Introduction
Dementia, which features memory loss and cognitive
decline, severely influences daily life (1). As a most common contributor to
dementia, Alzheimer's disease (AD) accounts for 50-75% of dementia
cases (2). AD, which is a common
progressive neurodegenerative disease, features the formation of
extracellular amyloid plaques as well as intracellular
neurofibrillary tangles (3). An
increasing number of researches have shown that the
neuroinflammation of AD has a close association with neurological
dysfunction, which is mediated by the progressive activation of
glial cells and the consequent overproduction of pro-inflammatory
cytokines (4,5). In AD, neuroinflammation acts as a
major element in disease advancement, which features the activities
of brain resident glial cells, particularly microglia (6). The over-activated microglia can
promote the release of inflammatory cytokines, chemokines, and
oxygen/nitrogen radicals, thereby exacerbating the neuronal damage
(7,8).
The dedicator of cytokinesis (DOCK) proteins, which
are the members of the family of atypical guanine exchange factors,
can trigger ρGTPases Rac1 and/or Cdc42 and serve as critical
players in cellular activities, such as cell migration, neuronal
polarization as well as neuroprotection (9). DOCK8, which belongs to DOCK family of
proteins, is highly expressed in B cells as well as T cells and has
been widely investigated in immune system (9,10).
In addition, DOCK8 exists in microglia and can regulate the
advancement of neurodegenerative diseases (11). The depletion of DOCK8 imparts
alleviative effects on the migration and function of immune cells
(12). DOCK2, another member of
DOCK family of proteins, is involved with the advancement of AD
(13,14). Nevertheless, the role that DOCK8
played in AD remains to be elucidated.
In the present study, Aβ1-42 (Aβ) was
used for the stimulation of BV2 cells to induce inflammatory
damage. The present study was performed to discuss the role of
DOCK8 in Aβ-induced BV2 cells and to elucidate its hidden
regulatory mechanism in alleviating hippocampal neuronal
damage.
Materials and methods
Cell culture and treatment
BV2 microglia cells and hippocampal HT22 cells
provided by Shanghai Hongshun Biotechnology Co., Ltd were
cultivated in modified Eagle's medium (MEM; Thermo Fisher
Scientific, Inc.) which contained 10% fetal bovine serum (FBS;
Guangzhou Perseco Biotechnology Co., Ltd.) and 1%
penicillin/streptomycin and were placed in a humid atmosphere at
37˚C in the presence of 5% CO2. To stimulate
inflammatory damage, 10 µM Aβ oligomers (GL Biochem) was
administered to BV2 cells for 24 h at 37˚C and the culture medium
was considered as conditioned medium (CM) (1). With the purpose of further
discovering the mechanism of DOCK8 in STAT3 signaling, STAT3
activator Colivelin (0.5 µM) was applied to BV2 cells with Aβ
induction (15). To activate HT22
cells, cells were plated in 96-well plates (1.5
x104/well) in serum-free culture medium were treated
with BV2 CM for 24 h at 37˚C (16).
Cell transfection
To deplete DOCK8 expression, 100 nM small
interfering RNAs (siRNA) specific to DOCK8 (siRNA-DOCK8-1,
CGGAAAAACCAAGGAAGTTCAGA; siRNA-DOCK8-2, CTCTGAAGTTTGAGATTGAAATT) as
well as its negative control (siRNA-NC, CCCGATTTCCGAGAATTCTCATTCA)
provided by Shanghai GeneChem Co., Ltd. were transfected into BV2
cells or HT22 cells seeded into 6-well plates (2x105
cells/well) using Lipofectamine® 3000 (Invitrogen;
Thermo Fisher Scientific, Inc.) for 48 h at 37˚C according to the
manufacturer's protocols. At 48 h following transfection, the
transfection efficacy was tested with reverse
transcription-quantitative polymerase chain reaction (RT-qPCR).
Immunofluorescence (IF) staining
Following Aβ induction, BV2 cells were subjected to
4% paraformaldehyde for 20 min at room temperature and 0.2% Triton
X-100 permeation for 20 min at room temperature. Subsequently, the
PBS-rinsed cells were blocked with 1% bovine serum albumin at room
temperature. The overnight exposure of cells to primary antibodies
targeting IBA-1 (ab178846; 1:500; Abcam) and CD11b (ab184308;
1:500; Abcam) was performed at 4˚C, followed by probing with rabbit
anti-mouse IgG H&L (ab6728; 1:1,000; Abcam) at 37˚C for 30 min.
After nuclear staining with DAPI (Shenzhen Ziker Biotechnology Co.,
Ltd.) for 10 min at room temperature, a fluorescence microscope
(Olympus Corporation) was used to capture images.
Enzyme-linked immunosorbent assay
(ELISA)
Using ELISA kits (Shanghai Xitang Biotechnology,
China), the levels of inflammatory cytokines TNF-α (cat. no.
F11630), IL-1β (cat. no. F10770) and IL-6 (cat. no. F10830) in cell
supernatants were detected according to the manufacturer's
protocols. Optical density (OD) value was resolved at λ=450 nm
using a microplate reader (Molecular Devices, LLC). The results
were calculated according to the standard curve.
Wound healing assay
The migrative capability of Aβ-induced BV2 cells was
assessed using wound healing assay. Initially, BV2 cells were
cultured in 6-well plates using serum-free medium until 95%
confluence was achieved. Thereafter, a wound in the cell monolayer
was created with a 10-µl pipette tip. The BV2 cells were rinsed
with PBS and cultured at 37˚C in the presence of 5% CO2.
At 0 and 24 h, images of the wound areas were captured by a light
microscope (Olympus Corporation). Image J software (Version 1.52r;
National Institutes of Health) was used to visualize the of
migrative cells.
Transwell assay
The invasive capability of Aβ-induced BV2 cells was
estimated using Transwell assay. The upper compartment of the
Transwell was coated with Matrigel (BD Biosciences) at 37˚C for 30
min and used for BV2 cells. Medium, with 10% FBS, was added to the
low chamber. Invaded BV2 cells were fixed with 4% paraformaldehyde
for 30 min and stained with 0.1% crystal violet for 20 min at room
temperature. The area of invaded cells was tracked using a light
microscope (Olympus Corporation).
Cell Counting Kit-8 (CCK-8) assay
BV2 cells in 96-well plates were cultured for 24 h
at 37˚C. CCK-8 reagent (10 µl; Beyotime Institute of Biotechnology)
was added and the cells cultured for another 2 h at 37˚C. A
microplate reader (Molecular Devices, LLC) was used to assess
absorbance at 450 nm.
Terminal-deoxynucleotidyl transferase
mediated nick end labeling (TUNEL)
The apoptosis level of BV2 cells was assessed using
TUNEL staining (Beyotime Institute of Biotechnology).
Paraformaldehyde (4%) and Triton X-100 (0.5%) was used to treat BV2
cells for 15 and 20 min respectively at room temperature.
Subsequently, the cells were labeled with TUNEL for 1 h at 37 ˚C.
The counterstaining of cells with 1 µg/ml DAPI was performed at
37˚C for 30 min in the dark. The observation of positive cells in
five randomly selected fields was performed under a florescent
microscope (Olympus Corporation).
RT-qPCR
Total RNA was isolated from sample cells placed in a
6-well plate (6x104 cells/well) with TRIzol®
reagent (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols and reverse transcribed into cDNA using
the RevertAid cDNA Synthesis kit (Beijing Zhijie Fangyuan
Technology Co., Ltd.) according to the manufacturer's protocols.
PCR reactions were performed using iTaq Universal SYBR Green kit
(Bio-Rad Laboratories, Inc.) on the MX3000p PCR system (Agilent
Technologies, Inc.) according to the manufacturer's protocols.
RT-qPCR was performed at 50˚C for 2 min and 95˚C for 2 min,
followed by 40 cycles at 95˚C for 15 sec and 60˚C for 1 min. The
calculation of relative gene expression was operated with the
2-ΔΔCq (17). The
primer sequences were: DOCK8 forward primer:
5'-GGCTGACAGATGAGGCTGG-3', reverse primer:
5'-TCAAAGTCCACTGGCTCGAC-3'; inducible nitric oxide synthase (iNOS)
forward primer: 5'-AGGGCCACCTCTACATTTGC-3', reverse primer:
5'-CCCAAGCCATCATTGGGAGT-3'; CD86 forward primer:
5'-TCAATGGGACTGCATATCTGCC-3', reverse primer:
5'-GCCAAAATACTACCAGCTCACT-3' or GAPDH forward primer:
5'-GCCTCCTCCAATTCAACCCT-3', reverse primer:
5'-CTCGTGGTTCACACCCATCA-3'. GAPDH was designated as a standard
internal control for relative gene expression. This experiment was
repeated three times.
