Introduction
Castleman disease (CD) was previously known as giant
lymphadenopathy, angiolymphatic hamartoma or angiofollic lymph node
hyperplasia. The etiology remains elusive and shares certain
clinical and pathological features with viral infection, tumors and
autoimmune disease (1). The
hyaline-vascular variant was mostly seen in unicentric CD and UCD
has no obvious symptoms or produces corresponding compression
symptoms due to enlarged lymph nodes pressing on adjacent tissues.
When accompanied by paraneoplastic pemphigus (PNP), the skin and
mucosa of the mouth may be damaged, and oral ulcers and rashes may
appear along with lung involvement. However, the clinical
manifestations of multicentric CD (MCD) are heterogeneous but not
specific, which may include multiple regional lymph node
enlargement, recurrent systemic symptoms (weight loss, fever,
fatigue), anemia, edema, hypoproteinemia, and involvement of
multiple important organs, such as lung, liver, spleen and kidney,
and the prognosis is worse than that of UCD. Hyaline-vascular
variant MCD (HV-MCD) is a relatively rare clinical type and
particularly those cases with pulmonary involvement are rarely
described in this subcategory (2).
However, previous case reports were lacking details of pulmonary
involvement. Thus, to improve the understanding of the clinical
characteristics of this rare type, the clinical data of 3 patients
admitted to the hospital were systematically and retrospectively
analyzed.
Case presentations
Case 1
A 16-year-old male patient was admitted to the local
hospital due to ‘abdominal mass accompanied by fever, oral ulcer
and shortness of breath for one and a half years, which had
worsened for three months’. In January 2008, the patient noticed an
abdominal mass without obvious inducement, accompanied by oral
ulcer, fever, shortness of breath, cough, expectoration, abdominal
pain and abdominal distension. In late March 2009, the patient
developed shortness of breath, obvious during activities, but no
chest tightness or chest pain. The patient was then admitted to the
First Affiliated Hospital of Guangxi Medical University (Nanning,
China) in June 2009. Physical examination on admission indicated
splenomegaly, a mass could be palpated in the abdomen, lymph nodes
of the size of soybeans could be palpated in the bilateral groins,
and rales of dry and wet could be heard in both lungs. Laboratory
tests indicated elevated white blood cells, IgM and reduced
albumin, while the globulin, C-reactive protein (CRP), IgG and IgA
levels were normal. Analysis of autoantibodies determined the
following: Histone (+/-), keratin antibody (+), anti-nuclear
antibody (+), anti-DS-DNA (+/-), anti-cardiolipin antibody (+) and
desmoglein 3 (+). T-lymphocyte subsets were as follows: Total T
cells, 78.2%; CD4+ cells, 39.4%; CD8+ cells, 33.4%; CD4/CD8 ratio,
1.18 (normal). IL-6 levels were 0.204 ng/l (normal range,
0.373-0.463 ng/l). Blood gas analysis was normal. Plain CT scan of
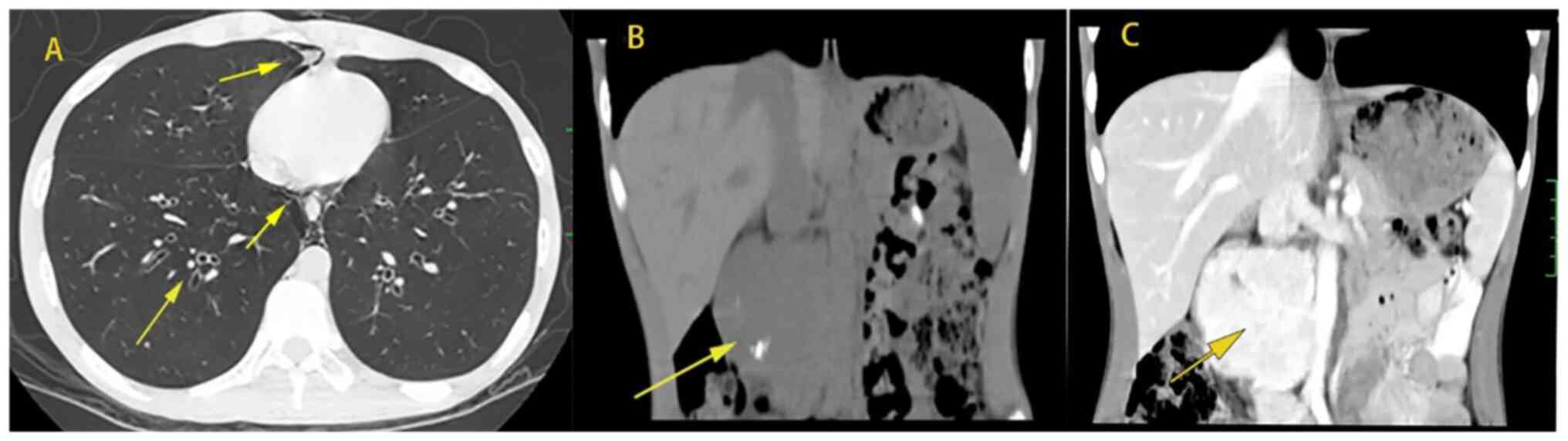
the chest and abdomen revealed the following: i) Huge
space-occupying lesion in the right posterior abdominal cavity; ii)
splenomegaly; iii) emphysema, infectious lesions in the anterior
segment of the right upper lobe, peripheral bronchiectasis,
hyperinflation, uneven lung density, high-ventilation and
low-ventilation areas mixed; iv) endogenous gas in the mediastinum
and posterior chest wall (Fig. 1).
