1. Introduction
The bioactive constituents from traditional Chinese
medicine are valued as promising novel drug discovery sources owing
to their unique chemical structures and potent efficacy in treating
human diseases. One such constituent of considerable interest is
celastrol. Celastrol (tripterine) is a pharmacologically active
pentacyclic triterpene extracted from Tripterygium wilfordii
Hook F. Celastrol has been reported to exhibit anti-tumor,
anti-inflammatory, anti-obesity and anti-oxidant activities. In
addition, celastrol provided substantial therapeutic efficacy
against infectious, neurodegenerative, cardiovascular and metabolic
diseases (1). However, its
clinical translation has yet to make substantial progress owing to
its poor aqueous solubility, low bioavailability and narrow
therapeutic window. Various strategies have been investigated so
far to deal with these challenges, including the usage of
water-soluble analogs, combination therapy and nanotechnology-based
formulations (1,2). In fact, celastrol's underlying
mechanisms of action have been vigorously investigated in several
disease models and various molecular targets or signaling pathways
responsible for these activities have been identified, including
heat shock protein 90 (Hsp90), heat shock factor 1, proteasomes,
NFκB, protein kinase B (AKT)/mammalian target of rapamycin (mTOR)
and AMP-activated protein kinase (AMPK) pathways (2). In addition, it has been reported that
the pharmacological activities of celastrol are attributable to its
ability to influence autophagy (3).
Autophagy is a highly conserved intracellular
catabolic process that transports cytoplasmic materials to
lysosomes for degradation. Mammalian cells may undergo various
types of autophagy, including chaperone-mediated autophagy,
microautophagy and macroautophagy (4,5).
Current research has mainly focused on macroautophagy (hereafter
referred to as ‘autophagy’ for simplicity). To maintain normal
cellular metabolism, autophagy occurs at a basal level to eliminate
damaged organelles and harmful protein aggregates. Autophagy
contributes to cell survival when environmental stresses are
encountered, such as nutrient deprivation, oxidative stress and
microbial infection. By contrast, prolonged constitutive defective
or excessive autophagy eventually causes autophagic cell death
induction (type II programmed cell death) (6). Thus, autophagy has a crucial role in
cellular physiology and dysregulations of this process may lead to
the pathogenesis of diverse human diseases (7). For instance, autophagy alteration has
been observed in the affected neurons of patients with
neurodegenerative disorders (8);
the essential autophagy gene Beclin1 was mono-allelically deleted
in human breast cancers and autophagy deficiency in mice made them
susceptible to benign hepatomas (9).
Based on these observations, autophagy modulation
has emerged as an attractive therapeutic strategy for diverse
diseases and the pharmacological functions of several drugs relate
to potent autophagic-regulatory effects. The present article
provides an overview of autophagy regulation by celastrol and
discusses the underlying molecular mechanisms that define
celastrol's effect on autophagy to outline the critical roles of
autophagy in the pharmacological activities of celastrol.
2. An overview of autophagy
Autophagy begins at the proposed site for
autophagosome formation termed phagophore assembly site in the
cytoplasm. Following initiation, the membranes possibly derived
from endomembrane compartments, such as endoplasmic reticulum (ER),
Golgi complex and mitochondria, begin to expand and form the
cup-shaped autophagic precursor structure called the phagophore,
which sequentially engulfs a portion of the cytoplasm and
elongates, resulting in the formation of double-membrane spherical
autophagosomes (10). Ultimately,
autophagosomes fuse with lysosomes to form autolysosomes, where the
cytoplasmic materials and the inner membranes are degraded by
lysosomal hydrolases and the breakdown products are released into
the cytoplasm to be recycled as an energy source or new
macromolecule building blocks (11).
Autophagy was once characterized as a bulk,
nonselective, degradative process. However, the identification of
multiple autophagy adaptor proteins, such as p62 and neighbor of
Brca1 gene 1 (NBR1), which simultaneously bind ubiquitinated
substrates and autophagy-specific ubiquitin-like system modifiers
on the surface of autophagosomes, has unraveled the selectivity in
autophagy (12). The selective
elimination of damaged or redundant mitochondria via autophagy is
termed as mitophagy. The interaction between ubiquitin kinase
phosphatase and tensin homolog-induced kinase 1 (PINK1) and E3
ubiquitin ligase Parkin activates mitophagy, leading to the
translocation of Parkin to the damaged mitochondrial membrane.
Subsequently, Parkin-mediated poly-ubiquitination on mitochondrial
outer-membrane proteins recruits autophagy adaptor protein p62 for
the clearance of mitochondria (13). Furthermore, adipocyte triglyceride
lipase on the lipid droplets acts as a selective autophagy receptor
associated with the autophagic component microtubule-associated
protein light chain 3 (LC3), culminating in lipophagy activation
and degradation of lipid droplets (14). Based on the type of the digested
organelle, other forms of selective autophagy have been named, such
as ER-phagy, ribophagy, perophagy and nucleophagy. Selective
autophagy greatly contributes to the efficient removal of
organelles or macromolecules and is deemed essential for organelle
quality control and homeostasis regulation (15).
A series of autophagy-related genes (Atgs) is
involved in autophagy regulation. The unc-51-like kinase (ULK)
complex, containing various Atg proteins, is required for autophagy
induction. The class III phosphatidylinositol 3 kinase (PI3K)-Atg14
complex has important roles in autophagosome expansion and
maturation, while also requiring two ubiquitin-like proteins (Atg12
and Atg8/LC3 conjugation systems). The member RAS oncogene family
protein 7 (Rab7), homotypic fusion and protein sorting protein and
soluble N-ethylmaleimide-sensitive factor attachment protein
receptors contribute to the fusion of autophagosomes with lysosomes
(16). Multiple signaling pathways
have been associated with autophagy, including the mTOR, PI3K/AKT,
AMPK and Hedgehog signaling pathways (17). Furthermore, there is strong
evidence supporting epigenetic modifications of both Atgs and
signaling molecule genes by DNA methylation and histone
modifications, as well as non-coding RNAs, which impact their
transcription and subsequently manage the autophagic process
(18).
3. Autophagy regulation by celastrol
Extensive experimentation has indicated that
autophagy modulation may be an important mechanism underlying the
anti-tumor therapeutic effect of celastrol (19-30).
Celastrol also alleviates inflammatory reactions and restrains the
immune response through autophagic pathways (31-40).
Furthermore, autophagy has a role when celastrol exerts
neuroprotective, anti-atherosclerosis, anti-pulmonary fibrosis and
anti-macular degeneration effects (41-48).
Further details on the autophagy modulation involved in the
multiple pharmacological activities of celastrol in vitro
and in vivo are listed in Tables I and II, respectively.
 | Table IAutophagy modulation by celastrol
in vitro. |
Table I
Autophagy modulation by celastrol
in vitro.
| Cell lines | Celastrol dosage
and treatment times | Autophagy
status | Related biological
effects | (Refs.) |
|---|
| Human prostate
cancer cell line LNCaP | 2 µmol/l, 3-24
h | ↑ | Atg5 and Atg7
mRNA↑; LC3II↑; p62↓; LC3 puncta↑; AR↓; miR-101↓; cell death↓ | (19) |
| Human prostate
cancer cell line LNCaP | 2 µmol/l, 3-24
h | ↑ | LC3II↑; p62↓; LC3