Western blotting
Total proteins were isolated from samples with RIPA
lysis buffer (Beyotime Institute of Biotechnology), after which was
the concentration quantification applying a bicinchoninic acid
(protein assay kit (Shanghai Yisheng Biotechnology Co., Ltd.). The
proteins (30 µg) were separated by 8% SDS-PAGE, transferred to PVDF
membranes and blocked by 5% non-fat milk at room temperature for 1
h. Subsequently, the overnight cultivation of membranes with
primary antibodies was performed at 4˚C, after which was the probe
with HRP-labeled rabbit anti-mouse secondary antibody (cat. no.
7074P2; 1:5,000; Cell Signaling Technology, Inc.) at room
temperature for 2 h. Finally, the visualization and analysis of
protein blots were performed with ECL (Yeasen Biotech) and ImageJ
(Version 1.52r; National Institutes of Health). GAPDH was used as
the loading control. The following primary antibodies were used:
anti-DOCK8 (cat. no. 39263S; 1:1,000), anti-CD86 (cat. no. 19589S;
1:1,000), anti-iNOS (cat. no. 13120S; 1:1,000), anti-NOD-like
receptor family pyrin domain containing 3 (NLRP3; cat. no. 15101S;
1:1,000), anti-apoptosis-associated speck-like protein containing a
caspase recruitment domain (ASC; cat. no. 67824T; 1:1,000),
anti-caspase1 (cat. no. 24232S; 1:1,000), anti-p-p65 (cat. no.
3033T; 1:1,000), anti-p65 (cat. no. 8242T; 1:1,000), anti-Bcl2
(cat. no. 3498T; 1:1,000), anti-Bax (cat. no. 14796S; 1:1,000),
anti-cleaved caspase3 (cat. no. 9664T; 1:1,000), anti-caspase3
(cat. no. 9662S; 1:1,000) and anti-GAPDH (cat. no. 5174T; 1:1,000)
antibodies were from by Cell Signaling Technology, Inc.
Anti-p-STAT3 (cat. no. ab76315; 1:2,000) and anti-STAT3 (cat. no.
ab68153; 1:2,000) antibodies were purchased from Abcam.
Statistical analysis
Data were given as mean ± standard deviation (SD)
and were analyzed using GraphPad Prism 8.0 software (Dotmatics).
One-way ANOVA and Tukey's post-hoc test was used for comparisons
between multiple groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
DOCK8 expression increases in
Aβ-induced BV2 cells
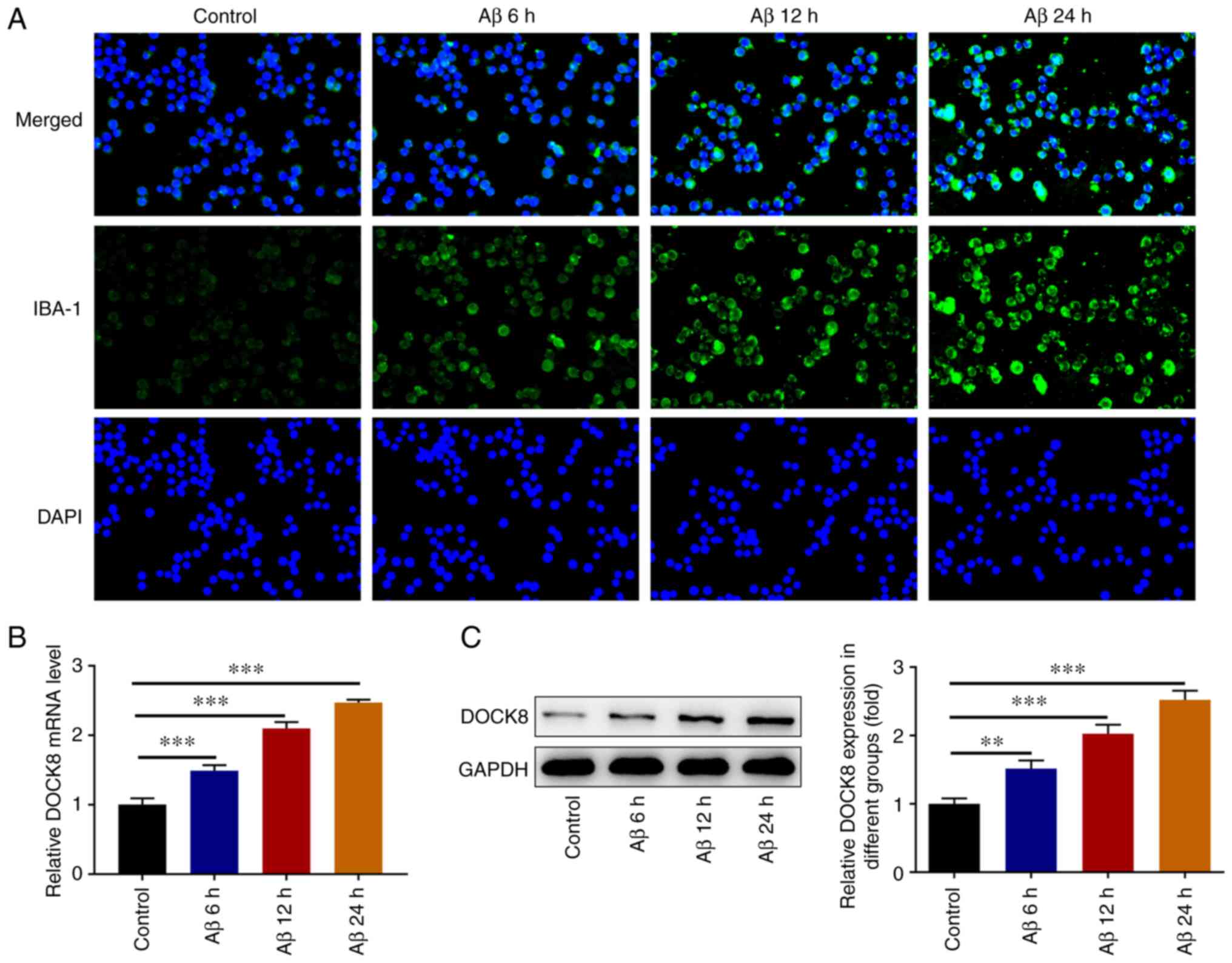
Initially, Aβ was used for the induction of BV2
cells and IF adopted to analyze IBA-1 expression. Compared with the
Control group, Aβ induction elevated IBA-1 expression in BV2 cells
in a time-dependent manner (Fig.
1A). Results obtained from RT-qPCR and western blotting
demonstrated that the mRNA and protein expression levels of DOCK8
in BV2 cells were enhanced by Aβ stimulation compared with the
Control group (Fig. 1B and
C). It was evident that DOCK8 was
increased in Aβ-induced BV2 cells.