Pulmonary function examination suggested the following: i) Severe
obstructive ventilation dysfunction; ii) peripheral resistance,
total airway resistance, peripheral elastic resistance all
increased. The bronchial dilation test indicated that the absolute
value of first second forced expiratory volume was increased by 70
ml. Right posterior abdominal mass resection was performed during
hospitalization. Postoperative pathology indicated CD of lymph node
HV. Oral mucosal biopsy revealed slight epithelial hyperplasia,
basal cell liquefaction, spinous layer release and vascular
hyperplasia in lamina propria, as well as lymphocyte infiltration.
The diagnosis was as follows: i) Multicentric transparent vascular
CD (left neck, right supraclavicular, bilateral axilla, inguinal
lymph nodes); ii) paraneoplastic pemphigus; and iii) bronchiolitis
obliterans (BO). No significant improvement was observed after
treatment with methylprednisolone, aminophylline and
bronchodilator. The symptoms were not improved after the treatment
with the Cyclophosphamide, Vincristine and Prednisone regimen
combined with interferon and the Cyclophosphamide, Doxorubicin,
Vinblastine and Prednisone (CHOP) regimen. The patient was
subsequently lost to follow-up.
Case 2
A 51-year-old male patient was admitted to the First
Affiliated Hospital of Guangxi Medical University (Nanning, China)
in November 2011 with ‘cervical lymph node enlargement and exertive
shortness of breath for 1 month’. In October 2011, the patient went
to a local hospital due to ‘inguinal hernia’. After physical
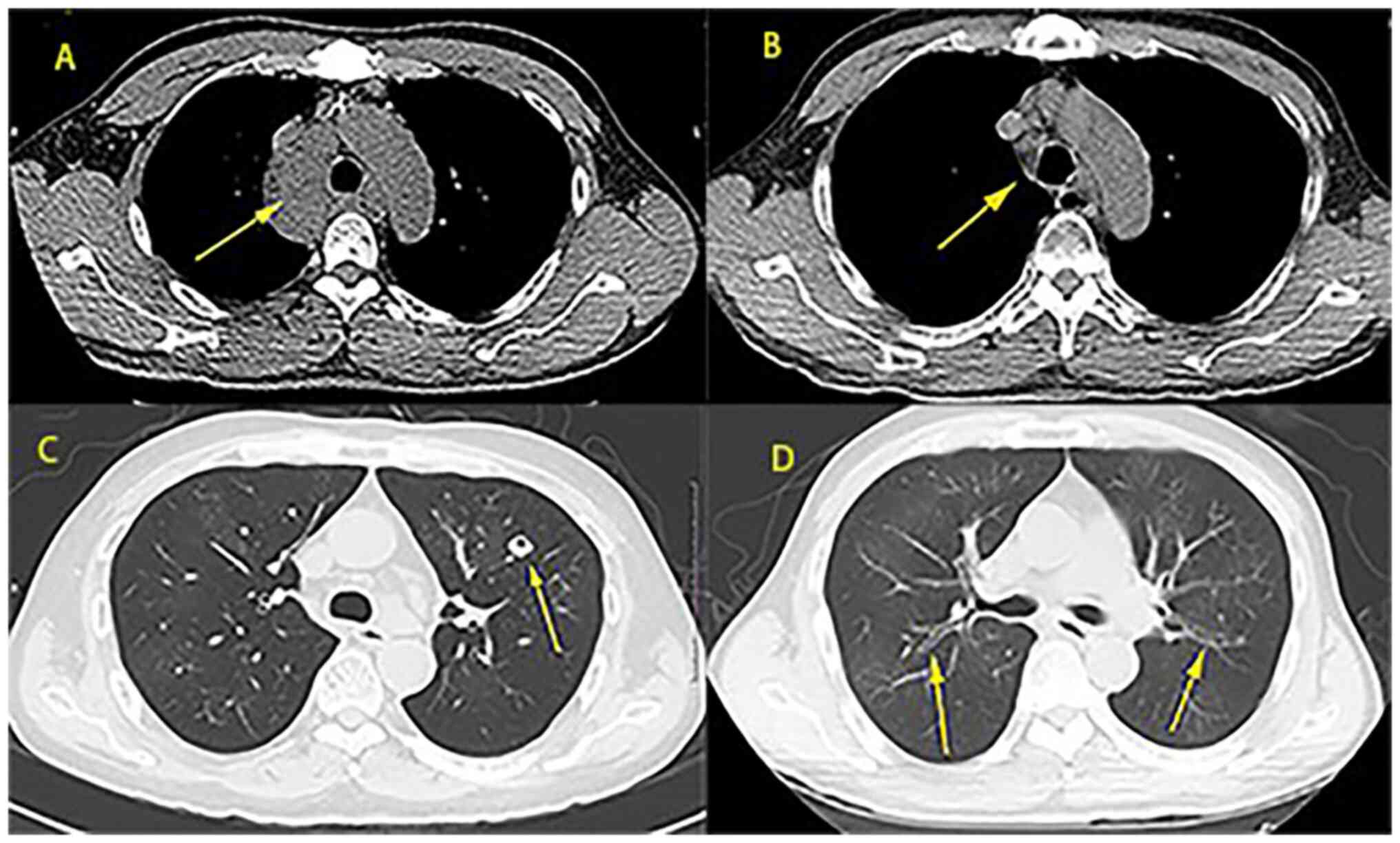
examination, cervical lymph node enlargement was found. Positron
emission tomography/CT examination revealed multiple nodules under
the left neck, bilateral clavicle region and mediastinum, with an
uneven increase in metabolism (Fig.
2A). Pathological examination of the left cervical lymph node
revealed HV of giant lymph node hyperplasia (CD). The patient
gradually developed weight loss, shortness of breath, blood sputum,
right chest pain and oral ulcer in late October. The patient then
came to the First Affiliated Hospital for further diagnosis and
treatment in November 2011. Physical examination revealed several
lymph nodes of soybean size in the left neck and supraclavicular
fossa. Dry and wet rales were found in both lungs. All results of
blood routine examination, including albumin, globulin, IgG and
IgA, and liver function and urine routine examination, were normal.
