Introduction
Acute kidney injury (AKI) induced by endotoxins is a
common cause of morbidity and mortality in critically ill patients
(1-3).
Previous studies have demonstrated that oxidative stress and
persistent inflammation can result in the necrosis and apoptosis of
renal tubular epithelial cells (RTECs) (4,5).
Recently, the pyroptosis of RTECs was observed in endotoxin-induced
AKI in vivo (6). Pyroptosis
is a type of programmed cell death that occurs when a cell bulks up
until the cell membrane bursts, causing the release of cellular
contents, which activate a potent inflammatory response (7,8). It
is characterized by caspase-1 and IL-1β activation, which is
associated with the release of a large number of pro-inflammatory
factors. Pyroptosis is a natural immune response in the body that
plays a critical role in combatting infections (9). Mitochondrial injury is related to
pyroptosis (10).
Endotoxin-induced AKI leads to mitochondrial fission and RTEC
pyroptosis (11).
The core of cellular energy metabolism is the
mitochondrion, which provides energy for cellular metabolism in the
form of adenosine triphosphate (ATP). Cellular stress leads to
mitochondrial damage and dysfunction, resulting in damage to the
electron respiration complex, mitochondrial oxygen consumption,
oxidative phosphorylation, decreased ATP synthesis and the
increased production of reactive oxygen species (ROS). These
processes induce programmed cell death (12). In addition, it has been shown that
the tendency of RTEC mitochondria to divide may induce dysfunction.
The inhibition of the latter is the key to endogenous protection
towards the treatment of AKI induced by endotoxemia (13).
Heme oxygenase (HO) is a rate-limiting enzyme used
in the conversion of heme into biliverdin and bilirubin. It
contains two isoforms, namely HO-1 and HO-2. HO-1 inhibits
inflammatory responses, reduces oxidative stress and improves cell
survival rates (14). In recent
years, HO-1 has been shown to exert a protective effect on
endogenous levels. HO-1 can inhibit inflammatory responses and
reduce oxidative stress to improve endotoxemia-induced organ damage
by regulating specific organelles, such as the mitochondria and the
endoplasmic reticulum (15,16).
PTEN-induced putative kinase 1 (PINK1) can clear dysfunctional
mitochondria by regulating mitochondrial autophagy (17). HO-1 regulates mitochondrial
fusion/fission via PINK1 to improve endotoxin-induced AKI in
vivo (11). However, whether
HO-1 regulates mitochondrial dysfunction through PINK1 in
vitro and inhibits pyroptosis in lipopolysaccharide
(LPS)-stimulated RTECs remains to be determined. In the present
study, it was hypothesized that HO-1 inhibits inflammation,
regulates mitochondrial function and inhibits focal prolapse,
thereby reducing the damage caused to RTECs by the LPS-mediated
induction of PINK1 expression.
Materials and methods
Animals
A total of 40 1-month-old Sprague-Dawley neonatal
rats were purchased, including 20 wild-type (WT) neonatal rats and
20 PINK1-knockout (PINK1KO) neonatal rats. The 1-month-old male
Sprague-Dawley neonatal rats (weighing 50-60 g) were provided by
the Laboratory Animal Center of the Nankai Clinical Institution of
Tianjin Medical University, Tianjin, China. The 1-month-old PINK1KO
neonatal rats (weighing 50-60 g) were purchased from Beijing Baiao
Saitu Gene Biotechnology Co. Ltd. (EGE-WL-008). PINK1 gene
conditional knockout in Sprague-Dawley rats was successfully
established using the CRISPR/Cas9 method (Data S1, and Figs. S1 and S2).
The WT and PINK1KO neonatal rats used in the
experiments were age- and weight-matched littermates. The neonatal
rats were kept alone in a cage at 23-25˚C and adapted to a 12-h
light-dark cycle, 60-65% humidity, with free access to food and
water.
The present study was approved by the Animal Ethical
and Welfare Committee of the Institute of Radiation Medicine,
Chinese Academy of Medical Sciences (no. IRM-DWLL-201907) and was
performed in accordance with the ARRIVE guidelines developed by the
National Center for the Replacement, Refinement, and Reduction of
Animals in Research. As the neonatal rats in this experiment were
sacrificed and part of the experiments were conducted in the animal
laboratory of the aforementioned institution, the ethics approval
for the use of animals was provided by this institution.
Cells and cell culture
The ex vivo primary culture of RTECs in WT
rats and PINK1KO rats was performed as previously described
(18). The neonatal rats (weighing
50-60 g) were anesthetized by the inhalation of 3% isoflurane until
the four toes of the rats were pinched with tweezers and the rats
had no reaction after pinching; the rats were then sacrificed by
cervical dislocation and disinfected with 75% alcohol for 3 min.
Their skin was cut open at the coastal ridge angle to expose the
kidneys. The kidneys were gently removed and the ureter and blood
vessels were cut; the kidneys were placed in medium containing
double resistance (cat. no. 12100-500; Beijing Solarbio Science
& Technology Co., Ltd.; 1% of two antibiotics, including
penicillin and streptomycin). Subsequently, the kidneys were cut
open, the pedicles were cut off, and the capsules were separated
and removed. The medulla of the kidneys was removed and the cortex
was cut and placed into a Petri dish containing double-resistance
PBS (Biosharp Life Sciences). The cortex was dissected under a
microscope (optical, Olympus Corporation) and the solution was
filtered using 100- and 80-micron filtering screens. The filtrate
containing the cells was obtained from sieve filtration, added to
50-ml centrifuge tubes, and centrifuged at 250 x g at room
temperature. The supernatant was discarded and the sample was
digested with collagenase for 20 min at 37˚C in a water bath (every
3-5 min and percussion). The sample was centrifuged (rotational
speed, 250 x g at room temperature; duration, 5 min), the
supernatant was discarded and whole medium was added at a 2:1
volume ratio. The mixture was discarded evenly with a straw to
terminate the digestion, and subsequently was centrifuged at a
rotational speed of 250 x g at room temperature for 5 min. The
number and density of the cells were observed under a microscope
(optical, Olympus Corporation) and subsequently divided into
culture plates. RTECs are identified by their morphology (Data S1,
and Figs. S3 and S4). The cells were cultured in an
incubator at 37˚C containing a 5% CO2. The solution was
changed every 2-3 days. The cells were seeded in 96-well culture
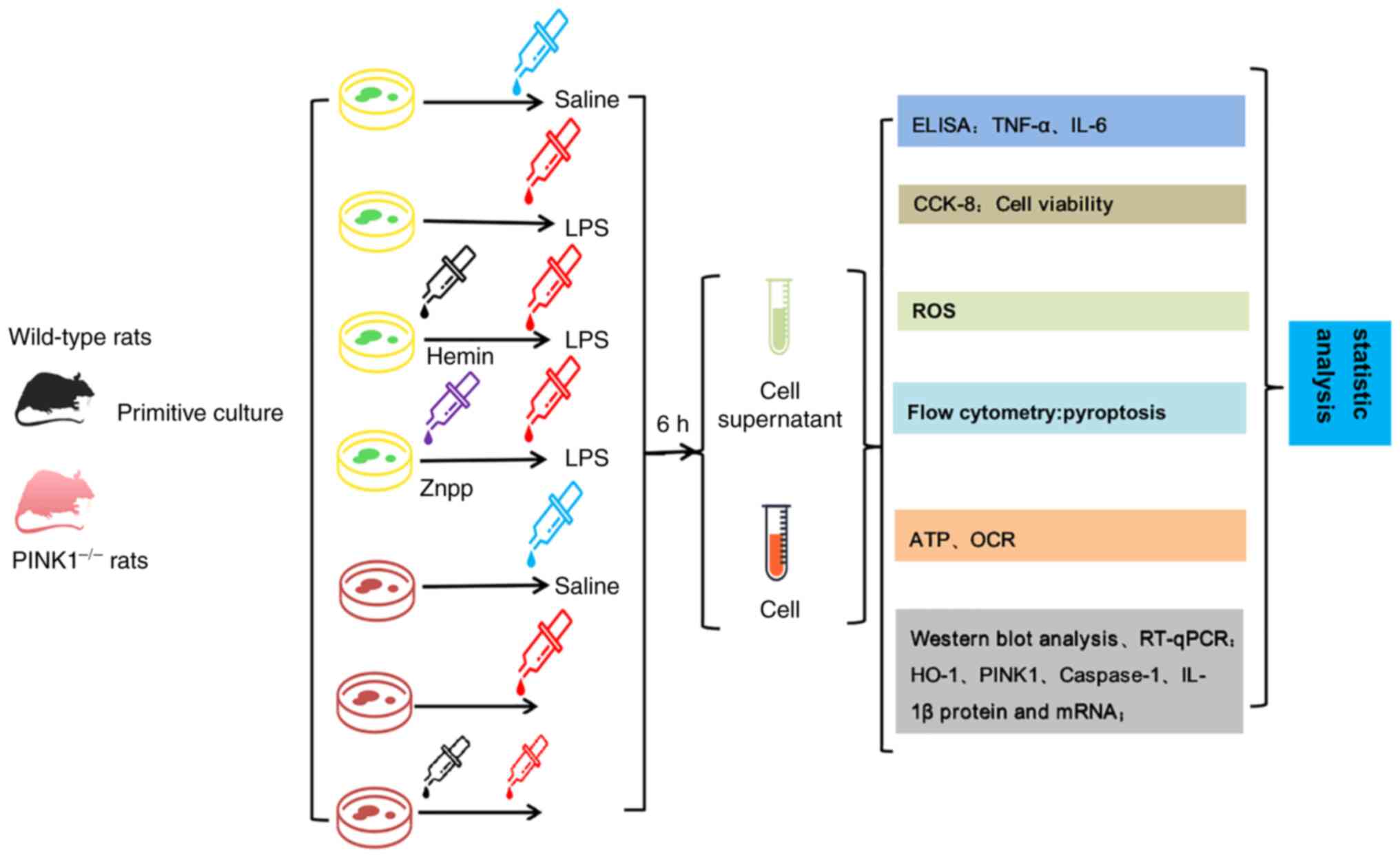
plates at a density of 4x104 cells/ml. The RTECs were
divided into seven groups (n=3) as follows: The WT control RTECs,
LPS, zinc protoporphyrin IX (Znpp) + LPS and Hemin + LPS groups.
