Introduction
Sarcoidosis is a chronic inflammatory disease
characterized by granuloma formation and a gradual loss of lung
function (1). Sarcoidosis can
affect any organ, but most commonly, it affects the lungs and the
intrathoracic lymph nodes (2). The
granuloma is distinguished by a core of epithelioid histiocytes and
multinucleate giant cells, interspersed by CD4+ T
lymphocytes with a Th1 immuno-phenotype, as well as a minority of
CD8+ T lymphocytes, fibroblasts and B lymphocytes
(3-5).
It is difficult to tailor treatment to specific disease phenotypes
or disease mechanisms. Thus, at present, the treatment relies
primarily on immunosuppression with steroids and/or steroid-sparing
drugs to palliate symptoms, switch off acute inflammation and
prevent end-organ disease (6).
Regulatory T cells (Tregs)/Th17 and Th1/Th2
paradigms play a role in the pathogenesis of sarcoidosis (7,8). The
population of Tregs act as the key controllers for limiting
immunopathology and maintaining immune homeostasis (9). The lymph nodes and granulomas of
sarcoidosis patients are flooded with Treg cells (10), whereas their presence in blood
demonstrates conflicting results. Even by the same team of
researchers, patients with sarcoidosis have been reported to have
various amounts of Tregs, which are sorted into higher, lower and
normal groups (9,11). These controversial numbers of Tregs
may be associated with the stage of the disease. Patients with
active sarcoidosis have increased numbers of Tregs, while patients
with inactive sarcoidosis and spontaneous remission have normal or
decreased numbers of Tregs (3,12-15).
Our previous superoxide dismutase A (SodA)-induced sarcoidosis
models showed changes in Treg numbers correlated with disease
phases (16,17). This demonstrates that, compared
with the function of Tregs in healthy individuals, the function of
Tregs in patients with sarcoidosis is severely impaired (3). Due to this impairment, Tregs fail to
inhibit the persistent and excessive inflammatory responses of
sarcotic lesions and immune homeostasis cannot be maintained
(9). In addition, without the
ability to actively regulate reactive T cells, Tregs can only
partially inhibit early granuloma formation and have no positive
effects on the formed granulomatous lesions (12).
In our previous study, patients and mice with
sarcoidosis both showed phosphoinositide 3-kinase (PI3K)/Akt
signaling pathway activation, affecting FoxP3 expression in Tregs
(14,16,17).
It has been reported that controlling PI3K signaling in Tregs is
critical for maintaining their homeostasis, function and stability
(18). Previously, we reported
that inhibition of PI3K/Akt signaling by BKM120 (an inhibitor of
pan-class I PI3K) or LY294002 (an inhibitor of PI3Kα, δ and β)
reverses the disturbance of Th1/Th17/Tregs and improves the
function of Tregs in SodA-induced sarcoidosis. Notably, BKM120 is
more effective compared with LY294002 in restoring the
immunosuppressive function of Tregs, which demonstrates that
PI3K/Akt signaling in Tregs may be associated with the PI3Kp110γ
subunit in SodA-induced sarcoidosis (17). Thus, understanding its intrinsic
effects in Sarcoidosis is necessary to formulate future therapies
that favor homeostasis of Tregs in the disease. Therefore, the
present study examined the impact of the effects of PI3Kp110γ/δ
inhibition on the function of Tregs in SodA-induced sarcoidosis,
especially at the chosen time point when the number of Tregs is
normal.
Materials and methods
Animals
First, six-week-old female C57BL/6J mice (n=30)
weighing 20-25 g were purchased from the Shanghai Lab. Animal,
Research Center. Mice were maintained under a specific
pathogen-free environment (temperature, 22±1˚C; relative humidity,
50±5%; and 12-h light/dark cycle). Water and food were supplied
ad libitum. All in vivo manipulations were performed
under protocols approved by the Institutional Animal Care and Use
Committee of Nanjing University Medical School (approval no.
SCXK-Jiangsu-2019-0056). Mice were acclimated for at least one week
before use. Mice were euthanized using cervical dislocation
following carbon dioxide inhalation according to according to the
American Veterinary Medical Association guidelines (19). Euthanasia was performed in a
dedicated box with CO2, using a gradual 30% vol/min
displacement rate. The percentage of CO2 and
O2 used was 6:4. Based on the humane end point criteria,
the distress of the animals was constantly monitored. The condition
of the mice were checked daily. Mice were sacrificed when they
developed hypothermia, inability to stand and wounds that did not
heal. None of the mice developed symptoms requiring sacrifice
during the experiment.
SodA-induced sarcoidosis model
Sarcoidosis was induced based on a method provided
by Swaisgood et al (20).
In brief, on the first day, mice were pretreated with 20 mg of
mycobacterial SodA peptide
(Ala-Ala-Ala-IIe-Ala-Gly-Ala-Phe-Gly-Ser-PheAsp-Lys-Phe-Arg;
GenBank accession no. DQ768096) dissolved in 0.25 ml of incomplete
Freund's adjuvant (MilliporeSigma) by subcutaneous injection.
Subsequently, 14 days later, the SodA-sensitized mice were
challenged with 6000 N-hydroxysuccinimide-activated Sepharose 4B
naked beads [in 0.5 ml of phosphate-buffered saline (PBS)]
covalently coupled to SodA by tail vein injection. After 28 days,
lung tissues were collected to establish sarcoidosis.
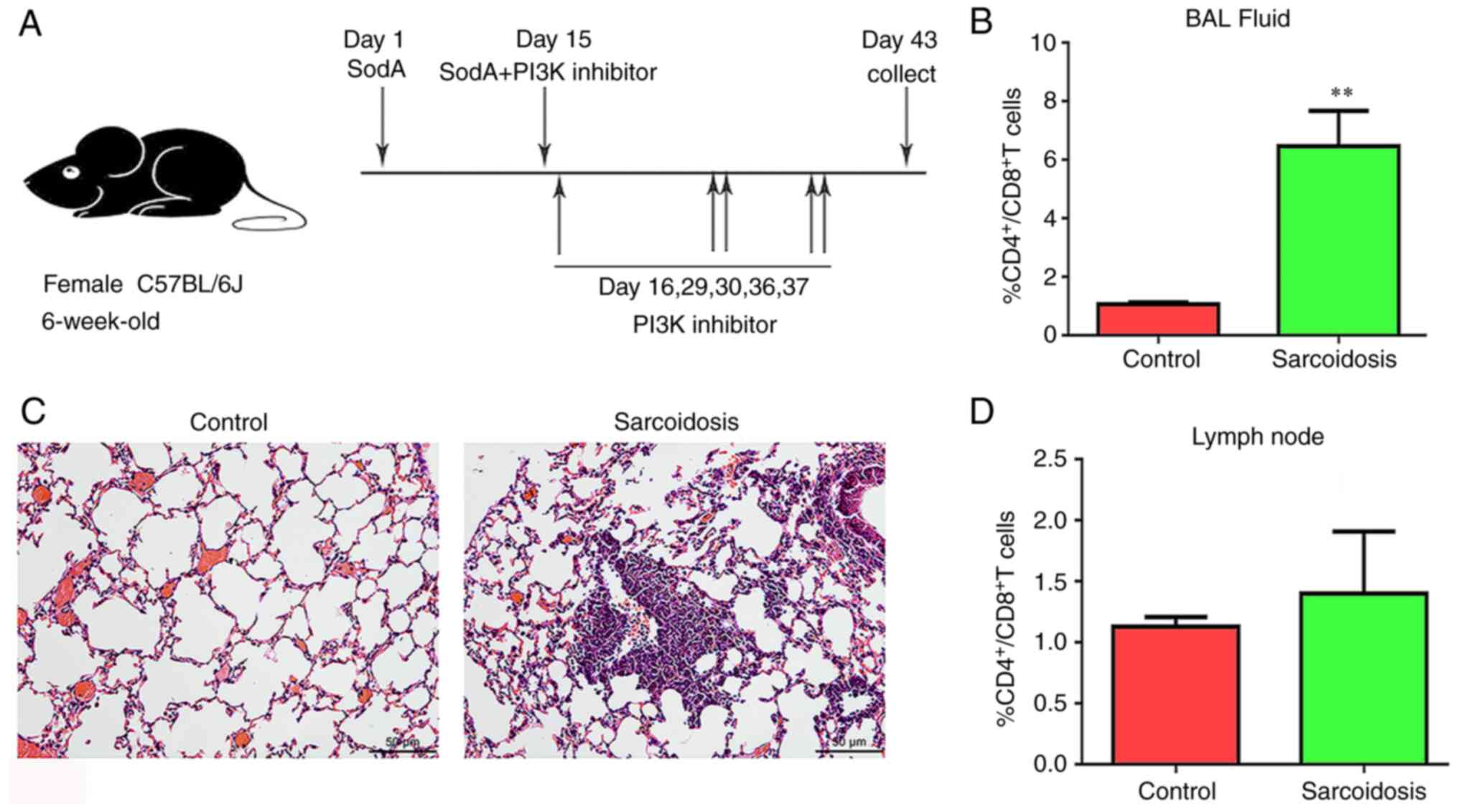
CAL-101/AS-605240 treatment
SodA-induced sarcoidosis mice were randomly divided
into five groups the day after tail vein injection. A total of 2
mg/kg of CAL-101 (PI3Kδ inhibitor; TargetMol Chemicals, Inc.) or
AS-605240 (PI3Kγ inhibitor; TargetMol Chemicals, Inc.) were
respectively given to the mice via airway on days 15, 16, 29, 30,
36 and 37 (Fig. 1A). DMSO was
applied to group sarcoidosis. As the positive control group,
dexamethasone was administered to mice by intraperitoneal injection
at 5 mg/kg. The other healthy mice were kept as the control
group.