puncta↑; miR-17-92a cluster↓; Atg7↑ | (49) |
| Ox-LDL-treated
human clear cell renal cell carcinoma cell line 786-O | 0.25-1 µmol/l, 24
h | ↑ | LC3II↑; p62↓;
p-mTOR↓; autophagic flux↑; LXRα↑; ABCA1↑; Vim↓; MMP2↓; lipophagic
vesicles↑; lipid accumulation↓ | (50) |
| Human glioblastoma
cell line U251N | 1-10 µmol/l, 3-24
h | ↓ | LC3II↑; p62↑;
autophagic flux↓; lysosome number↑; lysosomal integrity↓;
proteotoxic stress↑ | (21) |
| Human glioma cell
line U251 | 0.3-10 µmol/l, 3-24
h | ↑ | LC3 puncta↑;
LC3II↑; Beclin1↑; p62↑; lysosomal degradation ↓; cell death↓;
ROS/JNK signaling pathway↑; Akt/mTOR signaling pathway↓ | (22) |
| Human osteosarcoma
cell lines HOS, MG-63 | 2-3 µmol/l, 24
h | ↑ | LC3II↑; acidic
vesicular organelles↑; autophagic vacuoles↑; cell death↑; ROS↑;
p-JNK↑ | (23) |
| Human primary
osteosarcoma cell OS-718 | 1-1.5 µmol/l, 24
h | ↑ | LC3II↑; p-JNK↑;
ROS↑; cell proliferation↓ | (23) |
| Human primary
osteosarcoma cell OS-1227 | 4-6 µmol/l, 24
h | ↑ | LC3II↑; p-JNK↑;
ROS↑; cell proliferation↓ | (23) |
| Human osteosarcoma
cell line HOS | 3 µmol/l, 24 h | ↑ | LC3II↑; p62↓; Bip↑;
p-PERK↑ | (24) |
| Human pancreatic
ductal adenocarcinoma cell line MiaPaCa-2 | 0.5-4 µg/ml, 24-48
h | ↑ | LC3II↑, PTEN↑; LC3
puncta↑; autophagic vacuoles↑; acidic vesicular organelles↑;
apoptosis↓ | (25) |
| Human pancreatic
ductal adenocarcinoma cell line CFPAC-1 | 0.5-2 µg/ml, 24-48
h | ↑ | LC3II↑; PTEN↑; LC3
puncta↑; autophagic vacuoles↑; acidic vesicular organelles↑ | (25) |
| Human
hepatocellular carcinoma cell line Bel7402 | 0.625-2.5 µmol/l,
24 h | ↑ | Autophagic
vacuoles↑; LC3 puncta↑; LC3II↑ | (26) |
| Human
hepatocarcinoma cell line HepG2 | 4 µmol/l, 6-24
h | ↑ | LC3II↑; LC3
puncta↑; ROS↑; p- Akt↑; p-p70S6K↑; HIF-1a↑; BNIP3↑ | (27) |
| Human stomach
adenocarcinoma cell line AGS | 0.25-1 µmol/l,
0.25-24 h | ↑ | LC3 puncta↑;
LC3II↑; Atg5↑; Atg7↑; Beclin1↑; cell growth↓; p-AMPK↑; p-Akt↓;
p-mTOR↓; and p-p70S6K↓ | (28) |
| Human gastric
carcinoma cell line YCC-2 | 0.25-1 µmol/l,
0.25-24 h | ↑ | LC3II↑; Atg5↑;
Atg7↑; Beclin1↑; cell growth↓; p-AMPK↑; p-Akt↓; p-mTOR↓; p-
p70S6K↓ | (28) |
| Mouse pituitary
ACTH- secreting adenoma cell line AtT20 | 0.5, 1 µmol/l, 48
h | ↑ | LC3 puncta↑;
LC3II↑; p62↓; p-Akt↓; p-mTOR↓; cell viability↓; cell migration and
invasion↓ | (29) |
| Human colorectal
cancer cell lines HCT-116, SW480 | 1.25-5 µmol/l, 24
h | ↑ | LC3II↑; p62↓;
Beclin1↑; Beclin1- Bcl2 interaction↓; Atg4B and
Beclin1 mRNA↑; autophagic vesicles↑; Atg7↑; Nur77↓ | (30) |
| Human cervical
cancer cell line Hela | 1 µmol/l, 0.5-4 h;
0.25-2 µmol/l, 24 h | ↑ | LC3II puncta↑;
LC3II↑; autophagic flux↑; Ca2+ mobilization↑; cell
death↑; CaMKKβ↑; p-AMPK↑; p-p70S6K↓; ER stress and unfolded protein
response↑ | (51) |
| Human cervical
cancer cell line Hela | 1.2 µmol/l, 12
h | ↑ | LC3II↑; LC3
puncta↑; autophagy flux↑; paraptosis-related cytoplasmic
vacuolization↓; proteasome function↓; ER stress↑; Hsp90
function↓ | (52) |
| Human non-small
cell lung cancer cell lines H23, H292 | 0.8 µmol/l,
combined with 10 µmol/l afatinib, 12-24 h | ↓ | LC3II/LC3I ratio↓;
p62↑; NBR1↑; p62 aggregates↑; autophagic flux↓; lysosomal
activity↓ | (53) |
| Human lung cancer
cell line A549 | 1-4 µmol/l, 12
h | ↓ | LC3II↑; p62↑;
autophagy flux↓ | (54) |
| Human non-small
cell lung cancer cell line H1975 | 0.5-4 µmol/l, 24
h | ↑ | LC3 puncta↑;
Ca2+ mobilization↑; EGFR↑; Akt↑; apoptosis↑ | (55) |
| Human non-small
cell lung cancer cell line H1650 | 1 µmol/l, 24 h | ↑ | LC3 puncta↑;
Ca2+ mobilization↑; apoptosis↑ | (55) |
| Human lung
adenocarcinoma cell line HOP62 | 0.75 µmol/l,
combined with 25 µmol/l ellagic, acid 24 h | ↑ | LC3II↑; CIP2A↓;
cell proliferation↓ | (56) |
| Human breast cancer
cell line MCF-7 | 0.3 µmol/l,
combined with 10 µmol/l tamoxifen, 6-24 h | ↑ | LC3II/LC3I ratio↑;
p62↓; LC3 puncta↑; p-Akt↓; p-mTOR↑; cell death↑ | (57) |
| Human non-small
cell lung cancer cell line HCC827 | 1.25 µmol/l,
combined with 2.5 µmol/l erastin, 24 h | ↑ | LC3 puncta↑;
LC3II↑; autophagic flux↑; Beclin1↑; Atg5↑; Atg7↑; p62↑; ROS↑; cell
death↑; colocalization of p62 with TOM20↑; PINK1 and Parkin↑;
mitophagy↑; ubiquitinated mitochondrial proteins↑; p-DRP1↑;
mitochondrial fission↑ | (58) |
| TNFα-treated human
cervical cancer cell line Hela | 2-4 µmol/l, 1-6
h | ↑ | LC3II↑; LC3
puncta↑; IκBa↓; Nur77-p62 interaction↑; mitophagy↑ | (31) |
| TNFα-treated mouse
embryonic fibroblasts | 2 µmol/l, 1 h | ↑ | LC3 puncta↑,
mitophagy↑ | (31) |
| LPS-primed mouse
peritoneal macrophages | 0.25 µmol/l, 0.5
h | ↑ | LC3II↑; IL-1β
secretion↓; cleaved caspase-1↓; ASC oligomerization↓ | (33) |
| High
glucose-treated mouse podocytes | 1.5 µmol/l, 5
h | ↑ | LC3II↑; p62↓;
Beclin1↑; cell viability↑; proinflammatory cytokines (TNF-α, IL-1β
and IL-6) ↓; glucose uptake↑; HO-1↑ | (36) |
| Human rheumatoid
arthritis fibroblast-like synoviocytes MH7A | 1-2 µmol/l, 24
h | ↑ | LC3 puncta↑;
autophagic flux↑; Ca2+ mobilization↑; p-AMPK↑;
p-p70S6K↓; CaMKKβ↑; cell death↑ | (37) |
| Human rheumatoid
arthritis synovial fibroblasts | 1-2 µmol/l, 24
h | ↑ | LC3 puncta↑; LC3
II↑; autophagic flux↑; p-AMPK↑; p-p70S6K↓; CaMKKβ↑ | (37) |
| Primary
cardiomyocytes from collagen induced rheumatoid arthritis rats | 10 µmol/l, 48
h | ↓ | LC3II↓; Beclin1↓;
p62↑; TLR2↓; HMGB↓ | (38) |
| Primary SD rat
chondrocytes | 0.2 µmol/l, 24
h | ↑ | LC3II↑; p62↓;
mTOR↓; caspase-12↓, DDIT3↓ | (39) |
| IL-1β-treated
primary SD rat chondrocytes | 0.2 µmol/l, 24
h | ↑ | Autophagic
vacuoles↑; LC3II/LC3I ratio↑; Beclin1↑; p62↓; LC3 puncta↑;
apoptosis↓; pro-inflammatory cytokines (TNF-α and IL6) ↓;
p-IκBα/IκBα and p-p65/p65↓; nuclear p65↓ | (40) |
| Human cervical
cancer cell line HeLa cells stably expressing 3XFlag-TFEB | 0.25-1 µmol/l, 24
h | ↑ | LC3II↑; autophagy
flux↑; lysosomal contents↑; LAMP1↑; autophagy- lysosome-related
genes mRNA (Atg16L1, Atg12, Atp6V1H, CtsB, CtsD, CtsF, LAMP1, GLA,
MAP1LC3B, MCOLN1, SQSTM1) ↑; nuclear TFEB↑ | (59) |
| Mouse neuronal cell
line N2a transiently expressing P301L Tau | 0.05-0.2 µmol/l, 24
h | ↑ | Nuclear TFEB↑;
LC3II↑; autophagy flux↑; p-Tau aggregates↓ | (59) |
| Rotenone-treated
human dopaminergic neuronal cell line SH-SY5Y | 0.5 µmol/l, 1
h | ↑ | LC3II/LC3I ratio↑;
autophagic vacuoles↑; α-synuclein aggregates↓; SOD and GSH↑; ROS↓;
mitochondria membrane potential↑; cytochrome C release↓;
mitophagy↑; cell death↓ | (41) |
| α-synuclein-pulsed
monocyte derived dendritic cells | 0.25 µmol/l, 1
h | ↑ | Beclin1↑;
LC3II/LC3I ratio↑; p62↓; Rab5/ Beclin1/α-synuclein and Rab7/LC3/α-
synuclein puncta↑; Th1 and Th17↓; Th17/Treg ratio↓ | (42) |
| Human dopaminergic
neuronal cell line SH-SY5Y | 0.1-3 µmol/l, 2-24
h | ↑ | MPAK signaling
pathways (p-p38, p-ERK1/2, p-Akt1/2/3, p-p65 and p-JNK1/ 2/3) ↑;
LC3I↓; LC3II↑; p62↓; Beclin1↑; Ambra1↑; Vps34↑; Atg7↑; Atg12↑;
PINK1↑; DJ-1↑; LRRK2↓; mitophagy↑; MPP+neurotoxicity↓;
apoptosis↓ | (43) |
| ox-LDL treated
human vascular smooth muscle cells | 0.2 µmol/l, 24
h | ↑ | LC3II/LC3I ratio↑;
p62↓; autophagic flux↑; lipid accumulation↓ | (45) |
| Angiotensin
II-treated rat vascular smooth muscle cells | 0.05 µmol/l, 12
h | ↑ | LC3 puncta↑; LC3
II↑, p62↓; ROS↓; p-PI3K, p-Akt, p-mTOR and p-p70S6K↓;
senescence↓ | (46) |
| PDGF-BB-stimulated
human vascular smooth muscle cells | 0.05-0.2 µmol/l, 24
h | ↑ | autophagic
vacuoles↑; autophagic flux↑; LC3II/LC3I ratio↑; p62↓, Wnt5a↓,
p-PKC↓; p-mTOR↓; c-Myc↓; cell proliferation↓ | (47) |
|
H2O2-treated human
retinal pigment epithelial cell line ARPE-19 | 0.5-1 µmol/l, 0.5
h | ↑ | LC3II/LC3I ratio↑;
Beclin1↑; SIRT3↑ | (48) |
| CCCP-treated human
cervical cancer cell line Hela | 10 µmol/l, 1.5-12
h | ↓ | Parkin
mitochondrial recruitment↓; PINK1↓; p-Parkin↓; mitophagy↓;
mitochondrial clustering↓; PINK1- TOM20 interaction↓ | (60) |
| Table IIAutophagy modulation by celastrol
in vivo. |
Table II
Autophagy modulation by celastrol
in vivo.
