Introduction
TAB2 encodes the TGF-β activated kinase 1
(MAP3K7) binding protein 2 (TAB2), which can self-phosphorylate and
regulate chemokines and inflammatory signaling molecules.
Therefore, TAB2 gene defects are primarily associated with
inflammatory responses (1,2). Thienpont et al (3) demonstrated the presence of a 6q24-q25
deletion following array-based comparative genomic hybridization
detection in seven patients with congenital heart disease (CHD),
and further revealed that insufficient haploid dosage of
TAB2 in this region can lead to abnormal human heart
development. Mutations in TAB2 lead to congenital heart
defects, non-syndromic, 2 (CHTD2; OMIM#614980). TAB2 results
in varying types of cardiac abnormalities, such as mitral,
tricuspid valve prolapse and aortic stenosis, while some patients
may face sudden cardiac death (3,4). In
addition to cardiac structural abnormalities, patients with CHTD2
may present dysmorphic facial, joint and skin phenotypes (5,6).
Therefore, identifying the abnormal features caused by mutations of
TAB2 aids in clinical diagnosis, especially in prenatal
diagnosis and genetic counseling.
The present study performed trio-whole exome
sequencing (trio-WES) on a Chinese family with short stature and
CHD as the main clinical features. The patient's cardiac color
Doppler ultrasound showed insufficient mitral and mild
regurgitation. In addition, the patient exhibited growth
restriction and dysmorphic facial features. The results of genetic
testing indicated that a novel pathogenic mutation occurred in the
TAB2 gene; this may lead to protein non-expression, as
indicated by in vitro functional experiments. The present
study further expanded the pathogenic mutation spectrum of
TAB2.
Case report
A 3-year-old male child was the subject of the
present study (his mother is G1P1). Prenatal ultrasound diagnosis
at 24 weeks of pregnancy showed that the fetus had short legs.
Vaginal delivery occurred at 41+6 weeks of pregnancy,
there was no history of birth asphyxia, the Apgar score was unknown
and birth weight was 3,200 g. After birth, the motor milestone
development of the child was normal. At the age of 10 months, his
parents noticed that his growth development lagged behind those of
his peers. No special treatment was given, and the annual growth
rate was unknown. During the study period of the present study, the
child was hospitalized in The Department of Pediatric Endocrinology
and Metabolic Disease (Children's Hospital of Fudan University,
Anhui Hospital, Hefei, China) for the growth related treatment. The
physical examination conducted on admission showed that the child
was in good mental state. He had dysmorphic facial features with a
wide forehead and low ear position, with a height of 89.5 cm [-3
standard deviation (SD)] and a weight of 12.7 kg (-1.5 SD). His
bilateral scrotum was empty, and the testicles were not touched.
His limbs moved freely, with normal muscle tension of the limbs and
no bone deformities were observed. No obvious abnormalities were
detected during the auxiliary examination. Color Doppler
echocardiography showed mitral insufficiency with mild
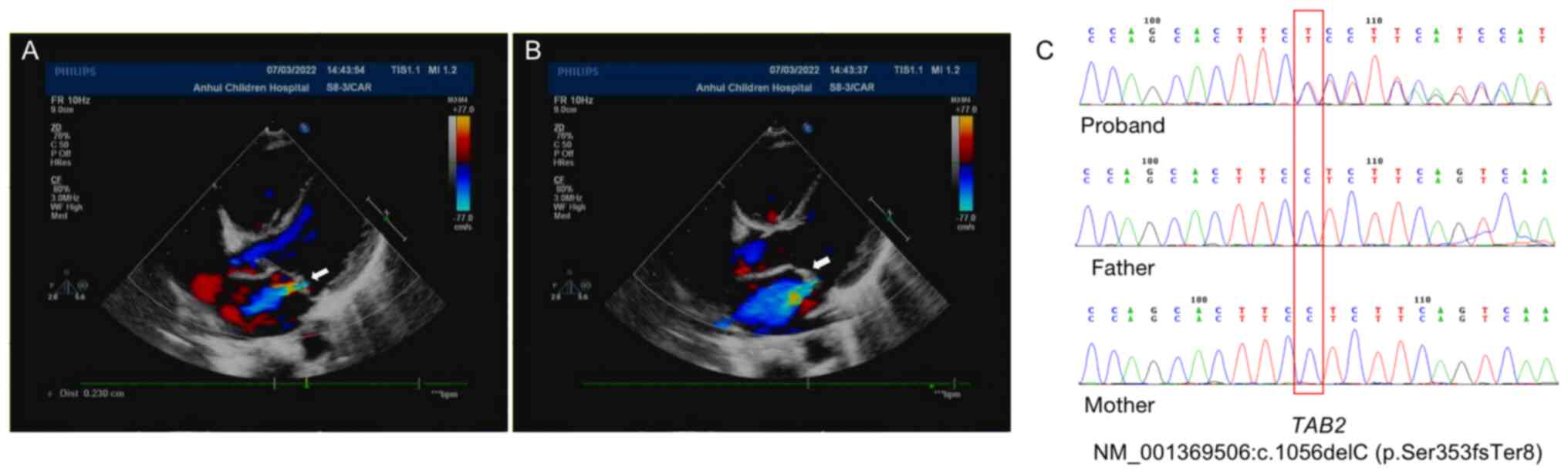
regurgitation; a closing gap of ~0.23 cm was observed, as shown in
Fig. 1A and B. The parents of the child were healthy,
and stated that they were non-consanguineously married, and had no
family history of short stature and CHD.