DOCK8 interference inhibits the
activation and inflammatory factors release of Aβ-induced BV2
cells
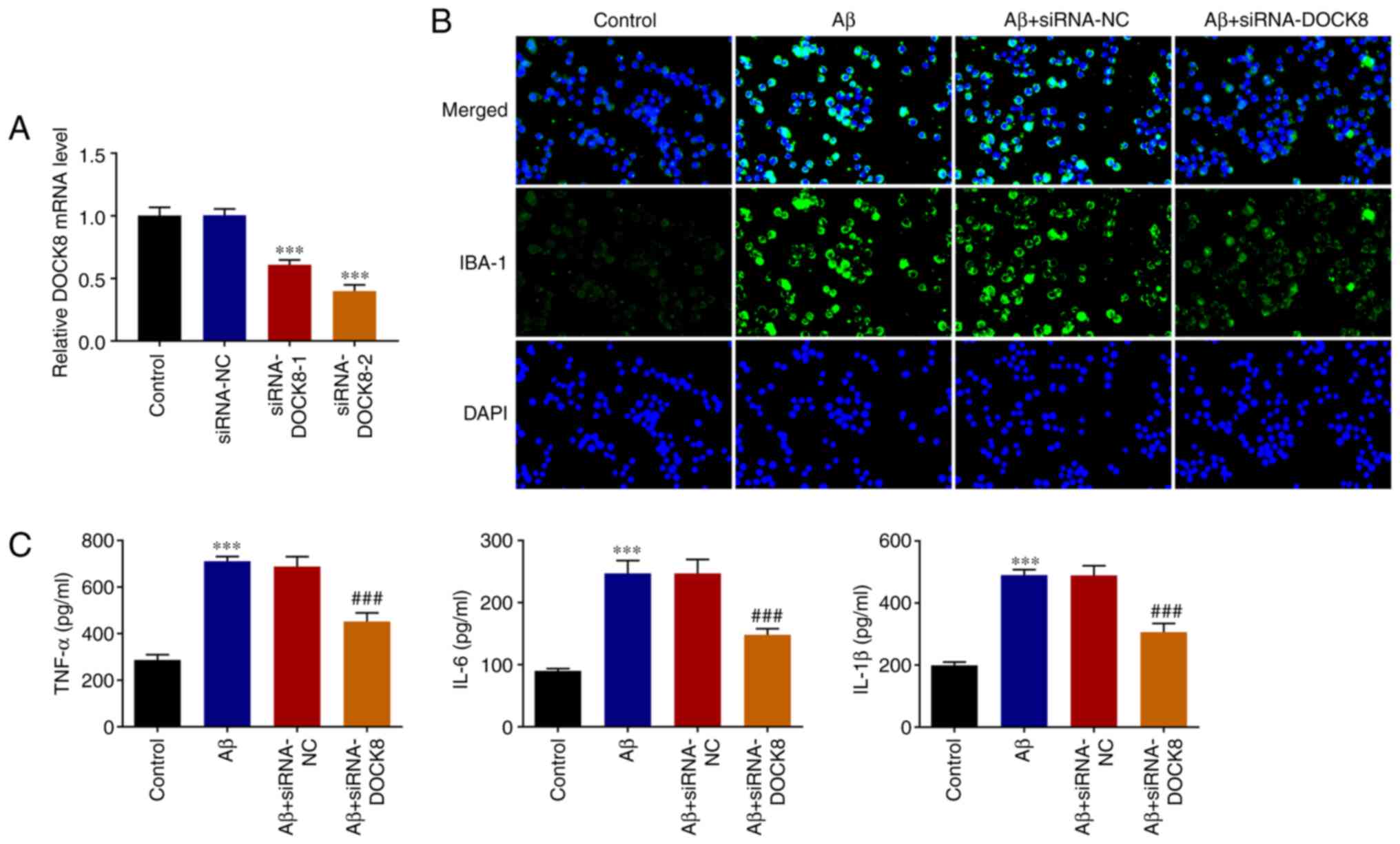
To decrease DOCK8 expression, siRNAs specific to
DOCK8 were transfected into BV2 cells and then RT-qPCR was used to
the test transfection efficacy. As shown in Fig. 2A, the expression of DOCK8 in BV2
cells declined after transfection with siRNA targeting DOCK8 when
compared to the Control group. Notably, DOCK8 had lower expression
in siRNA-DOCK8-2 group than that in siRNA-DOCK8-1 group, thus
siRNA-DOCK8-2 was adopted the following experiments. Compared with
the Control group, Aβ stimulation markedly increased IBA-1
expression, which was then decreased by DOCK8 interference
(Fig. 2B). In addition, the
increased levels of TNF-α, IL-6 and IL-1β in BV2 cells caused by Aβ
induction were decreased after decreasing DOCK8 expression
(Fig. 2C), suggesting that DOCK8
silence imparted a suppressive effect on the inflammatory response
of Aβ-induced BV2 cells.
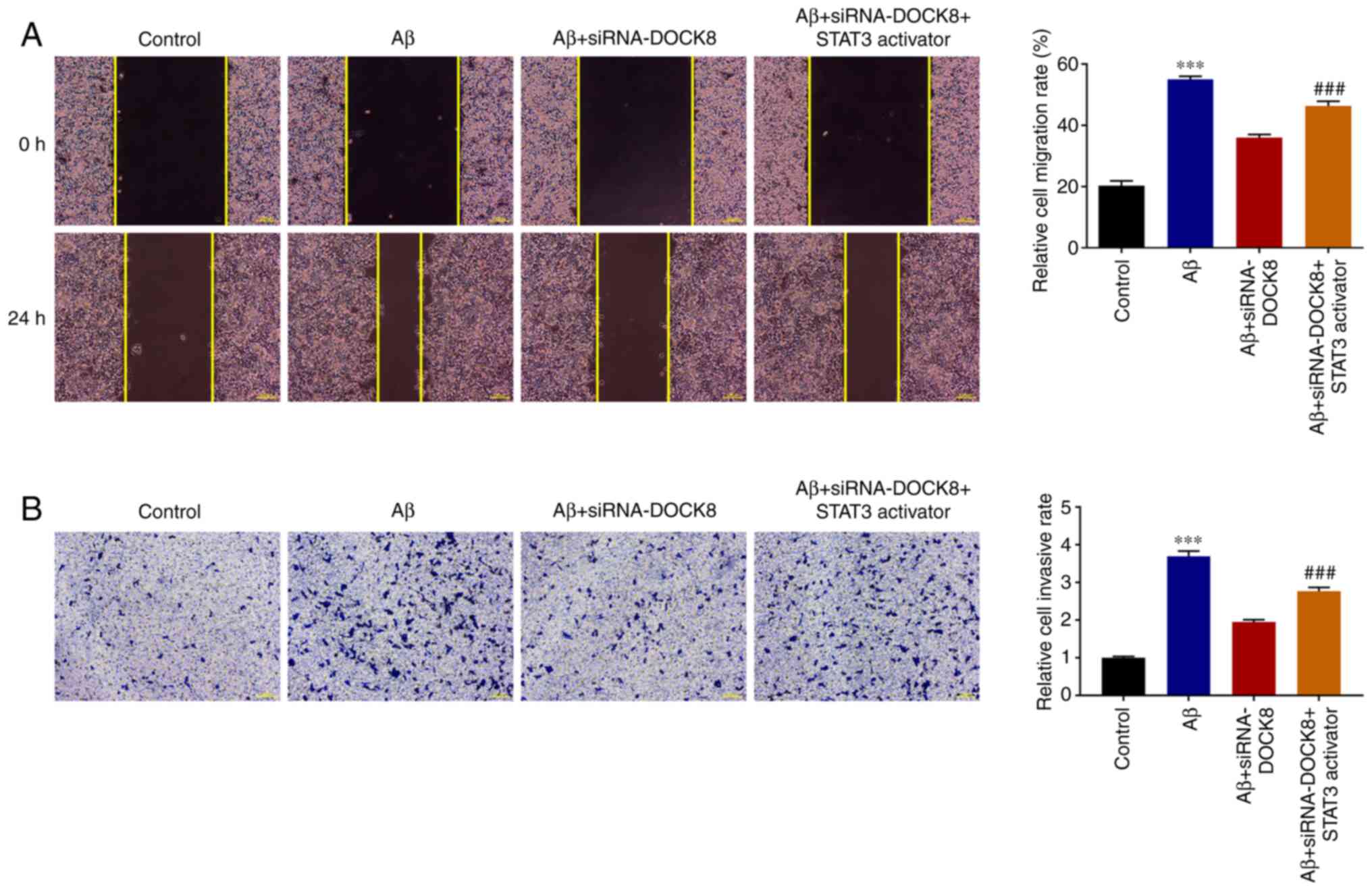
DOCK8 interference inhibits the
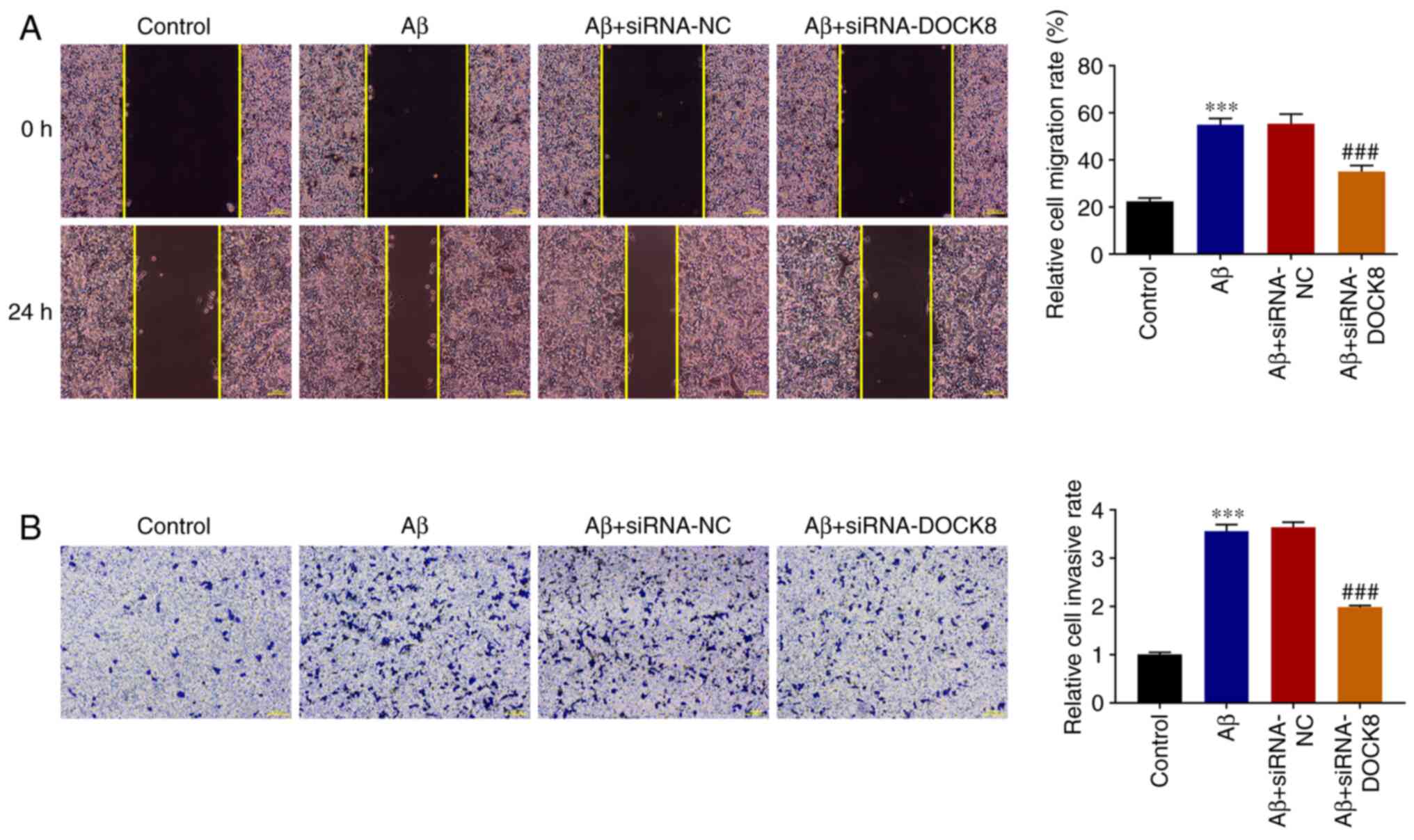
migration and invasion of Aβ-induced BV2 cells
In contrast to the Control group, the migrative
ability of BV2 cells was markedly increased by Aβ stimulation.