| Tissue type | Celastrol dosage
and treatment times | Autophagy
status | Related biological
effects | (Refs.) |
|---|
| Xenografted human
clear cell renal cell carcinoma from high- fat diet-fed BALB/c nude
mice | 1 mg/kg/d, 28
d | ↑ | Plasma TC, TG, LDL
and VLDL↓; LC3↑; ABCA1↑; LXRα↑; E-cadherin↑; Vim↓; MMP2↓; p62↓;
tumor growth↓; lipid droplet↓ | (50) |
| Xenografted human
glioma from BALB/c-nude mice | 2-4 mg/kg/d, 14
d | ↑ | LC3↑; p-JNK↑;
p-Akt↓; p-mTOR↓; tumor growth↓ | (22) |
| Xenografted human
osteosarcoma from BALB/c- nude mice | 1-2 mg/kg/d, 7
d | ↑ | LC3II↑; p-JNK↑;
tumor volume↓; cell viability↓ | (23) |
| Xenografted
pituitary ACTH- secreting adenoma from C57/ B6J mice | 2 mg/kg/2d, 14
d | ↑ | LC3II↑; tumor
volumes and weights↓; cell viability↓; G0/G1 phase arrest↑ | (29) |
| Xenografted human
colorectal cancer from BALB/C nude mice | 1.25-2.5 mg/kg/2d,
21 d | ↑ | LC3II↑; Atg7↑;
Nur77↓; tumor growth↓ | (30) |
| Xenografted human
lung adenocarcinoma from BALB/C nude mice | 1 mg/kg/d, combined
with 40 mg/kg/d ellagic acid, 22 d | ↑ | LC3II↑; CIP2A↓;
tumor growth↓ | (56) |
| Xenografted human
non- small cell lung cancer from BALB/c nude mice | 1 mg/kg/d, combined
with 5 mg/kg/d erastin, 21 d | ↑ | LC3II↑; Parkin↑;
tumor volumes and weights↓ | (58) |
| Liver tissue from
LPS- and D- GalN-injected C57BL/6 mice | 0.2 or 0.5 mg/kg,
12 h | ↑ | LC3II↑; LC3
puncta↑ | (31) |
| Liver tissue from
high-fat diet- fed C57BL/6 mice | 0.1 mg/kg/d, 14
d | ↑ | LC3II↑; LC3
puncta↑ | (31) |
| Serum of
LPS-induced septic shock mouse model | 1 mg/kg, 4 h | ↑ | IL-1β↓; NLRP3
inflammasome activity↓ | (33) |
| Colon tissues of
DSS-induced colitis mouse model | 1.0 mg/kg/d, 7
d | ↑ | IL-1β↓; caspase-1↓;
ASC↓; COX2↓; NLRP3 inflammasome activity↓; IL-1β, IL-6, TNF-α,
IL-17A and IFNg mRNA↓; colitis↓ | (33) |
| Colon tissues of
dextran sodium sulphate-induced colitis rat model | 1 mg/kg/d, combined
with 20 mg/kg/d CP- 456773, 16 d | ↑ | Beclin1↑; p62↓;
p-AMPKα↑; p-mTOR↓; HSP-90↓; NLRP3 mRNA↓; TNF-α, IL-6, IL-1β and
IL-18↓ | (32) |
| Proximal colons of
IL-10 deficient mice | 2 mg/kg/d, 7 d | ↑ | LC3II/LC3I ratio↑;
LC3 puncta↑; PI3K↓; Akt1↓; mTOR↓; p70S6K↓; colitis↓;
proinflammatory cytokines (IL-1β, IL-17A, TNF-α and IFN-γ) and
chemokines (CXCL-1 and CXCL-2) ↓ | (34) |
| Renal tissues of
diabetic nephropathy rat model | 1.5 mg/kg/d, 28
d | ↑ | LC3II↑; mTOR↓;
PI3K↓; Akt mRNA↓; p-Akt↓ | (35) |
| Synovial joints of
adjuvant- induced arthritis rat model | 1 mg/kg/d, 36
d | ↑ | Ca2+↑;
LC3B↑; Vim↓; arthritic score and hind paw volume↓ | (37) |
| Myocardium of
collagen induced rheumatoid arthritis rat model | 0.2 mg/kg/d, 6
d | ↓ | Inflammatory
cytokines (TNF-α, IL-6 and IL-1β) ↓; apoptosis↓; Bcl-2↑; Bax↓;
cleaved caspase-3↓ | (38) |
| Articular cartilage
of anterior cruciate ligament transection SD rat model | 0.5-1 mg/kg/d, 84
d | ↑ | Beclin1↑; p62↓;
apoptosis↓; articular cartilage degeneration↓; cleaved caspase-3↓;
IL6↓; p-p65↓ | (40) |
| Frontal cortex of
C57 mice | 1-2 mg/kg/d, 7
d | ↑ | LC3II↑; LAMP1↑;
cathepsin B↑; cathepsin D↑; nuclear TFEB↑ | (59) |
| Brain of P301S tau
mice | 1-2 mg/kg/d, 7
d | ↑ | LC3II↑; p62↓;
cathepsin B↑; cathepsin D↑; nuclear TFEB↑; p-tau aggregates↓ | (59) |
| Brain of 3xTg
mice | 1, 2 mg/kg/d, 270
d | ↑ | LC3II↑, p62↓; p-Tau
aggregates↓ | (59) |
| Striatum of PD
mouse model | 3 mg/kg/d, 3 d | ↑ | PINK1↑; DJ-1↑;
dopaminergic nerve terminal degeneration↓ | (43) |
| Lung tissue from
bleomycin- induced pulmonary fibrosis male Wistar albino rat
model | 5 mg/kg/81 h, 28
d | ↑ | Beclin 1↑; Vps34↑;
Atg5↑; Atg7↑; Atg 12↑; Atg16L↑; LC3II↑; Atg3↑; autophagic
vacuoles↑; p62↓; PI3K↓, Akt↓; mTOR↓; fibrosis↓ | (44) |
| Injured femoral
artery of C57BL/6 mice | 2 mg/kg/day, 28
d | ↑ | Autophagic
vacuoles↑; LC3II/LC3I ratio↑; p62↓; Wnt5a↓; p-PKC↓; p-mTOR↓;
c-Myc↓; neointimal hyperplasia↓ | (47) |
Autophagy is involved in the
anti-tumor activities of celastrol
Autophagy modulation is intimately related to the
anti-tumor effects of celatrol; however, the role of autophagy in
tumors is complex and may either act as a tumor suppressor or
display opposing pro-oncogenic functions in a context and cell
type-dependent manner (61). In
prostate cancer cells, celastrol treatment suppressed cell
viability and upregulated autophagic activity, which was indicated
by elevated Atg5 and Atg7 expression, increased autophagosome
formation and promoted degradation of the canonical autophagy
substrate p62. However, autophagy impairment significantly
potentiated the cytotoxic effects of celastrol. These results
demonstrate that autophagy is a cytoprotective mechanism for
prostate cancer cell survival (19,49).
Of note, a celastrol pyrazine derivative (11i) displayed potent
anti-breast cancer activity and induced remarkable autophagy,
whereas autophagy blockage markedly increased breast cancer cell
viability, suggesting that induced autophagy may serve a pro-death
function (20). As a defining
morphological hallmark of clear cell renal cell carcinoma,
excessive cytoplasmic lipid droplets were selectively delivered for
lysosomal degradation via celastrol-induced lipophagy, thereby
markedly facilitating cholesterol efflux, impairing the
epithelial-mesenchymal transition process and finally contributing
to the inhibition of clear cell renal cell carcinoma cell
migration, invasion and tumor growth (50).
Through interference with both autophagic and
proteasomal protein degradation quality-control pathways, celastrol
promoted the accumulation of polyubiquitinated aggregates as well
as p62 and exerted widespread proteotoxicity via a thiol-dependent
mechanism in glioblastoma cells (21). However, the exposure of glioma
cells to celastrol markedly increased LC3 puncta formation and
improved LC3 and Beclin1 expression, while it also unexpectedly
upregulated p62 expression. Application of autophagy inhibitor
moderately reinforced the inhibitory effect of celastrol on cell
viability. These results suggest that celastrol may induce
autophagosome accumulation, accompanied by the partial blockage of
lysosomal degradation function, and that celastrol-mediated
autophagy has a role in promoting cell survival. In addition,
celastrol simultaneously induced apoptosis in glioma cells, whereas
apoptosis suppression strengthened autophagy and vice versa.
Hence, the relationship between autophagy and apoptosis caused by
celastrol may be antagonistic in glioma cells (22). Celastrol also triggered both
autophagy and apoptosis in osteosarcoma cell lines and primary
cells. Celastrol-induced apoptosis was moderately diminished by an
autophagy inhibitor, whereas apoptosis blockage rendered the cells
particularly susceptible to autophagic death. This observation
indicates that autophagy aids the pro-death function and that the
cells may switch between the two responses following celastrol
treatment (23,24). Similarly, autophagy and apoptosis
were simultaneously induced by celastrol in pancreatic ductal
adenocarcinoma cells (25),
hepatocellular carcinoma cells (26,27),
gastric carcinoma cells (28),
pituitary adrenocorticotropic hormone-secreting adenoma cells
(29), and colorectal cancer cells
(30). However, the celastrol
dosage required to stimulate autophagy was lower than that for the
apoptotic threshold in hepatocellular carcinoma cells, and
autophagy occurred at an earlier phase than apoptosis (27). The celastrol-induced apoptosis in
pancreatic cancer cells and colorectal cancer cells was upgraded
with autophagy inhibition, which enhanced the therapeutic effect of
celastrol. Thus, autophagy may be activated after celastrol
treatment as an adaptive mechanism for cell survival to protect
against apoptosis (25,30). However, autophagy induction by
celastrol treatment likely sensitized the gastric cancer cells to
apoptosis, thereby distinctly inhibiting cell proliferation and
migration (28). Although the
relationship between celastrol-induced autophagy and apoptosis was
not fully addressed, autophagy and apoptosis may act
synergistically to increase pituitary adenoma cell mortality rates
(29).
Multidrug resistance (MDR) frequently develops in
certain tumor cells after repeated chemotherapy treatments and MDR
tumor cells exhibit apparent insensitivity toward drug-induced
apoptosis. Celastrol has the potency to effectively induce
autophagic cell death as an alternative cell death mechanism,
re-sensitize resistant cancer cells or produce a synergistic effect
in combination with chemotherapeutic agents by either stimulating
or inhibiting autophagy for the treatment of MDR cancer (51,53-55).
Taxol-resistant cervical cancer cells displayed an Atg7-dependent
increased autophagosome formation and enhanced autophagic flux on
celastrol treatment. As autophagy inhibition completely abolished
the celastrol-mediated cytotoxicity, it is indicated that
autophagic cell death was ultimately induced. Concomitantly,
celastrol produced obvious collateral sensitivity of MDR cells and
reversed the taxol-resistant phenotype in vitro, as well as
suppressed tumor growth and metastasis in vivo (51). In addition, another study indicated
that treatment with celastrol simultaneously induced autophagy
accompanied by apoptosis and an atypical cell death mode termed
‘paraptosis’ in cervical cancer cells, which was morphologically
characterized by extensive cytoplasmic vacuolization derived from
dilated ER and mitochondria (62).