Genetic testing
The family was tested using trio-WES. The work
described in the present case report was approved by the Ethics
Committee of the Children's Hospital of Fudan University Anhui
Hospital (Hefei, China), and informed consent was obtained from the
parents of the patient. Approximately 3 ml of peripheral blood was
collected from the proband as well as his parents, DNA was
extracted using the genomic DNA extraction kit (Tiangen Biochemical
Technology Inc.). A DNA library was constructed using the xGen
Exome Research Panel V2 Kit (Integrated DNA Technologies Inc.). The
paired-end sequencing (150 bp) was run on the Hiseq 4000 platform
(Illumina Inc.). The off-line data were filtered for quality
control and screened using the Burrows-Wheeler Aligner sequence
alignment method (refer to hg19 version) (7). The target sequence mutation sites
were identified using the GATK v4.1.9 software (8) and annotated according to the public
variation databases (dbSNP build 155, www.ncbi.nlm.nih.gov/snp/; ExAC v0.3.1, https://gnomad.broadinstitute.org/; and 1000
Genomes, www.internationalgenome.org/). Multiple biohazard
prediction software were used to analyze the suspected variation to
the TAB2 gene (SIFT build 37, https://sift.bii.a-star.edu.sg/; Polyphen-2 v2.3,
http://genetics.bwh.harvard.edu/pph2/; and
MutationTaster 2021, www.mutationtaster.org/).
The results of the genetic testing for the child,
but not his parents, revealed a heterozygous base deletion mutation
TC>T in TAB2 exon 4, resulting in a frameshift mutation
after encoded amino acid 352, NM_001369506: c.1056delC,
(p.Ser353fsTer8), suggesting that the frameshift mutation observed
may be a de novo mutation of TAB2. The mutation was
not included in the public database. According to the guidelines of
the American College of Medical Genetics and Genomics, the mutation
is rated as pathogenic, and the evidence rating is
PVS1+PM2+PM6(9). Sanger sequencing
was performed using genomic DNA isolated from the blood of the
patient and his parents. Direct sequencing was performed on the ABI
3730XL Automatic Sequencer (Thermo Fisher Scientific, Inc.).
Amplification was performed using the following conditions: 1 cycle
of 95˚C for 2 min, followed by 39 cycles of 95˚C for 30 sec, 60˚C
for 30 sec, 72˚C for 40 sec and a final extension at 72˚C for 5
min. Sanger sequencing confirmed the existence of this mutation
(Fig. 1C).
Plasmid construction and protein
expression
The pcDNA3.1 vector (Tsingke Biotech) was used to
construct a TAB2-wild-type (WT) plasmid using Phanta®
Max Super-Fidelity DNA Polymerase (cat. no. P505; Vazyme Biotech
Co., Ltd.), according to the manufacturer's instructions
(amplification primers: Forward,
5'-CTTGGTACCGAGCTCGGATCCATGGCCCAAGGAAGCCAC-3'; and Reverse,
5'-TGCTGGATATCTGCAGAATTCTCAGAAATGCCTTGGCATCTC-3'). The TAB2-mutant
(MUT) plasmid was constructed using a Mut Express MultiS Fast
Mutagenesis Kit V2 (cat. no. C215; Vazyme Biotech Co., Ltd.)