IgM and creatinine were elevated and the endogenous creatinine
clearance rate was reduced. T-cell subsets were as follows: CD4+T
cells, 28.10%; CD8+T cells, 41.20%; CD4/CD8, 0.68; total T cells,
75.50% (normal). IL-6 levels were 0.501 ng/l (normal). Blood gas
analysis revealed a high PCO2. Chest CT revealed
multiple mediastinal lymphadenopathies (Fig. 2C). The diagnosis was multicenter
clear vascular CD (left lower neck, bilateral clavicular region,
mediastinal lymph node). After four cycles of chemotherapy on the
CHOP regimen, no significant reduction of mediastinal lymph nodes
was observed. The fifth and sixth cycles of chemotherapy were
changed to the Fluorouracil, Cisplatin and Docetaxel (FCD) regimen.
In December 2011, chest CT indicated that the mediastinal multiple
lymph node enlargement was slightly smaller than previously
(Fig. 2A and C). In January 2013, the patient began to
develop oral ulcer, which was diagnosed as ‘pemphigus’ at the
Affiliated Stemmatological Hospital of Guangxi Medical University
(Nanning, China). The patient was treated with hydrocortisone, but
there was no obvious improvement of oral ulcer. The patient stopped
taking hormones in November 2013 and gradually developed
discomfort, such as shortness of breath after activity. The patient
returned to the hospital for treatment due to cough, sputum and
shortness of breath after activity. During hospitalization, lung
function examination was performed and the result was as follows:
i) Severe obstructive ventilate dysfunction; ii) severe peripheral
airway obstruction; and iii) severe diffuse dysfunction. The
bronchial dilation test indicated that 20 min after inhalation of
Ventolin, the FEV1 increased by 16.9%, and the absolute value of
the FEV1 only increased by 90 ml. Antibody against desmoglein 3 (+)
was detected. Chest CT indicated patchy, cable-like high-density
and ground-glass shadows in both lungs, and multiple enlarged lymph
node shadows were seen in the mediastinum. The diagnosis was as
follows: i) CD; ii) paraneoplastic pemphigus; iii) interstitial
pneumonia. The patient was given piperacillin sodium tazobactam,
levofloxacin and fluconazole for anti-infective treatment. After
discharge, the patient voluntarily switched to itraconazole but
still had mild symptoms. In November 2013, the patient was admitted
to the hospital, as cough, sputum and shortness of breath gradually
worsened. Reexamination by chest CT indicated that the mediastinal
lymph nodes had disappeared (Fig.
2B and D), but the lung lesion
was irreversible with cavitation and was susceptible to respiratory
infection.
Case 3
A 63-year-old female was admitted with left chest
and left lower abdominal pain for >1 month. The patient
complained of paroxysmal dull pain in the left chest and the left
lower abdomen with no obvious cause in early January 2012, with
oral ulcer, fever, nausea, vomiting, diarrhea and pain gradually
aggravated, which affected the patient's sleep. At the local
hospital, abdominal ultrasound was conducted but no obvious
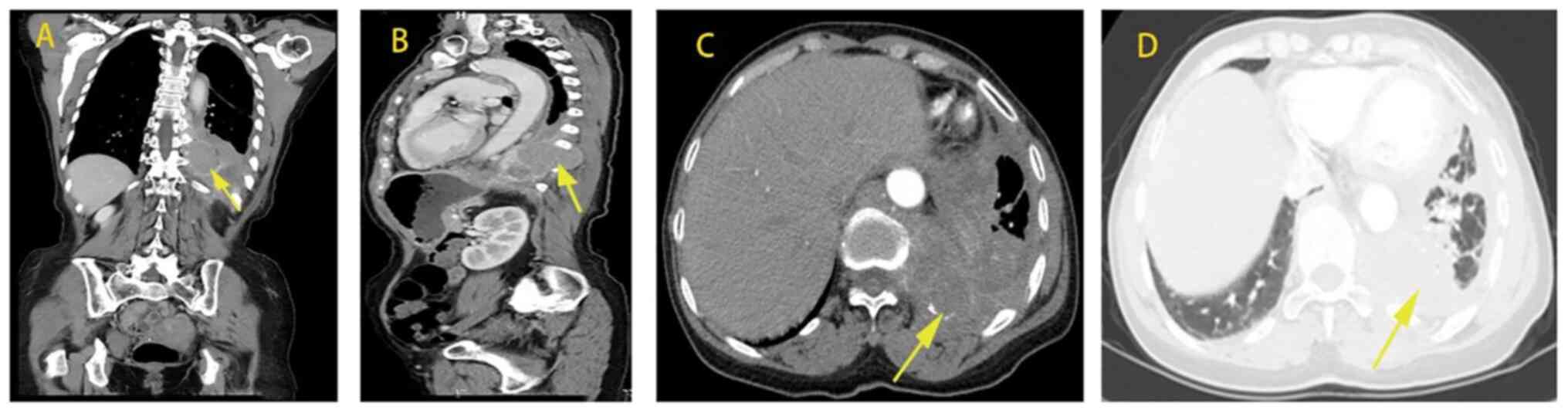
abnormity was seen. Chest and abdominal CT indicated the following:
Patchy high-density shadow in both lungs, peripheral bronchiole
wall thickened, bronchiole dilatation with secretion retention,
soft tissue mass shadow protruding into the left thoracic cavity
near the spine of the left lower lung, irregular thickening and
envelopment of the left pleura, as well as lymph node shadow of the
mediastinal tracheal bulge (Fig.