The PINK1KO RTECs were divided into the control, LPS and Hemin +
LPS groups. Normal culture was performed in the control group and 1
µg/ml LPS was added to each medium in the LPS, Znpp + LPS and Hemin
+ LPS groups to stimulate the RTECs. The RTECs in the Znpp + LPS
and Hemin + LPS groups were treated with 10 µM of the HO-1
inhibitor, ZnPP (Sigma-Aldrich; Merck KGaA), and 20 µM of the HO-1
promoter, hemin (Sigma-Aldrich; Merck KGaA), for 30 min prior to
LPS (Beijing Solarbio Science & Technology Co., Ltd.)
stimulation. A flow diagram of the experiment is presented in
Fig. 1.
Cell viability
The RTECs were incubated overnight in 96-well plates
at a density of 4x104 cells/ml at 37˚C in a cell culture
incubator containing 5% CO2. The medium was replaced
with DMEM (cat. no. 8122059; Gibco; Thermo Fisher Scientific, Inc.)
containing 1% FBS (cat. no. 212619; Biological Industries), which
was synchronously treated for 24 h. Each group was provided with
the corresponding treatment. Following incubation at 37˚C in a cell
culture incubator containing 5% CO2 for 24 h, 10 µl Cell
Counting Kit-8 (CCK-8; 0.5 g/l; cat. no. BS350B, Biosharp Life
Sciences) solution was added to each well, which was incubated at
37˚C for 4 h. The liquid in the plate was removed by centrifugation
at 320 x g at room temperature for 10 min. Subsequently, dimethyl
sulfoxide (Beijing Solarbio Science & Technology Co., Ltd.)
(100 µl/well) was added and mixed, and the optical density (OD) at
490 nm was determined using a microplate reader (Hidex), indicating
cell viability.
Enzyme-linked immunosorbent assay
(ELISA)
ELISA kits were used to detect the levels of the
inflammatory factors, IL-6 (cat. no. EK306; LiankeBio) and TNF-α
(cat. no. CSB-E11987BC; Cusabio Technology, LLC) in the cell
supernatant. The experimental procedures adhered to the
instructions described in the manufacturer's protocol.
Determination of pyroptosis using flow
cytometry
The RTECs were washed twice using cold PBS buffer,
and a 1x106 cells/ml cell suspension was prepared. The
cell suspension (100 µl) was added to the test tube and mixed
gently with fluorescence-labeled Annexin V nucleic acid dye 660
Caspase-1 Assay (cat. no. 9122; ImmunoChemistry Technologies,
LLC.). The Annexin V nucleic acid dye was placed in the dark at
room temperature for 15 min. The cells were initially washed with a
buffer solution and the supernatant was removed when Annexin
V-biotin was used for testing. Subsequently, Annexin V-fluorescein
isothiocyanate (0.5 µg) was dissolved in 100 µl buffer solution,
added to the tube containing the cells, and mixed gently. The
buffer solution (400 µl) was added to each test tube, and the
results were measured using a flow cytometer (FACSCalibur II, BD
Biosciences) within 1 h. The proportion of FL4-H in each cell
sample was analyzed using CellQuest 6.1 software (BD
Biosciences).
Extraction of mitochondria from
RTECs
The RTECs were removed, placed into the pre-cooled
medium I, and washed twice. A total of 3 ml pre-cooled medium I was
added to the Petri dishes. The pre-cooled homogenate (5 ml) was
moved to a glass tube. In the ice bath, a 160 x g electric
homogenizer was used thrice. The homogenate was pre-cooled in a
50-ml centrifuge tube, and 4 ml precooled medium I was added,
followed by centrifugation at 4˚C at 40 x g for 10 min. The
precipitate was discarded, and the supernatant was centrifuged at
320 x g at 4˚C for 10 min. The supernatant was subsequently removed
and 5 ml medium I (including 250 µl fatty acid-free BSA) were added
to the precipitate; the resulting solution was further centrifuged
at 4˚C at 5,400 x g for 10 min. The supernatant was removed,
precipitated and 2 ml medium II were added to the sample, including
40 µl BSA. The resulting mixture was incubated at 4˚C and
centrifuged at 1,400 x g for 10 min. A total of 300 µl II suspended
medium was precipitated. Of note, the entire process was performed
on ice, and the reagent, centrifuge tube, and homogenizer tube were
pre-cooled. The reagent preparation included the following: Medium
I (0.12 M KCl, 20 mM HEPES, 5 mM MgCl2, 1 mM EDTA, pH
7.4); medium II (0.3 M sucrose, 2.0 mM HEPES, 0.1 mM EDTA, pH 7.4);
fatty acid-free BSA: 0.1 g/ml (cat. No. G007-1-1, Nanjing Jiancheng
Bioengineering Institute).
Detection of mitochondrial ROS
production
The fluorescent probe, dihydroethidium (DHE), was
used for labeling ROS. The underlying principle is as follows: DHE
freely enters the mitochondria through the mitochondrial membrane
and is oxidized by mitochondrial ROS to form ethidium, which can
bind to the chromosomal DNA and produce fluorescence. Briefly, the
cells were cultured in 96-well plates until the cultures were
confluent. Subsequently, the cell suspension was incubated with 10
µM DHE at 37˚C for 30 min. A Chameleon microplate reader (Hidex)
was used to monitor the DHE fluorescence at an excitation
wavelength of 480 nm and an emission wavelength of 530 nm. The
results were reported as the differences from the initial
fluorescence. Based on the production of fluorescence and the
change in its activity, the amount and change in the mitochondrial
ROS content could be determined.
Determination of mitochondrial
ATP
According to the instructions of the ATP detection
kit (cat. No. A016-1; Nanjing Jiancheng Bioengineering Institute),
an enzymolysis reaction was performed as follows: 130 µl solution A
was added to a tube followed by 100 µl of sample and mixed; the
reaction was performed at 37˚C for 10 min. Subsequently, 750 µl
reagent were added to the tube and mixed with the sample. The tube
was centrifuged at 1,000-1,800 x g at room temperature for 10 min,
and the supernatant containing phosphorus was obtained. A standard
phosphorus application solution (100 µl) with a concentration of 1
M/ml was added to the standard tube and 100 µl supernatant were
added to the ABCDE tube. The phosphorus fixative was added to the
ABCDE tube, mixed, and cooled to room temperature in a water bath
at 45˚C for 20 min. At 660 nm, the optical diameter was 1 cm. The
following formula was used to calculate the ATPase activity: ATPase
activity (U/mg prot)=(measured OD value-control OD value)/standard
OD value x concentration of standard substance (1 M/ml) x dilution
ratio of samples in the reaction system x 6/concentration of
protein sample to be measured (mg prot/ml).
Mitochondrial oxygen consumption
Mitochondrial oxygen consumption was monitored using
the MitoXpress oxygen-sensitive probe (Cayman Chemical Company)
according to the manufacturer's protocols. Briefly, 10 µl
phosphorescent oxygen probe were added to each well. The wells were
sealed with 100 µl 19HS mineral oil (Cayman Chemical
Company). The plate was measured kinetically for 30 min to ensure
that the fluorescent signal was stable. Time-resolved fluorescence
measurements were performed at 380 nm excitation and 650 nm
emission with a delay of 30 µs and a gate time of 100 µs using a
fluorescence microplate reader (Infinite M200, Tecan Group,
Ltd.).
Western blot analysis
The concentration of protein from the RTECs was
determined using a BCA protein quantitative kit (Thermo Fisher
Scientific, Inc.), and the amount of protein loading was
calculated. An appropriate amount of RIPA buffer (Beyotime
Institute of Biotechnology) was used for lysis and the sample was
incubated for 24 h. The protein was extracted according to the
manufacturer's instructions (Thermo Fisher Scientific, Inc.). the
amount of protein loaded per lane was 40 µg. Electrophoresis was
performed using a 12% SDS-PAGE gel. Following PVDF membrane
transfer, the membrane was incubated with 5% skimmed milk powder at
37˚C for 2 h. After washing, the membrane was incubated with the
following antibodies at 4˚C overnight: HO-1 (1:500; cat. no.
ab13248; Abcam), PINK1 (1:600; cat. no. ab23707; Abcam), caspase-1
(1:800; cat. no. 22915-1-AP; ProteinTech Group, Inc.), IL-1β
(1:1,000; cat. no. ab9722; Abcam), and β-actin (1:1,000; cat. no.