(amplification primer: Forward,
5'-CCAGCACTTCTCTTCAGTCAATAGCCAGACCTTAA-3'; and Reverse,
5'-ACTGAAGAGAAGTGCTGGAGGTTCGAGGTCCAG-3'). The amplification
conditions were as follows: Pre-denaturation at 95˚C for 3 min,
denaturation at 95˚C for 15 sec, annealing at 60˚C for 15 sec,
extension at 72˚C for 1 min (35 cycles in total), and a final
extension at 72˚C for 10 min. The plasmids were then transfected
into human 293T cells (The Cell Bank of Type Culture Collection of
The Chinese Academy of Sciences) using Lipofectamine 2000 (Thermo
Fisher Scientific, Inc.). After the cells were fully lysed in cell
lysis buffer (Beyotime Institute of Biotechnology), 80 µg of
protein was extracted and analyzed using western blotting (WB).
Ultramicro spectrophotometer (NanoDrop 2000; Thermo Fisher
Scientific, Inc.) was used to determine the protein concentration,
and 10% SDS-PAGE gel (Epizyme, Inc.) was used to concentrate and
isolate 80 µg of protein. After using polyvinylidene difluoride to
transfer the film, a rapid sealing solution (cat. no. P0220;
Beyotime Institute of Biotechology) was used at 20˚C for 15 min.
The primary antibodies TAB2 (cat. no. A9867; ABclonal Biotech Co.,
Ltd.) and β-actin (cat. no. 3700; Cell Signaling Technology, Inc.)
were used at a dilution ratio of 1:1,000. The secondary antibodies
used were HRP goat anti-rabbit IgG (H+L) (cat. no. AS014; ABclonal
Biotech Co., Ltd.), anti-mouse IgG and HRP-linked antibody (cat.
no. 7076; Cell Signaling Technology, Inc.) at a dilution ratio of
1:5,000. The WB bands were analyzed using BeyoECL (cat. no.
P0018FS; Beyotime Institute of Biotechnology) and ImageJ software
(v1.80; National Institutes of Health), and GraphPad Prism 8
(GraphPad Software, Inc.) was used to conduct the statistical
analyses and create the figures. Each experiment was repeated
thrice. P<0.05 was considered to indicate a statistically
significant difference.
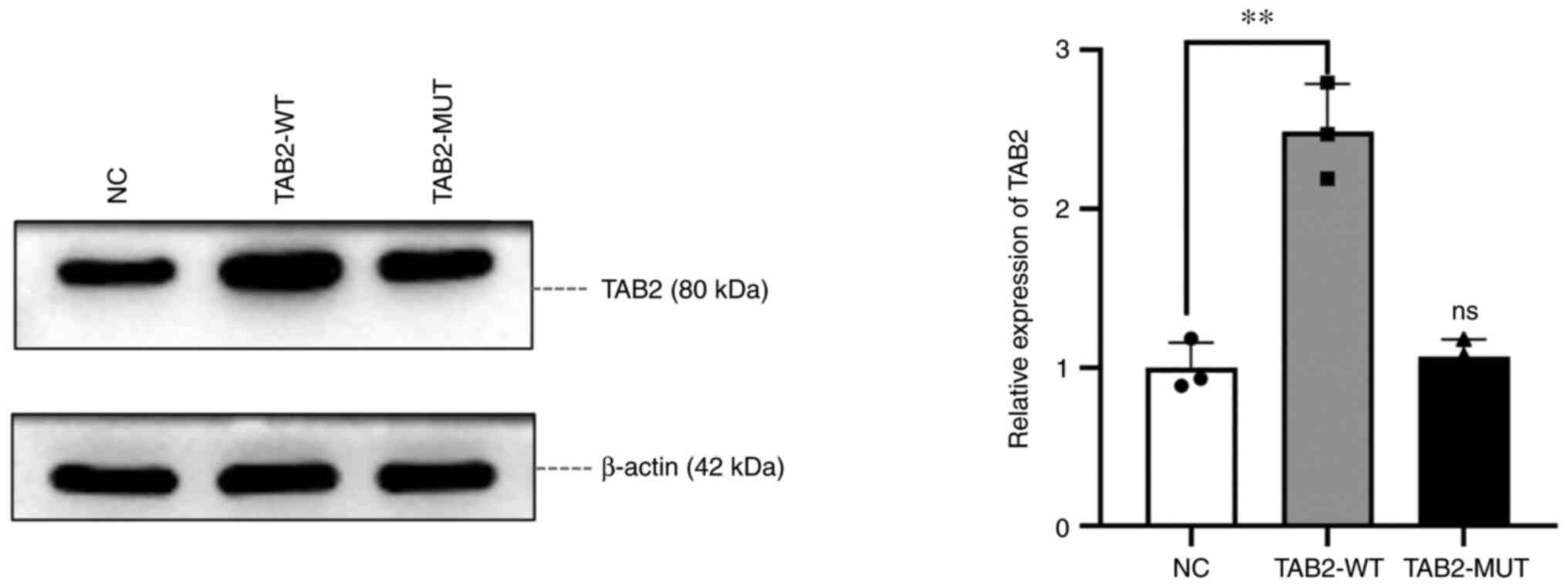
WB experiment results showed that the protein
expression in the TAB2-WT group was 2.48 times higher compared with
that in the blank vector group (NC group) (P=0.0016), whereas no
significant difference in the expression was observed in the
TAB2-MUT group (P=0.57), indicating that the mutation led to the
non-expression of TAB2 (Fig.