3). Subsequently, a series of symptoms, including cough with a
small amount of white foam-like sputum, decreased appetite and
fatigue occurred. For further diagnosis and treatment, the patient
was admitted to the First Affiliated Hospital of Guangxi Medical
University (Nanning, China) in February 2012. Physical examination
indicated right supraclavicular lymph node enlargement. Complete
blood routine examination revealed reduced hemoglobin and a high
platelet count; CRP, globulin and IgG were elevated, while albumin
was reduced, and IgM and IgA were normal. Autoantibodies were as
follows: Anti-RO-52 antibody (+), desmoglein 3 (+) and the
remaining indices were negative. IL-6 levels were 0.354 ng/l. No
abnormality of renal function, routine urine and complement was
observed. Pulmonary function examination was as follows: i) Mixed
ventilatory dysfunction with mild limitation; ii) mild peripheral
airway obstruction. Chest CT revealed a patchy high-density shadow
in both lungs, irregular thickening of the left pleura with
envelopment and lymph node shadow in the mediastinal trachea. Fiber
bronchoscopy indicated chronic bronchitis, a small amount of lung
tissue, clear alveoli and no tumor. B-mode ultrasound indicated
multiple enlarged lymph nodes in the left neck and right
supraclavicular region and multiple hypoechoic masses in the
bilateral axilla and inguinal region (lymph node sonography).
Percutaneous lung penetration of the lower left lung mass:
Microscopic observation revealed fibrous connective tissue,
hyalinosis and small focal-like chronic inflammatory cell
infiltration, with no histological evidence of granuloma or
carcinoma. Microscopic examination of lymph nodes in the left neck
indicated structural destruction of lymph nodes, atrophy of
lymphatic follicles, hyperplasia of interstitial fibrous tissue
with hyalinosis and hyalinosis in the vascular wall, which was
consistent with giant lymph node hyperplasia (clear vascular type).
The diagnosis was as follows: i) HV-MCD (left neck, right
supraclavicular, bilateral axilla, inguinal lymph nodes); ii)
paraneoplastic pemphigus; and iii) BO. After receiving
anti-inflammatory, analgesic and other symptomatic supportive
treatment, it was suggested that the patient consults the thoracic
surgery department to evaluate whether surgical treatment may be
performed, but the patient refused and asked to be discharged when
the condition did not improve and soon died of respiratory
failure.
Summary of the three cases
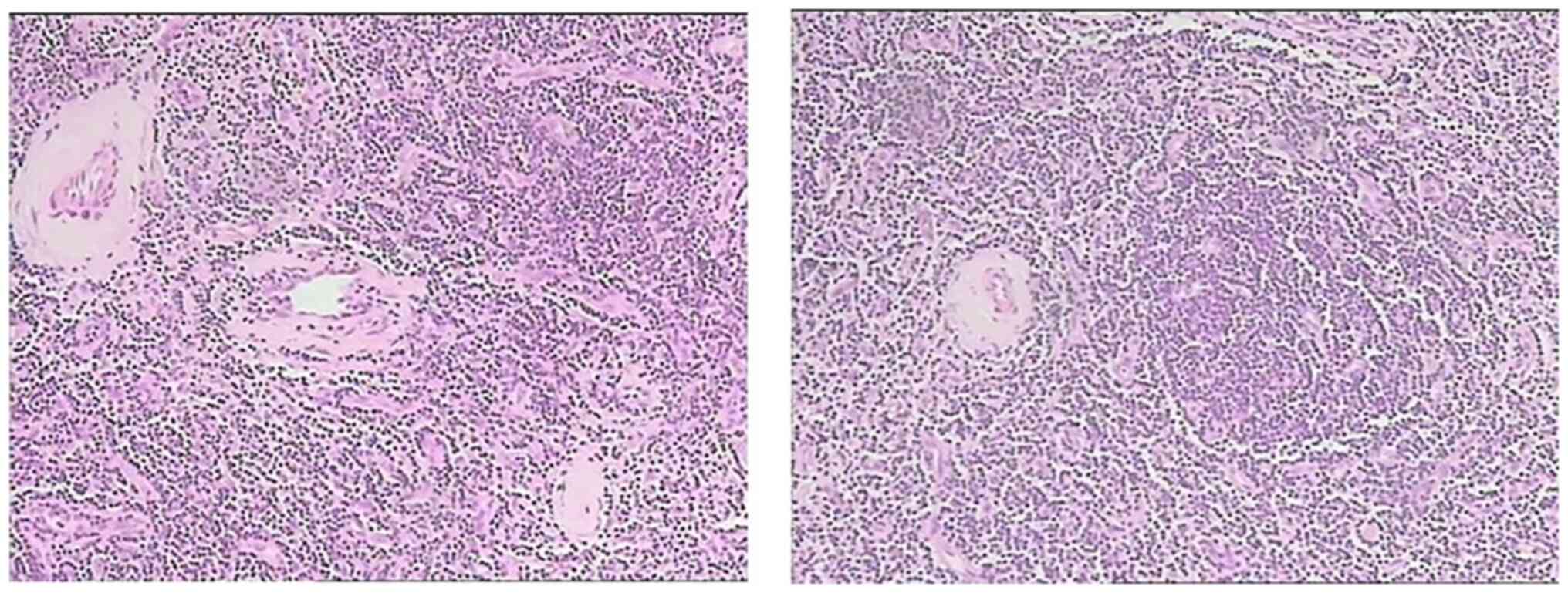
All of the three cases were pathologically diagnosed
with HV (Fig. 4) and were
HIV-negative with no smoking history. In two of the three patients,
the condition included mediastinum involvement and their clinical
manifestations comprised common symptoms of respiratory diseases,
such as cough, expectoration, shortness of breath and chest pain.