Nevertheless, DOCK8 silencing imparted suppressive effects on the
migration of Aβ-induced BV2 cells, as evidenced by reduced
migrative ability in the Aβ + siRNA-DOCK8 group compared with the
Aβ + siRNA-NC group (Fig. 3A).
Additionally, the enhanced invasion of Aβ-induced BV2 cells
subsequently declined following siRNA DOCK8 expression (Fig. 3B). The above results demonstrated
that DOCK8 interference inhibited the migration and invasion of
Aβ-induced BV2 cells.
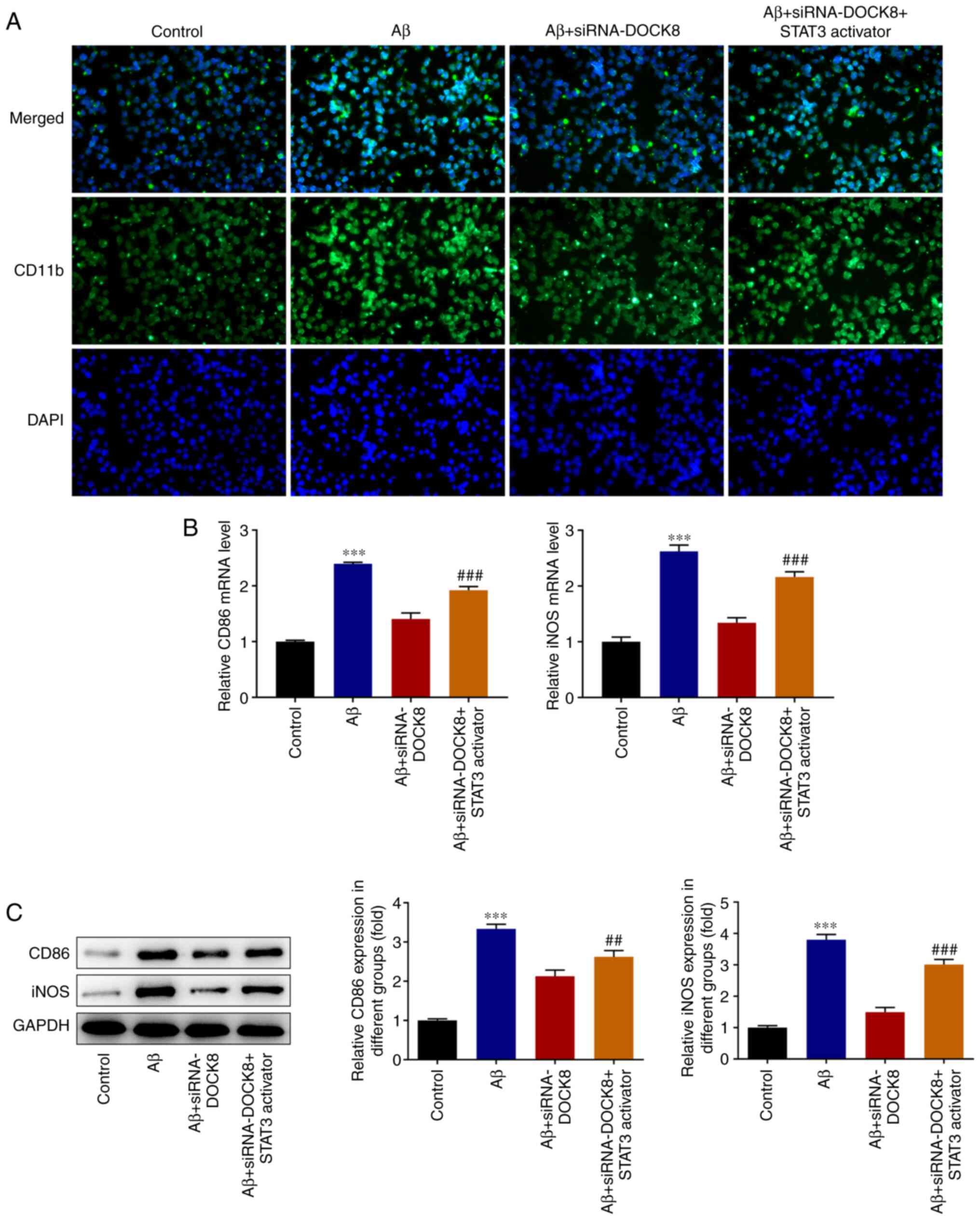
DOCK8 interference inhibits the
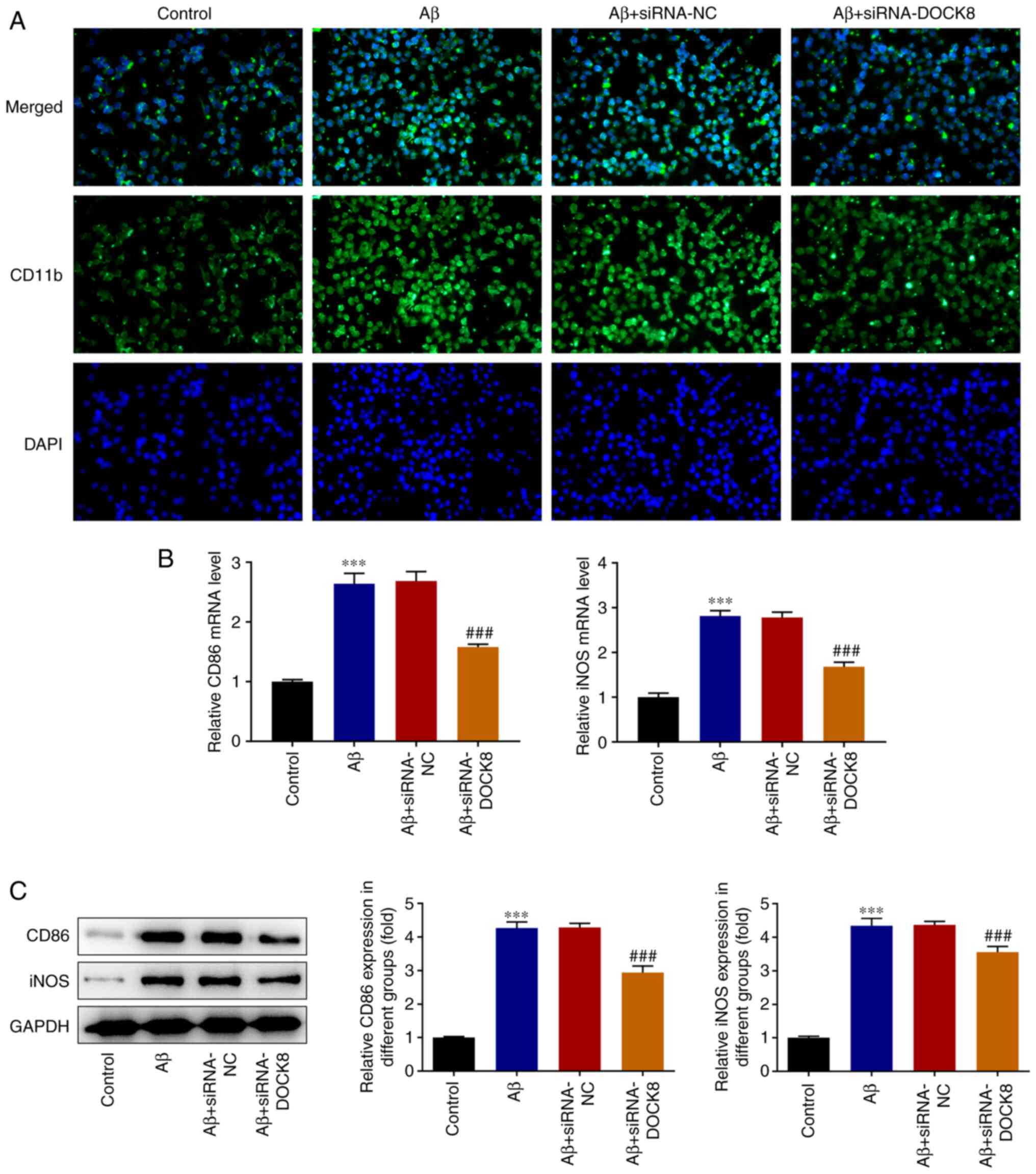
polarization of Aβ-induced BV2 cells to M1 cells
Results from IF staining showed that Aβ stimulation
conspicuously increased the level of CD11b compared with that in
the Control group while DOCK8 deficiency showed opposite effects on
this protein, evidenced by reduced CD11b content in Aβ +
siRNA-DOCK8 in comparison with that in Aβ + siRNA-NC group
(Fig. 4A). Elsewhere, the
expression levels of iNOS and CD86 in BV2 cells assessed with
RT-qPCR and western blotting were notably elevated by Aβ induction
and then reduced following interfering DOCK8 expression (Fig. 4B and C). Collectively, DOCK8 interference
inhibited the polarization of Aβ-induced BV2 cells to M1 cells.