2).
Discussion
The present case study investigated a Chinese child
with unexplained growth restriction and CHD. The height and weight
of the patient in infancy were lower compared with those of his
peers; however, his developmental motor milestones did not appear
to be abnormal relative to his peers. Until 3-years old, there was
obvious growth restriction. In addition, the patient had mild
mitral valve prolapse and mild to moderate mitral regurgitation at
the age of two. At the age of three, the patient still showed
mitral insufficiency and semi moderate regurgitation as determined
by color Doppler echocardiography. A novel frameshift mutation of
TAB2, p.Ser353fsTer8, was discovered through genetic
testing. The parents were WT at this locus; thus, the mutation
identified may be a de novo mutation. In vitro
function experiments showed that this mutation led to the
non-expression of the TAB2 protein, suggesting that the heart
structural defects and short stature may be caused by the
insufficient haploid dosage of TAB2.
So far, to the best of our knowledge, a total of 62
cases related to TAB2 mutation in 31 families have been
reported worldwide (3-6,10-16).
The main clinical manifestations of these cases are valve
abnormalities (such as valve stenosis and mitral or aortic
insufficiency), dilated cardiomyopathy (DCM), connective tissue
diseases (such as growth restriction and hyperactivity of joints in
children) and abnormal facial features (increased forehead width,
wider eye distances, ptosis and lower ear positions). It is worth
noting that when CHTD2 is associated with TAB2 deficiency,
it is a non-syndromic CHD by definition (3). However, statistical studies have
demonstrated that <30% of affected patients show isolated CHD
characteristics (10,11). The clinical phenotype spectrum of
CHTD2 patients with the syndrome expressed is not scattered.
Excluding cardiac structural abnormalities, the clinical phenotypes
are mainly concentrated in connective/skeletal tissue and facial
features, with a small sample of patients exhibiting hypotonia and
hearing loss (11). The reported
TAB2 mutation is primarily located in exon 2 (NM_015093),
and the majority of the mutant types are loss of function
mutations, such as nonsense or frameshift mutations (3-6,10-16).
The phenotypical differences between individuals appear not to be
associated with the mutation sites or types, and carriers within
the same family may exhibit different phenotypes. For example,
Westphal et al (10)
reported that both the patient and his father carried the
TAB2 mutation c.878del, but his father did not show physical
characteristics. The patient in the present study was initially
treated for growth restriction, and no other abnormal
manifestations were revealed during the clinical examination,
except for cardiac structural abnormalities and very slight facial
abnormalities. The results of genetic testing suggested that CHDT2
is caused by the novel frameshift mutation of TAB2.
Therefore, multidisciplinary consultation, in conjunction with
cardiovascular medicine department is necessary; furthermore,
improved methodologies for the growth abnormalities (such as short
stature) should also be formulated.
TAB2 is located on autosomal 6q25.1, and the
encoded TAB2 protein has 693 amino acids. TAB2 plays an
important role in interleukin-1 signaling pathway and embryonic
development of heart tissue (3).