Furthermore, all of the three patients had refractory oral ulcer,
systemic symptoms such as emaciation and fever, splenomegaly and
local compression symptoms, such as local lymph node enlargement or
abdominal pain. With regard to laboratory results, anemia was more
common (2/3 patients). Furthermore, elevated erythrocyte
sedimentation rate (ESR), C-reactive protein (CRP) and
immunoglobulin, as well as declined albumin may be seen in certain
cases. Chest CT mainly indicated bronchiectasis and mediastinal
lymph node enlargement, with flocculent, patchy and stripey
high-density shadow. Blood gas analysis revealed hypoxemia and
pulmonary function examination showed obstructive ventilation
dysfunction; 1 case had mixed ventilation dysfunction with mild
restrictive ventilation dysfunction. The bronchial dilation test
was normal. Physical examination and other auxiliary examinations
confirmed the polycentric diagnosis of CD (Table I, Table II, Table III and Table IV).
 | Table IGeneral information and clinical
manifestations of the 3 cases of hyaline-vascular variant
multicentric Castleman disease. |
Table I
General information and clinical
manifestations of the 3 cases of hyaline-vascular variant
multicentric Castleman disease.
| Case no. | Age, years | Sex | Occupation | Clinical
features | Involved regions |
|---|
| 1 | 16 | Male | Student | Fever, weight loss,
shortness of breath, cough, sputum, abdominal pain, abdominal
distention, oral ulcer, splenomegaly | Oral mucosa,
abdominal cavity, bilateral inguinal lymph nodes |
| 2 | 51 | Male | Teacher | Neck lymph node
enlargement, weight loss, shortness of breath, bloody sputum, chest
pain, oral ulcer | Bilateral neck,
supraclavicular fossa, mediastinum, abdomen |
| 3 | 63 | Female | Worker | Chest pain, abdominal
pain, oral ulcer | Left neck, left
supraclavicular fossa, mediastinum |
| Table IILaboratory test results of the 3 cases
of hyaline-vascular variant multicentric Castleman disease. |
Table II
Laboratory test results of the 3 cases
of hyaline-vascular variant multicentric Castleman disease.
| Parameter (normal
range) | Case 1 | Case 2 | Case 3 |
|---|
| WBC (3.5-9.5),
x109/l | 10.31 | 7.6 | 9.1 |
| HGB (130-150),
g/l | 119 | 146 | 103 |
| PLT (125-350),
x109/l | 329 | 286 | 523 |
| N% (0.4-0.75) | 0.67 | 0.548 | 0.696 |
| L% (0.2-0.5) | 0.251 | 0.208 | 0.145 |
| ESR (0-15), mm/h | NA | NA | 84 |
| CRP (0-5), mg/l | 1.8 | N/A | 31.13 |
| ALB (40-55), g/l | 27.5 | 44.5 | 29.2 |
| GLB (20-40), g/l | 26.5 | 27.5 | 41.5 |
| A/G (1.2-2.4) | 1.02 | 1.8 | 0.7 |
| C3 (0.79-1.52),
g/l | 1.15 | NA | 1.35 |
| C4 (0.16-0.38),
g/l | 0.34 | NA | 0.37 |
| IgM (0.840-1.32),
g/l | 2.52 | 1.693 | 1.16 |
| IgG (8-18), g/l | 11.69 | Normal | 20.27 |
| IgA (0.9-4.5),
g/l | 1.73 | Normal | 4.03 |
| Creatinine
(59-104), µmol/l | Normal | Normal | Normal |
| RF (0-12.5),
IU/ml | 2.6 | NA | 5.3 |
| Autoantibodies | Histone (+/-), AKA
(+) ANA (+), anti-ds-DNA (+/-), ACA (+) | NA | Ro-52 (+) |
| Tumor markers | Normal | NA | Normal |
| T-cell subsets | | | |
|
Total T, %
(62.6-76.8) | 78.2 | 75.50 | NA |
|
CD4+T, %
(30-46) | 39.4 | 28.10 | NA |
|
CD8+T, %
(19.2-33.6) | 33.4 | 41.20 | NA |
|
CD4/CD8
(0.95-2.13) | 1.18 | 0.68 | NA |
| Table IIIInstrumental examination results of
the 3 cases of hyaline-vascular variant multicentric Castleman
disease. |
Table III
Instrumental examination results of
the 3 cases of hyaline-vascular variant multicentric Castleman
disease.
| Examination | Case 1 | Case 2 | Case 3 |
|---|
| ECG | Sinus
arrhythmia | Sinus rhythm, T
wave changes | Normal |
| Abdominal
ultrasound | Right abdominal
mass of substance | Liver inner
gallbladder wall thickened, echo enhancement, gallbladder
stone | Normal |
| Urinary
ultrasound | UE | Left kidney
cyst | Normal |
| Others | Abdominal CT plain
and enhanced: i) Huge space-occupying lesion in the right posterior
abdominal cavity; ii) splenomegaly | Positron emission
tomography/CT: Multiple nodules under the left neck, bilateral
clavicle area and mediastinum, with uneven and increased metabolism
masses (lymph nodes ranging from 3 to 7 mm), gallbladder stones and
small left renal cyst considered | Uterorectal
ultrasound: Depression and liquid dark area, 20x13 cm. Conclusion:
Pelvic effusion |
| Table IVResults of blood gas analysis,
respiratory function and bronchodilation test of the 3 cases of
hyaline-vascular variant multicentric Castleman disease. |
Table IV
Results of blood gas analysis,
respiratory function and bronchodilation test of the 3 cases of
hyaline-vascular variant multicentric Castleman disease.
| Parameter | Case 1 | Case 2 | Case 3 |
|---|
| FEV1, % | 23.3 | 18.8 | 69.4 |
| FVC, % | 67.2 | 48.6 | 77.6 |
| FEV1/FVC, % | 28.96 | 31.5 | 73.32 |
| VC, % | 67.1 | 46.9 | 77.1 |
| Bronchial dilation
test | Absolute value of
FEV1 was increased by 70 ml | Absolute value of
FEV1 was increased by 90 ml | Negative |
| Diffusing
capacity | UE | 0.49 | 1.62 |
| Dispersion
rate | UE | 0.33 | 1.46 |
| RV/TLC, % | UE | 78.01 | 46.18 |
| PH | 7.434 | 7.352 | UE |
| PO2,
mmHg | 78.4 | 120a | UE |
| PCO2,
mmHg | 42.7 | 46.5 | UE |
|
HCO3- | 28 | 24.1 | UE |
Discussion
The main pathological type of UCD is HV. Except for
local tumor compression symptoms, there are generally no systemic
symptoms. Chest CT typically displays isolated, well-bordered,
enlarged lymph nodes or localized nodular masses, with no other
pulmonary involvement except for the masses (3-8).