TA-09; Abcam). The following morning, the membrane was washed with
TBS-Tween-20 (TBST) to remove the excess primary antibodies and
incubated with a secondary rabbit or goat IgG antibody (1:3,000;
cat. no. zb-2301, ZSGB Institute of Biotechnology), for 1 h at room
temperature. Following additional washing with TBST, the membrane
was incubated with ECL reagents (Beyotime Institute of
Biotechnology) for 2-3 min at room temperature, and the signal
corresponding to protein expression was developed in a darkroom on
a film. The experiment was repeated thrice and the ratio of the
gray values of the target protein bands to those of β-actin bands
was analyzed. The area and integrated optical density of the bands
were analyzed using Image-Pro Plus 6.0 software (Media Cybernetics,
Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
A high-purity RNA kit (Roche Diagnostics) was used
for the isolation of total RNA from the RTECs and a
spectrophotometer [Runqee (Shanghai) Instruments Technology Co.,
Ltd.)] was used to quantify the absorbance at 260 nm. Subsequently,
5 µl total RNA were reverse transcribed; cDNA was synthetized using
the PrimeScript RT Reagent Kit (no. 6110A, Takara Bio, Inc.) as
follows: 4 µl dNTP Mix, 2 µl Primer Mix and 7 µl RNA template were
added to the tube and placed on the vibrator for full mixing,
incubated at 70˚C for 10 min, and then rapidly washed on in ice for
2 min. Subsequently, 5X RT Buffer (4 µl), DTT (2 µl) and HiFiScript
(1 µl) were added for full mixing, and incubated at 50˚C for 15
min, and then at 85˚C for 5 min. The mixture was then stored at
-80˚C to prevent degradation. The cDNA was subjected to PCR using
an ABI Prism 7000 sequence detector system (Applied Biosystems;
Thermo Fisher Scientific, Inc.). PCR was conducted under the
following conditions: 95˚C for 30 sec; 95˚C for 5 sec, 60˚C for 34
sec (40 cycles); and 95˚C, 5 sec, and 60˚C for 60 sec. The primer
sequences used are listed in Table
I (Mr. Yan-Fang Liu, research fellow from Tianjin Yishengyuan
Biotechnology Co. Ltd. designed these sequences). The threshold
cycle was obtained from triplicate samples and averaged. β-actin as
an internal control for normalization. The calculations were based
on the ΔΔCq method using the equation R (ratio)=2-ΔΔCq
(19).
 | Table IThe primer sequences used in the
present study. |
Table I
The primer sequences used in the
present study.
| Gene | Forward | Reverse |
|---|
| β-actin |
5'-CGCGAGTACAACCTTCTTGC-3' |
5'-ATACCCACCATCACACCCTG-3' |
| HO-1 |
5'-GACAGAGTTTCTTCGCCAGA-3' |
5'-GCCACGGTCGCCAACAGGAA-3' |
| IL-1β |
5'-TGTGGCAGCTACCTATGTCTT-3' |
5'-AGTGCAGCTGTCTAATGGGAA-3' |
|
Caspase1 |
5'-ACACCCACTCGTACACGTCTT-3' |
5'-TTGTCATCTCCAGAGCTGTGA-3' |
| PINK1 |
5'-TACCGCTTCTTCCGCCAGTC-3' |
5'-CGCCTGCTTCTCCTCGATCA-3' |
Statistical analysis
SPSS (IBM Corp.) statistical software (version 23.0)
was used for statistical analysis. The measurement data of the
normal distribution are expressed as mean ± standard deviation. The
sample was repeated three times. Comparisons between groups were
performed using one-way analysis of variance (ANOVA) followed by
Tukey's post hoc test. Mitochondrial oxygen consumption was
analyzed using two-way ANOVA followed by Bonferroni's post hoc
test. Prism 8.3.0 software (GraphPad Software, Inc.) was used to
plot the graphs. P<0.05 was considered to indicate a
statistically significant difference.
Results
HO-1 regulates PINK1 in LPS-stimulated
RTECs
Successful PINK1KO was established by the
identification of the tail genotypes of F1 generation rats and the
determination of PINK1 protein and mRNA expression (Figs. S1 and S2). PINK1 protein and mRNA were barely
expressed in the PINK1KO RTECs (Fig.
2A, C and E). No significant differences were noted
in the protein and mRNA expression levels of PINK1 between the LPS
and hemin pre-treatment groups in the PINK1KO RTECs, which
indicated that the PINK1 gene was knocked out. To confirm the
association between HO-1 and PINK1 in LPS-stimulated RTECs, the
expression levels of these proteins were determined in
LPS-stimulated RTECs. The HO-1 and PINK1 protein and mRNA
expression levels were upregulated when the RTECs were exposed to
LPS. However, the HO-1 and PINK1 protein and mRNA expression levels
were downregulated following pre-treatment of the cells with the
HO-1 inhibitor, Znpp. The HO-1 and PINK1 protein and mRNA
expression levels were upregulated following treatment of the cells
with the HO-1 promoter and pre-treatment with hemin, which
indicated that HO-1 promoted PINK1 expression (Fig. 2).
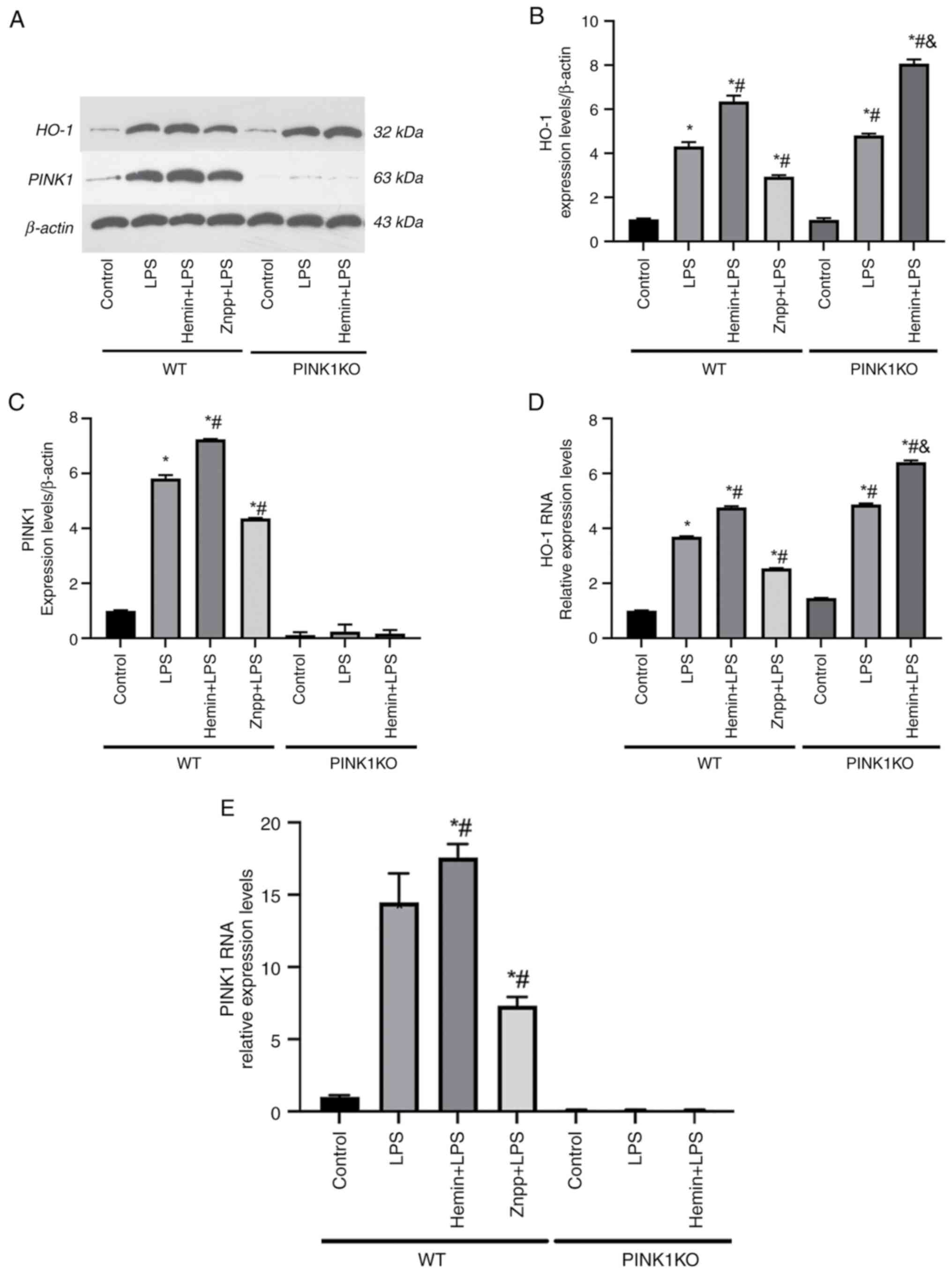 | Figure 2HO-1 regulates PINK1 in
LPS-stimulated RTECs. (A) Representative western blots of HO-1 and
PINK1. (B) Determination of the expression levels of HO-1 in RTECs.
(C) Determination of the expression levels of PINK1 in RTECs. (D)
Detection of HO-1 mRNA using reverse transcription-quantitative
PCR. (E) mRNA expression levels of PINK1 in RTECs. The data are
expressed as the mean ± SD, n=3. *P<0.05 vs. the WT
control group, #P<0.05 vs. the WT LPS group,
&P<0.05 vs. the PINK1KO LPS group. HO-1, heme
oxygenase-1; PINK1, PTEN-induced putative kinase 1; LPS,
lipopolysaccharide; RTECs, renal tubular epithelial cells; SD,
standard deviation; WT, wild-type; KO, knockout; Znpp, zinc
protoporphyrin IX. |
Effects of HO-1/PINK1 on RTEC
viability
The effects of HO-1/PINK1 on RTEC viability were
examined using CCK-8 assay (Fig.
3). In the WT RTECs, cell viability was decreased following
exposure to LPS and even further decreased following pre-treatment
with Znpp. By contrast, cell viability was increased following
pre-treatment of the cells with hemin, which indicated that HO-1
promoted cell viability. However, the viability of the PINK1KO
RTECs did not increase following pre-treatment with hemin, which
indicated that HO-1 promoted cell viability via PINK1.