Histopathology and
immunofluorescence
Right upper lung tissues were fixed in 4%
paraformaldehyde at 4˚C overnight, dehydrated and cut into 4-µm
thick sections after being embedded in paraffin. Tissue sections
were routinely stained with hematoxylin and eosin (H&E) for
conventional morphological evaluation under the light microscope
(Olympus Corporation). The specific methods were as follows: First,
the sections were placed in an oven and heated at 60˚C for 1 h. The
following steps are all performed at room temperature; the paraffin
sections were immersed in xylene solution for 10 min, and then
immersed in a new xylene solution for another 10 min; Then the
tissue sections were immersed in 100, 95, 90, 80, 75, and 50%
alcohol solution for 2 min, respectively. Finally, the tissue
sections were immersed in ddH2O for 5 min. The sections
were placed in hematoxylin and stained for 15 min. After staining,
the sections were washed with ddH2O for 5 min to turn
blue. Then the sections were placed in 1% hydrochloric acid ethanol
solution for about 5 sec, as soon as the sections turned red, they
were placed in ddH2O to restore the blue color. After
this, the rinsed sections were soaked in 50, 70 and 80% alcohol for
3 min in turn, and then counterstained in 0.5% eosin ethanol
solution for 1-3 min. The excess red color was washed off with 95%
alcohol, and the sections soaked in 100% alcohol for 5 min, then
the excess ethanol washed off with ddH2O, and the
sections soaked in xylene for 5 min, and new xylene replaced for
another 5 min. Finally the neutral gum was diluted with xylene to
the appropriate viscosity and the slide sealed.
Immunofluorescence was performed by using p-PI3K p85
antibody (cat. no. 4228S; Cell Signaling Technology, Inc.), p-Akt
(ser473) rabbit monoclonal antibody (cat. no. 4060; Cell Signaling
Technology, Inc.), Alexa Fluor 488 goat anti-rabbit IgG (cat. no.
FMS-MSAF48801; Nanjing Fcmacs Biotechnology Co., Ltd.) and Alexa
Fluor 647 goat anti-mouse IgG (cat. no. FMS-RBAF64701; Nanjing
Fcmacs Biotechnology Co., Ltd.). The dilution ratio of all primary
antibodies was 1:500 and the dilution ratio of secondary antibodies
was 1:1,000. The specific method was as follows: The prepared
paraffin sections of lung tissue are placed on a shelf and heated
in a 60˚C oven for 30 min. Tissue sections were soaked in xylene
for 20 min and then soaked in 100, 95, 90, 80, 75 and 50% alcohol
solutions for 3 min, respectively. The sections were soaked in
ddH2O for 5 min, then re-soaked after changing the water and
repeated three times. 0.5% TritonX-100 was prepared with PBS,
preheated in advance, and added into the tissue section for 10 min,
so that the antibody could fully enter the cell for reaction. The
antigen repair solution was heated in the microwave oven until it
boiled slightly and the sections were put into the heated antigen
repair solution. The microwave oven was turned to medium heat for 2
min, then to medium heat for 2 min, then to medium heat for 7 min.
After the heating stopped, the antigen recovery solution was
returned to room temperature. The sections are placed flat on a
shelf in a box of water to keep the culture moist and prevent the
sections from drying out. Then, ~200 µl of blocking solution (PBS
containing 5% BSA) was dropped on and around the tissue surface and
incubated at room temperature for 1 h. Then, 100 µl diluted primary
antibody solution was added and incubated at 4˚C overnight. Dilute
fluorescent secondary antibody (100 µl) was added and incubated at
room temperature and dark for 1.5 h. DAPI was diluted with PBS at a
dilution ratio of 1:1,500 and 100 µl was added to each section and
incubated at room temperature and away from light for 10-15 min.
Finally, the sections were sealed with anti-fluorescence quencher.
Fluorescence images were obtained using confocal microscopy (cat.
no. FV3000; Olympus Corporation). All analysis was performed by a
pathologist who was independent of this study, blindly.
Western blotting
The total protein from lung tissues was collected
using chilled RIPA lysis buffer (Beyotime Institute of
Biotechnology), and protein concentration was measured according to
the BCA kit (Takara, Bio, Inc.) instruction method. Equal lysates
(50 µg) were resolved on 10% SDS-PAGE and transferred to PVDF
membranes with transfer buffer (25 mM Tris-base, 0.2 M Glycine, 20%
Methanol, pH 8.5). Membranes were blocked in 5% bovine serum
albumin (BSA) for 2 h at room temperature and then subjected to
primary antibodies against PI3 Kinase p85α (6G10) mouse monoclonal
antibody (cat. no. 13666; Cell Signaling Technology, Inc.),
Human/Mouse/Rat Akt pan specific antibody (cat. no. 4691; Cell
Signaling Technology, Inc.), p-PI3 Kinase p85 antibody (cat. no.
4228S; Cell Signaling Technology, Inc.), p-Akt (ser473) rabbit
monoclonal antibody (cat. no. 4060; Cell Signaling Technology,
Inc.) and GAPDH Human/Mouse/Rat polyclonal antibody (cat. no.
BS65529, Bioworld Technology). Membranes were incubated overnight
at 4˚C with primary antibodies. The membranes were then incubated
with HRP-labeled goat anti-rabbit IgG(H+L) (A0208, Beyotime
Institute of Biotechnology) or HRP-labeled goat anti-mouse IgG(H+L)
(A0216, Beyotime Institute of Biotechnology) for 2 h at room
temperature, depending on the species. All primary and secondary
antibodies were diluted 1:1,000. For visualization,
chemiluminescence was performed with ECL Western Blotting Substrate
(Tanon Science and Technology Co., Ltd.). Quantification of western
blots was performed with Image J (ImageJ 1.49; National Institutes
of Health).
Fluorescence-activated cell sorting
analysis (FACS)
To obtain bronchoalveolar lavage fluid (BALF), the
lungs were flushed via the trachea using 1 ml PBS. BALF was
recollected, and cells were stained using 0.25 µl fluorescein
isothiocyanate-conjugated anti-mouse CD4 (Miltenyi Biotec GmbH) and
0.625 µl APC conjugated anti-mouse CD8a (eBioscience; Thermo Fisher
Scientific, Inc.). The antibody was added and incubated for 0.5 h.