DOCK8 interference inhibits
STAT3/NLRP3/NF-κB signal in Aβ-induced BV2 cells
Compared with the Control group, Aβ induction
remarkably increased the expression of p-STAT3, NLRP3, ASC,
caspase1 and p-p65. Nonetheless, DOCK8 depletion decreased the
contents of the above proteins in Aβ-induced BV2 cells when
compared to the Aβ + siRNA-NC group (Fig. 5). Notably, both Aβ induction and
DOCK8 silencing had no significant effects on the contents of STAT3
and p65. The above results suggested that DOCK8 interference
inhibited the STAT3/NLRP3/NF-κB signaling pathway in Aβ-stimulated
BV2 cells.
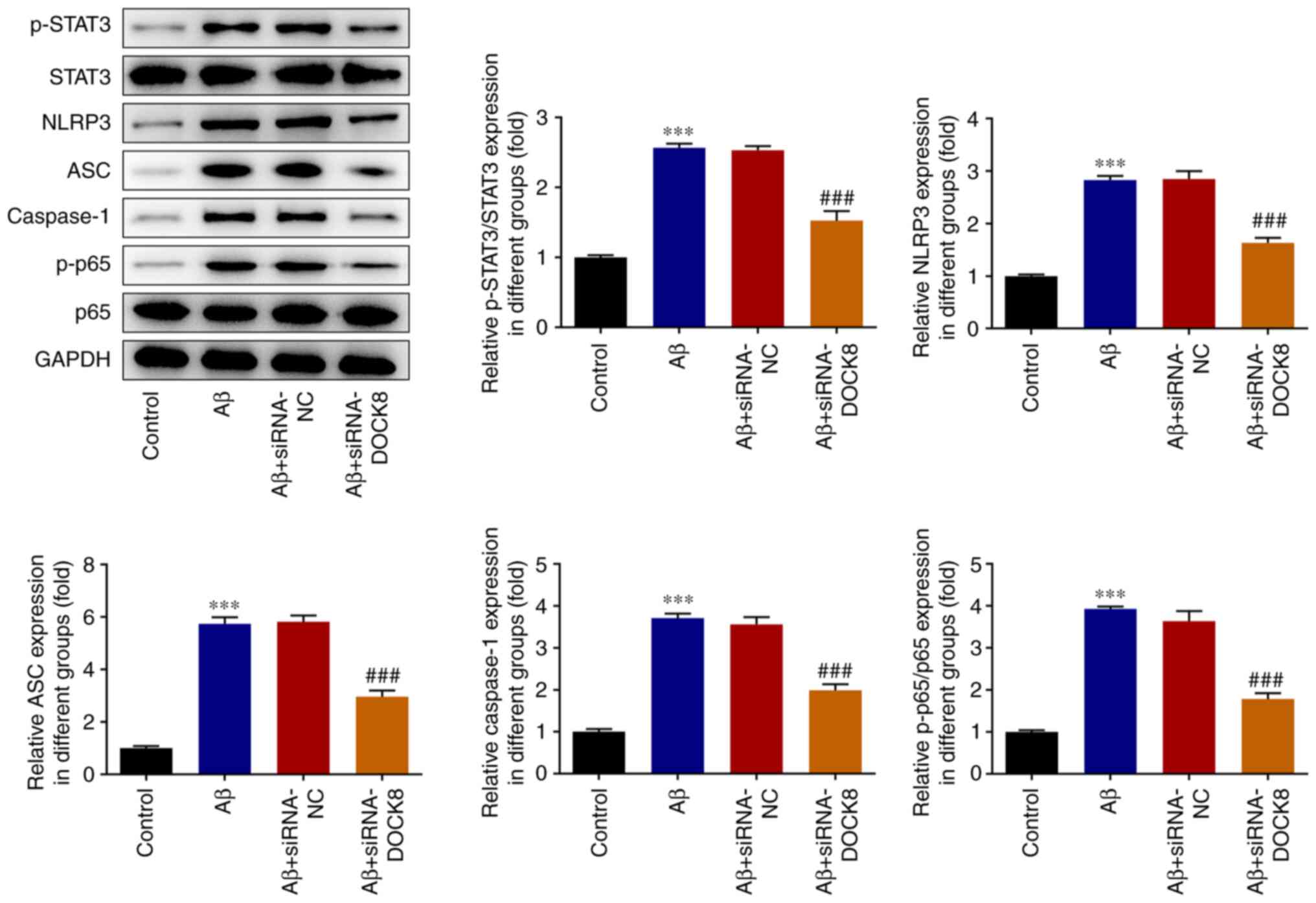 | Figure 5DOCK8 interference inhibits
STAT3/NLRP3/NF-κB signaling in Aβ-induced BV2 cells. The expression
levels of STAT3, p-STAT3, NLRP3, ASC, caspase1, p-p65 and p65 were
detected using western blotting. ***P<0.001 vs.
control; ###P<0.001 vs. Aβ + siRNA-NC. DOCK8,
dedicator of cytokinesis 8; NLRP3, NLR family pyrin domain
containing 3; Aβ, amyloid β; p-, phosphorylated; siRNA, short
interfering RNA; NC, negative control. |
DOCK8 interference inhibits the
activation and release of inflammatory factors in Aβ-induced BV2
cells via inactivation of STAT3/NLRP3/NF-κB pathway
To further investigate the mechanism of DOCK8 in
STAT3 signal, Colivelin, which is an activator of STAT3, was
administered to BV2 cells. Results obtained from western blotting
showed that the decreased contents of p-STAT3, NLRP3, ASC, caspase1
and p-p65 in Aβ-induced BV2 cells caused by DOCK8 interference were
partially elevated after administration with Colivelin (Fig. 6A). Compared with the Aβ group, the
expression of IBA-1 was decreased after depleting DOCK8 expression
and was subsequently rescued by Colivelin administration (Fig. 6B). Furthermore, DOCK8 interference
decreased the levels of TNF-α, IL-1β and IL-6 in Aβ-induced BV2
cells compared with the Aβ group while Colivelin exhibited opposite
effects on these inflammatory cytokines, as testified by increased
levels of TNF-α, IL-1β and IL-6 in Aβ + siRNA-DOCK8+STAT3 activator
group (Fig. 6C). To conclude,
DOCK8 interference suppressed the activation and inflammation of
Aβ-induced BV2 cells by blocking STAT3/NLRP3/NF-κB pathway.