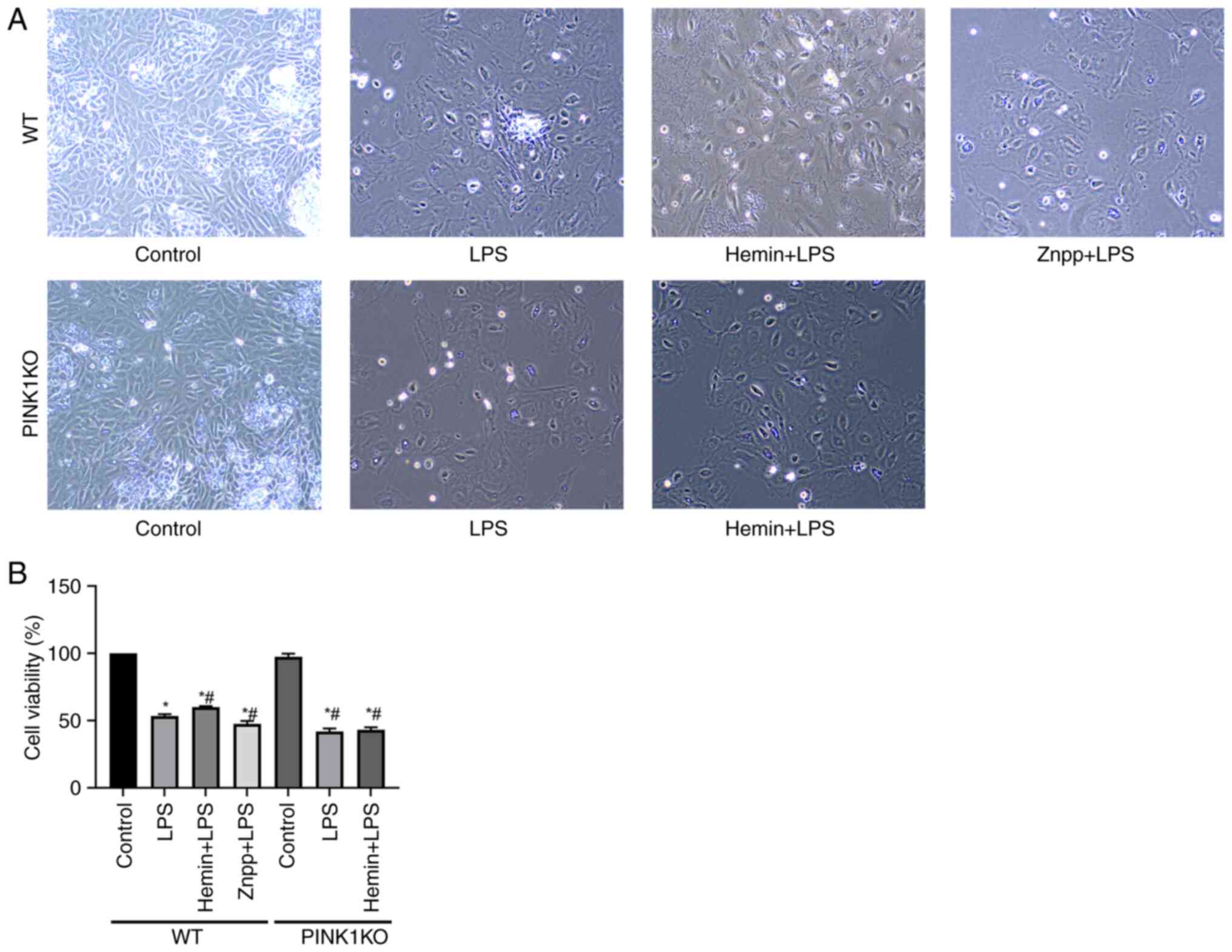 | Figure 3Effects of HO-1/PINK1 on RTEC
viability. (A) Images of the activated state of cells observed
under a microscope (magnification, x100). (B) Bar charts indicate
the detection of cell viability. The data are expressed as the mean
± SD, n=3. *P<0.05 vs. the WT control group,
#P<0.05 vs. the WT LPS group. HO-1, heme oxygenase-1;
PINK1, PTEN-induced putative kinase 1; RTEC, renal tubular
epithelial cell; SD, standard deviation; WT, wild-type; KO,
knockout; LPS, lipopolysaccharide; Znpp, zinc protoporphyrin
IX. |
Effects of HO-1/PINK1 on inflammatory
factors expressed in RTECs
RTEC injury is caused by the excessive release of
inflammatory cytokines. To assess the anti-inflammatory effects of
HO-1/PINK1, the expression levels of IL-6 and TNF-α were determined
in the cell supernatant of RTECs (Fig.
4). In WT RTECs, the expression levels of IL-6 and TNF-α were
increased following exposure of the cells to LPS. Furthermore, the
expression levels of IL-6 and TNF-α were increased and decreased
following pre-treatment of the cells with Znpp and hemin,
respectively, indicating that HO-1 inhibited inflammatory cytokine
release. However, the expression levels of IL-6 and TNF-α did not
decrease following pre-treatment of PINK1KO RTECs with hemin,
indicating that HO-1 inhibited inflammatory cytokine release via
PINK1.
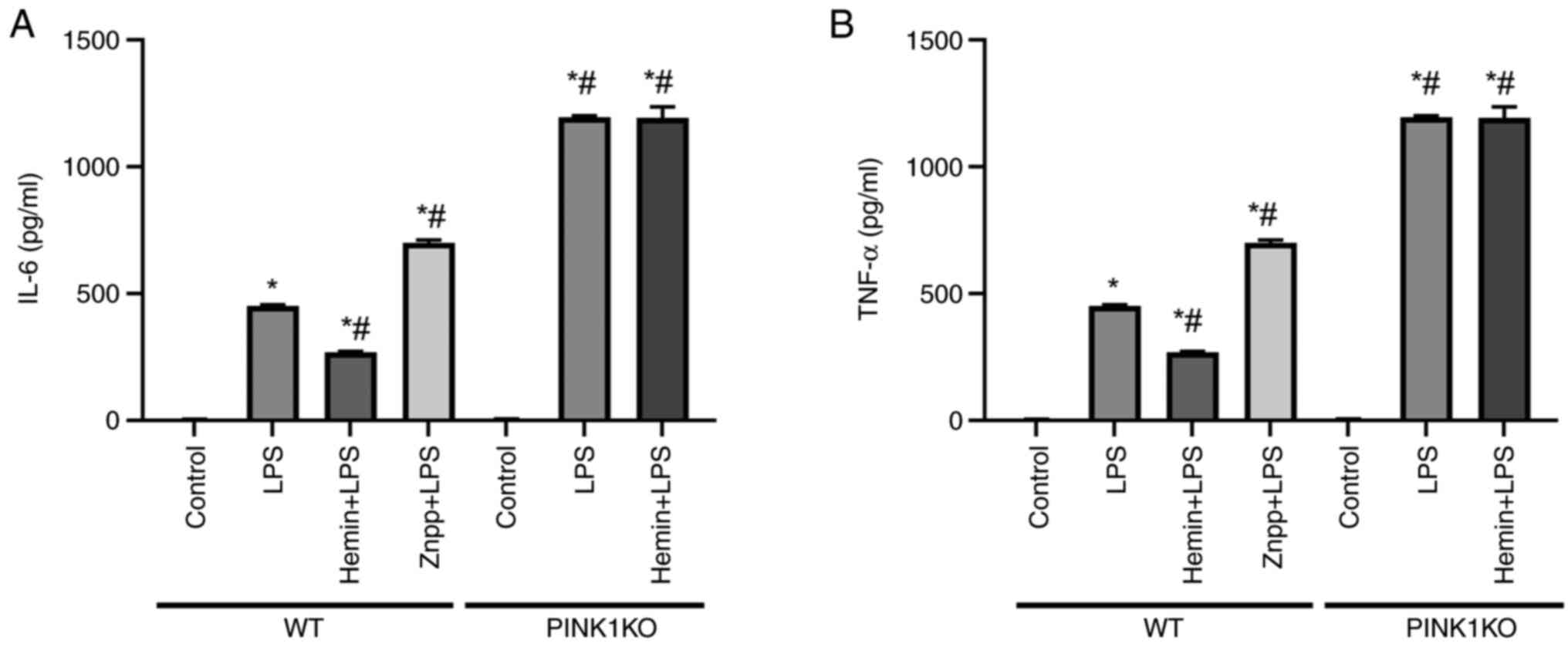 | Figure 4Effects of HO-1/PINK1 on the
expression levels of inflammatory factors released in RTECs. (A)
The levels of IL-6 in the cell supernatant. (B) The levels of TNF-α
in the cell supernatant. The data are expressed as the mean ± SD,
n=3. *P<0.05 vs. the WT control group,
#P<0.05 vs. the WT LPS group. HO-1, heme oxygenase-1;
PINK1, PTEN-induced putative kinase 1; RTECs, renal tubular
epithelial cells; SD, standard deviation; WT, wild-type; KO,
knockout; LPS, lipopolysaccharide; Znpp, zinc protoporphyrin
IX. |
Effects of HO-1/PINK1 on the
pyroptosis of RTECs
The induction of pyroptosis was investigated in
RTECs using flow cytometry (Fig.
5). In the WT RTECs, the rate of pyroptosis was increased
following stimulation of the cells with LPS. The rate of pyroptosis
was increased following pre-treatment of the cells with Znpp and
decreased following pre-treatment with hemin. However, the rate of
pyroptosis did not decrease following pre-treatment of PINK1KO
RTECs with hemin. This was consistent with the results of the flow
cytometry experiments (Fig. 5A and
B).