Mononuclear cells (MNCs) were obtained by grinding the mediastinal
lymph nodes and then suspending them in RPMI 1640 (Thermo Fisher
Scientific, Inc.). For MNCs staining, the following antibodies were
used: FITC CD4 monoclonal antibody (cat. no. GK1.5), PE CD8a
monoclonal antibody, APC CD25 monoclonal antibody (cat. no.
PC61.5), APC IFNγ monoclonal antibody (cat. no. XMG1.2), PE IL-4
monoclonal antibody (cat. no. 11B11), PE FoxP3 monoclonal antibody
(cat. no. FJK-16s) and PE IL-17A monoclonal antibody (cat. no.
eBio17B7; all from eBioscience; Thermo Fisher Scientific, Inc.).
For CD4+ and CD8+ cells, the staining was performed as above. For
Th1, Th2, and Th17 cells, we first stimulated at 37˚C for 4 h using
a Stimulation Cocktail, then added 0.25 ul CD4 antibody and
incubated at 4˚C for 0.5 h. The membrane was then broken according
to the fixed membrane breaking kit instructions (cat. no.
FMS-FP0100; fmacs Inc.). Finally, 0.625 µl of the corresponding
IFN-γ, IL-4, and IL-17A antibodies were added and incubated at 4˚C
for 0.5 h. For Treg cells, 0.25 µl of CD4 antibody was first added
and 0.625 µl of CD25 antibody and incubated for 0.5 h 4˚C. Membrane
rupture was then performed according to the instructions of the
membrane rupture kit (eBioscience; Thermo Fisher Scientific, Inc.).
Finally, Foxp3 antibody was added at 4˚C for 0.5 h. Tests were
performed using a FACSCalibur system (BD Biosciences). Flow
cytometry data were analyzed using the FlowJo software (V10.6;
FlowJo LLC.).
Suppression assays
Spleens were gently ground into homogenate and
filtered using 200 mesh filters to prepare MNCs. The MNCs were
incubated in PBS, PH 7.2, containing 0.5% BSA and 2 mM EDTA.
Isolation of CD4+CD25+ T cells was performed
with microbeads magnetic separation provided by magnetic cell
sorting (Miltenyi Biotec GmbH) from each group.
CD4+CD25- T cells (Teff) were collected from
the control group. To investigate the suppression capacity,
CD4+CD25- T cells were first labelled with
carboxyfluorescein diacetate succinimidyl ester (CFSE) dye
(Invitrogen; Thermo Fisher Scientific, Inc.). Then CFSE+
T cells were co-cultured with CD4+CD25+ T
cells in RPMI 1640 containing 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.) in a 1:1 or 2:1 ratio under the stimulation
of IL-2 (1 µg/106 cells) and anti-CD3/CD28 antibody (1
µg/106 cells). The cells were cultured in 24-well plates
for 72 h at 37˚C. CFSE+ T cells were detected using FACS
as described above.
Relative mRNA expressions
detection
Shock-frozen lung tissue was homogenized in RNAiso
Reagent (Takara Bio, Inc.). RNA was isolated using RNAiso Reagent
(Takara Bio, Inc.) according to the manufacturer's instructions.
Equal amounts of RNA were converted to cDNA using the Primerscript
RT reagent kit (Takara Bio, Inc.) according to the manufacturer's
protocol. Levels of mRNA were determined using Power SYBR Green PCR
Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.)
with a StepOnePlus™ Real-Time PCR System (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The thermocycling conditions are
listed in Table SII. Relative
expression levels of mRNA were normalized to GAPDH. All of the
values were expressed as 2-ΔΔCq
(21). Each sample was analyzed in
triplicate. The primers pairs are listed in Table SI.
Statistical analysis
Data were analyzed using GraphPad Prism 8
(Dotmatics). Comparisons between cohorts were performed using an
unpaired t-test. The t-test was used to calculate statistical
differences between two populations. For multiple group
comparisons, one-way ANOVA was used followed by Tukey's post-hoc
analysis for group size ≥3. Data are expressed as the mean ± SEM
(standard error of the mean). P<0.05 was considered to indicate
a statistically significant difference.
Results
Establishment of experimental
sarcoidosis with overactivated PI3K signaling
Histological examination confirmed the presence of a
granulomatous group in the mouse model consisting of epithelioid
cells and lymphocytes. This phenomenon is similar to granulomas in
patients with sarcoidosis (Fig.
1B). Tested by FACS, the ratios of
CD4+/CD8+ T cells in BALF in group
sarcoidosis were increased significantly compared with the control
group (P=0.0012). At the same time, no significant difference was
observed in those in lymph nodes (Fig.
1C and D).
CAL-101/AS-605240 ameliorates
pulmonary granuloma in sarcoidosis
Similar to those in group dexamethasone, granulomas
in the lungs were significantly alleviated after treatment of
CAL-101 (Fig. 2A). Notably, the
alleviation of granulomas was more obvious in group CAL-101
compared with that in group AS-605240. To determine the mechanisms
of these therapeutic effects of CAL-101/AS-605240, the balance of
Th1/Th17/Tregs was then tested among groups at the selected time
point (28 days following tail vein injection) when the number of
Tregs was normal in disease phase. CD4+IFNγ+
T lymphocytes decreased after treatment of AS-605240 compared with
the control, sarcoidosis, dexamethasone or CAL-101 groups (P=0.001,
P<0.0001, P=0.00125 and P=0.0003, respectively) (AS-605240 vs.
CAL-101) (Fig. 2B). Meanwhile,
both CD4+IL-4+ and
CD4+IL-17+ T lymphocytes showed no changes
among groups (Fig. 2C and D). On day 53 after sensitization, when
the number of Tregs showed no difference between group sarcoidosis
and group normal, neither CAL-101 nor dexamethasone changed the
Tregs ratio, whereas AS-605240 significantly decreased Tregs
compared with those in the sarcoidosis group (P=0.0039; Fig. 2E). When examining
Th1/Th2/Th17/Tregs related mRNA markers, CAL-101 was much more
effective in normalizing relative expression levels of RORγt,
FoxP3, IL-6 and IL-23 in sarcoidosis (P=0.0002, P=0.0293, P=0.0031
and P=0.0019, respectively) (Fig.
3C, D, F and G).
Compared with AS-605240, CAL-101 was more effective in regulating
the mRNA expression of Th1, Th2, Th17 and Treg (Fig. 3A, B, E and
H). Collectively, AS-605240 showed
obvious effects on decreasing Th1 and Tregs in SodA-induced
sarcoidosis, suggesting the important role of PI3Kγ, rather than
PI3Kδ, in modulating Th1 and Tregs differentiation in SodA-induced
sarcoidosis.
