DECIPHER reaveled that among the 78
patients, showing an overalapping region, 12 of them reported
muscular hypotonia (Fig. 4A), with
2 of these patients reported joint laxity (Fig. 4B), similar clinical features
referred by the father of the fetus. Furthermore, among the 78
patients showing a complete overlapping deletion with our case, we
found a patient with ventriculomegaly (ID 395656) and none with
dysplasia of the corpus callosum. Despite all the mentioned
literature regarding the 3q and 12p deletions we noticed that on
NIH MedLine Plus website (https://medlineplus.gov/) does not report any
information about the identified deletions. We recognize several
limitations of the study. First of all, it was not possible to
perform medical examination of the fetus after pregnancy
termination. Moreover patients with abnormalities of the corpus
callosum may have severe intellectual impairment such as cerebral
palsy, hydrocephalus, spasticity, severe learning disabilities,
autism or seizures, all features not verifiable in a prenatal
period. Another limitation it was that the couple decided to
terminate the pregnancy without performing magnetic resonance
imaging or neurosonography, precluding a better clinical phenotype
definition and the correlation with the detected chromosomal
aberrations.
Although a phenotype-genotype association to
specific genes is not possible, we have speculated a possible
association considering the published literature referred to these
chromosomal aberrations. Due to the complexity of involved
chromosomal imbalances, specific 3q13.13q21.2 deletion might
contribute to the corpus callosum and ventriculomegaly, while
12p13.33 deletion could lead to muscular hypotonia, and joint
laxity observed in the father of fetus. Remarkably this region
contains ERC1 gene appearing as a strong candidate for the
aforementioned clinical features, since it was previously reported
to be associate with muscle organization (30), and DECIPHER analysis reveald ERC1
gene as the only gene in the 12p deleted region with %HI value
under 10%, suggesting is strong haploinsufficiency status.
This case report is expected to provide a reference
for clinicians facing with prenatal diagnosis and genetic
counseling in pregnant women with diagnosis of 3q13q21.2 deletions
or 12p13.33 microdeletion. Clinicians should consider 3q deletion
syndrome when they are exploring a diagnosis of fetus with corpus
callosum abnormalities or ventriculomegaly and the syndrome should
be confirmed by cytogenetic karyotype together with aCHG analysis.
Unfortunately, in prenatal period few data are collected regarding
neurological development of the fetus and only pediatric
neurologists can evaluate neurological features after birth.
An accurate characterization of the fetal
chromosomal defects has implications in the couple decision
regarding the continuing of the pregnancy or elective abortion and
brings important information for the future reproductive options in
order to give birth to a healthy baby.
Supplementary Material
DECIPHER analysis reveals 81 OMIM
genes at the region 3q13.13q21.2.
DECIPHER analysis reveals 9 OMIM genes
at the region of 12p13.33.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the ArrayExpress repository under
the following accession number E-MTAB-12413 deposited at BioStudies
platform (https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-12413).
Authors' contributions
FL, KM, MF were involved in conceptualization and
writing the original draft. LSC, RR and FM were involved in
experiments. KM and MF performed data analysis. KM, MF and AM
confirm the authenticity of all the raw data. AM and CG were
involved in design, methodology and correction of the manuscript.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the local ethical
committee of Artemisia SPA (approval no. #2022-0054-001; June 01,
2022). The protocols used in this study adhere to the tenets of the
Declaration of Helsinki.
Patient consent for publication
Written informed consent was obtained from subjects
involved in the study.
Competing interests
All authors are full-time employees of Artemisia
SPA. ALTAMEDICA is a branch of Artemisia SPA involved in Human
Genetics and Fetal-Maternal Medical Sciences.
References
|
1
|
Ramieri V, Tarani L, Costantino F, Basile
E, Liberati N, Rinna C, Cascone P and Colloridi F: Microdeletion 3q
syndrome. J Craniofac Surg. 22:2124–2128. 2011.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Molin AM, Andrieux J, Koolen DA, Malan V,
Carella M, Colleaux L, Cormier-Daire V, David A, de Leeuw N,
Delobel B, et al: A novel microdeletion syndrome at 3q13.31
characterised by developmental delay, postnatal overgrowth,
hypoplastic male genitals, and characteristic facial features. J
Med Genet. 49:104–109. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Tanaka-Arakawa MM, Matsui M, Tanaka C,
Uematsu A, Uda S, Miura K, Sakai T and Noguchi K: Developmental
changes in the corpus callosum from infancy to early adulthood: A
structural magnetic resonance imaging study. PLoS One.
10(e0118760)2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hofman J, Hutny M, Sztuba K and Paprocka
J: Corpus callosum agenesis: An insight into the etiology and
spectrum of symptoms. Brain Sci. 10(625)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Hanna RM, Marsh SE, Swistun D, Al-Gazali
L, Zaki MS, Abdel-Salam GM, Al-Tawari A, Bastaki L, Kayserili H,
Rajab A, et al: Distinguishing 3 classes of corpus callosal
abnormalities in consanguineous families. Neurology. 76:373–382.
2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Edwards TJ, Sherr EH, Barkovich AJ and
Richards LJ: Clinical, genetic and imaging findings identify new
causes for corpus callosum development syndromes. Brain.
137:1579–1613. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Raybaud C: The corpus callosum, the other
great forebrain commissures, and the septum pellucidum: Anatomy,
development, and malformation. Neuroradiology. 52:447–477.