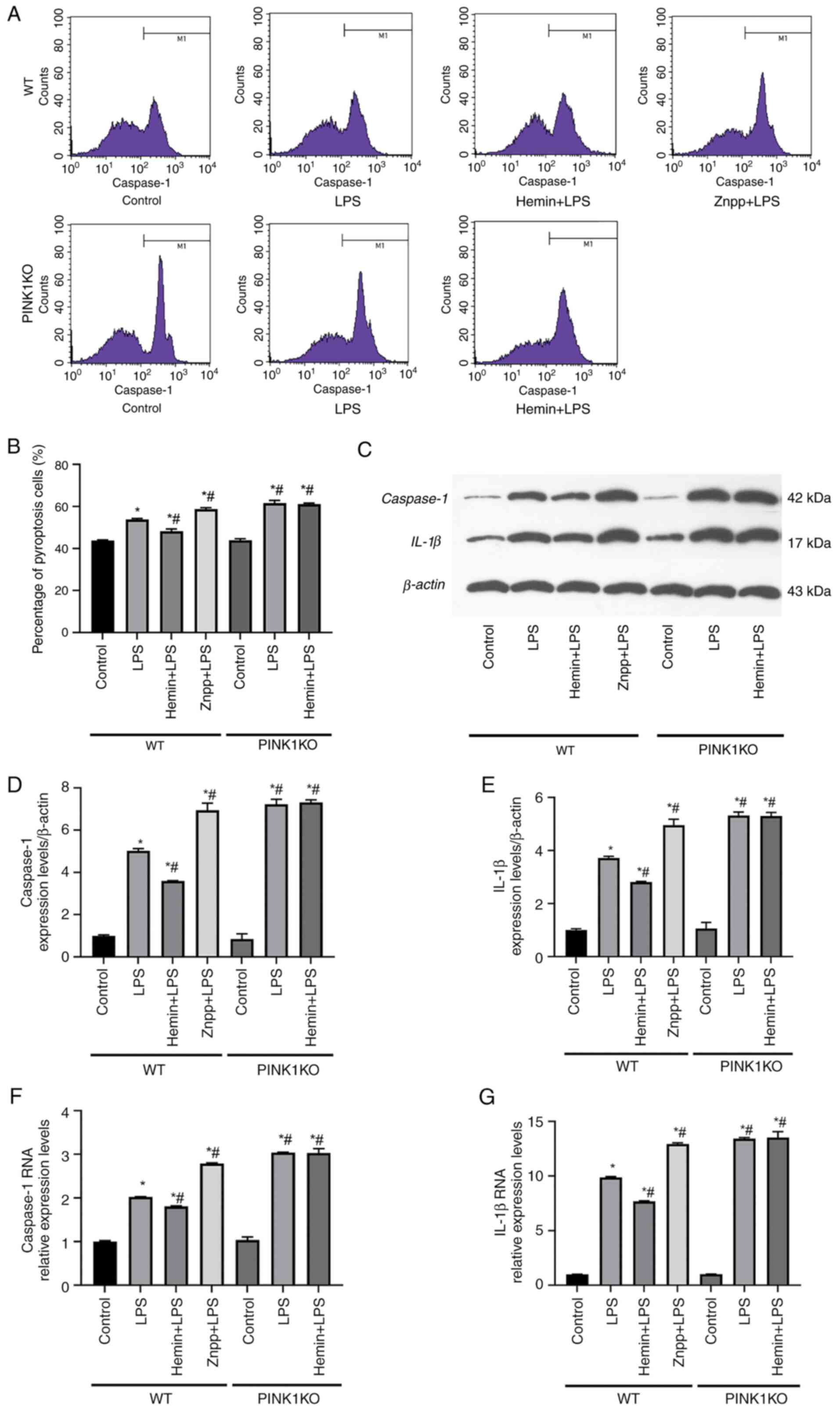 | Figure 5Effects of HO-1/PINK1 on the
induction of the pyroptosis of RTECs. (A) Representative images of
flow cytometry. (B) Percentage of pyroptosis-mediated RTEC damage.
(C-E) Western blot analysis of the protein expression levels of
caspase-1 and IL-1β in RTECs. (F and G) Reverse
transcription-quantitative PCR results indicating the relative mRNA
expression levels of caspase-1 and IL-1β. The data are expressed as
the mean ± SD, n=3. *P<0.05 vs. the WT control group,
#P<0.05 vs. the WT LPS group. HO-1, heme oxygenase-1;
PINK1, PTEN-induced putative kinase 1; RTECs, renal tubular
epithelial cells; SD, standard deviation; WT, wild-type; KO,
knockout; LPS, lipopolysaccharide; Znpp, zinc protoporphyrin
IX. |
In addition, the protein and mRNA expression levels
of caspase-1 and IL-1β were increased following stimulation of the
cells with LPS. Furthermore, the protein and mRNA expression levels
of caspase-1 and IL-1β were significantly increased following
pre-treatment of the cells with Znpp compared with those noted in
the LPS group. In contrast to these findings, the protein and mRNA
expression levels of caspase-1 and IL-1β were decreased following
pre-treatment of the cells with hemin. However, no significant
differences were noted between the LPS and Hemin + LPS groups in
the PINK1KO RTECs, which indicated that HO-1 did not inhibit the
protein or mRNA expression levels of caspase-1 and IL-1β when PINK1
was knocked out. On the whole, these data indicated that HO-1
inhibited pyroptosis via PINK1.
Effects of HO-1/PINK1 on mitochondrial
ROS in RTECs
To confirm the effects of HO-1/PINK1 on the
mitochondrial function, the levels of mitochondrial ROS were
detected in the RTECs (Fig. 6). In
the WT RTECs, mitochondrial ROS levels increased following exposure
to LPS compared with the control group. Furthermore, the ROS levels
were increased and decreased following pre-treatment of the cells
with Znpp and hemin respectively, compared with the LPS group,
indicating that HO-1 reduced mitochondrial ROS levels. However, the
ROS levels did not decrease following pre-treatment of PINK1KO
cells with hemin, indicating that HO-1 inhibited mitochondrial ROS
production via PINK1.
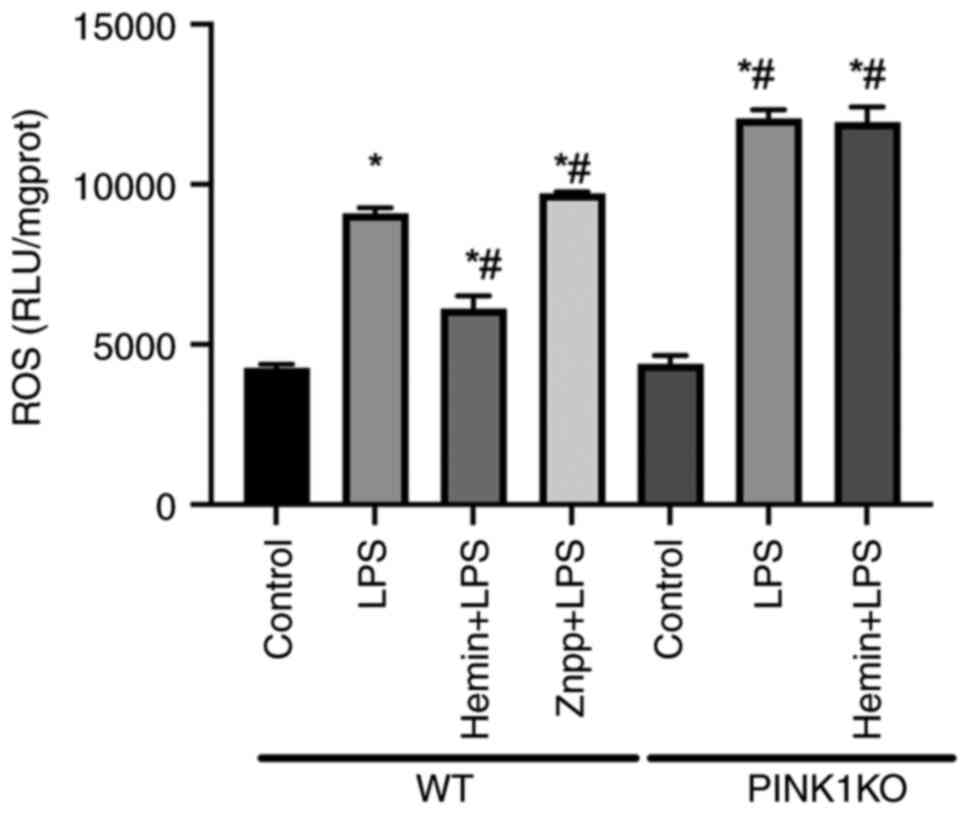 | Figure 6Effects of HO-1/PINK1 on
mitochondrial ROS levels in RTECs. The mitochondrial ROS levels
were assessed in RTECs. The data are expressed as the mean ± SD,
n=3. *P<0.05 vs. the WT control group,
#P<0.05 vs. the WT LPS group. HO-1, heme oxygenase-1;
PINK1, PTEN-induced putative kinase 1; ROS, reactive oxygen
species; RTECs, renal tubular epithelial cells; SD, standard
deviation; WT, wild-type; KO, knockout; LPS, lipopolysaccharide;
Znpp, zinc protoporphyrin IX. |
Effects of HO-1/PINK1 on mitochondrial
ATP production in RTECs
To confirm the effects of HO-1/PINK1 on
mitochondrial function, the mitochondrial ATP levels in RTECs were
examined (Fig. 7). In the WT
RTECs, the mitochondrial ATP levels were decreased following
exposure of the cells to LPS. Furthermore, the mitochondrial ATP
levels were decreased and increased following pre-treatment of the
cells with Znpp and hemin, respectively, compared with the LPS
group, which indicated that HO-1 promoted mitochondrial ATP
production. However, the mitochondrial ATP levels were not
increased following pre-treatment of PINK1KO RTECs with hemin,
indicating that HO-1 promoted mitochondrial ATP production via
PINK1.