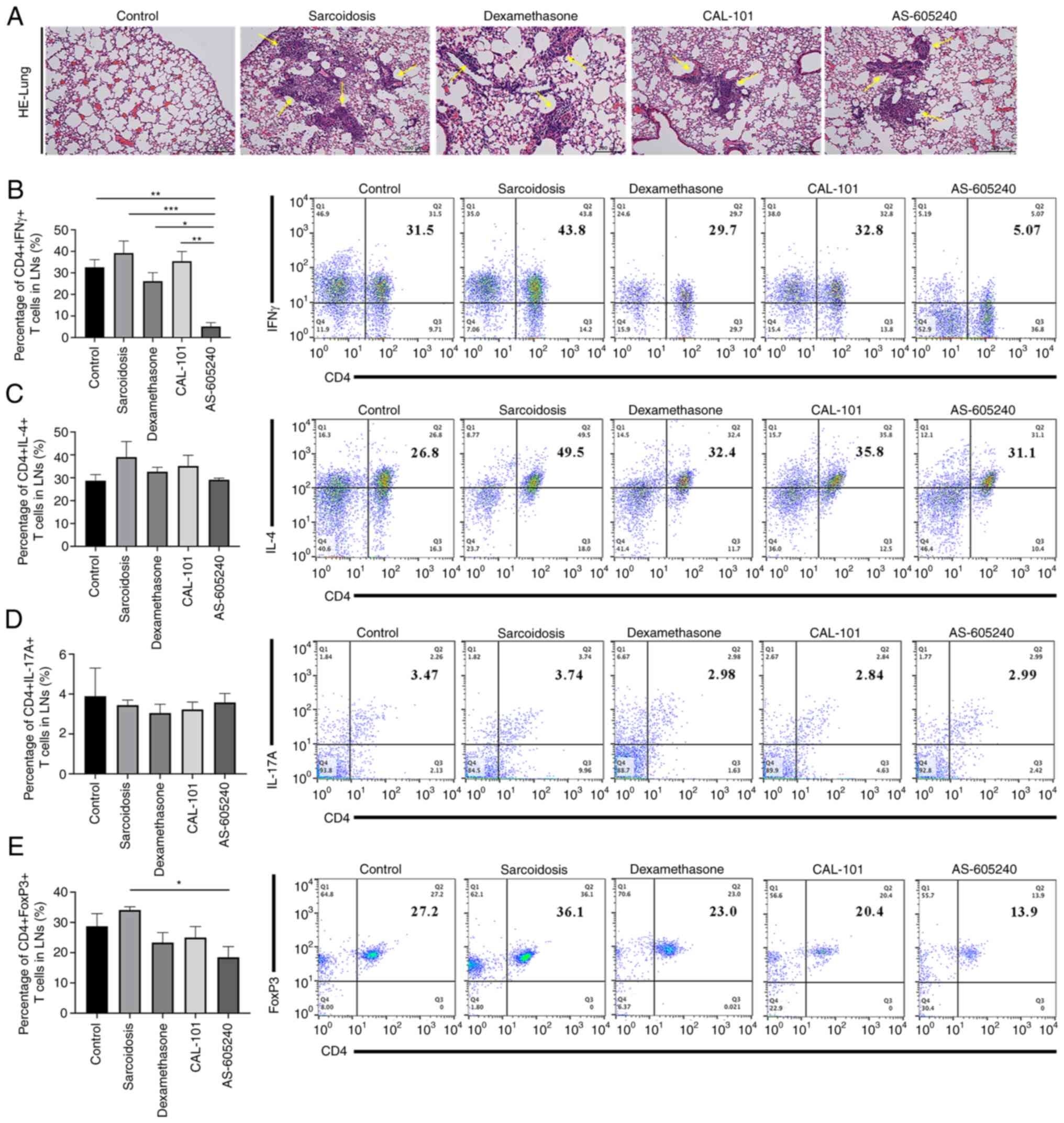 | Figure 2Histomorphology and cytological
characteristics of SodA-induced sarcoidosis after treatments with
CAL-101, AS-605240 or dexamethasone. (A) Histological images of
lungs stained with hematoxylin and eosin. The yellow arrows
indicate the pulmonary sarcoidosis granuloma. Magnification, x100.
(B-E) The frequencies of (B) Th1, (C) Th2, (D) Th17 and (E) Tregs
in lymph nodes from each group (n=6). *P<0.05,
**P<0.01, ***P<0.0002. SodA,
superoxidase A; IFNγ, interferon-gamma; IL-4, interleukin 4; IL-17,
interleukin; FoxP3, forkhead box protein P3. |
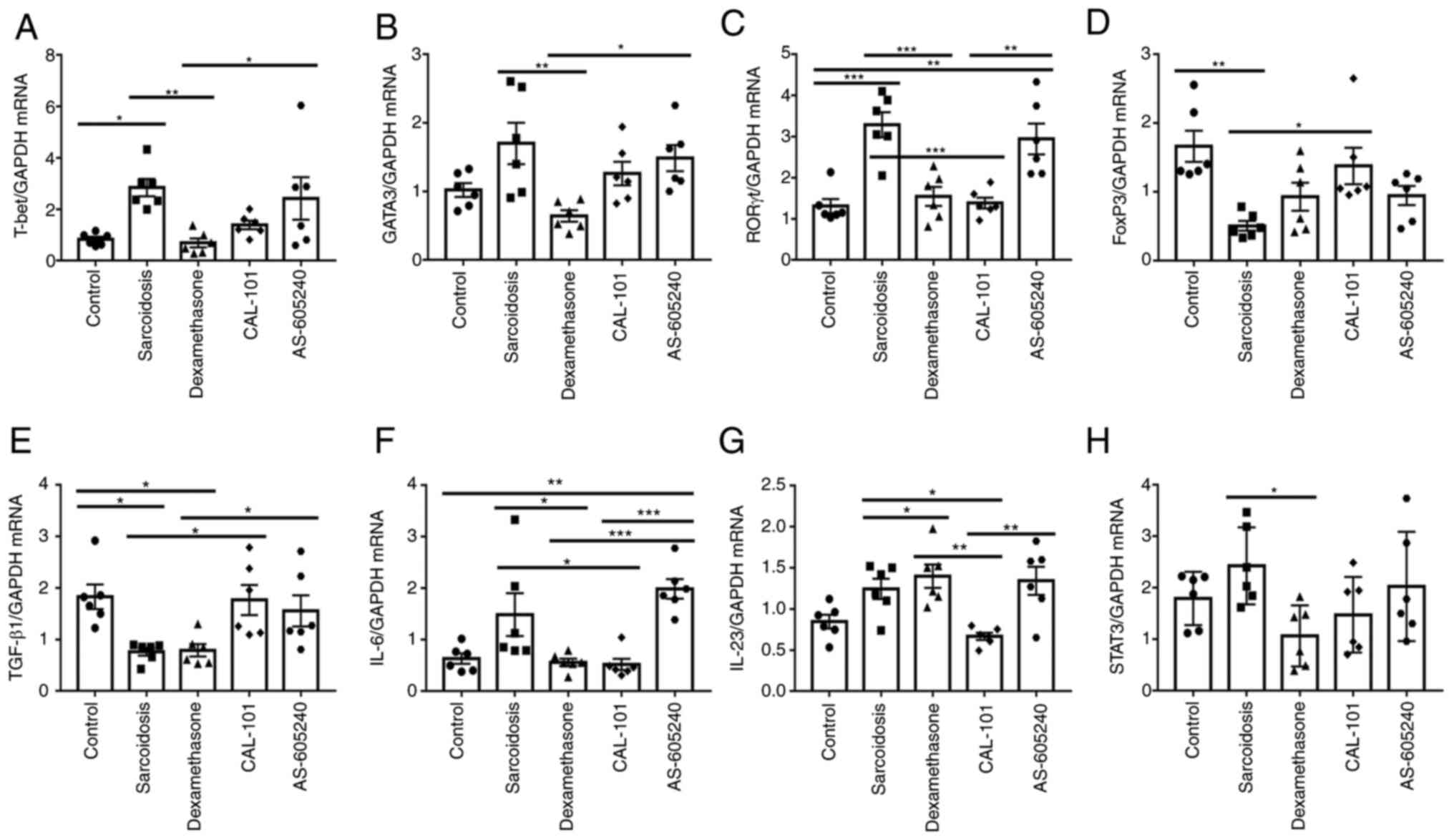 | Figure 3Th1, TH2, Th17 and Tregs-related
mRNAs expression levels in each group. (A-H) reverse
transcription-quantitative PCR was performed to determine relative
expression levels of (A) T-bet, (B) GATA3, (C)
RORγt, (D) FoxP3, (E) TGF-β, (F) IL-6,
(G) IL-23 and (H) STAT3 (n=6). GAPDH served as
internal control and three times repeats were performed.
*P<0.05, **P<0.01,
***P<0.0002. FoxP3, forkhead box protein P3; IL,
interleukin. |
PI3K inhibitors suppress activated
PI3K/Akt signaling in both lungs and Tregs
The immunofluorescent assay demonstrated that there
was increased p-PI3K and p-Akt expression in granulomas of the
group sarcoidosis compared with control. Administration of
CAL-101/AS-605240, as well as dexamethasone, significantly
suppressed these activations, while CAL-101 showed a more profound
effect compared with AS-605240 (Fig.