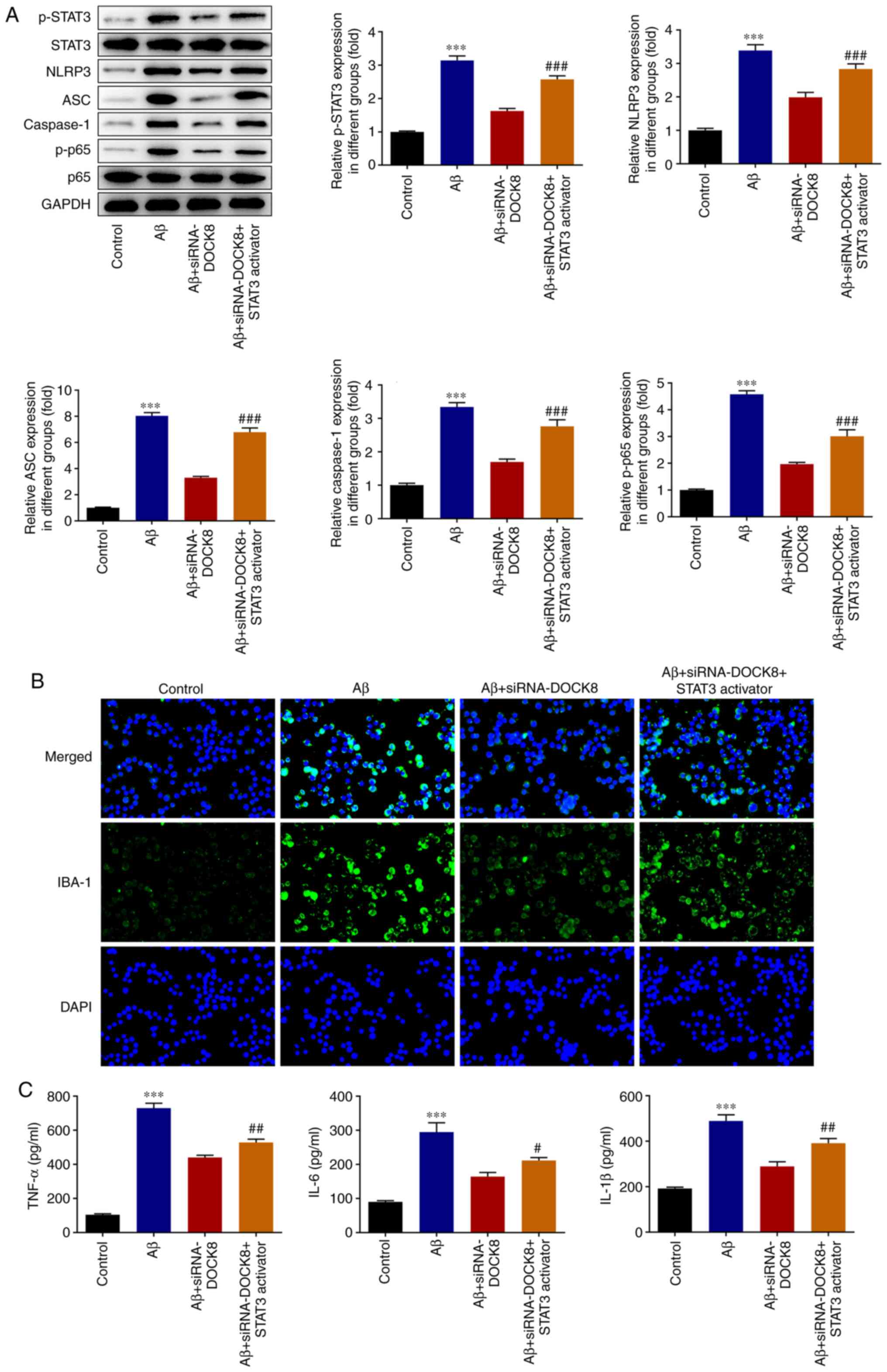 | Figure 6DOCK8 interference inhibits the
activation and inflammatory factors release of Aβ-induced BV2 cells
by suppressing STAT3/NLRP3/NF-κB signaling. (A) The expression
levels of STAT3, p-STAT3, NLRP3, ASC, caspase1, p-p65 and p65 were
detected using western blotting. (B) The expression of IBA-1 was
detected using immunofluorescence staining. Magnification, x200.
(C) The levels of inflammatory cytokines were detected using ELISA.
***P<0.001 vs. control; #P<0.05,
##P<0.01, ###P<0.001 vs. Aβ +
siRNA-DOCK8. DOCK8, dedicator of cytokinesis 8; Aβ, amyloid β;
NLRP3, NLR family pyrin domain containing 3; p-, phosphorylated;
siRNA, short interfering RNA; NC, negative control; IBA-1, ionized
calcium binding adapter molecule-1. |
DOCK8 interference inhibits the
migration and invasion of Aβ-induced BV2 cells by suppressing
STAT3/NLRP3/NF-κB pathway
In comparison with the Control group, Aβ induction
markedly increased the migrative ability of BV2 cells, which then
declined following transfection of cells with siRNA targeting DOCK8
(Fig. 7A). Nevertheless, the
decreased migration in Aβ-induced BV2 cells was quickly enhanced by
Colivelin administration, revealing that DOCK8 depletion suppressed
the migrative capability of Aβ-induced BV2 cells via inhibiting
STAT3 signal. Similarly, the reduced invasive ability of Aβ-induced
BV2 cells was also increased after treating cells with Colivelin
(Fig. 7B).
DOCK8 interference inhibits the
polarization of Aβ-induced BV2 cells to M1 cells by restraining
STAT3/NLRP3/NF-κB pathway
Evidently, the reduced CD11b level in Aβ-induced BV2
cells that resulted from DOCK8 interference was elevated by
Colivelin compared with that in Aβ + siRNA-DOCK8 group (Fig. 8A). Similarly, DOCK8 deficiency
decreased the contents of CD86 and iNOS in Aβ-induced BV2 cells
compared with the Aβ group and these were subsequently increased by
Colivelin treatment (Fig. 8B and
C). In summary, DOCK8 silencing
repressed the polarization of Aβ-induced BV2 cells to M1 cells via
blocking the STAT3/NLRP3/NF-κB pathway.
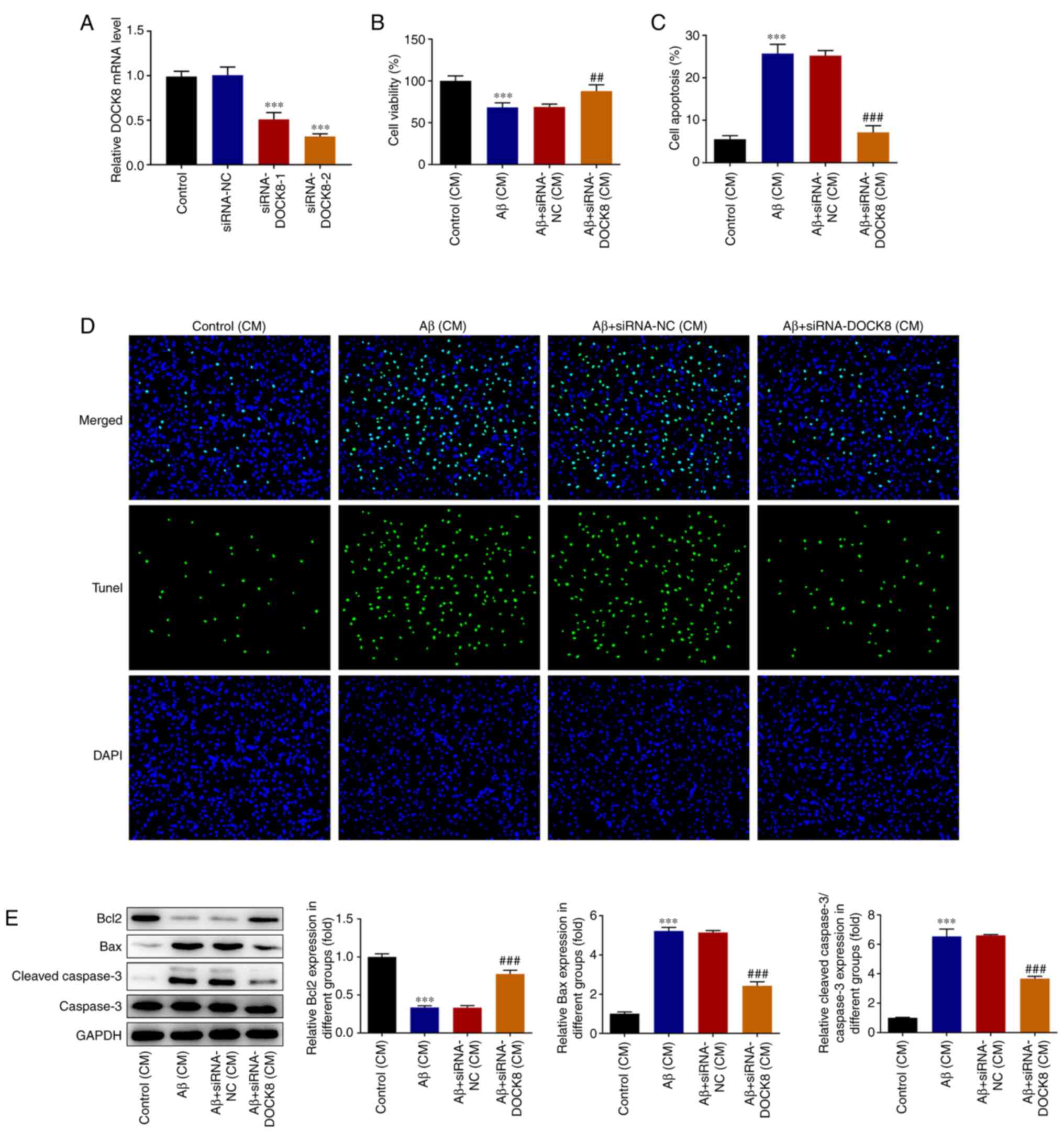
DOCK8 interference inhibits the
neuronal activity damage and apoptosis of hippocampal HT22 cells
induced by neuroinflammatory release in BV2 cells
As observed from Fig.