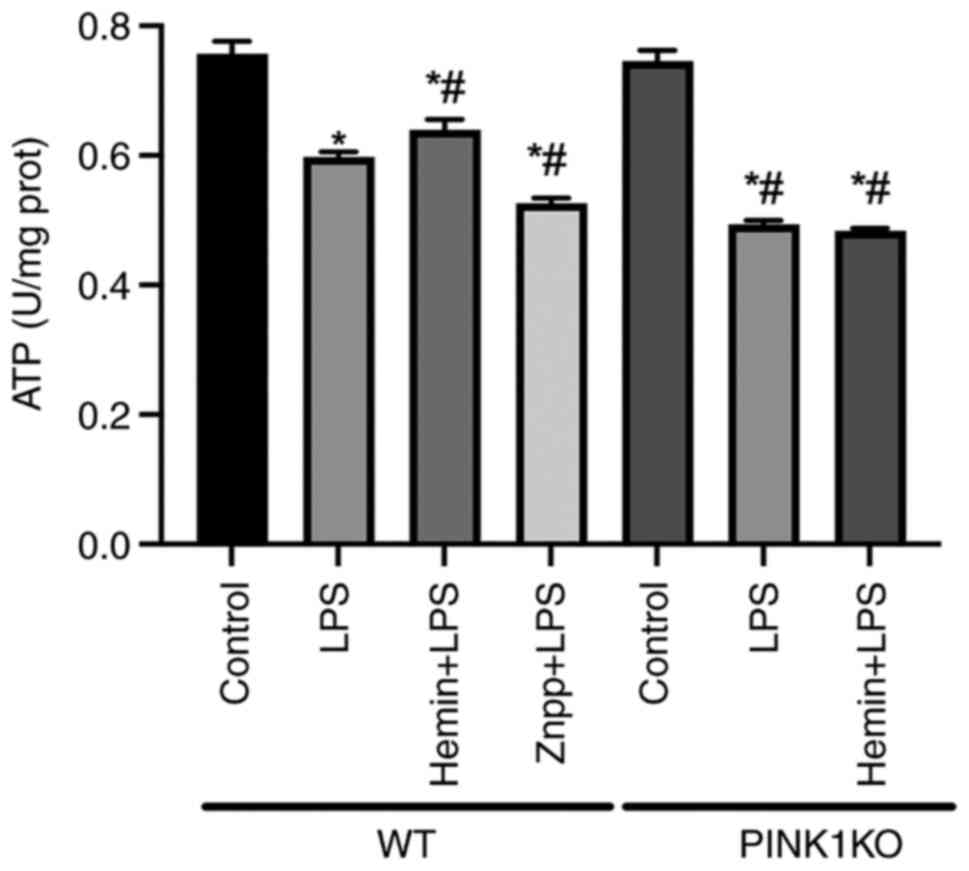 | Figure 7Effects of HO-1/PINK1 on
mitochondrial ATP levels in RTECs. Determination of the
mitochondrial ATP levels in RTECs. The data are expressed as the
mean ± SD, n=3. *P<0.05 vs. the WT Control group,
#P<0.05 vs. the WT LPS group. HO-1, heme oxygenase-1;
PINK1, PTEN-induced putative kinase 1; ATP, adenosine triphosphate;
RTECs, renal tubular epithelial cells; SD, standard deviation; WT,
wild-type; KO, knockout; LPS, lipopolysaccharide; Znpp, zinc
protoporphyrin IX. |
Effects of HO-1/PINK1 on the
mitochondrial respiratory function of RTECs
To confirm the effects of HO-1/PINK1 on
mitochondrial function, the respiratory function of RTECs was
investigated (Fig. 8). In the WT
RTECs, the mitochondrial oxygen consumption was decreased following
exposure of the cells to LPS. Furthermore, the mitochondrial oxygen
consumption was decreased and increased following Znpp and hemin
pre-treatment, respectively compared with the LPS group, which
indicated that HO-1 promoted mitochondrial oxygen consumption.
However, no significant difference was noted in the mitochondrial
oxygen consumption between the LPS and Hemin + LPS groups in the
PINK1KO RTECs. Collectively, these data indicated that HO-1
promoted mitochondrial oxygen consumption via PINK1.
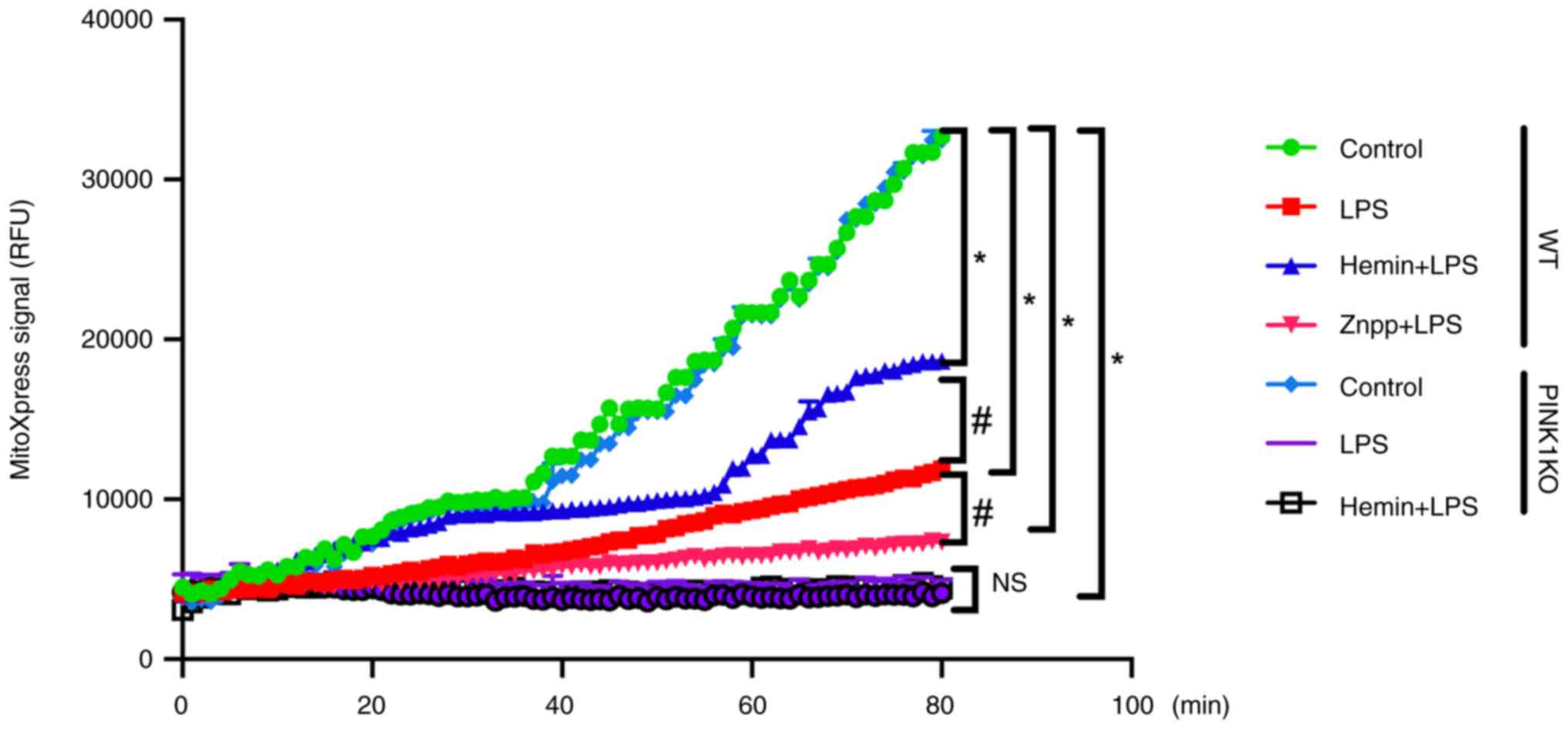 | Figure 8Effects of HO-1/PINK1 on the
mitochondrial respiratory function in RTECs. The data are expressed
as the mean ± SD, n=3. *P<0.05 vs. the WT control
group, #P<0.05 vs. the WT LPS group. NS, not
significant; HO-1, heme oxygenase-1; PINK1, PTEN-induced putative
kinase 1; RTECs, renal tubular epithelial cells; SD, standard
deviation; WT, wild-type; KO, knockout; LPS, lipopolysaccharide;
Znpp, zinc protoporphyrin IX. |
Discussion
The primary culture of RTECs can be achieved by
sieve centrifugation. The separation of the glomerulus and renal
tubules can be performed by grinding of the renal tissue and
filtering using a mesh, and the exclusion of glomerulus cells can
be performed using 80- and 100-mesh screens, which can obtain more
RTECs and make them purer (20).
Under a microscope (optical), the RTECs exhibit a multilateral
cobblestone shape, and the cells are closely connected (21). In the present study, primary
cultured RTECs were also multilateral cobble-like under a
microscope (optical), and the cells were closely connected; thus,
it was confirmed that the cultured cells were RTECs.
AKI caused by endotoxemia is relatively common in
clinical practice (2,22,23).