However, the fact that autophagy inhibition significantly enhanced
rather than prevented paraptosis confirmed that autophagy was
possibly a survival mechanism in celastrol-treated cells (52). In afatinib-resistant lung cancer
cells, the combination of afatinib and celastrol impaired
autophagic activity by inhibiting lysosomal enzymatic activities,
leading to increased expression of autophagy substrates, such as
LC3II, p62 and NBR1. Of note, the combination induced paraptosis
and a subsequent cell death phenomenon, indicating a potential
interdependent and interactive relationship (53). Autophagy flux suppression mediated
by celastrol also restored sensitivity to tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis
in TRAIL-resistant lung cancer cells. A combined regimen of
celastrol and TRAIL markedly promoted the cytotoxic effects of
TRAIL, which in turn yielded an improved outcome relative to that
with celastrol or TRAIL treatment alone (54). On the contrary, celastrol was
observed to act as an autophagy inducer to promote oncogenic
epidermal growth factor receptor protein elimination, which is
responsible for activating downstream cell survival signaling,
thereby combating gefitinib-resistant lung cancer cells by inducing
apoptosis (55). Furthermore,
cotreatment with celastrol potentiated the inhibitory effects of
ellagic acid against lung cancer cell proliferation and tumor
growth by elevating autophagic cell death (56). Celastrol plus the ERα antagonist
tamoxifen markedly upregulated the apoptotic rate when compared
with tamoxifen alone in ER-positive breast cancer cells.
Furthermore, increased protein expression of LC3II and decreased
p62 levels were recorded, indicating an enhancement of autophagy.
Although the relationship between increased apoptosis and induced
autophagy by the combination treatment was not addressed, either a
collaboration or a backup mechanism seems probable (57). In addition, cotreatment with
celastrol and erastin, a ferroptosis inducer, initiated
Atg5/Atg7-mediated autophagy, as well as significantly enhanced the
expression and mitochondrial translocation of PINK1 and Parkin,
thereby markedly inducing mitochondrial degradation by selective
mitophagy and exerting a powerful anti-lung cancer effect (58).
Collectively, these findings provided substantial
evidence that autophagy is a target of action of celastrol and that
altered autophagic activities contribute to the anti-tumor
properties of celastrol.
Autophagy is involved in the
anti-inflammatory and immunomodulatory activities of celastrol
Regarding the anti-inflammatory functions, celastrol
has the ability to impair B-cell development and suppress T-cell
proliferation and T-helper 17-cell differentiation, while
facilitating the generation of regulatory T cells (63,64).
A connection between autophagy and anti-inflammatory activities of
celastrol has been determined, as autophagy may affect the systemic
inflammatory responses by modulating inflammatory cytokine
secretion or directly removing the exogenous and endogenous
inflammation sources, such as bacterial pathogens, protein
aggregates or damaged organelles (65). Celastrol reversed the pathological
states associated with inflammatory diseases, including acute
hepatic inflammatory injury and obesity, by specifically triggering
mitophagy (31). The damaged
mitochondria release reactive oxygen species (ROS) and function as
an important inflammatory source that initiates the assembly and
activation of the NOD-like receptor family pyrin domain containing
3 (NLRP3) inflammasomes, a high molecular protein complex
consisting of NLRP3, apoptosis-associated speck-like protein
containing a CARD (ASC) and caspase-1, and is implicated in several
inflammatory disorders (66). In
lipopolysaccharide (LPS)-primed murine peritoneal macrophages,
pretreatment with celastrol markedly promoted autophagy, thereby
attenuating NLRP3 inflammasome activation by blocking ASC
oligomerization and NLRP3-ASC interaction and subsequently
interrupting interleukin (IL)-1β secretion and caspase-1 activation
in a dose-dependent manner. In vivo experiments have
demonstrated that autophagy inhibition ameliorated
celastrol-mediated NLRP3 inflammasome suppression in LPS-induced
septic shock or dextran sodium sulfate (DSS)-induced colitis mouse
models, thereby indicating that autophagy may be responsible for
the protection against NLRP3-related diseases exerted by celastrol
(33). Another study validated
that the dual administration of celastrol and NLRP3 inhibitor
CP-456773 markedly inhibited Hsp90 expression to couple with NLRP3,
which resulted in autophagy induction to increase NLRP3
degradation, which decreased caspase-1 activation and IL-1β and
IL-18 release, restrained the pyroptosis process and provided
significant therapeutic benefits for DSS-induced colon injury
(32). Celastrol also markedly
inhibited colonic proinflammatory cytokine and chemokine
production, and decreased neutrophil infiltration of proximal colon
tissues during experimental colitis in IL-10-deficient mice. The
amelioration of inflammation was attributed to the upregulation of
intestinal autophagy (34).
Podocyte injury is a primary feature of diabetic
nephropathy and a critical role of the inflammatory mechanism was
identified in driving the loss of kidney function during disease
progression (67). With autophagy
deficiency observed in high glucose-administered podocytes, as well
as in the renal tissues of diabetic rats, it has been suggested
that autophagy is implicated in the pathogenesis of diabetic
nephropathy and is a potential therapeutic target for diabetic
nephropathy treatment (35,36).
Indeed, celastrol treatment restored blunted autophagy and
antagonized high glucose-invoked pro-inflammatory cytokine
secretion, insulin resistance and cytotoxic damage in podocytes.
However, the protective effect of celastrol on podocytes was
abrogated when the autophagy pathway was inhibited (36). It was found that celastrol
consistently and effectively promoted autophagy in diabetic
nephropathy rats, thereby protecting them from glomerular basement
membrane thickening and significantly relieving renal injury
(35).
Rheumatoid arthritis is a chronic, progressive
inflammatory autoimmune disease that predominantly affects the
synovial joints (68). Of note,
autophagy has a significant role in the mechanism of the
anti-arthritis properties of celastrol. In fact, celastrol
treatment triggered autophagic cell death to eliminate synovial
fibroblast over-proliferation through the enhancement of autophagy
flux, as it did in apoptosis-resistant fibroblasts. In the
adjuvant-induced arthritis rat model, celastrol administration
significantly induced autophagy, suppressed the proliferation and
epithelial-mesenchymal transition of fibroblasts, downregulated a
panel of inflammatory- and autoimmunity-associated genes and
ameliorated the arthritis phenotype in the joint tissues (37). Evidence suggests that patients with
rheumatoid arthritis have a higher cardiovascular disease risk than
the general population and that systemic inflammation is closely
linked to cardiac dysfunction (69). Autophagy was significantly
upregulated in cultured primary cardiomyocytes as well as
myocardial tissues of collagen-induced rheumatoid arthritis rat
models, whereas celastrol treatment simultaneously ameliorated
cardiomyocyte injury, suppressed the expression of inflammatory
cytokines and inhibited cardiomyocyte autophagy at the same time.
However, the autophagy inducer rapamycin attenuated the
cardioprotective effect of celastrol toward rheumatoid
arthritis-induced cardiotoxicity, highlighting that
celastrol-mediated autophagy suppression accounts for the
alleviation of cardiomyocyte injury (38). Osteoarthritis is another most
common type of arthritis, mainly characterized by progressive
articular cartilage degeneration. During this process, large
amounts of inflammatory cytokines, such as IL-1β, are released into
the osteoarthritis cartilage, which induces chondrocyte apoptosis
(70). Autophagy deficiency is
always accompanied by an increase in apoptotic chondrocytes,
whereas celastrol treatment resulted in obvious reversal effects
(39). By restoring autophagy
activation, celastrol inhibited the IL-1β-stimulated inflammatory
response, counteracted chondrocyte apoptosis and achieved a
promising therapeutic effect against osteoarthritis in vitro
and in vivo (40).
Taken together, these results suggested that
autophagy regulation is a specific mechanism for celastrol to
confer its anti-inflammatory and immunomodulatory effects.
Autophagy is involved in the
neuroprotective activities of celastrol
As the two most common neurodegenerative diseases,
Alzheimer's disease (AD) and Parkinson's disease (PD) are
frequently pathologically defined by the abnormal accumulation of
misfolded tau or α-synuclein protein aggregates in the affected
neurons, respectively, which probably arises from autophagy
impairment to a certain extent (71). Although much controversy remains
regarding the role of these aggregates, it is well recognized that
their appearance may alter neuronal function, cause neurotoxicity
and ultimately lead to cell death (72). The accelerated autophagic removal
of related toxic aggregates may potentially mitigate disease
severity, considering that autophagy controls the degradation of
protein aggregates and that autophagy failure represents the
primary underlying pathology of neurodegenerative diseases. In a
mouse model of AD, improved cognitive deficits and reduction of
hyperphosphorylated tau aggregate in the brain tissues were
observed after the oral administration of celastrol, which reveals
the essential role of autophagy in its anti-AD effect. Furthermore,
celastrol treatment specifically promoted autophagy flux and
lysosomal biogenesis, accompanied by hyperphosphorylated Tau
aggregate degradation in neuronal cell lines, exhibiting a
significant increase in the expression of multiple autophagic and
lysosomal associated genes. Of note, inhibition of autophagy and
lysosomal pathways compromised celastrol-mediated tau reduction,
which further verifies that autophagy is required for decreased
hyperphosphorylated tau levels in response to celastrol (59). Similarly, celastrol activated
autophagic pathways in a dose- and time-dependent pattern and
markedly protected dopaminergic neurons from cell injury, with a
particular focus on reducing rotenone-induced α-synuclein
aggregation in a cellular model of rotenone-induced PD (41). In addition to inducing autophagy
with upregulated Beclin1 expression, decreased p62 expression and
enhanced LC3II conversion, celastrol was able to promote the
interaction between autophagic pathway components and fibrillar
α-synuclein for modulating the trafficking pathways in
α-synuclein-pulsed monocyte-derived dendritic cells, which possibly
facilitated α-synuclein processing, thereby minimizing the
availability of antigenic peptides for antigen presentation and
counteracting the specific T-cell immune response to α-synuclein in
PD treatment (42). Mitophagy
induction is probably another pathway of the neuroprotective effect
of celastrol in PD. By clearing the dysfunctional mitochondria for
quality control, celastrol was able to prevent the release of ROS
and apoptosis-promoting factors caused by rotenone or
1-methyl-4-phenylpyridinium, thereby ameliorating oxidative stress
and further blocking cell death (41,43).