9A, hippocampal HT22 cells transfected with siRNA-DOCK8-1/2
presented significantly downregulated DOCK8 expression compared
with the siRNA-NC group. SiRNA-DOCK8-2 was selected to perform the
subsequent experiments due to the lower DOCK8 expression in HT22
cells. As Fig. 9B showed, Aβ
stimulation reduced the viability of HT22 cells compared with the
Control group. Nonetheless, the declined viability of Aβ-induced
HT22 cells was rapidly revived after the cells were transfection
with siRNA-DOCK8. Apoptosis was appraised by TUNEL and the results
demonstrated that Aβ stimulation conspicuously enhanced the
apoptosis level of HT22 cells compared with the Control group,
which was subsequently notably diminished by DOCK8 interference
(Fig. 9C and D). Moreover, Aβ stimulation decreased
Bcl2 content but markedly increased the contents of Bax and cleaved
caspase3 compared with the Control group, while DOCK8 depletion
exhibited opposite effects on these proteins, evidenced by elevated
Bcl2 content as well as diminished contents of Bax and cleaved
caspase3 in Aβ + siRNA-DOCK8 group (Fig. 9E).
Discussion
Extensive studies evidence that neuroinflammation
acts as a predominant player in the pathogenesis of AD as well as
other neurodegenerative disorders (4,18,19).
Accumulated Aβ deposition can be a marker of AD and the aggregation
of Aβ peptide can trigger microglia (19). In addition, the triggered microglia
can stimulate inflammatory response including the secretion of
inflammatory factors, thereby contributing to brain damage
(20). IBA-1, which is a
microglia/macrophage-specific marker, has been broadly used for
microglial assessment (21-23).
IBA-1 is increased in activated microglia (24,25),
which indicates that the upregulation of IBA-1 could serve as a
hallmark for microglial activation (26,27).
Therefore, the present study used Aβ to stimulate BV2 microglia
cells to induce inflammatory damage in vitro and then used
IF staining to resolve the expression of IBA-1. It was discovered
that Aβ induction elevated the level of IBA-1, suggesting the
activation of BV2 cells, which was consistent with the findings in
aforementioned studies.
DOCK8, which is located on chromosome 9p24.3, is
reported to be expressed in microglia (28). Previous researches have verified
that DOCK8 exhibits the feature of autoimmunity and serves as an
indispensable role in immune surveillance (29,30).
In addition, DOCK8 has been widely considered in neurodegenerative
diseases. Taking glaucoma as an instance, DOCK8 exhibits existence
in microglia and mediates microglial activity in the process of
neurodegeneration (11).
Additionally, Xu et al (31) suggest that DOCK8 is conspicuously
increased in patients suffering from multiple sclerosis. DOCK8 is
acknowledged to be a critical regulator in cell migration,
invasion, survival, inflammation as well as polarization. For
example, the interference of DOCK8 can suppress the migrative
ability of PDGF-induced Schwann cell precursor (32). A previous study demonstrated that
depleted DOCK8 contributed to decreased polarization (33). Although numerous researches have
been performed (11,31-33),
the role that DOCK8 played in AD has not yet been elucidated. In
the present study, the mRNA and protein expression levels of DOCK8
in BV2 cells were notably increased following Aβ stimulation,
revealing the upregulation of DOCK8 in AD. After silencing DOCK8, a
series of functional experiments were performed. The increased
expression of IBA-1 in Aβ-induced BV2 cells was downregulated
following silencing DOCK8, revealing that DOCK8 deficiency
suppressed the activation of BV2 cells with Aβ stimulation. In
addition, DOCK8 silencing also imparted suppressive effects on the
inflammation, migration and invasion of Aβ-stimulated BV2 cells.
Elsewhere, the decreased contents of CD11b, CD86 and iNOS in
DOCK8-silenced BV2 cells with Aβ stimulation implied that DOCK8
interference repressed the polarization of Aβ-induced BV2 cells to
M1 cells.
On the basis of size similarity, antigenic as well
as structural relatedness, STAT3 is identified as belonging to the
STAT family (34). It is reported
that STAT3 possesses an anti-inflammatory property and can mediate
crucial cellular activities, such as cell growth, apoptosis and
transcription of inflammatory genes (35,36).
Additionally, the dysregulation of STAT3 is involved with the
advance of numerous malignancies and neurodegenerative diseases
(37). STAT3 signaling can be
activated in Aβ-induced microglia (38), which can further activate the
expression of NLRP3 and NF-κB signaling, ultimately promoting the
release of inflammation (39,40).
Evidence indicates that DOCK8 can activate STAT3 signaling in B
cells (41). The same finding has
been reported by Keles et al (38); that DOCK8 can interact with STAT3
and regulates its activation in T cells. The results of the present
study showed that the enhanced contents of p-STAT3, NLRP3, ASC,
caspase1 and p-p65 in Aβ-induced BV2 cells were decreased following
interfering DOCK8 expression, implying that DOCK8 knockdown could
inhibit the STAT3/NLRP3/NF-κB signaling pathway in Aβ-induced BV2
cells. The suppressive effects of DOCK8 silencing on the
activation, inflammation, migration, invasion and polarization of
Aβ-induced BV2 cells were reversed by Colivelin, indicating that
DOCK8 interference repressed the malignant behaviors of Aβ-induced
BV2 cells by blocking STAT3/NLRP3/NF-κB signaling. Furthermore, it
was found that DOCK8 deficiency promoted the viability but
repressed the apoptosis of Aβ-induced HT22 cells, implying that
DOCK8 silencing helped to protect against hippocampal neuronal
damage. Further mechanistic studies are needed to elucidate the
interaction between Aß stimulation, DOCK8 and any subsequent
pathway in the future experiments, which is a limitation of the
present study.
In conclusion, the present study was the first, to
the best of the authors' knowledge, to discuss the regulatory role
DOCK8 in AD and uncover its detailed mechanism, which lays the
foundation for the study of DOCK8 in neurodegenerative diseases.
However, there were also some limitations. For example, the role of
DOCK8 in animal model and in clinic was not addressed in the
present study; more studies need to be performed in the future.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by Special Funds for
Fundamental Research Expenses of Central Universities (grant nos.
2017KFYXJJ084 and 2018KFYYXJJ113).
Availability of data and material
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Authors' contributions
XZ and QW designed the study and performed the
experiments. JH and DX performed the experiments and analyzed the
data. XZ and SZ interpreted the data and drafted the manuscript. QW
revised the manuscript for important intellectual content. All
authors have read and approved the final manuscript. XZ and QW
confirm the authenticity of all the raw data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cui Y, Wang Y, Zhao D, Feng X, Zhang L and
Liu C: Loganin prevents BV-2 microglia cells from Abeta1-42
-induced inflammation via regulating TLR4/TRAF6/NF-κB axis. Cell
Biol Int. 42:1632–1642. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Lane CA, Hardy J and Schott JM:
Alzheimer's disease. Eur J Neurol. 25:59–70. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
De-Paula VJ, Radanovic M, Diniz BS and
Forlenza OV: Alzheimer's disease. Subcell Biochem. 65:329–352.
2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Calsolaro V and Edison P:
Neuroinflammation in Alzheimer's disease: Current evidence and
future directions. Alzheimers Dement. 12:719–732. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Varnum MM and Ikezu T: The classification
of microglial activation phenotypes on neurodegeneration and
regeneration in Alzheimer's disease brain. Arch Immunol Ther Exp
(Warsz). 60:251–266. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Unger MS, Li E, Scharnagl L, Poupardin R,
Altendorfer B, Mrowetz H, Hutter-Paier B, Weiger TM, Heneka MT,
Attems J and Aigner L: CD8(+) T-cells infiltrate Alzheimer's
disease brains and regulate neuronal- and synapse-related gene
expression in APP-PS1 transgenic mice. Brain Behav Immun. 89:67–86.