Among the common pathogenic bacteria that cause endotoxemia,
Gram-negative bacteria are the most common and cause endotoxemia by
secreting LPS (24). Therefore,
LPS was selected in the present study to induce endotoxemia in
RTECs. An LPS concentration of 1 µg/ml can lead to RTEC injury
(25). Therefore, the present
study used an LPS concentration of 1 µg/ml to prepare the
LPS-induced RTEC injury model. The changes in sex hormone levels
may have an effect on endotoxemia. Male rats are affected at
different levels by endotoxins compared with female rats. It has
been shown that in the presence of toxins and hematic disease
caused by shock, male rats are more likely to develop
immunosuppression, which leads to their inability to tolerate AKI.
In contrast to male rats, female rats can tolerate AKI due to the
increased levels of estrogen (26). Therefore, although the present
study involved mainly in vitro experiments, male rats were
selected in order to reduce the effect of estrogen on RTECs. The
results indicated that the viability of the RTECs was decreased and
the expression levels of the inflammatory cytokines, IL-6 and
TNF-α, were increased following exposure of the cells to LPS. These
findings indicated the successful establishment of the model used
in the present study.
RTEC injury plays a crucial role in the pathogenesis
of AKI (5,27). RTEC injury is mainly manifested by
cell necrosis and apoptosis; the pyroptosis of RTECs has been
recently discovered (6,28). Therefore, RTEC pyroptosis was
observed in the present study. Pyroptosis is a form of programmed
necrotizing death that is primarily mediated by caspase-1 and leads
to the activation of IL-1β (8,9,29,30).
Therefore, the expression levels of caspase-1 and IL-1β were
detected in the present study. The data indicated that the
expression levels of caspase-1 and IL-1β were increased along with
the rate of pyroptosis of the RTECs following their exposure to
LPS, which indicated that LPS induced the pyroptosis of RTECs.
Mitochondria are considered the core organelles of
cellular energy metabolism and consequently, mitochondrial
dysfunction is a main cause of pyroptosis (31). Mitochondria produce ATP through a
complex of electron respiratory chains, which provide energy for
cells (12). Mitochondrial damage
leads to mitochondrial dysfunction; consequently, ATP synthesis
decreases, ROS production increases and the mitochondrial oxygen
consumption rate decreases, leading to cell necrosis, apoptosis and
pyroptosis (10,32,33).
In vivo research has indicated that the mitochondrial
fission of endotoxemia induces the pyroptosis of RTECs and causes
AKI (11). In the present study,
ATP production and ROS levels were increased following the
stimulation of isolated RTECs with LPS, whereas the mitochondrial
oxygen consumption rate was decreased, indicating mitochondrial
dysfunction.
HO-1 exerts endogenous protective effects and can
protect multiple organs against damage caused by endotoxemia
(16,34,35).
The protection from toxin release by endogenous HO-1 expression in
AKI was also confirmed in a previous in vivo study by the
authors (11). The HO-1 mRNA and
protein levels in the kidneys have been shown to be upregulated in
rats following stimulation by lipopolysaccharide (34). The HO-1 mRNA and protein levels
RTECs were upregulated following stimulation with LPS in the
present study. This type of protection is not only reflected in its
effect on mitochondrial dynamics. By contrast, HO-1 exerts
antiapoptotic, antioxidant and anti-inflammatory functions, and
assists in maintaining cellular homeostasis and function by playing
a critical role in the process (36-39).
In the present study, it was found that the upregulation of HO-1
expression inhibited the production of inflammatory factors, while
it increased ATP synthesis, decreased ROS levels, increased
mitochondrial oxygen consumption, inhibited pyroptosis, and
increased the viability of RTECs. The inhibition of HO-1 expression
exerted the opposite effects. These results suggested that HO-1
inhibited the inflammatory response, improved mitochondrial
function, inhibited pyroptosis, and attenuated the injury to
LPS-stimulated RTECs. Previous in vivo studies have shown
that HO-1 regulates mitochondrial fusion/fission through PINK1,
inhibits pyroptosis and improves endotoxemia in AKI; PINK1 can
regulate mitochondrial autophagy, remove damaged mitochondria and
improve mitochondrial function (40-42).
The present study investigated the induction of pyroptosis,
mitochondrial dysfunction, and the upregulation of PINK1 expression
in LPS-stimulated RTECs in vitro. When HO-1 was upregulated,
the expression levels of PINK1 were also upregulated, whereas when
the expression of HO-1 was inhibited, the expression of PINK1 was
downregulated; this indicated that HO-1 expression regulated PINK1
and that HO-1 was involved in the regulation of mitochondrial
function. When PINK1 expression was knocked out, HO-1 expression
was upregulated and its effect in regulating mitochondrial function
was weakened. This further demonstrated that HO-1 regulated
mitochondrial function, inhibited pyroptosis, and enhanced the
viability of RTECs via PINK1.
The present study is based on previous findings,
which mainly discussed the influence of the HO-1/PINK1 pathway on
mitochondrial fusion/division in vivo (11). The innovation of the present study
was the in vitro culture of RTECs, and the verification of
the influence of the HO-1/PINK1 pathway on mitochondrial function.
The present study has several limitations, however. Firstly, it was
observed that AKI was induced only 6 h following stimulation of the
cells with LPS; therefore, additional time points are required to
further explore the induction of this disease. Secondly, the
effects of various concentrations of LPS on RTECs need to be
determined. Thirdly, mitochondrial function was only noted in
RTECs; however, mitochondrial morphology needs to be determined.
Fourthly, mitochondrial dysfunction inhibiting pyroptosis needs to
be investigated using a mitochondrial dysfunction inhibitor.
Lastly, in order to illustrate the HO-1-mediated regulation of
PINK1 more efficiently, PINK1 overexpression should be verified. In
the PINK1KO RTECs, the setting of the LPS + ZnPP group will provide
more information. In addition, only HO-1 inhibitors were used to
alter the expression of HO-1, which would be more effective to
verify the results if the changes were made at the gene level by
using siRNA or overexpression plasmids. Previous research has
demonstrated that electroacupuncture can inhibit the inflammatory
response and oxidative stress in sepsis and improve acute kidney
injury (43). Whether
electroacupuncture can attenuate RTEC injury through HO-1/PINK1 is
worthy of further investigation. Additional studies are required to
investigate the mechanisms underlying the HO-1/PINK1 interaction in
endotoxin-stimulated RTECs.
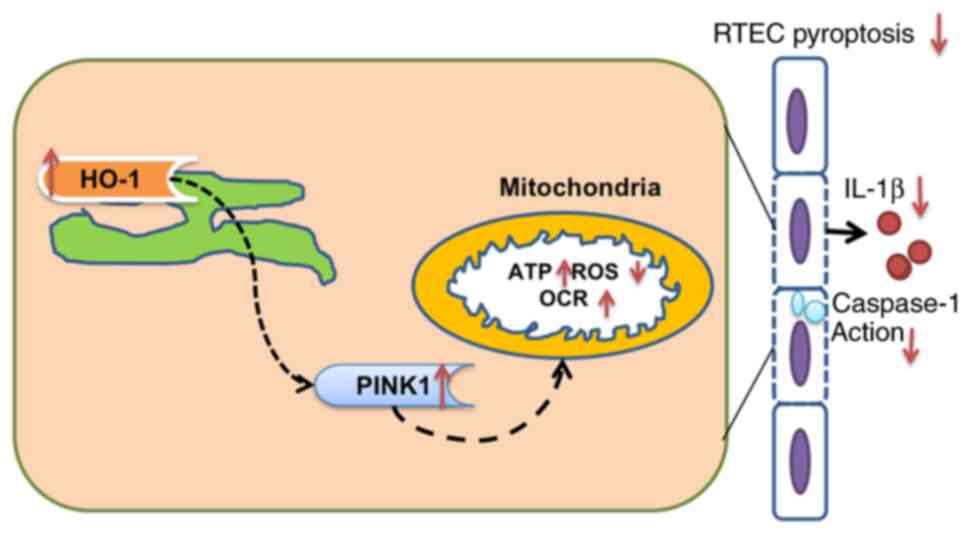
In conclusion, the present study demonstrated that
HO-1 inhibited the inflammatory response, improved mitochondrial
function, and inhibited the pyroptosis of LPS-stimulated RTECs,
which may be related to PINK1 (Fig.
9).
Supplementary Material
Identification of rat tail genotypes
in F0 generation rats. The primers used were as follows:
EGE-WL-008-WT-F1/EGE-WL-008-Mut-R; E5L8-003, E5L8-006, E5L8-007,
E5L8-024, E5L8-029, E5L8-030, E5L8-057, E5L8-060, E5L8-064,
E5L8-067 and E5L8-086 were F0 generation positive rats.
Genotype identification of rat tail in
F1 generation rats. The primers used were as follows:
EGE-WL-008-WT-F1/EGE-WL-008-WT-R1 and
EGE-WL-008-WT-F1/EGE-WL-008-Mut-R; 1E5L8-017, 1E5L8-023, 1E5L8-028,
1E5L8-032, 1E5L8-039 and 1E5L8-040 were F1-positive rats.
Immunocytochemistry for the expression
of cytokeratin 18 (x40 magnification).
The typical cobblestone appearance of
renal tubular epithelial cells (x40 magnification).
Supplementary Materials and
methods
Primer sequence information.
Acknowledgements
The authors would like to thank Mr. Yan-Fang Liu
(research fellow from Tianjin Yishengyuan Biotechnology Co. Ltd.)
for providing technical assistance.
Funding
Funding: The present study was financially supported by the
Inner Mongolia Natural Science Foundation (grant no.
2021MS08061).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HBL, YSM and XZZ contributed to the preparation of
the manuscript and the overall study design. QZ, XDL and JNS
contributed to data analysis. LNH, JNW, YG and DDF performed the
experiments. JBY and YS contributed to the overall study design and
performed a critical review of the manuscript. All authors have
read and approved the final version of the manuscript and confirm
the authenticity of all the raw data.