PD mouse model studies further confirmed the pivotal role of
mitophagy in improving motor symptoms and diminishing dopaminergic
neuronal degeneration (43).
Overall, these findings have demonstrated that the
neuroprotective effects of celastrol are intimately associated with
autophagy modulation.
Autophagy is involved in other
pharmacological activities of celastrol
Overexpression of p62 protein in lung tissues
represents insufficient autophagy during severe pulmonary fibrotic
responses (73). Of note,
celastrol was proven to be effective in the treatment of
bleomycin-induced experimental rat pulmonary fibrosis via the
induction of protective autophagy. Celastrol treatment
significantly elicited the initiation, elongation and maturation of
autophagosomes, resulting in enhanced p62 degradation, thereby
reducing collagen deposition and slowing down pulmonary fibrosis
progression (44).
Celastrol serves an anti-atherosclerosis role by
triggering autophagy in vascular smooth muscle cells (VSMCs). On
the one hand, celastrol-mediated autophagy significantly impeded
lipid storage induced by oxidized low-density lipoprotein in VSMCs,
thereby inhibiting VSMC-derived foam cell formation and the
resultant atherosclerotic plaque development (45). On the other hand,
celastrol-stimulated autophagy functioned as an intracellular ROS
scavenging system to specifically inhibit angiotensin II-mediated
ROS production, which ultimately counteracted VSMC senescence
(46). Furthermore, autophagy was
found to be involved in alleviating the overgrowth and migration of
VSMCs by celastrol. Celastrol treatment markedly activated VSMC
autophagy in both in vitro and in vivo intimal
hyperplasia models, as manifested by accumulated autophagosomes,
promoted autophagic flux, increased LC3II/LC3I ratio and decreased
p62 expression (47). C-myc, a key
transcription factor that induces the continuous proliferation of
VSMCs, combined with p62 to form a conjugate during
celastrol-induced autophagy, which was then delivered to lysosomes
for degradation, and thus exerted inhibitory effects on abnormal
VSMC proliferation and vascular neointimal hyperplasia (47).
In a human retinal pigment epithelial cell line,
incubation with hydrogen peroxide led to a significant induction of
apoptosis and oxidative stress, as well as the inhibition of cell
survival and autophagy, whereas this phenomenon was obviously
antagonized upon celastrol cotreatment. Celastrol-induced autophagy
markedly impeded ROS production and protected against hydrogen
peroxide-induced cell damage by increasing the LC3II/LC3I ratio and
Beclin1 expression, suggesting the therapeutic potential of
celastrol in age-related macular degeneration via autophagy
modulation (48).
Therefore, these findings confirm that autophagy
modulation acts as a crucial pathway for celastrol to alleviate the
pathogenic symptoms of multiple diseases, such as pulmonary
fibrosis, atherosclerosis, neointimal hyperplasia and macular
degeneration.
4. Mechanisms underlying autophagy
modulation by celastrol
The autophagy regulation by celastrol includes
epigenetic pathways through a series of microRNAs (miRNAs/miRs),
which are short non-coding RNAs that bind to specific mRNAs,
leading to their degradation or translational repression (74). By negatively regulating the
expression of autophagy-related genes or regulators, such as Atg4D,
Rab5A and Stathmin1, miR-101 functions as a potent autophagy
initiation and maturation inhibitor (75). Decreased miR-101 expression was
confirmed upon celastrol treatment, which rescued the suppressive
effect on the dynamic process of autophagy in prostate cancer cells
(19). Analogously, celastrol
treatment led to downregulation of two miR-17-92a cluster members,
miR-20a and miR-17a, which targeted Atg7 to inhibit autophagy,
resulting in autophagy induction in prostate cancer cells (49). Furthermore, the ligand-regulated
transcription factor androgen receptor (AR) that selectively
transactivates the miR-101 and miR-17-92a cluster was degraded in
the same sample mentioned above during celastrol-induced autophagy
and AR knockdown decreased, whereas the ectopic expression of AR
enhanced the expression of these miRNAs in the presence of
celastrol (19,49). Therefore, downregulation of AR
caused by celastrol was proposed to mediate the transcription
reduction of its downstream target miRNAs, which subsequently
abolished the negative regulation of autophagy, leading to
autophagy activation.
Celastrol may regulate autophagy via a mechanism
involving transcription factor EB (TFEB). TFEB, the transcription
factor that binds to a promoter motif coordinating the
transcription of multiple autophagic and lysosomal genes, is known
as the key regulator of the autophagy pathway (76). Celastrol increased the nuclear
translocation of TFEB from the cytosol in a neuronal cell line and
in mouse brains. The participation of the mammalian target of
rapamycin complex 1 (mTORC1) in the activation of TFEB by celastrol
has been characterized. By inhibiting the activity of mTORC1
kinase, celastrol inhibited the phosphorylation of TFEB that caused
the dissociation of TFEB with the tyrosine
3-monooxygenase/tryptophan 5-monooxygenase activation protein and
the rapid transport of TFEB to the nucleus, consequently enhancing
autophagy and lysosomal biogenesis (59,77).
In the mTORC1, the highly conserved serine/threonine kinase mTOR is
the central catalytic subunit of the multiprotein complex and may
phosphorylate multiple autophagy proteins, which results in a
blockade of autophagy-initiating kinase ULK1 and functions as a
negative autophagy mediator (78).
Transcriptomics data and network pharmacology analysis predicted
that mTOR is the direct therapeutic target of celastrol against
osteoarthritis (79), and a
subsequent study validated that the promotion of chondrocyte
autophagy by celastrol occurs as a result of the reduction in mTOR
levels (39). In addition,
numerous signaling pathways converging at mTOR involve
celastrol-mediated autophagy. After treatment with celastrol, AMPK
was phosphorylated by the upstream kinases, including liver kinase
B1 complex, Ca2+/calmodulin dependent protein kinase
kinase 2 (CaMKK2) and TGF-β-activated kinase 1, leading to its
activation and the subsequent inhibition of the phosphorylation of
the downstream target mTOR, resulting in increased autophagic
activity in gastric cancer cells and gastric tumors of xenografted
mice (28,80). AMPK/mTOR pathway activation was
also implicated in the pro-autophagic effects of the celastrol and
CP-456773 combination on the colon tissues of the rat colitis
model, as both increased phosphorylated AMPK and decreased
phosphorylated mTOR were observed in comparison to the control
(32). Furthermore, the effect of
celastrol on autophagy is wingless/integrase 1 (Wnt)5α-dependent,
which is a secretory glycoprotein belonging to the Wnt family.
Celastrol significantly reduced Wnt5α expression and attenuated the
phosphorylation and activation of protein kinase C and mTOR,
thereby promoting autophagy of VSMCs (47). The activated form of AKT,
phosphorylated AKT, was significantly suppressed in pituitary
adenoma cells, breast cancer cells and glioma cells exposed to
celastrol, which resulted in decreased expression of phosphorylated
mTOR and induction of autophagy (22,29,57).
However, in bleomycin-induced rat lung tissues, IL-10-deficient
mouse colon tissues and diabetic nephropathy rat renal tissues,
celastrol treatment reduced the expression of PI3K along with its
downstream effectors, AKT and mTOR, and upregulated autophagy,
suggesting that inactivation of the PI3K/AKT/mTOR pathway may
represent a mechanism through which celastrol induces autophagy
(34,35,44).
ROS may act as crucial upstream intracellular
transducers that sustain autophagy (81). Previous evidence indicates that
sirtuin 3 (SIRT3), a histone/protein deacetylase, contributes to
the effect of celastrol on autophagy through the modulation of ROS
production. Celastrol treatment markedly promoted autophagy and
impeded the expression of SIRT3 and ROS production in hydrogen
peroxide-treated retinal pigment epithelial cells, whereas SIRT3
depletion reversed the effects of celastrol on ROS production and
autophagy (48). The autophagy
regulation by celastrol was also proceeded via the ROS/c-Jun
N-terminal kinase (JNK) signaling pathway. A sharp decrease in the
mitochondrial membrane potential due to mitochondrial dysfunction
occurred after glioma and osteosarcoma cells were exposed to
celastrol, which promoted the generation of an excessive amount of
ROS. ROS phosphorylated the stress-activated signaling molecule JNK
that positively regulated autophagy and mediated autophagy
activation (22,23). Celastrol stimulated hepatocarcinoma
cell autophagy by ROS to activate AKT/p70 ribosomal S6 kinase
signaling, which further promoted hypoxia-inducible factor-1α
(HIF-1α) translation and sequentially elevated the expression of
the HIF-1α-target gene Bcl2/adenovirus E1B 19kD-interacting protein
3(27). The damage to mitochondria
in lung cancer cells was markedly augmented after cotreatment with
celastrol and erastin, which resulted in marked ROS accumulation,
followed by autophagy upregulation. In the meantime, high levels of
ROS enhanced p38 phosphorylation and increased its interaction with
dynamin-related protein 1, a protein that mediated mitochondrial
fission, thereby promoting mitophagy induction (58).
In addition to ROS accumulation, increased
cytoplasmic calcium release from the internal stores appeared in
osteosarcoma cells and hepatocarcinoma cells following treatment
with celastrol, with ER stress induction and subsequent unfolded
protein response signaling activation to induce the
autophagy-related protein expression and the assembly of autophagic
structures (24,26). Celastrol also stimulated
calcium-mediated autophagy via the activation of the
CaMKKβ-AMPK-mTOR signaling cascade in cervical cancer cells and
synovial fibroblasts. The calcium homeostasis perturbation by
celastrol may be ascribed to its inhibitory activity toward the
sarcoplasmic reticulum (SR)/ER Ca2+-ATPase pump, which
transports calcium ions back into the SR/ER lumen from the
cytoplasm and is necessary for the release of calcium into the
cytosol (37,51).