Abnormalities of Laboratory parameters are uncommon in HV-UCD and
lung function is generally normal (9,10).
Blood gas analysis indicated hypoxemia. Most of the pulmonary
functions were obstructive ventilation dysfunction, and mosaic sign
and other signs of BO could be seen on chest CT. In a study of 14
patients diagnosed with PNP, Mimouni et al (11) found 12 patients with HV-UCD,
suggesting that PNP may be a sign of the presence of HV-UCD. In
that study, 10 patients progressed to BO and eventually died. In
certain patients, PNP may be partially improved after resection of
the primary tumor. Therefore, for HV-UCD, surgical resection of the
local mass is advocated at present and may improve most symptoms
and abnormalities of test results. If surgery is not possible,
irradiation, embolization or neoadjuvant therapy with rituximab or
siltuximab/tocilizumab (if evidence of acute inflammatory state is
present) should be considered (12-14).
The most common pathological type of MCD is the
plasma cell type (PC). PC-MCD is frequently accompanied by systemic
symptoms, such as fever, night sweats, fatigue, loss of appetite,
weight loss, organ enlargement, diffuse polyadenosis and edema.
Furthermore, the disease is associated with a number of
abnormalities of laboratory parameters, including thrombocytopenia,
anemia, leukocytosis, hypoproteinemia, hyperglobulinemia, positive
autoantibodies, abnormal renal function and increases in
acute-phase proteins, such as CRP, ESR, fibrinogen and IL-6. Lung
involvement is more common than in HV-UCD, with respiratory
symptoms including cough, sputum and shortness of breath. Hypoxemia
is commonly found in the blood gas analysis and pulmonary function
analysis indicates mixed ventilation disorder (15-18),
suggesting interstitial lung damage and small airway lesions, and
the pathological features of BO and lymphocytic interstitial
pneumonia have also been confirmed in the literature (19). CT findings include lymphadenopathy,
often with diffuse hilar and mediastinal lymphadenopathy, and
pulmonary parenchyma changes, including centrilobular nodules,
bronchovesicular bundle and septal thickening, cysts and pleural
effusion. Since MCD involves multiple sites and is a diffuse
lesion, the feasibility of surgical resection is low. Two small
studies reported that ~50% of MCD patients achieved complete
remission after receiving four drugs combined with chemotherapy
(20,21). Steroids are used primarily to
induce remission in acute situations, but lasting remission is rare
and therefore not recommended for maintenance therapy. Currently,
rituximab has been used in certain CD20-positive patients with MCD,
either alone or after failure of other treatments, and >50% of
patients have achieved remission with mild side effects (22-26).
Rituximab-based therapy markedly improved 5-year OS for Human
herpes virus type 8 (HHV8)-MCD from 33 to 90% (27). Siltuximab, an anti-IL-6 antibody,
is the only Food and Drug Administration-approved treatment for
idiopathic MCD.
The present study reported on 3 rare cases of
HV-MCD. Although not all of the patients had masses in the lungs,
they all had respiratory symptoms, such as shortness of breath,
cough and sputum, chest pain and other symptoms of concomitant PNP,
as well as discomfort at the location of the masses, fever,
emaciation, splenomegaly and further systemic symptoms. Laboratory
tests revealed elevated IgM or IgG and positive autoimmune
antibodies. These changes are common laboratory abnormalities in
PC-MCD. Blood gas analysis indicated hypoxemia, 2 patients with
varying degrees of obstructed ventilation dysfunction and 1 patient
with restricted mixed ventilation dysfunction. These lung functions
suggested small airway lesions and possible interstitial damage.
The most common characteristic of CD accompanied by PNP is
stomatitis (28,29). Stomatitis is manifested as mucosal
erosion and ulceration, which is the most common in HV-UCD. As a
result, the 3 cases of HV-MCD in the present study had
characteristics of obstructive or mixed ventilation dysfunction,
hypoxemia and carbon dioxide increases, as well as fever,
emaciation, increases in immunoglobulin, autoimmune antibody
abnormalities and likeliness of concurrent PNP. Therefore, it is
evident that HV-MCD has the clinical features of HV-UCD and PC-MCD.
From the treatment effect aspect, regardless of the type of CD, BO
is one of the main causes of death, while surgical resection or
chemotherapy has no obvious effect, leading to the death of
patients due to respiratory failure, resulting in poor
prognosis.