4A and B). Specifically,
CAL-101 normalized p-PI3K and downregulated p-Akt (P<0.0001). By
contrast, AS-605240 only downregulated p-PI3K but was less
effective in suppressing p-Akt compared with CAL-101 (P<0.0001),
suggesting the unique role of the PI3Kδ inhibitor in regulating
PI3K/Akt signaling in the granuloma. On the other hand,
CD4+CD25+ T cells also demonstrated high PI3K
expression levels and Akt activity levels (P<0.0001). Upon
CAL-101/AS-605240 treatment, the abnormal activation of PI3K/Akt
signaling in CD4+CD25+ cells was also
significantly recovered, with a stronger effect of CAL-101 compared
with AS-605240 (P<0.05; Fig. 4C
and D).
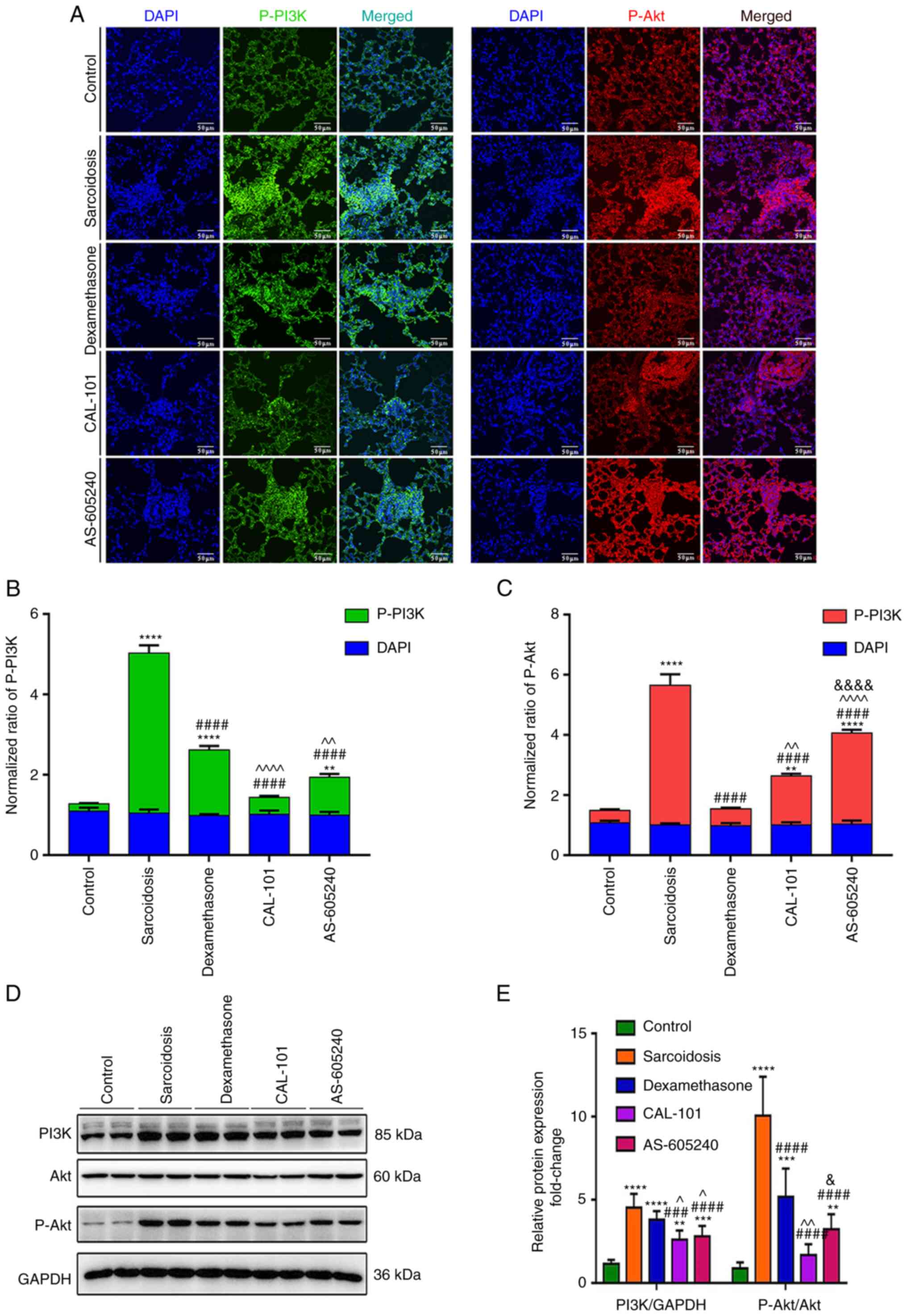 | Figure 4CAL-101/AS-605240 rescues the
overactivation of the PI3K/AKT pathway in SodA-induced sarcoidosis.
(A) Representative images showed the expression levels of p-PI3K
(green) and p-AKT (red) in granulomas in each group. Magnification,
x100. Summary of the mean fluorescence intensities of (B) p-PI3K,
(C) p-AKT (top) and DAPI (bottom) in each group. (D) Western blot
analysis (E) detecting expression levels of PI3K, p-PI3K, AKT and
p-AKT in CD4+CD25+ T cells purified by MACS
in each group. **P<0.01, ***P<0.002,
****P<0.0001 vs. the control group;
###P<0.002, ####P<0.0001 vs. the
sarcoidosis group; ^P<0.05, ^^P<0.01,
^^^^P<0.001 compared with the dexamethasone group;
&P<0.05,
&&&&P<0.001 vs. the CAL-101 group.
SodA, superoxidase A; PI3K, Phosphoinositide 3-kinases; p-,
phosphorylated-; MACS, magnetic cell sorting. |
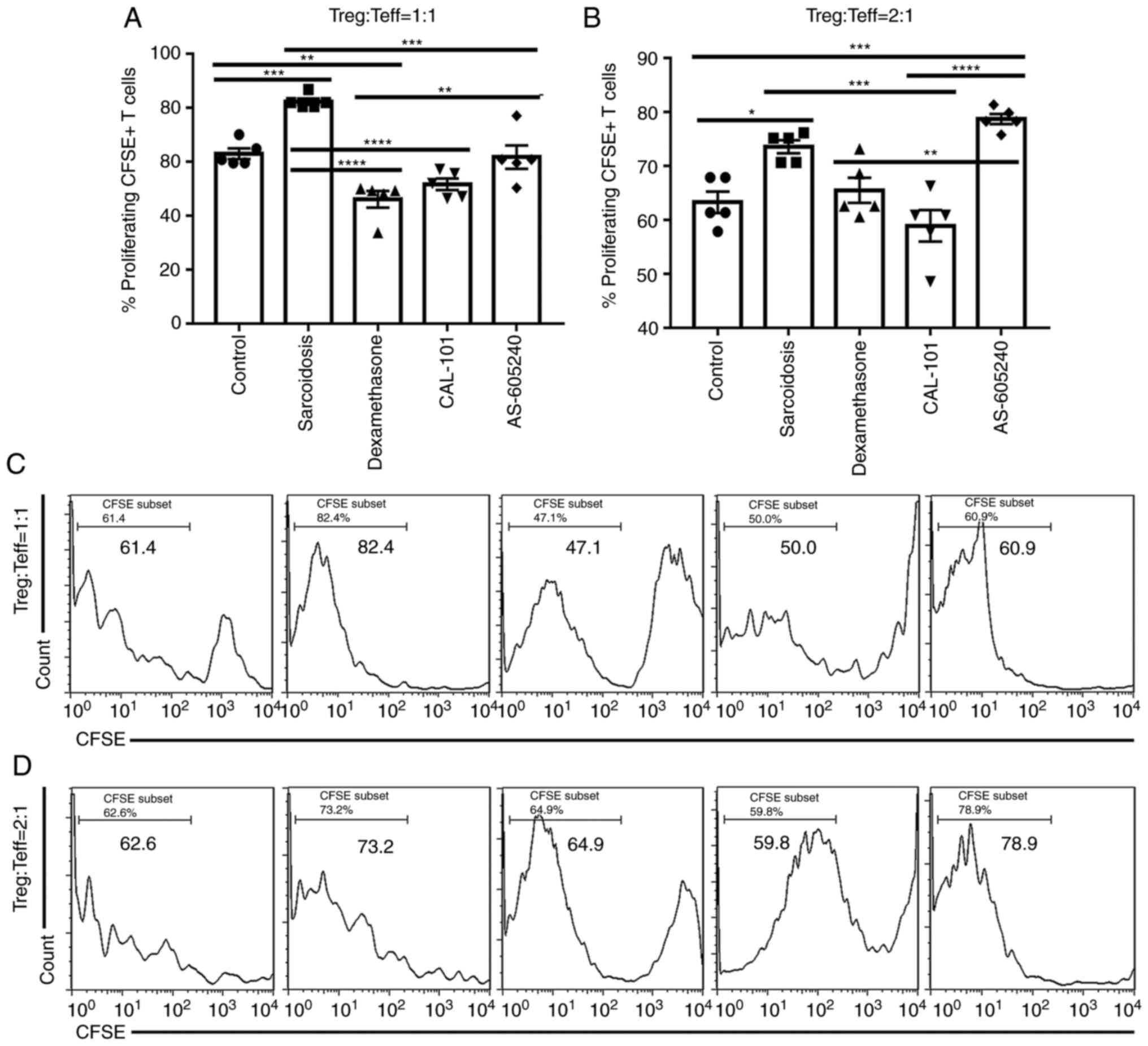
Inhibition of PI3K signaling rescues
the suppressive function of Tregs in sarcoidosis
CAL-101 and AS-605240 inhibited the abnormal
activation of PI3K/Akt signaling in Tregs, especially the former.