2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Libotte F, Fabiani M, Margiotti K, Viola
A, Mesoraca A and Giorlandino C: Prenatal diagnosis of combined
maternal 4q interstitial deletion and paternal 15q
microduplication. Genes (Basel). 12(1626)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Pilu G and Hobbins JC: Sonography of fetal
cerebrospinal anomalies. Prenat Diagn. 22:321–330. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Wang J, Zhang Z, Li Q, Zhu H, Lai Y, Luo
W, Liu S, Wang H and Hu T: Prenatal diagnosis of chromosomal
aberrations by chromosomal microarray analysis in foetuses with
ventriculomegaly. Sci Rep. 10(20765)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Griffiths PD, Reeves MJ, Morris JE, Mason
G, Russell SA, Paley MN and Whitby EH: A prospective study of
fetuses with isolated ventriculomegaly investigated by antenatal
sonography and in utero MR imaging. AJNR Am J Neuroradiol.
31:106–111. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Alluhaybi AA, Altuhaini K and Ahmad M:
Fetal ventriculomegaly: A review of literature. Cureus.
14(e22352)2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ravnan JB, Tepperberg JH, Papenhausen P,
Lamb AN, Hedrick J, Eash D, Ledbetter DH and Martin CL: Subtelomere
FISH analysis of 11 688 cases: An evaluation of the frequency and
pattern of subtelomere rearrangements in individuals with
developmental disabilities. J Med Genet. 43:478–489.
2006.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Firth HV, Richards SM, Bevan AP, Clayton
S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM and Carter
NP: DECIPHER: Database of chromosomal imbalance and phenotype in
humans using ensembl resources. Am J Hum Genet. 84:524–533.
2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Mirzaa G, Parry DA, Fry AE, Giamanco KA,
Schwartzentruber J, Vanstone M, Logan CV, Roberts N, Johnson CA,
Singh S, et al: De novo CCND2 mutations leading to stabilization of
cyclin D2 cause
megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome.
Nat Genet. 46:510–515. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Ozcelik T, Rosenthal A and Francke U:
Chromosomal mapping of brain-derived neurotrophic factor and
neurotrophin-3 genes in man and mouse. Genomics. 10:569–575.
1991.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Awad S, Al-Dosari MS, Al-Yacoub N, Colak
D, Salih MA, Alkuraya FS and Poizat C: Mutation in PHC1 implicates
chromatin remodeling in primary microcephaly pathogenesis. Hum Mol
Genet. 22:2200–2213. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Nagao M, Ogata T, Sawada Y and Gotoh Y:
Zbtb20 promotes astrocytogenesis during neocortical development.
Nat Commun. 7(11102)2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Nielsen JV, Thomassen M, Mollgard K,
Noraberg J and Jensen NA: Zbtb20 defines a hippocampal neuronal
identity through direct repression of genes that control projection
neuron development in the isocortex. Cereb Cortex. 24:1216–1229.
2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Xie Z, Ma X, Ji W, Zhou G, Lu Y, Xiang Z,
Wang YX, Zhang L, Hu Y, Ding YQ and Zhang WJ: Zbtb20 is essential
for the specification of CA1 field identity in the developing
hippocampus. Proc Natl Acad Sci USA. 107:6510–6515. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Sokoloff P, Giros B, Martres MP, Bouthenet
ML and Schwartz JC: Molecular cloning and characterization of a
novel dopamine receptor (D3) as a target for neuroleptics. Nature.
347:146–151. 1990.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Leyser M, Dias BL, Coelho AL, Vasconcelos
M and Nascimento OJ: 12p deletion spectrum syndrome: A new case
report reinforces the evidence regarding the potential relationship
to autism spectrum disorder and related developmental impairments.
Mol Cytogenet. 9(75)2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Fanizza I, Bertuzzo S, Beri S, Scalera E,
Massagli A, Sali ME, Giorda R and Bonaglia MC: Genotype-phenotype
relationship in a child with 2.3 Mb de novo interstitial
12p13.33-p13.32 deletion. Eur J Med Genet. 57:334–338.
2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Rincic M, Rados M, Kopic J, Krsnik Z and
Liehr T: 7p21.3 together with a 12p13.32 deletion in a patient with
microcephaly-does 12p13.32 locus possibly comprises a candidate
gene region for microcephaly? Front Mol Neurosci.
14(613091)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Thevenon J, Callier P, Andrieux J, Delobel
B, David A, Sukno S, Minot D, Mosca Anne L, Marle N, Sanlaville D,
et al: 12p13.33 microdeletion including ELKS/ERC1, a new locus
associated with childhood apraxia of speech. Eur J Hum Genet.
21:82–88. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
El Hayek L, Tuncay IO, Nijem N, Russell J,
Ludwig S, Kaur K, Li X, Anderton P, Tang M, Gerard A, et al: KDM5A
mutations identified in autism spectrum disorder using forward
genetics. Elife. 9(e56883)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Silva IM, Rosenfeld J, Antoniuk SA, Raskin
S and Sotomaior VS: A 1.5Mb terminal deletion of 12p associated
with autism spectrum disorder. Gene. 542:83–86. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Han JY and Park J: Variable phenotypes of
epilepsy, intellectual disability, and schizophrenia caused by
12p13.33-p13.32 terminal microdeletion in a korean family: A case
report and literature review. Genes (Basel).
12(1001)2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Rodan AR and Jenny A: WNK kinases in
development and disease. Curr Top Dev Biol. 123:1–47.
2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Luderman LN, Michaels MT, Levic DS and
Knapik EW: Zebrafish Erc1b mediates motor innervation and
organization of craniofacial muscles in control of jaw movement.
Dev Dyn: Jun 16, 2022 (Epub Ahead of Print).
|