Other regulatory pathways of autophagy in response
to celastrol have been explored. Celastrol obviously enhanced heme
oxygenase-1 expression in podocytes under high-glucose conditions
and antagonized high glucose-induced autophagy pathway deficiency
(36). Celastrol-triggered
autophagy in colorectal cancer cells was associated with the
decreased expression of nuclear receptor-77 (Nur77), a
transcription factor in the nucleus negatively regulating the
transcription of Atg7 which was essential for autophagosome
formation (30). Another study
demonstrated that celastrol promoted the mitochondrial
translocation of Nur77 and its ubiquitination by E3 ubiquitin
ligase, named tumor necrosis factor receptor-associated factor 2.
The subsequent interaction between Nur77 and p62 served to prime
the ubiquitin-labeled mitochondria for autophagic degradation
(31). However, celastrol was also
identified as a mitophagy inhibitor in response to mitochondrial
damage by blocking Parkin recruitment into the mitochondria and
simultaneously by disrupting the association between PINK1 and
TOM20, a component of the translocase of the outer mitochondrial
membrane machinery (60).
In VSMCs and clear cell renal cell carcinoma cells,
celastrol-mediated upregulation of autophagy involved the upstream
activation of transcription factor liver X receptor α (LXRα), as
LXRα reduction reversed the promotion of autophagic flux induced by
celastrol (45,50). In addition, the participation of
the toll-like receptor 2 (TLR2)/high mobility group box 1 (HMGB1)
signaling pathway in the regulation of autophagy by celastrol has
been confirmed. Through the attenuation of TLR2 and HMGB1 protein
expression, celastrol was able to suppress rheumatoid
arthritis-induced autophagy in cardiomyocytes (38). Overall, these results demonstrated
the complexity through which celastrol regulated autophagy via
multiple targets or signaling pathways (Fig. 1).
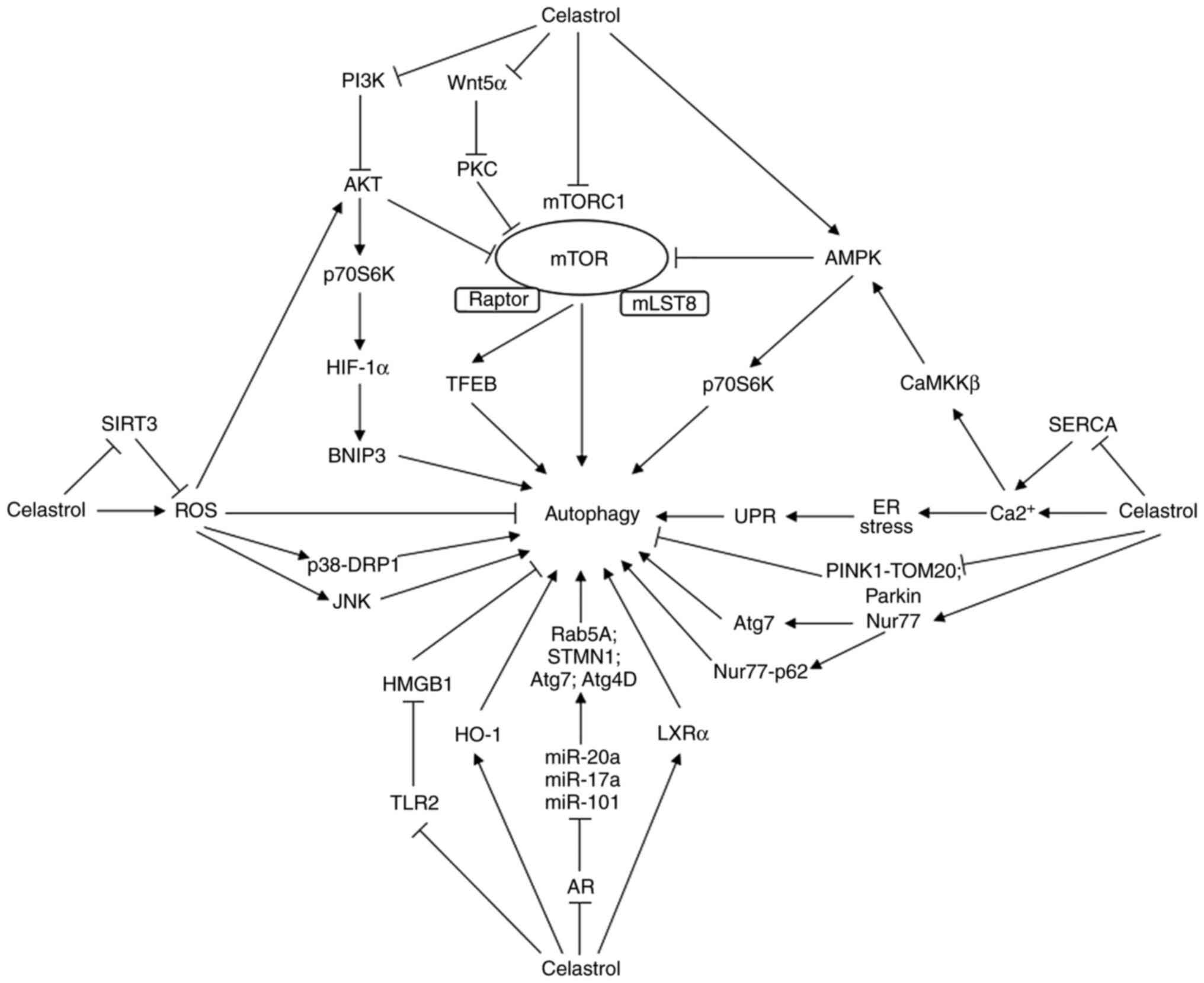 | Figure 1Schematic diagram depicting the
potential mechanisms underlying celastrol modulating autophagy.
Through either positive or negative regulation of a variety of
effectors, including AR, mTOR, ROS, Ca2+, Nur77, LXRα,
TLR2, HO-1, Parkin and PINK1, celastrol influences the downstream
molecular machineries or signaling pathways, resulting in autophagy
activation or suppression. AR, androgen receptor; miRs, microRNAs;
Rab, member RAS oncogene family protein; STMN1, stathmin1; Atg,
autophagy-related gene; HO-1, heme oxygenase-1; TLR2, toll-like
receptor 2; HMGB1, high mobility group box 1; ROS, reactive oxygen
species; SIRT3, sirtuin 3; DRP1, dynamin-related protein 1; JNK,
c-Jun NH2-terminal kinase; mTORC1, mammalian target of rapamycin
complex 1; mLST8, mammalian target of rapamycin associated protein;
Wnt, Wingless/integrase 1; PKC, protein kinase C; TFEB,
transcription factor EB; PI3K, phosphatidylinositol 3 kinase; Akt,
protein kinase B; p70S6K, p70 ribosomal S6 kinase; HIF-1α,
hypoxia-inducible factor-1α; BNIP3, Bcl2/adenovirus E1B
19kD-interacting protein3; AMPK, AMP-activated protein kinase;
Ca2+, calcium; SERCA, sarcoplasmic/endoplasmic reticulum
Ca2+-ATPase; CaMKKβ, Ca2+/calmodulin
dependent protein kinase kinase β; ER, endoplasmic reticulum; UPR,
unfolded protein response; PINK1, phosphatase and tensin
homolog-induced kinase 1; TOM20, translocase of the outer membrane
20; Nur77, nuclear receptor-77; LXRα, liver X receptor α. |
5. Conclusion and future perspectives
Celastrol has been regarded as one of the bioactive
components identified from certain traditional Chinese herbs that
is most likely to be developed as a modern drug thanks to its
multiple biological activities and high efficacy in animal and cell
culture disease models. Hence, elucidating the molecular mechanisms
behind the action of celastrol is in high demand. Studies have
indicated that celastrol alters autophagic activities through
diverse mechanisms, which take part in an intricate network that
modulates the crosstalk and interplay with other metabolic
processes, highlighting that autophagy may be a specific action
target for celastrol to accomplish its multiple pharmacological
activities. Based on the available data, celastrol mostly functions
as an autophagy inducer, except that celastrol was observed to
suppress autophagy in lung cancer cells (53,54),
glioblastoma cells (21), cervical
cancer cells (60) and
cardiomyocytes (38). This
discrepancy may be explained by different cell phenotypes,
pathological status and the particular stimulus of celastrol. To
date, studies have mainly addressed the effects of celastrol on
autophagy in certain cellular models with pharmacologic
activators/inhibitors such as rapamycin, bafilomycin A1 and
3-methyladenine; however, more specific gene deletion or
overexpression methods may confirm whether autophagy is the main
mechanism of celastrol activity. Furthermore, the lack of relevant
in vivo data has limited the determination of the exact role
of autophagy. The systemic evaluation of the in vivo
activities will further validate the conclusions. Furthermore,
whether autophagy is implicated in other pharmacological activities
of celastrol remains elusive. Although autophagy is a general
house-keeping process to maintain cellular homeostasis, how to
improve the specific targeting and selectivity of celastrol to
pathological tissues while minimizing undesired negative effects on
normal tissues will be a major issue when using celastrol as an
autophagy modulator.
The ubiquitin-proteasome system and autophagy
comprise the main proteolytic mechanisms in eukaryotic cells
(82). The inhibitory effect of
celastrol on the chymotrypsin-like activity of the purified 20S
proteasome and 26S proteasome has been confirmed in several types
of cultured tumor cells and tumor tissues (83). Autophagy is compensatorily
activated with the inhibition of the ubiquitin-proteasome system,
whereas the proteasomal flux is impaired if autophagy is disrupted
(84). Prospective studies are
required to clarify the presence of causality between the
regulation of autophagy and the ubiquitin-proteasome system
mediated by celastrol, as well as the possible molecular machinery
involved. A common feature of these investigations is the focus on
the regulatory activities on macroautophagy, while the impact on
the other two forms of autophagy has remained largely elusive.
Questions still remain as to whether microphagy or
chaperone-mediated autophagy directly have a functional role in
response to celastrol.
In conclusion, celastrol possesses potent
autophagic-regulatory effects with a broad potential
pharmacological utility. In addition, celastrol may serve as a
promising agent for studying autophagy. Further advances in
unveiling the detailed molecular mechanisms and the specific
functions of celastrol-mediated autophagy may provide effective
strategies for drug development, as well as valuable therapeutic
approaches for the treatment of certain autophagy-related
diseases.
Acknowledgements
Not applicable.
Funding
Funding: The present review was supported by the Applied Basic
Research Program of Science and Technology Department of Shanxi
Province (grant no. 202103021224295), the Research Project of
Shanxi Provincial Health and Family Planning Commission (grant no.