Although it is not clear how TAB2 defects lead to cardiac
structural or functional abnormalities mainly involving valves,
studies have demonstrated that TAB2 mutations or copy number
deletions lead to insufficient haploid dose, reduce the binding
ability with TAK1 (MAP3K7 coding) or TRAF6 protein, block
TAK1 protein activation, lead to NF-κB signal error regulation and
slow the necrosis of cardiomyocytes (1,17).
In addition to regulating the NF-κB signaling pathway, Yin et
al (18) proposed a new
regulatory mechanism, suggesting that TAB2 protein plays an
indispensable role in mediating TAK1-RIPK1 interaction. When the
TAB2 protein is deleted, it can promote RIPK1 kinase activation and
induce RIPK1 kinase dependent apoptosis by destroying TAK1-RIPK1
interaction. Therefore, different individuals may have early- or
late-onset cardiac structure issues or dysfunctions, which may be
associated with the activation of TAK1-RIPK1 or the NF-κB signaling
pathway regulation mediated by TAB2 defects.
At present, there is no effective treatment for
CHTD2 resulting in individuals with CHD, DCM or cardiac
dysfunction. This necessitates targeted treatment for the cause of
these diseases. Understanding the pathogenesis of the disease is
important for developing treatment strategies. The mechanism of the
aforementioned TAB2-related cardiac phenotype expression may
be caused by promoting the activation of RIPK1 kinase (17,18).
Researchers inactivated Ripk1 in a mouse model with the
TAB2 gene knocked out; this largely reversed the
pathological changes, such as severe cardiac contractile
dysfunction, ventricular dilatation, and cardiac hypertrophy in
mice (18). Inhibiting the RIPK1
protein or regulating the activity of the TAK1-RIPK1 pathway are
potential strategies for the treatment of TAB2-related
cardiac phenotypes (19). However,
the effectiveness of growth hormone therapy on TAB2-related
growth restriction requires further clinical study. The patient
mentioned in this case report was treated using growth hormones,
but longer-term follow-ups are required to ascertain its
therapeutic value.
In summary, the present study reported a Chinese
child with atypical facial features, short stature and CHD. The
genetic testing results showed that the novel frameshift mutation
of TAB2 caused CHTD2. Functional experiments in vitro
showed that the mutant p.Ser353fsTer8 may lead to protein
non-expression. The current study adds additional information on
the TAB2 mutation spectrum and emphasizes that the
occurrence of growth restriction and cardiac disorder may be
associated with TAB2 defects. For patients with confirmed
CHTD2, the present study recommends multidisciplinary consultation
and regular follow-ups for abnormalities in the growth,
development, hearing, vision and connective tissue of the patient.
However, the feasibility of using human growth hormone to treat
TAB2-related growth restriction still needs to be evaluated.
Acknowledgements
Not applicable.
Funding
Funding: The funding for this research was provided by the
Scientific Research Fund of Anhui Medical University (grant no.
2019xkj080).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are not publicly available due to data privacy
considerations but are available from the corresponding author on
reasonable request. TAB2 mutation information from this
study has been deposited in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/submitters/508986).
Authors' contributions
DQ, XW and YC designed the experiments. DQ, XW, XX,
YW and YZ collected and analyzed the clinical data. XW, JG and YC
were responsible for the protein experiments. XX, YW and YZ
analyzed the genetic data. JG and YC confirm the authenticity of
all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The study was conducted according to the guidelines
of the World Medical Association (Declaration of Helsinki) and
approved by the Ethics Committee of the Children's Hospital of
Fudan University Anhui Hospital (approval no. EYLL-2018-020).
Patient consent for publication
The parents of patients looked at in this case
signed and provided informed consent for participation in the
study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xia ZP, Sun L, Chen X, Pineda G, Jiang X,
Adhikari A, Zeng W and Chen ZJ: Direct activation of protein
kinases by unanchored polyubiquitin chains. Nature. 461:114–119.
2009.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Shim JH, Xiao C, Paschal AE, Bailey ST,
Rao P, Hayden MS, Lee KY, Bussey C, Steckel M, Tanaka N, et al:
TAK1, but not TAB1 or TAB2, plays an essential role in multiple
signaling pathways in vivo. Genes Dev. 19:2668–2681.