To determine whether these inhibitions induced immune homeostasis
in SodA-induced sarcoidosis, CD4+CD25+ cells
(Tregs) were co-cultured with CFSE-labeled Teff in the presence of
IL-2 and anti-CD3/CD28. As shown in Fig. 5, the proliferated CFSE+
T cells were significantly increased in group sarcoidosis compared
with the control group [P=0.0007 (1:1) and P=0.0131 (2:1),
respectively], demonstrating an impaired suppressive function of
Tregs in sarcoidosis. Similar to those in group dexamethasone,
CAL-101 induced significant decreases of CFSE+ T cells
when CD4+CD25+ T cells were incubated with
Teff at a ratio of 1:1 or 2:1 [P<0.0001 (1:1), and P=0.0004
(2:1), respectively] (Fig. 5A and
B). Notably, AS-605240 was less
effective than CAL-101 in inhibiting proliferation of CFSE+ T cells
in sodium-induced sarcoidosis when CD4+CD25+ T cells were incubated
in a 1:1 ratio with Teff cells (P=0.0003; Fig. 5A and C). Furthermore, AS-605240 still showed no
effects on rescuing the function of Tregs when
CD4+CD25+ T cells were incubated with Teff at
a high ratio of 2:1 (Fig. 5B and
D). By contrast, the effects of
CAL-101 on suppressing CFSE+ T cells proliferation were
relatively steady.
Discussion
Altogether these results demonstrated that
inhibition of PI3Kp110δ/p110γ by transtracheal CAL-101/AS-605240
administration ameliorated pulmonary granuloma in SodA-induced
sarcoidosis. Therapeutic effects could be attributed to the
inhibition of PI3K/Akt signaling both in granulomas and Tregs,
particularly in rescuing Treg suppression. CAL-101 was more
effective compared with AS-605240 in helping maintain the
homeostasis of both Th1 and Tregs and in overcoming the aberrantly
activated Akt in lungs and Tregs, which underlined that PI3Kδ
played a more important role compared with PI3Kγ in immune
modulation in SodA-induced sarcoidosis.
Class I PI3K is a heterodimeric enzyme consisting of
a regulatory unit and a catalytic unit (p110α, p110β, p110γ or
p110δ) (22,23). The expression of the p110δ and
p110γ subunits is mainly restricted to leukocytes, whereas p110α
and p110β are expressed by all cell types (9). Clinical trials with PI3K inhibitors
in breast cancer, leukemia and lymphoma are showing encouraging
results (24-27),
highlighting the potential of PI3K inhibitors in cancer
immunotherapy. PI3K/Akt signaling is usually dysregulated and
increased, having multifaceted regulatory functions in the immune
system, producing an autoimmune phenotype in immune disorders
(28,29). Aberrantly activated PI3K pathway
leads to loss of tolerance, aberrant immune activation and
production of autoantibody (19,30).
Although p110γ and p110δ are preferentially expressed in immune
cells, p110γ/p110δ inhibitors targeting these isoforms are reported
to inhibit inflammatory activity in autoimmune and inflammatory
diseases models by affecting the adaptive and innate immune
response, such as collagen-induced arthritis, ovalbumin-induced
asthma, microbiota-dependent colitis and systemic lupus
erythematosus models (31,32), leading to potential therapeutic
effects in multiple inflammatory and autoimmune diseases.
Furthermore, clinical trials have also shown promising effects of
PI3Kδ/γ inhibitors on immune disorders such as asthma, Sjögren's
syndrome and allergic rhinitis (33-35),
showing significant potential of PI3K inhibitors for clinical
use.
Despite this, not all PI3K inhibitors can produce a
good response in treatment. The effect of PI3K inhibitors targeting
various subtypes differ in different autoimmune diseases. To the
best of our knowledge, there are few studies on the use of PI3K
inhibitors in the treatment of sarcoidosis. In the treatment of
other autoimmune diseases, it has been revealed that not all
PI3Kγ/δ inhibitors can produce a good response in treatment
(36-38).
In the experimental autoimmune encephalomyelitis (EAE) mouse model,
the PI3Kδ-selective inhibitor does not affect the progression of T
cell-mediated autoimmune inflammation or disease severity (36). By contrast, PI3Kγ inhibitor
significantly alleviates the severity of EAE, and significantly
reduces leukocyte infiltration in the central nervous system
(37). For the experimental
epidermolysis bullosa acquisita, a PI3Kβ-selective inhibitor is
more effective compared with a PI3Kδ-selective one in inhibiting
neutrophil-driven inflammation (38). Whether other selective PI3Kγ/δ
inhibitors have the same therapeutic effect remains to be verified,
and further experiments will be conducted in the future. Thus,
discussing the most effective PI3K subtypes in treating sarcoidosis
is reasonable.
In sarcoidosis, PI3K signaling is one of the most
aberrantly activated pathways. Our early study demonstrated that
inhibition of PI3K/Akt signaling by BKM120 or LY294002 can
ameliorate granuloma (14,17). Previous results implied that
co-inhibiting the PI3Kγ subunit is more efficient in augmenting
therapies when using non-selective PI3K inhibitors (17). The current study aimed to determine
the selective PI3K inhibitors that would optimize therapy. Although
the distinction is not absolute, PI3Kγ is functionally dominant in
myeloid cells (19). It plays a
key role in chemokine-mediated recruitment and activation of innate
immune cells at sites of inflammation, whereas PI3Kδ is highly
expressed in leukocytes and is crucial in antigen receptor and
cytokine-mediated B and T cell development, differentiation and
function (39). They may be
capable of modulating the Th1/Tregs/Th17 paradigm due to their
diverse roles in diverse immune functions. In that case, the
present study tested whether the presence of PI3Kδ or PI3Kγ
inhibition by CAL-101 or AS-605240 would benefit SodA-induced
sarcoidosis on the selected time point when the number of Tregs was
normal. The results turned out to be more complex than anticipated.
When PI3Kδ inhibitor induced more profound therapeutic effects
compared with the PI3Kγ inhibitor, PI3Kδ and PI3Kγ may work
together to generate a functional output in SodA-induced
sarcoidosis.
Overall, studies suggest that sarcoidosis is
considered a model of persistent inflammation caused by
abnormalities in Th1/Th2 and Treg/Th17 paradigms and impaired the
function of Tregs (7,8). The homeostatic ratio of circulating
Tregs/Th17 inversely trends with sarcoidosis activity, decreasing
in the relapsed patients and increasing back to normal ranges in
non-active sarcoidosis or with treatment with corticosteroids
(15,40). In addition, the increase in the
function of Tregs is associated with improvements in clinical
measures of sarcoidosis (1,11).