2022133), the Scientific and Technological Innovation Programs of
Higher Education Institutions in Shanxi (grant no. 2021L356) and
Research Fund from the Toxicity and Effect Innovation Team in
Shanxi University of Chinese Medicine (grant no. 2022TD1016).
Availability of data and materials
Not applicable.
Authors' contributions
YW conceived and supervised the present study. CZ
prepared the first draft of the manuscript. WW, CD, HL, KZ, ZL, YC
and SL reviewed the manuscript for important intellectual content.
All authors have read and approved the final manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hou W, Liu B and Xu H: Celastrol:
Progresses in structure-modifications, structure-activity
relationships, pharmacology and toxicology. Eur J Med Chem.
189(112081)2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Chen SR, Dai Y, Zhao J, Lin L and Wang Y
and Wang Y: A mechanistic overview of triptolide and celastrol,
natural products from Tripterygium wilfordii Hook F. Front
Pharmacol. 9(104)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang SF, Wu MY, Cai CZ, Li M and Lu JH:
Autophagy modulators from traditional Chinese medicine: Mechanisms
and therapeutic potentials for cancer and neurodegenerative
diseases. J Ethnopharmacol. 194:861–876. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wei YM, Wang YH, Xue HQ, Luan ZH, Liu BW
and Ren JH: Triptolide, a Potential autophagy modulator. Chin J
Integr Med. 25:233–240. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Denton D and Kumar S: Autophagy-dependent
cell death. Cell Death Differ. 26:605–616. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Levine B and Kroemer G: Biological
functions of autophagy genes: A disease perspective. Cell.
176:11–42. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Nixon RA: The role of autophagy in
neurodegenerative disease. Nat Med. 19:983–997. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
White E: The role for autophagy in cancer.
J Clin Invest. 125:42–46. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Yang Y, Zheng L, Zheng X and Ge L:
Autophagosomal membrane origin and formation. Adv Exp Med Biol.
1208:17–42. 2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kaushal GP, Chandrashekar K, Juncos LA and
Shah SV: Autophagy function and regulation in kidney disease.
Biomolecules. 10(100)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Shaid S, Brandts CH, Serve H and Dikic I:
Ubiquitination and selective autophagy. Cell Death Differ.
20:21–30. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Geisler S, Holmstrom KM, Skujat D, Fiesel
FC, Rothfuss OC, Kahle PJ and Spinger W: PINK1/Parkin-mediated
mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol.
12:119–131. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Martinez-Lopez N, Garcia-Macia M, Sahu S,
Athonvarangkul D, Liebling E, Merlo P, Cecconi F, Schwartz GJ and
Singh R: Autophagy in the CNS and periphery coordinate lipophagy
and lipolysis in the brown adipose tissue and liver. Cell Metab.
23:113–127. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Li W, He P, Huang Y, Li YF, Lu J, Li M,
Kurihara H, Lou Z, Meng T, Onishi M, et al: Selective autophagy of
intracellular organelles: Recent research advances. Theranostics.
11:222–256. 2021.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Dikic I and Elazar Z: Mechanism and
medical implications of mammalian autophagy. Nat Rev Mol Cell Biol.
19:349–364. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Fleming A, Bourdenx M, Fujimaki M,
Karabiyik C, Krause GJ, Lopez A, Martín-Segura A, Puri C, Scrivo A,
Skidmore J, et al: The different autophagy degradation pathways and
neurodegeneration. Neuron. 110:935–966. 2022.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hu LF: Epigenetic regulation of autophagy.
Adv Exp Med Biol. 1206:221–236. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Guo J, Huang X, Wang H and Yang H:
Celastrol induces autophagy by targeting AR/miR-101 in prostate
cancer cells. PLoS One. 10(e0140745)2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Feng Y, Zhang B, Lv J, Zhang P, Mao Q, Lin
F, Zhao J, Fu X, Yang Y, Li Z, et al: Scaffold hopping of celastrol
provides derivatives containing pepper ring, pyrazine and oxazole
substructures as potent autophagy inducers against breast cancer
cell line MCF-7. Eur J Med Chem. 234(114254)2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Boridy S, Le PU, Petrecca K and Maysinger
D: Celastrol targets proteostasis and acts synergistically with a
heat-shock protein 90 inhibitor to kill human glioblastoma cells.
Cell Death Dis. 5(e1216)2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Liu X, Zhao P, Wang X, Wang L, Zhu Y, Song
Y and Gao W: Celastrol mediates autophagy and apoptosis via the
ROS/JNK and Akt/mTOR signaling pathways in glioma cells. J Exp Clin
Cancer Res. 38(184)2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Li HY, Zhang J, Sun LL, Li BH, Gao HL, Xie
T, Zhang N and Ye ZM: Celastrol induces apoptosis and autophagy via
the ROS/JNK signaling pathway in human osteosarcoma cells: An in
vitro and in vivo study. Cell Death Dis. 6(e1604)2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chen Y, Ou Y, Tao Y, Liu H, Yin H, Zhong
S, Yu H, Zhao Z and He B: Effect and mechanisms of celastrol on the
apoptosis of HOS osteosarcoma cells. Oncol Rep. 40:2260–2268.
2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhao X, Gao S, Ren H, Huang H, Ji W and
Hao J: Inhibition of autophagy strengthens celastrol-induced
apoptosis in human pancreatic cancer in vitro and in vivo models.
Curr Mol Med. 14:555–563. 2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ren B, Liu H, Gao H, Liu S, Zhang Z,
Fribley AM, Callaghan MU, Xu Z, Zeng Q and Li Y: Celastrol induces
apoptosis in hepatocellular carcinoma cells via targeting
ER-stress/UPR. Oncotarget. 8:93039–93050. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Han X, Sun S, Zhao M, Cheng X, Chen G, Lin
S, Guan Y and Yu X: Celastrol stimulates hypoxia-inducible factor-1
activity in tumor cells by initiating the ROS/Akt/p70S6K signaling
pathway and enhancing hypoxia-inducible factor-1α protein
synthesis. PLoS One. 9(e112470)2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Lee HW, Jang KS, Choi HJ, Jo A, Cheong JH
and Chun KH: Celastrol inhibits gastric cancer growth by induction
of apoptosis and autophagy. BMB Rep. 47:697–702. 2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cai Z, Qian B, Pang J, Tan ZB, Zhao K and
Lei T: Celastrol induces apoptosis and autophagy via the AKT/mTOR
signaling pathway in the pituitary ACTH-secreting adenoma cells.
Curr Med Sci. 42:387–396. 2022.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang W, Wu Z, Qi H, Chen L, Wang T, Mao
X, Shi H, Chen H, Zhong M, Shi X, et al: Celastrol upregulated ATG7
triggers autophagy via targeting Nur77 in colorectal cancer.
Phytomedicine. 104(154280)2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Hu M, Luo Q, Alitongbieke G, Chong S, Xu
C, Xie L, Chen X, Zhang D, Zhou Y, Wang Z, et al: Celastrol-induced
Nur77 interaction with TRAF2 alleviates inflammation by promoting
mitochondrial ubiquitination and autophagy. Mol Cell. 66:141–153
e6. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Saber S, Abd El-Kader EM, Sharaf H,
El-Shamy R, El-Saeed B, Mostafa A, Ezzat D and Shata A: Celastrol
augments sensitivity of NLRP3 to CP-456773 by modulating HSP-90 and
inducing autophagy in dextran sodium sulphate-induced colitis in
rats. Toxicol Appl Pharmacol. 400(115075)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yu X, Zhao Q, Zhang X and Zhang H, Liu Y,
Wu X, Li M, Li X, Zhang J, Ruan X and Zhang H: Celastrol
ameliorates inflammation through inhibition of NLRP3 inflammasome
activation. Oncotarget. 8:67300–67314. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhao J, Sun Y, Shi P, Dong JN, Zuo LG,
Wang HG, Gong JF, Li Y, Gu LL, Li N, et al: Celastrol ameliorates
experimental colitis in IL-10 deficient mice via the up-regulation
of autophagy. Int Immunopharmacol. 26:221–228. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Nie Y, Fu C, Zhang H, Zhang M, Xie H, Tong
X, Li Y, Hou Z, Fan X and Yan M: Celastrol slows the progression of
early diabetic nephropathy in rats via the PI3K/AKT pathway. BMC
Complement Med Ther. 20(321)2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhan X, Yan C, Chen Y, Wei X, Xiao J, Deng
L, Yang Y, Qiu P and Chen Q: Celastrol antagonizes high
glucose-evoked podocyte injury, inflammation and insulin resistance
by restoring the HO-1-mediated autophagy pathway. Mol Immunol.
104:61–68. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wong VKW, Qiu C, Xu SW, Law BYK, Zeng W,
Wang H, Michelangeli F, Dias IRSR, Qu YQ, Chan TW, et al: Ca(2+)
signalling plays a role in celastrol-mediated suppression of
synovial fibroblasts of rheumatoid arthritis patients and
experimental arthritis in rats. Br J Pharmacol. 176:2922–2944.
2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lu X, Gong S, Wang X, Hu N, Pu D, Zhang J,
Wang Y, Luo J, An Q, Ju B and He L: Celastrol exerts
cardioprotective effect in rheumatoid arthritis by inhibiting
TLR2/HMGB1 signaling pathway-mediated autophagy. Int Arch Allergy
Immunol. 182:1245–1254. 2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Dai S, Fan J, Zhang Y, Hao Z, Yu H and
Zhang Z: Celastrol promotes chondrocyte autophagy by regulating
mTOR expression. Chin Med J (Engl). 135:92–94. 2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Feng K, Chen H and Xu C:
Chondro-protective effects of celastrol on osteoarthritis through
autophagy activation and NF-κB signaling pathway inhibition.
Inflamm Res. 69:385–400. 2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Deng YN, Shi J, Liu J and Qu QM: Celastrol
protects human neuroblastoma SH-SY5Y cells from rotenone-induced
injury through induction of autophagy. Neurochem Int. 63:1–9.
2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Ng L, Wang X, Yang C, Su C, Li M and
Cheung AKL: Celastrol downmodulates alpha-Synuclein-specific T cell
responses by mediating antigen trafficking in dendritic cells.
Front Immunol. 13(833515)2022.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Lin MW, Lin CC, Chen YH, Yang HB and Hung
SY: Celastrol inhibits dopaminergic neuronal death of Parkinson's
disease through activating mitophagy. Antioxidants (Basel).