In the three cases of the present study, abnormal
lung function was due to PNP. Wang et al (30) found that a specific B-cell clone
exists in HV-CD, which may produce antibodies to identify antigen
expressed in epithelium, and patients were prone to having
concurrent PNP and oral ulcer. Antibodies against bronchial
epithelial proliferation caused airway occlusion and thus led to
the formation of occlusive bronchiolitis. After the removal of
diseased tissue, the antibody concentration declined. Thus, tumor
removal brought about a good prognosis. In certain studies, lung
lesions of PC-MCD other than mass were confirmed by pathological
biopsy as interstitial fibrous thickening, plasma-cell infiltration
and alveolar collapse. Reichard et al (31) found a karyotype change of
chromosome 7p15 double-allele containing IL-6 locus in PC-CD, which
may be related to the imbalance of IL-6 cytokines. Mihara et
al (32) found that IL-6
promoted the migration of inflammatory cells and the production of
antibodies by B cells. Therefore, lung lesions of PC-MCD may be
caused by interstitial destruction and fibrosis may be caused by
excessive infiltration of neutrophils and plasma cells due to
increased IL-6 secretion. It has been suggested that anti-IL-6
therapy has an important role in the treatment strategy of PC-MCD.
In these three cases, IL-6 was not elevated.
A total of four cases of HV-MCD involving the lungs
were reported previously (2,33),
but chest CT, blood gas analysis and lung function results were not
described in detail. HV-MCD is a rare type of CD. All of the 3
cases in the present study had abnormal lung function, suggesting
that abnormal lung function due to pemphigus paraneoplasia of
HV-MCD is an important clinical feature. Attention should be paid
when hypoxemia, obstructive ventilation dysfunction and lung
shadow, accompanied by fever, emaciation, splenomegaly and other
systemic symptoms, e.g., anemia, hypoalbuminemia, increased
immunoglobulin, CRP and other laboratory abnormalities, or
refractory oral mucosa and skin lesions are observed in a patient
to rule out whether it is caused by CD. Timely examination of lymph
nodes, chest and abdomen should be performed. If a mass or enlarged
lymph node is present, early biopsy should be performed.
HV-MCD not only presents similarly to HV-UCD, which
is easily complicated with PNP and BO, but also manifests as a lung
shadow and is accompanied by systemic symptoms such as fever,
emaciation and splenomegaly, as well as abnormalities of laboratory
parameters, such as anemia, hypoalbuminemia, elevated
immunoglobulin and elevated CRP, which are also common in PC-MCD.
While the primary focus disappears after treatment, the small
airway lesion is irreversible and affects the quality of life, and
more severe cases can lead to death. Thus, the prognosis of HV-MCD
is poor. Since HV-MCD is a rare disease and is easily misdiagnosed
by respiratory physicians, if a patient presents with respiratory
symptoms, lymphadenectasis, elevated immunoglobulin, hypoxemia,
obstructive ventilation function disturbance and small airway
lesion, and no improvement is seen after conventional therapy,
general check-up and lymph node biopsy are required to clarify the
diagnosis.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by grants from the Natural
Science Foundation of China (grants no. NSFC81760010 and 82060364)
and the Science and Technology Department of Guangxi Zhuang
Autonomous Foundation of Guangxi Key Research and Development
Program (grant no. GuikeAB20238025).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JC, YQ and JZ performed the research. JC, WZ and JZ
designed the study. JC, YQ, WZ, MP and JZ contributed essential
reagents or tools. MP analysed the data. JC analyzed the data and
wrote the manuscript. All authors have read and approved the final
manuscript. JZ and JC confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee
associated with the Faculty of Medicine at The First Affiliated
Hospital of Guangxi Medical University (Guangxi, China; no.
2023-E065-01). Written informed consent was provided by the
patients.
Patient consent for publication
Written informed consent was obtained from the adult
patients for publication of identifying images or other personal or
clinical details of participants. When referring to individuals
younger than the age of 18, consent for publication was obtained
from the parents.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Oksenhendler E, Boutboul D, Fajgenbaum D,
Mirouse A, Fieschi C, Malphettes M, Vercellino L, Meignin V, Gérard
L and Galicier L: The full spectrum of Castleman disease: 273
patients studied over 20 years. Br J Haematol. 180:206–216.
2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Biçakçioğlu P, Gülhan SS, Acar LN,
Ağaçkiran Y, Özkara Ş, Kaya S and Karaoğlanoğlu N: Intrathoracic
Castleman disease. Turk J Med Sci. 44:197–202. 2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Dispenzieri A and Fajgenbaum DC: Overview
of Castleman disease. Blood. 135:1353–1364. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Haap M, Wiefels J, Horger M, Hoyer A and
Müssig K: Clinical, laboratory and imaging findings in Castleman's
disease-The subtype decides. Blood Rev. 32:225–234. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
van Rhee F, Oksenhendler E, Srkalovic G,
Voorhees P, Lim M, Dispenzieri A, Ide M, Parente S, Schey S,
Streetly M, et al: International evidence-based consensus
diagnostic and treatment guidelines for unicentric Castleman
disease. Blood Adv. 4:6039–6050. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kligerman SJ, Auerbach A, Franks TJ,
Franks TJ and Galvin JR: Castleman disease of the thorax: Clinical,
radiologic, and pathologic correlation: From the radiologic
pathology archives. Radiographics. 36:1309–1332. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhang X, Rao H, Xu X, Li Z, Liao B, Wu H,
Li M, Tong X, Li J and Cai Q: Clinical characteristics and outcomes
of Castleman disease: A multicenter study of 185 Chinese patients.
Cancer Sci. 109:199–206. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hill AJ, Tirumani SH, Rosenthal MH,
Shinagare AB, Carrasco RD, Munshi NC, Ramaiya NH and Howard SA:
Multimodality imaging and clinical features in Castleman disease:
Single institute experience in 30 patients. Br J Radiol.
88(20140670)2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ota H, Kawai H and Matsuo T: Unicentric
castleman's disease arising from an intrapulmonary lymph node. Case
Rep Surg. 2013(289089)2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sarana B, Jaal J, Tamm H and Laisaar T:
Resection of unicentric interlobar Castleman disease with following
adjuvant radiotherapy. SAGE Open Med Case Rep.