Ethics approval and consent to
participate
The present study was approved by the Animal Ethical
and Welfare Committee of the Institute of Radiation Medicine,
Chinese Academy of Medical Sciences (no. IRM-DWLL-201907) and was
performed in accordance with the ARRIVE guidelines developed by the
National Center for the Replacement, Refinement, and Reduction of
Animals in Research.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lai TS, Wang CY, Pan SC, Huang TM, Lin MC,
Lai CF, Wu CH, Wu VC and Chien KL: National Taiwan University
Hospital Study Group on Acute Renal Failure (NSARF). Risk of
developing severe sepsis after acute kidney injury: A
population-based cohort study. Crit Care. 17(R231)2013.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Weng L, Zeng XY, Yin P, Wang LJ, Wang CY,
Jiang W, Zhou MG and Du B: China Critical Care Clinical Trials
Group (CCCCTG). Sepsis-related mortality in China: A descriptive
analysis. Intensive Care Med. 44:1071–1080. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Poston JT and Koyner JL: Sepsis associated
acute kidney injury. BMJ. 364(k4891)2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Messaris E, Memos N, Chatzigianni E,
Kataki A, Nikolopoulou M, Manouras A, Albanopoulos K,
Konstadoulakis MM and Bramis J: Apoptotic death of renal tubular
cells in experimental sepsis. Surg Infect (Larchmt). 9:377–388.
2008.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Priante G, Gianesello L, Ceol M, Del Prete
D and Anglani F: Cell death in the kidney. Int J Mol Sci.
20(3598)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ye Z, Zhang L, Li R, Dong W, Liu S, Li Z,
Liang H, Wang L, Shi W, Malik AB, et al: Caspase-11 mediates
pyroptosis of tubular epithelial cells and septic acute kidney
injury. Kidney Blood Press Res. 44:465–478. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bergsbaken T, Fink SL and Cookson BT:
Pyroptosis: Host cell death and inflammation. Nat Rev Microbiol.
7:99–109. 2009.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Man SM, Karki R and Kanneganti TD:
Molecular mechanisms and functions of pyroptosis, inflammatory
caspases and inflammasomes in infectious diseases. Immunol Rev.
277:61–75. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Jorgensen I, Rayamajhi M and Miao EA:
Programmed cell death as a defence against infection. Nat Rev
Immunol. 17:151–164. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sedlackova L and Korolchuk VI:
Mitochondrial quality control as a key determinant of cell
survival. Biochim Biophys Acta Mol Cell Res. 1866:575–587.
2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li HB, Zhang XZ, Sun Y, Zhou Q, Song JN,
Hu ZF, Li Y, Wu JN, Guo Y, Zhang Y, et al: HO-1/PINK1 regulated
mitochondrial fusion/fission to inhibit pyroptosis and attenuate
septic acute kidney injury. Biomed Res Int.
2020(2148706)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Pfanner N, Warscheid B and Wiedemann N:
Mitochondrial protein organization: From biogenesis to networks and
function. Nat Rev Mol Cell Biol. 20:267–284. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Supinski GS, Schroder EA and Callahan LA:
Mitochondria and critical illness. Chest. 157:310–322.
2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bolisetty S, Zarjou A and Agarwal A: Heme
oxygenase 1 as a therapeutic target in acute kidney injury. Am J
Kidney Dis. 69:531–545. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Suliman HB, Keenan JE and Piantadosi CA:
Mitochondrial quality-control dysregulation in conditional
HO-1-/- mice. JCI Insight. 2(e89676)2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Chen X, Wang Y, Xie X, Chen H, Zhu Q, Ge
Z, Wei H, Deng J, Xia Z and Lian Q: Heme oxygenase-1 reduces
sepsis-induced endoplasmic reticulum stress and acute lung injury.
Mediators Inflamm. 2018(9413876)2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z,
He L, Tan J, Liu Y, Liu H, et al: PINK1-PRKN/PARK2 pathway of
mitophagy is activated to protect against renal
ischemia-reperfusion injury. Autophagy. 14:880–897. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ding W, Yousefi K and Shehadeh LA:
Isolation, characterization, and high throughput extracellular flux
analysis of mouse primary renal tubular epithelial cells. J Vis
Exp. (57718)2018.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Mattila PM, Nietosvaara YA, Ustinov JK,
Renkonen RL and Häyry PJ: Antigen expression in different
parenchymal cell types of rat kidney and heart. Kidney Int.
36:228–233. 1989.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Van Kooten C, Lam S and Daha MR:
Isolation, culture, characterization and use of human renal tubular
epithelial cells. J Nephrol. 14:204–210. 2001.PubMed/NCBI
|
|
22
|
Shankar-Hari M, Phillips GS, Levy ML,
Seymour CW, Liu VX, Deutschman CS, Angus DC and Rubenfeld GD:
Developing a new definition and assessing new clinical criteria for
septic shock: For the third international consensus definitions for
sepsis and septic shock (sepsis-3). JAMA. 315:775–787.
2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Cecconi M, Evans L, Levy M and Rhodes A:
Sepsis and septic shock. Lancet. 392:75–87. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Plotnikov EY, Brezgunova AA, Pevzner IB,
Zorova LD, Manskikh VN, Popkov VA, Silachev DN and Zorov DB:
Mechanisms of LPS-induced acute kidney injury in neonatal and adult
rats. Antioxidants (Basel). 7(105)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Quoilin C, Mouithys-Mickalad A, Duranteau
J, Gallez B and Hoebeke M: Endotoxin-induced basal respiration
alterations of renal HK-2 cells: A sign of pathologic metabolism
down-regulation. Biochem Biophys Res Commun. 423:350–354.
2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Tang YQ and Li L: Development strategy and
application of animal model of sepsis. Chin J Exp Surg.
12:1433–1434. 2006.(In Chinese).
|
|
27
|
Peerapornratana S, Manrique-Caballero CL,
Gómez H and Kellum JA: Acute kidney injury from sepsis: Current
concepts, epidemiology, pathophysiology, prevention and treatment.
Kidney Int. 96:1083–1099. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Chen F, Lu J, Yang X, Xiao B, Chen H, Pei
W, Jin Y, Wang M, Li Y, Zhang J, et al: Acetylbritannilactone
attenuates contrast-induced acute kidney injury through its
anti-pyroptosis effects. Biosci Rep. 40(BSR20193253)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li
P, Hu L and Shao F: Inflammatory caspases are innate immune
receptors for intracellular LPS. Nature. 514:187–192.
2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Broz P: Immunology: Caspase target drives
pyroptosis. Nature. 526:642–643. 2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Friedman JR and Nunnari J: Mitochondrial
form and function. Nature. 505:335–343. 2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yu W, Sheng M, Xu R, Yu J, Cui K, Tong J,
Shi L, Ren H and Du H: Berberine protects human renal proximal
tubular cells from hypoxia/reoxygenation injury via inhibiting
endoplasmic reticulum and mitochondrial stress pathways. J Transl
Med. 11(24)2013.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Murphy MP and Hartley RC: Mitochondria as
a therapeutic target for common pathologies. Nat Rev Drug Discov.
17:865–886. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Yu JB, Zhou F, Yao SL, Tang ZH, Wang M and
Chen HR: Effect of heme oxygenase-1 on the kidney during septic
shock in rats. Transl Res. 153:283–287. 2009.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yin H, Li X, Yuan B, Zhang B, Hu S, Gu H,
Jin X and Zhu J: Heme oxygenase-1 ameliorates LPS-induced acute
lung injury correlated with downregulation of interleukin-33. Int
Immunopharmacol. 11:2112–2117. 2011.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Abraham NG, Lin JH, Schwartzman ML, Levere
RD and Shibahara S: The physiological significance of heme
oxygenase. Int J Biochem. 20:543–558. 1988.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Gozzelino R, Jeney V and Soares MP:
Mechanisms of cell protection by heme oxygenase-1. Annu Rev
Pharmacol Toxicol. 50:323–354. 2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kozakowska M, Dulak J and Józkowicz A:
Heme oxygenase-1-more than the cytoprotection. Postepy Biochem.
61:147–158. 2015.PubMed/NCBI(In Polish).
|
|
39
|
Cai ZY, Sheng ZX and Yao H: Pachymic acid
ameliorates sepsis-induced acute kidney injury by suppressing
inflammation and activating the Nrf2/HO-1 pathway in rats. Eur Rev
Med Pharmacol Sci. 21:1924–1931. 2017.PubMed/NCBI
|
|
40
|
Bayne AN and Trempe J: Mechanisms of
PINK1, ubiquitin and parkin interactions in mitochondrial quality
control and beyond. Cell Mol Life Sci. 76:4589–4611.
2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Lin Q, Li S, Jiang N, Shao X, Zhang M, Jin
H, Zhang Z, Shen J, Zhou Y, Zhou W, et al: PINK1-parkin pathway of
mitophagy protects against contrast-induced acute kidney injury via
decreasing mitochondrial ROS and NLRP3 inflammasome activation.
Redox Biol. 26(101254)2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Leites EP and Morais VA: Mitochondrial
quality control pathways: PINK1 acts as a gatekeeper. Biochem
Biophys Res Commun. 500:45–50. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Yu JB, Shi J, Zhang Y, Gong LR, Dong SA,
Cao XS, Wu LL and Wu LN: Electroacupuncture ameliorates acute renal
injury in lipopolysaccharide-stimulated rabbits via induction of
HO-1 through the PI3K/Akt/Nrf2 pathways. PLoS One.
10(e0141622)2015.PubMed/NCBI View Article : Google Scholar
|