9(37)2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Divya T, Sureshkumar A and Sudhandiran G:
Autophagy induction by celastrol augments protection against
bleomycin-induced experimental pulmonary fibrosis in rats: Role of
adaptor protein p62/SQSTM1. Pulm Pharmacol Ther. 45:47–61.
2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Shi Y, Jiang S, Zhao T, Gong Y, Liao D and
Qin L: Celastrol suppresses lipid accumulation through LXRα/ABCA1
signaling pathway and autophagy in vascular smooth muscle cells.
Biochem Biophys Res Commun. 532:466–474. 2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Xu XJ, Zhao WB, Feng SB, Sun C, Chen Q, Ni
B and Hu HY: Celastrol alleviates angiotensin II mediated vascular
smooth muscle cell senescence via induction of autophagy. Mol Med
Rep. 16:7657–7664. 2017.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Shi YN, Liu LP, Deng CF, Zhao TJ, Shi Z,
Yan JY, Gong YZ, Liao DF and Qin L: Celastrol ameliorates vascular
neointimal hyperplasia through Wnt5a-involved autophagy. Int J Biol
Sci. 17:2561–2575. 2021.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Du Z, Zhang W, Wang S, Zhang J, He J, Wang
Y, Dong Y and Huo M: Celastrol protects human retinal pigment
epithelial cells against hydrogen peroxide mediated oxidative
stress, autophagy, and apoptosis through sirtuin 3 signal pathway.
J Cell Biochem. 120:10413–10420. 2019.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Guo J, Mei Y, Li K, Huang X and Yang H:
Downregulation of miR-17-92a cluster promotes autophagy induction
in response to celastrol treatment in prostate cancer cells.
Biochem Biophys Res Commun. 478:804–810. 2016.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Zhang CJ, Zhu N, Long J, Wu HT, Wang YX,
Liu BY, Liao DF and Qin L: Celastrol induces lipophagy via the
LXRα/ABCA1 pathway in clear cell renal cell carcinoma. Acta
Pharmacol Sin. 42:1472–1485. 2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Xu SW, Law BYK, Qu SLQ, Hamdoun S, Chen J,
Zhang W, Guo JR, Wu AG, Mok SWF, Zhang DW, et al: SERCA and
P-glycoprotein inhibition and ATP depletion are necessary for
celastrol-induced autophagic cell death and collateral sensitivity
in multidrug-resistant tumor cells. Pharmacol Res.
153(104660)2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Wang WB, Feng LX, Yue QX, Wu WY, Guan SH,
Jiang BH, Yang M, Liu X and Guo DA: Paraptosis accompanied by
autophagy and apoptosis was induced by celastrol, a natural
compound with influence on proteasome, ER stress and Hsp90. J Cell
Physiol. 227:2196–2206. 2012.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Dai CH, Zhu LR, Wang Y, Tang XP, Du YJ,
Chen YC and Li J: Celastrol acts synergistically with afatinib to
suppress non-small cell lung cancer cell proliferation by inducing
paraptosis. J Cell Physiol. 236:4538–4554. 2021.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Nazim UM, Yin H and Park SY: Autophagy
flux inhibition mediated by celastrol sensitized lung cancer cells
to TRAIL induced apoptosis via regulation of mitochondrial
transmembrane potential and reactive oxygen species. Mol Med Rep.
19:984–993. 2019.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Xu SW, Law BY, Mok SW, Leung EL, Fan XX,
Coghi PS, Zeng W, Leung CH, Ma DL and Liu Land Wong VK: Autophagic
degradation of epidermal growth factor receptor in
gefitinib-resistant lung cancer by celastrol. Int J Oncol.
49:1576–1588. 2016.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Duan J, Zhan JC, Wang GZ, Zhao XC, Huang
WD and Zhou GB: The red wine component ellagic acid induces
autophagy and exhibits anti-lung cancer activity in vitro and in
vivo. J Cell Mol Med. 23:143–154. 2019.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Wang L, Tang L, Yao C, Liu C and Shu Y:
The synergistic effects of celastrol in combination with tamoxifen
on apoptosis and autophagy in MCF-7 Cells. J Immunol Res.
2021(5532269)2021.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Liu M, Fan Y, Li D, Han B, Meng Y, Chen F,
Liu T, Song Z, Han Y, Huang L, et al: Ferroptosis inducer erastin
sensitizes NSCLC cells to celastrol through activation of the
ROS-mitochondrial fission-mitophagy axis. Mol Oncol. 15:2084–2105.
2021.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Yang C, Su C, Iyaswamy A, Krishnamoorthi
SK, Zhu Z, Yang S, Tong BC, Liu J, Sreenivasmurthy SG, Guan X, et
al: Celastrol enhances transcription factor EB (TFEB)-mediated
autophagy and mitigates Tau pathology: Implications for Alzheimer's
disease therapy. Acta Pharm Sin B. 12:1707–1722. 2022.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Zhang C, Wang R, Liu Z, Bunker E, Lee S,
Giuntini M, Chapnick D and Liu X: The plant triterpenoid celastrol
blocks PINK1-dependent mitophagy by disrupting PINK1's association
with the mitochondrial protein TOM20. J Biol Chem. 294:7472–7487.
2019.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Kocaturk NM, Akkoc Y, Kig C, Bayraktar O,
Gozuacik D and Kutlu O: Autophagy as a molecular target for cancer
treatment. Eur J Pharm Sci. 134:116–137. 2019.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Kessel D: Apoptosis, paraptosis and
autophagy: Death and survival pathways associated with photodynamic
therapy. Photochem Photobiol. 95:119–125. 2019.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Kusy S, Ghosn EE, Herzenberg LA and Contag
CH: Development of B cells and erythrocytes is specifically
impaired by the drug celastrol in mice. PLoS One.
7(e35733)2012.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Zhang J, Shan J, Chen X, Li S, Long D and
Li Y: Celastrol mediates Th17 and Treg cell generation via
metabolic siganling. Biochem Biophys Res Commun. 497:883–889.
2018.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Deretic V: Autophagy in inflammation,
infection, and immunometabolism. Immunity. 54:437–453.
2021.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Gurung P, Lukens JR and Kanneganti TD:
Mitochondria: diversity in the regulation of the NLRP3
inflammasome. Trends Mol Med. 21:193–201. 2015.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Sawaf H, Thomas G, Taliercio JJ, Nakhoul
G, Vachharajani TJ and Mehdi A: Therapeutic advances in diabetic
nephropathy. J Clin Med. 11(378)2022.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Cush JJ: Rheumatoid arthritis: Early
diagnosis and treatment. Rheum Dis Clin North Am. 48:537–547.
2022.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Zhou Z, Miao Z, Luo A, Zhu D, Lu Y, Li P,
Feng X, Tan W and Wang F: Identifying a marked inflammation
mediated cardiac dysfunction during the development of arthritis in
collagen-induced arthritis mice. Clin Exp Rheumatol. 38:203–211.
2020.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Hosseinzadeh A, Kamrava SK, Joghataei MT,
Darabi R, Shakeri-Zadeh A, Shahriari M, Reiter RJ, Ghaznavi H and
Mehrzadi S: Apoptosis signaling pathways in osteoarthritis and
possible protective role of melatonin. J Pineal Res. 61:411–425.
2016.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Fujikake N, Shin M and Shimizu S:
Association between autophagy and neurodegenerative diseases. Front
Neurosci. 12(255)2018.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Wei YM and Han B: Beclin1 decreases the
RIPA-insoluble fraction of amyotrophic lateral sclerosis-linked
SOD1 mutant via autophagy. Neurosci Lett. 690:106–111.
2019.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Zhang L, Cheng S, Jiang X, Zhang J, Meng
P, Tang Q, Qin X, Wang B, Chen C and Zou Z: Pregnancy exposure to
carbon black nanoparticles exacerbates bleomycin-induced lung
fibrosis in offspring via disrupting LKB1-AMPK-ULK1 axis-mediated
autophagy. Toxicology. 425(152244)2019.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Zhou K, Chang Y, Han B, Li R and Wei Y:
MicroRNAs as crucial mediators in the pharmacological activities of
triptolide (Review). Exp Ther Med. 21(499)2021.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Frankel LB, Wen J, Lees M, Høyer-Hansen M,
Farkas T, Krogh A, Jäättelä M and Lund AH: microRNA-101 is a potent
inhibitor of autophagy. EMBO J. 30:4628–4641. 2011.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Martini-Stoica H, Xu Y, Ballabio A and
Zheng H: The autophagy-lysosomal pathway in neurodegeneration: A
TFEB perspective. Trends Neurosci. 39:221–234. 2016.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Martina JA, Chen Y, Gucek M and
Puertollano R: MTORC1 functions as a transcriptional regulator of
autophagy by preventing nuclear transport of TFEB. Autophagy.
8:903–914. 2012.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Zhu Z, Yang C, Iyaswamy A, Krishnamoorthi
S, Sreenivasmurthy SG, Liu J, Wang Z, Tong BC, Song J, Lu J, et al:
Balancing mTOR signaling and autophagy in the treatment of
Parkinson's disease. Int J Mol Sci. 20(728)2019.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Dai S, Wang H, Wang M, Zhang Y, Zhang Z
and Lin Z: Comparative transcriptomics and network pharmacology
analysis to identify the potential mechanism of celastrol against
osteoarthritis. Clin Rheumatol. 40:4259–4268. 2021.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Li Y and Chen Y: AMPK and autophagy. Adv
Exp Med Biol. 1206:85–108. 2019.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Gao L, Loveless J, Shay C and Teng Y:
Targeting ROS-mediated crosstalk between autophagy and apoptosis in
cancer. Adv Exp Med Biol. 1260:1–12. 2020.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Pohl C and Dikic I: Cellular quality
control by the ubiquitin-proteasome system and autophagy. Science.
366:818–822. 2019.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Yang H, Landis-Piwowar KR, Chen D, Milacic
V and Dou QP: Natural compounds with proteasome inhibitory activity
for cancer prevention and treatment. Curr Protein Pept Sci.
9:227–239. 2008.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Ji CH and Kwon YT: Crosstalk and interplay
between the ubiquitin-proteasome system and autophagy. Mol Cells.
40:441–449. 2017.PubMed/NCBI View Article : Google Scholar
|