5(2050313x17744481)2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Mimouni D, Anhalt GJ, Lazarova Z, Aho S,
Kazerounian S, Kouba DJ, Mascaro JM Jr and Nousari HC:
Paraneoplastic pemphigus in children and adolescents. Br J
Dermatol. 147:725–732. 2002.PubMed/NCBI View Article : Google Scholar
|
|
12
|
van Rhee F and Munshi NC: Castleman
disease. Hematol Oncol Clin North Am. 32:xiii–xiv. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Boutboul D, Fadlallah J, Chawki S, Fieschi
C, Malphettes M, Dossier A, Gérard L, Mordant P, Meignin V,
Oksenhendler E and Galicier L: Treatment and outcome of unicentric
castleman disease: A retrospective analysis of 71 cases. Br J
Haematol. 186:269–273. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Larroche C, Cacoub P, Soulier J,
Oksenhendler E, Clauvel JP, Piette JC and Raphael M: Castleman's
disease and lymphoma: Report of eight cases in HIV-negative
patients and literature review. Am J Hematol. 69:119–126.
2002.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wang ST, Wang QP, Li J, Zhang T, Zhang L
and Mao YY: Amyloidosis secondary to intrapulmonary Castleman
disease mimicking pulmonary hyalinizing granuloma-like clinical
features: A rare case report. Medicine (Baltimore).
98(e15039)2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang H, Zhang JQ, Zhong XN, Liu GN, Li
MH, Bai J, He ZY and Deng JM: Two case reports of Castleman disease
with pulmonary involvement. Int J Tuberc Lung Dis. 19:362–365.
2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Hwangbo Y, Cha SI, Lee YH, Lee SY, Seo H,
Oh S, Kim M, Choi SH, Park TI and Shin KM: A case of multicentric
castleman's disease presenting with follicular bronchiolitis.
Tuberc Respir Dis (Seoul). 74:23–27. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Nabeya D, Yoshimatsu Y and Fujiwara H: A
case of acute progressive diffuse interstitial lung disease
preceding idiopathic multicentric Castleman disease. Respir Med
Case Rep. 31(101216)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Johkoh T, Müller NL, Ichikado K, Nishimoto
N, Yoshizaki K, Honda O, Tomiyama N, Naitoh H, Nakamura H and
Yamamoto S: Intrathoracic multicentric Castleman disease: CT
findings in 12 patients. Radiology. 209:477–481. 1998.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Herrada J, Cabanillas F, Rice L, Manning J
and Pugh W: The clinical behavior of localized and multicentric
Castleman disease. Ann Intern Med. 128:657–662. 1998.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chronowski GM, Ha CS, Wilder RB,
Cabanillas F, Manning J and Cox JD: Treatment of unicentric and
multicentric Castleman disease and the role of radiotherapy.
Cancer. 92:670–676. 2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Marcelin AG, Aaron L, Mateus C, Gyan E,
Gorin I, Viard JP, Calvez V and Dupin N: Rituximab therapy for
HIV-associated Castleman disease. Blood. 102:2786–2788.
2003.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ide M, Kawachi Y, Izumi Y, Kasagi K and
Ogino T: Long-term remission in HIV-negative patients with
multicentric Castleman's disease using rituximab. Eur J Haematol.
76:119–123. 2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ocio EM, Sanchez-Guijo FM, Diez-Campelo M,
Castilla C, Blanco OJ, Caballero D and San Miguel JF: Efficacy of
rituximab in an aggressive form of multicentric Castleman disease
associated with immune phenomena. Am J Hematol. 78:302–325.
2005.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Corbellino M, Bestetti G, Scalamogna C,
Calattini S, Galazzi M, Meroni L, Manganaro D, Fasan M, Moroni M,
Galli M and Parravicini C: Long-term remission of Kaposi
sarcoma-associated herpesvirus-related multicentric Castleman
disease with anti-CD20 monoclonal antibody therapy. Blood.
98:3473–3475. 2001.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kofteridis DP, Tzagarakis N, Mixaki I,
Maganas E, Xilouri E, Stathopoulos EN, Eliopoulos GD and Gikas A:
Multicentric Castleman's disease: Prolonged remission with anti
CD-20 monoclonal antibody in an HIV-infected patient. AIDS.
18:585–586. 2004.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Bower M, Newsom-Davis T, Naresh K,
Merchant S, Lee B, Gazzard B, Stebbing J and Nelson M: Clinical
features and outcome in HIV-Associated multicentric castleman's
disease. J Clin Oncol. 29:2481–2486. 2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Anhalt GJ: Paraneoplastic pemphigus. J
Investig Dermatol Symp Proc. 9:29–33. 2004.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lim JM, Lee SE, Seo J, Kim DY, Hashimoto T
and Kim SC: Paraneoplastic pemphigus associated with a malignant
thymoma: A case of persistent and refractory oral ulcerations
following thymectomy. Ann Dermatol. 29:219–222. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang L, Bu D, Yang Y, Chen X and Zhu X:
Castleman's tumours and production of autoantibody in
paraneoplastic pemphigus. Lancet. 363:525–531. 2004.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Reichard KK, Robinett S and Foucar MK:
Clonal cytogenetic abnormalities in the plasma cell variant of
Castleman disease. Cancer Genet. 204:323–327. 2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Mihara M, Hashizume M, Yoshida H, Suzuki M
and Shiina M: IL-6/IL-6 receptor system and its role in
physiological and pathological conditions. Clin Sci (Lond).
122:143–159. 2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Luo JM, Li S, Huang H, Cao J, Xu K, Bi YL,
Feng RE, Huang C, Qin YZ, Xu ZJ and Xiao Y: Clinical spectrum of
intrathoracic Castleman disease: A retrospective analysis of 48
cases in a single Chinese hospital. BMC Pulm Med.
15(34)2015.PubMed/NCBI View Article : Google Scholar
|