2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Mosher KI and Wyss-Coray T: Microglial
dysfunction in brain aging and Alzheimer's disease. Biochem
Pharmacol. 88:594–604. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kang SY, Jung HW, Lee MY, Lee HW, Chae SW
and Park YK: Effect of the semen extract of Cuscuta chinensis on
inflammatory responses in LPS-stimulated BV-2 microglia. Chin J Nat
Med. 12:573–581. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Namekata K, Kimura A, Kawamura K, Harada C
and Harada T: Dock GEFs and their therapeutic potential:
Neuroprotection and axon regeneration. Prog Retin Eye Res. 43:1–16.
2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kearney CJ, Randall KL and Oliaro J: DOCK8
regulates signal transduction events to control immunity. Cell Mol
Immunol. 14:406–411. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Namekata K, Guo X, Kimura A, Arai N,
Harada C and Harada T: DOCK8 is expressed in microglia, and it
regulates microglial activity during neurodegeneration in murine
disease models. J Biol Chem. 294:13421–13433. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Biggs CM, Keles S and Chatila TA: DOCK8
deficiency: Insights into pathophysiology, clinical features and
management. Clin Immunol. 181:75–82. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Moon MY, Kim HJ, Li Y, Kim JG, Jeon YJ,
Won HY, Kim JS, Kwon HY, Choi IG, Ro E, et al: Involvement of small
GTPase RhoA in the regulation of superoxide production in BV2 cells
in response to fibrillar Abeta peptides. Cell Signal. 25:1861–1869.
2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Cimino PJ, Yang Y, Li X, Hemingway JF,
Cherne MK, Khademi SB, Fukui Y, Montine KS, Montine TJ and Keene
CD: Ablation of the microglial protein DOCK2 reduces amyloid burden
in a mouse model of Alzheimer's disease. Exp Mol Pathol.
94:366–371. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Jiang CQ, Ma LL, Lv ZD, Feng F, Chen Z and
Liu ZD: Polydatin induces apoptosis and autophagy via STAT3
signaling in human osteosarcoma MG-63 cells. J Nat Med. 74:533–544.
2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Jian M, Kwan JS, Bunting M, Ng RC and Chan
KH: Adiponectin suppresses amyloid-β oligomer (AβO)-induced
inflammatory response of microglia via AdipoR1-AMPK-NF-κB signaling
pathway. J Neuroinflammation. 16(110)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Latta CH, Brothers HM and Wilcock DM:
Neuroinflammation in Alzheimer's disease; A source of heterogeneity
and target for personalized therapy. Neuroscience. 302:103–111.
2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Heneka MT, Carson MJ, El Khoury J,
Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T,
Vitorica J, Ransohoff RM, et al: Neuroinflammation in Alzheimer's
disease. Lancet Neurol. 14:388–405. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Heppner FL, Ransohoff RM and Becher B:
Immune attack: The role of inflammation in Alzheimer disease. Nat
Rev Neurosci. 16:358–372. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Imai Y, Ibata I, Ito D, Ohsawa K and
Kohsaka S: A novel gene iba1 in the major histocompatibility
complex class III region encoding an EF hand protein expressed in a
monocytic lineage. Biochem Biophys Res Commun. 224:855–862.
1996.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Maneu V, Noailles A, Megias J,
Gómez-Vicente V, Carpena N, Gil ML, Gozalbo D and Cuenca N: Retinal
microglia are activated by systemic fungal infection. Invest
Ophthalmol Vis Sci. 55:3578–3585. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Shi FJ, Xie H, Zhang CY, Qin HF, Zeng XW,
Lou H, Zhang L, Xu GT, Zhang JF and Xu GX: Is Iba-1 protein
expression a sensitive marker for microglia activation in
experimental diabetic retinopathy? Int J Ophthalmol. 14:200–208.
2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Muhammad T, Ikram M, Ullah R, Rehman SU
and Kim MO: Hesperetin, a citrus flavonoid, attenuates LPS-induced
neuroinflammation, apoptosis and memory impairments by modulating
TLR4/NF-κB signaling. Nutrients. 11(648)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Park T, Chen H, Kevala K, Lee JW and Kim
HY: N-Docosahexaenoylethanolamine ameliorates LPS-induced
neuroinflammation via cAMP/PKA-dependent signaling. J
Neuroinflammation. 13(284)2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhu SH, Liu BQ, Hao MJ, Fan YX, Qian C,
Teng P, Zhou XW, Hu L, Liu WT, Yuan ZL and Li QP: Paeoniflorin
suppressed high glucose-induced retinal microglia MMP-9 expression
and inflammatory response via inhibition of TLR4/NF-κB pathway
through upregulation of SOCS3 in diabetic retinopathy.
Inflammation. 40:1475–1486. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hopperton KE, Mohammad D, Trepanier MO,
Giuliano V and Bazinet RP: Markers of microglia in post-mortem
brain samples from patients with Alzheimer's disease: A systematic
review. Mol Psychiatry. 23:177–198. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Dobryakova YV, Kasianov A, Zaichenko MI,
Stepanichev MY, Chesnokova EA, Kolosov PM, Markevich VA and
Bolshakov AP: Intracerebroventricular administration of
(192)IgG-Saporin alters expression of microglia-associated genes in
the dorsal but not ventral hippocampus. Front Mol Neurosci.
10(429)2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Aydin SE, Kilic SS, Aytekin C, Kumar A,
Porras O, Kainulainen L, Kostyuchenko L, Genel F, Kütükcüler N,
Karaca N, et al: DOCK8 deficiency: Clinical and immunological
phenotype and treatment options-a review of 136 patients. J Clin
Immunol. 35:189–198. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang Z, Bao Y, Zhou L, Ye Y, Fu W and Sun
C: DOCK8 Serves as a prognostic biomarker and is related to immune
infiltration in patients with HPV positive head and neck squamous
cell carcinoma. Cancer Control.
28(10732748211011951)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Xu X, Han L, Zhao G, Xue S, Gao Y, Xiao J,
Zhang S, Chen P, Wu ZY, Ding J, et al: LRCH1 interferes with
DOCK8-Cdc42-induced T cell migration and ameliorates experimental
autoimmune encephalomyelitis. J Exp Med. 214:209–226.
2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Miyamoto Y, Torii T, Kawahara K, Tanoue A
and Yamauchi J: Dock8 interacts with Nck1 in mediating Schwann cell
precursor migration. Biochem Biophys Rep. 6:113–123.
2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ham H, Guerrier S, Kim J, Schoon RA,
Anderson EL, Hamann MJ, Lou Z and Billadeau DD: Dedicator of
cytokinesis 8 interacts with talin and Wiskott-Aldrich syndrome
protein to regulate NK cell cytotoxicity. J Immunol. 190:3661–3669.
2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Hillmer EJ, Zhang H, Li HS and Watowich
SS: STAT3 signaling in immunity. Cytokine Growth Factor Rev.
31:1–15. 2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Takeda K, Kaisho T, Yoshida N, Takeda J,
Kishimoto T and Akira S: Stat3 activation is responsible for
IL-6-dependent T cell proliferation through preventing apoptosis:
Generation and characterization of T cell-specific Stat3-deficient
mice. J Immunol. 161:4652–4660. 1998.PubMed/NCBI
|
|
36
|
Kortylewski M and Yu H: Role of Stat3 in
suppressing anti-tumor immunity. Curr Opin Immunol. 20:228–233.
2008.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Egwuagu CE: STAT3 in CD4+ T
helper cell differentiation and inflammatory diseases. Cytokine.
47:149–156. 2009.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Keles S, Charbonnier LM, Kabaleeswaran V,
Reisli I, Genel F, Gulez N, Al-Herz W, Ramesh N, Perez-Atayde A,
Karaca NE, et al: Dedicator of cytokinesis 8 regulates signal
transducer and activator of transcription 3 activation and promotes
TH17 cell differentiation. J Allergy Clin Immunol. 138:1384–1394
e1382. 2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zhu H, Jian Z, Zhong Y, Ye Y, Zhang Y, Hu
X, Pu B, Gu L and Xiong X: Janus kinase inhibition ameliorates
ischemic stroke injury and neuroinflammation through reducing NLRP3
inflammasome activation via JAK2/STAT3 pathway inhibition. Front
Immunol. 12(714943)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Dai R, Jiang Q, Zhou Y, Lin R, Lin H,
Zhang Y, Zhang J and Gao X: Lnc-STYK1-2 regulates bladder cancer
cell proliferation, migration, and invasion by targeting
miR-146b-5p expression and AKT/STAT3/NF-kB signaling. Cancer Cell
Int. 21(408)2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Pal M, Bao W, Wang R, Liu Y, An X,
Mitchell WB, Lobo CA, Minniti C, Shi PA, Manwani D, et al:
Hemolysis inhibits humoral B-cell responses and modulates
alloimmunization risk in patients with sickle cell disease. Blood.
137:269–280. 2021.PubMed/NCBI View Article : Google Scholar
|