In our previous study, inhibition of PI3Ks by BKM120 or LY294002
was show to significantly improve pulmonary granuloma, ameliorate
both Th1/Th2 and Tregs/Th17 disorders and restore the suppressive
function of Tregs in SodA-induced sarcoidosis at the time point
when there was a Tregs/Th17 imbalance (17). Furthermore, the present study
showed the effects of CAL-101 and AS-605240 on rescuing the
suppressive functions of Tregs at the selected time point with
normal Tregs ratios. However, notably, AS-605240 adversely affected
the balance between Th1 and Treg in a less potent manner compared
with CAL-101. These findings are in line with the key role of the
Th1/Tregs/Th17 paradigm in the pathogenesis of sarcoidosis
(7,8,15,40),
though the question of how PI3K inhibitors slow the development of
granuloma in this setting remains unsolved.
Mechanistic studies highlight the potential
importance of the PI3K/Akt signaling on the activation,
differentiation and function of both Tregs and T effector cells,
while PI3K signaling has been identified as a nodal control point
for Tregs homeostasis and stability (18,41,42).
Tregs seem to respond more easily to cancer immunotherapy compared
with other T cell populations, even though the mechanism of action
is still unclear (43,44). Inhibition of PI3Kγ and PI3Kδ both
restrict the expansion of alloreactive T cells in a mouse allograft
transplantation model. However, PI3Kδ inhibition instead of
concurrent p110γ deficiency detrimentally affects the function of
Tregs (41). In addition, PI3Kγ
inhibition may compensate for the negative effect of PI3Kδ
inhibition on long-term allograft survival (41). PI3Kγ and PI3Kδ inhibition may
synergistically modulate immune responses. While selective
inhibition of only PI3Kδ is weakly anti-inflammatory, dual
inhibition of PI3Kγ and δ has superior anti-inflammatory effects
(45,46). Moreover, PI3Kδ/γ inhibition yields
an anti-inflammatory signature distinct from pan-PI3K inhibition
and known anti-inflammatory drugs (46). Consistent with the findings of
these reports, the present study demonstrated that PI3Kδ inhibition
could overcome the aberrantly activated Akt in the lungs and Tregs,
thus maintaining the function of Tregs. By contrast, PI3Kγ
inhibition was less effective in recovering PI3K/Akt signaling in
granuloma and Tregs but even dampened Th1/Tregs balance.
Considering that BKM120 was previously found to have a more
profound effect on granuloma improvement compared with
LY294002(17), synergistic effects
of PI3Kγ and PI3Kδ may occur in immune modulation in SodA-induced
sarcoidosis.
In conclusion, these results indicated that
pharmacological inhibition of PI3Kδ by CAL-101 may be an effective
immunoregulatory strategy in treating diseases with deficient
suppressive functions of Tregs, such as sarcoidosis. Moreover, it
appears that inhibition of PI3Kδ is more efficient compared with
PI3Kγ, despite the possibility that synergism of these two PI3K
subunits may occur. Although it remains unknown whether dual
inhibition of PI3Kγ and PI3Kδ has an improved effect on immune
modulation under the distinct pathophysiological processes, PI3Kγ/δ
inhibitors may be developed into a new therapeutic principle for
sarcoidosis.
Supplementary Material
Primer sequences.
Thermocycling conditions.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by the National Natural Science
Foundation of China (grant nos. 81100386 and 81400046) and the
Independent and Open Grant of Jiangsu Key Laboratory of Molecular
Medicine and Innovation and Entrepreneurship Training Program for
College Students (grant nos. G201910284011 and 202010284016Z).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ participated in data interpretation and wrote the
manuscript. QD performed the experiments and analyzed the data. JS,
SZ, BZ, SL, YZ, YiW, XL, XJ and DL collected and assembled data. JD
analyzed the data and improved the language of the manuscript. YaW
contributed to the design of the study, received funding and
supervised the study. YoW reviewed and edited the manuscript and
provided administrative support for the study. QD and XZ confirm
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved under a
project license (approval no. SCXK-Jiangsu-2019-0056) granted by
the Institutional Animal Care and Use Committee of Nanjing
University, in compliance with institutional guidelines for the
care and use of animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Broos CE, van Nimwegen M, Hoogsteden HC,
Hendriks RW, Kool M and van den Blink B: Granuloma formation in
pulmonary sarcoidosis. Front Immunol. 4(437)2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hunninghake GW, Costabel U, Ando M,
Baughman R, Cordier JF, du Bois R, Eklund A, Kitaichi M, Lynch J,
Rizzato G, et al: ATS/ERS/WASOG statement on sarcoidosis. American
thoracic society/european respiratory society/world association of
sarcoidosis and other granulomatous disorders. Sarcoidosis Vasc
Diffuse Lung Dis. 16:149–173. 1999.PubMed/NCBI
|
|
3
|
Oswald-Richter KA, Richmond BW, Braun NA,
Isom J, Abraham S, Taylor TR, Drake JM, Culver DA, Wilkes DS and
Drake WP: Reversal of global CD4+ subset dysfunction is associated
with spontaneous clinical resolution of pulmonary sarcoidosis. J
Immunol. 190:5446–5453. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Cinetto F and Agostini C: Advances in
understanding the immunopathology of sarcoidosis and implications
on therapy. Expert Rev Clin Immunol. 12:973–988. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Iannuzzi MC, Rybicki BA and Teirstein AS:
Sarcoidosis. N Engl J Med. 357:2153–2165. 2007.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Gurram RK, Kujur W, Maurya SK and Agrewala
JN: Caerulomycin A enhances transforming growth factor-β
(TGF-β)-Smad3 protein signaling by suppressing interferon-γ
(IFN-γ)-signal transducer and activator of transcription 1 (STAT1)
protein signaling to expand regulatory T cells (Tregs). J Biol
Chem. 289:17515–17528. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ten Berge B, KleinJan A, Muskens F, Hammad
H, Hoogsteden HC, Hendriks RW, Lambrecht BN and Van den Blink B:
Evidence for local dendritic cell activation in pulmonary
sarcoidosis. Respir Res. 13(33)2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Patterson KC and Chen ES: The pathogenesis
of pulmonary sarcoidosis and implications for treatment. Chest.
153:1432–1442. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Miyara M, Amoura Z, Parizot C, Badoual C,
Dorgham K, Trad S, Kambouchner M, Valeyre D, Chapelon-Abric C,
Debré P, et al: The immune paradox of sarcoidosis and regulatory T
cells. J Exp Med. 203:359–370. 2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Idali F, Wahlström J, Müller-Suur C,
Eklund A and Grunewald J: Analysis of regulatory T cell associated
forkhead box P3 expression in the lungs of patients with
sarcoidosis. Clin Exp Immunol. 152:127–137. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Prasse A, Zissel G, Lützen N, Schupp J,
Schmiedlin R, Gonzalez-Rey E, Rensing-Ehl A, Bacher G, Cavalli V,
Bevec D, et al: Inhaled vasoactive intestinal peptide exerts
immunoregulatory effects in sarcoidosis. Am J Respir Crit Care Med.
182:540–548. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Taflin C, Miyara M, Nochy D, Valeyre D,
Naccache JM, Altare F, Salek-Peyron P, Badoual C, Bruneval P,
Haroche J, et al: FoxP3+ regulatory T cells suppress early stages
of granuloma formation but have little impact on sarcoidosis
lesions. Am J Pathol. 174:497–508. 2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Rappl G, Pabst S, Riemann D, Schmidt A,
Wickenhauser C, Schütte W, Hombach AA, Seliger B, Grohé C and Abken
H: Regulatory T cells with reduced repressor capacities are
extensively amplified in pulmonary sarcoid lesions and sustain
granuloma formation. Clin Immunol. 140:71–83. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ding J, Dai J, Cai H, Gao Q and Wen Y:
Extensively disturbance of regulatory T cells-Th17 cells balance in
stage II pulmonary sarcoidosis. Int J Med Sci. 14:1136–1142.
2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zissel G and Müller-Quernheim J: Cellular
players in the immunopathogenesis of sarcoidosis. Clin Chest Med.