2005.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Thienpont B, Zhang L, Postma AV, Breckpot
J, Tranchevent LC, Van Loo P, Møllgård K, Tommerup N, Bache I,
Tümer Z, et al: Haploinsufficiency of TAB2 causes congenital heart
defects in humans. Am J Hum Genet. 86:839–849. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Chen J, Yuan H, Xie K, Wang X, Tan L, Zou
Y, Yang Y, Pan L, Xiao J, Chen G and Liu Y: A novel TAB2 nonsense
mutation (p.S149X) causing autosomal dominant congenital heart
defects: A case report of a Chinese family. BMC Cardiovasc Disord.
20(27)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ackerman JP, Smestad JA, Tester DJ,
Qureshi MY, Crabb BA, Mendelsohn NJ and Ackerman MJ: Whole exome
sequencing, familial genomic triangulation, and systems biology
converge to identify a novel nonsense mutation in TAB2-encoded
TGF-beta activated kinase 1 in a child with polyvalvular Syndrome.
Congenit Heart Dis. 11:452–461. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ritelli M, Morlino S, Giacopuzzi E,
Bernardini L, Torres B, Santoro G, Ravasio V, Chiarelli N,
D'Angelantonio D, Novelli A, et al: A recognizable systemic
connective tissue disorder with polyvalvular heart dystrophy and
dysmorphism associated with TAB2 mutations. Clin Genet. 93:126–133.
2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009.PubMed/NCBI View Article : Google Scholar
|
|
8
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The Genome Analysis Toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Westphal DS, Mastantuono E, Seidel H,
Riedhammer KM, Hahn A, Vill K and Wagner M: There is more to it
than just congenital heart defects-The phenotypic spectrum of
TAB2-related syndrome. Gene. 814(146167)2022.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Engwerda A, Leenders EKSM, Frentz B,
Terhal PA, Löhner K, de Vries BBA, Dijkhuizen T, Vos YJ, Rinne T,
van den Berg MP, et al: TAB2 deletions and variants cause a highly
recognisable syndrome with mitral valve disease, cardiomyopathy,
short stature and hypermobility. Eur J Hum Genet. 29:1669–1676.
2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Permanyer E, Laurie S, Blasco-Lucas A,
Maldonado G, Amador-Catalan A, Ferrer-Curriu G, Fuste B, Perez ML,
Gonzalez-Alujas T, Beltran S, et al: A single nucleotide deletion
resulting in a frameshift in exon 4 of TAB2 is associated with a
polyvalular syndrome. Eur J Med Genet. 63(103854)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Vasilescu C, Ojala TH, Brilhante V, Ojanen
S, Hinterding HM, Palin E, Alastalo TP, Koskenvuo J, Hiippala A,
Jokinen E, et al: Genetic basis of severe childhood-onset
cardiomyopathies. J Am Coll Cardiol. 72:2324–2338. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Caulfield TR, Richter JE Jr, Brown EE,
Mohammad AN, Judge DP and Atwal PS: Protein molecular modeling
techniques investigating novel TAB2 variant R347X causing
cardiomyopathy and congenital heart defects in multigenerational
family. Mol Genet Genomic Med. 6:666–672. 2018.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Liu H, Giguet-Valard AG, Simonet T,
Szenker-Ravi E, Lambert L, Vincent-Delorme C, Scheidecker S, Fradin
M, Morice-Picard F, Naudion S, et al: Next-generation sequencing in
a series of 80 fetuses with complex cardiac malformations and/or
heterotaxy. Hum Mutat. 41:2167–2178. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hanson J, Brezavar D, Hughes S,
Amudhavalli S, Fleming E, Zhou D, Alaimo JT and Bonnen PE: TAB2
variants cause cardiovascular heart disease, connective tissue
disorder, and developmental delay. Clin Genet. 101:214–220.
2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Cheng A, Dinulos MBP, Neufeld-Kaiser W,
Rosenfeld J, Kyriss M, Madan-Khetarpal S, Risheg H, Byers PH and
Liu YJ: 6q25.1 (TAB2) microdeletion syndrome: Congenital heart
defects and cardiomyopathy. Am J Med Genet A. 173:1848–1857.
2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yin H, Guo X, Chen Y, Zeng Y, Mo X, Hong
S, He H, Li J, Steinmetz R and Liu Q: TAB2 deficiency induces
dilated cardiomyopathy by promoting RIPK1-dependent apoptosis and
necroptosis. J Clin Invest. 132(e152297)2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Mifflin L, Ofengeim D and Yuan J:
Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic
target. Nat Rev Drug Discov. 19:553–571. 2020.PubMed/NCBI View Article : Google Scholar
|