36:549–560. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang B, Zhao F, Mao H, Ma W, Zhang Y,
Zhang X, Ding J, Gao Q and Wen Y: Interleukin 33 ameliorates
disturbance of regulatory T cells in pulmonary sarcoidosis. Int
Immunopharmacol. 64:208–216. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhang B, Dai Q, Jin X, Liang D, Li X, Lu
H, Liu Y, Ding J, Gao Q and Wen Y: Phosphoinositide
3-kinase/protein kinase B inhibition restores regulatory T cell's
function in pulmonary sarcoidosis. J Cell Physiol. 234:19911–19920.
2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Huynh A, DuPage M, Priyadharshini B, Sage
PT, Quiros J, Borges CM, Townamchai N, Gerriets VA, Rathmell JC,
Sharpe AH, et al: Control of PI(3) kinase in Treg cells maintains
homeostasis and lineage stability. Nat Immunol. 16:188–196.
2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Greaves SA, Peterson JN, Strauch P, Torres
RM and Pelanda R: Active PI3K abrogates central tolerance in
high-avidity autoreactive B cells. J Exp Med. 216:1135–1153.
2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Swaisgood CM, Oswald-Richter K, Moeller
SD, Klemenc JM, Ruple LM, Farver CF, Drake JM, Culver DA and Drake
WP: Development of a sarcoidosis murine lung granuloma model using
mycobacterium superoxide dismutase A peptide. Am J Respir Cell Mol
Biol. 44:166–174. 2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Abdel-Magid AF: Potential of PI3Kβ
inhibitors in the treatment of cancer and other diseases. ACS Med
Chem Lett. 8:778–780. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Rathinaswamy MK and Burke JE: Class I
phosphoinositide 3-kinase (PI3K) regulatory subunits and their
roles in signaling and disease. Adv Biol Regul.
75(100657)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Scott WJ, Hentemann MF, Rowley RB, Bull
CO, Jenkins S, Bullion AM, Johnson J, Redman A, Robbins AH, Esler
W, et al: Discovery and SAR of novel
2,3-dihydroimidazo[1,2-c]quinazoline PI3K inhibitors:
Identification of copanlisib (BAY 80-6946). ChemMedChem.
11:1517–1530. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lannutti BJ, Meadows SA, Herman SE,
Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM,
Deininger M, et al: CAL-101, a p110delta selective
phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell
malignancies, inhibits PI3K signaling and cellular viability.
Blood. 117:591–594. 2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Furet P, Guagnano V, Fairhurst RA,
Imbach-Weese P, Bruce I, Knapp M, Fritsch C, Blasco F, Blanz J,
Aichholz R, et al: Discovery of NVP-BYL719 a potent and selective
phosphatidylinositol-3 kinase alpha inhibitor selected for clinical
evaluation. Bioorg Med Chem Lett. 23:3741–3748. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Burris HA III, Flinn IW, Patel MR, Fenske
TS, Deng C, Brander DM, Gutierrez M, Essell JH, Kuhn JG, Miskin HP,
et al: Umbralisib, a novel PI3Kδ and casein kinase-1ε inhibitor, in
relapsed or refractory chronic lymphocytic leukaemia and lymphoma:
An open-label, phase 1, dose-escalation, first-in-human study.
Lancet Oncol. 19:486–496. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Patel RK and Mohan C: PI3K/AKT signaling
and systemic autoimmunity. Immunol Res. 31:47–55. 2005.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Adefemi F, Fruman DA and Marshall AJ: A
case for phosphoinositide 3-kinase-targeted therapy for infectious
disease. J Immunol. 205:3237–3245. 2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Franks SE, Getahun A and Cambier JC: A
precision B cell-targeted therapeutic approach to autoimmunity
caused by phosphatidylinositol 3-kinase pathway dysregulation. J
Immunol. 202:3381–3393. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Winkler DG, Faia KL, DiNitto JP, Ali JA,
White KF, Brophy EE, Pink MM, Proctor JL, Lussier J, Martin CM, et
al: PI3K-δ and PI3K-γ inhibition by IPI-145 abrogates immune
responses and suppresses activity in autoimmune and inflammatory
disease models. Chem Biol. 20:1364–1374. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Steinbach EC, Kobayashi T, Russo SM,
Sheikh SZ, Gipson GR, Kennedy ST, Uno JK, Mishima Y, Borst LB, Liu
B, et al: Innate PI3K p110δ regulates Th1/Th17 development and
microbiota-dependent colitis. J Immunol. 192:3958–3968.
2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Juarez M, Diaz N, Johnston GI, Nayar S,
Payne A, Helmer E, Cain D, Williams P, Devauchelle-Pensec V, Fisher
BA, et al: A phase 2 randomized, double-blind, placebo-controlled,
proof-of-concept study of oral seletalisib in primary Sjögren's
syndrome. Rheumatology (Oxford). 60:1364–1375. 2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Horak F, Puri KD, Steiner BH, Holes L,
Xing G, Zieglmayer P, Zieglmayer R, Lemell P and Yu A: Randomized
phase 1 study of the phosphatidylinositol 3-kinase δ inhibitor
idelalisib in patients with allergic rhinitis. J Allergy Clin
Immunol. 137:1733–1741. 2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Perry MWD, Bjorhall K, Bold P, Brűlls M,
Börjesson U, Carlsson J, Chang HA, Chen Y, Eriksson A, Fihn BM, et
al: Discovery of AZD8154, a dual PI3Kγδ inhibitor for the treatment
of asthma. J Med Chem. 64:8053–8075. 2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Stark AK, Davenport ECM, Patton DT,
Scudamore CL, Vanhaesebroeck B, Veldhoen M, Garden OA and Okkenhaug
K: Loss of phosphatidylinositol 3-kinase activity in regulatory T
cells leads to neuronal inflammation. J Immunol. 205:78–89.
2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Li H, Park D, Abdul-Muneer PM, Xu B, Wang
H, Xing B, Wu D and Li S: PI3Kγ inhibition alleviates symptoms and
increases axon number in experimental autoimmune encephalomyelitis
mice. Neuroscience. 253:89–99. 2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zillikens H, Kasprick A, Osterloh C, Gross
N, Radziewitz M, Hass C, Hartmann V, Behnen-Härer M, Ernst N, Boch
K, et al: Topical application of the PI3Kβ-selective small molecule
inhibitor TGX-221 Is an effective treatment option for experimental
epidermolysis bullosa acquisita. Front Med (Lausanne).
8(713312)2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Hawkins PT and Stephens LR: PI3K
signalling in inflammation. Biochim Biophys Acta. 1851:882–897.
2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Liu Y, Qiu L, Wang Y, Aimurola H, Zhao Y,
Li S and Xu Z: The circulating Treg/Th17 cell ratio is correlated
with relapse and treatment response in pulmonary sarcoidosis
patients after corticosteroid withdrawal. PLoS One.
11(e0148207)2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Uehara M, McGrath MM, Ohori S, Solhjou Z,
Banouni N, Routray S, Evans C, DiNitto JP, Elkhal A, Turka LA, et
al: Regulation of T cell alloimmunity by PI3Kγ and PI3Kδ. Nat
Commun. 8(951)2017.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Gamper CJ and Powell JD: All PI3Kinase
signaling is not mTOR: Dissecting mTOR-dependent and independent
signaling pathways in T cells. Front Immunol. 3(312)2012.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Lim EL and Okkenhaug K: Phosphoinositide
3-kinase δ is a regulatory T-cell target in cancer immunotherapy.
Immunology. 157:210–218. 2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Vanhaesebroeck B, Perry MWD, Brown JR,
André F and Okkenhaug K: PI3K inhibitors are finally coming of age.
Nat Rev Drug Discov. 20:741–769. 2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Randis TM, Puri KD, Zhou H and Diacovo TG:
Role of PI3Kdelta and PI3Kgamma in inflammatory arthritis and
tissue localization of neutrophils. Eur J Immunol. 38:1215–1224.
2008.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Williams O, Houseman BT, Kunkel EJ,
Aizenstein B, Hoffman R, Knight ZA and Shokat KM: Discovery of dual
inhibitors of the immune cell PI3Ks p110delta and p110gamma: A
prototype for new anti-inflammatory drugs. Chem Biol. 17:123–134.
2010.PubMed/NCBI View Article : Google Scholar
|