Introduction
Inflammatory bowel disease (IBD) is a chronic
inflammatory and immune-related disease with unknown pathogenesis,
and it is typically categorized into two subtypes: Crohn's disease
(CD) and ulcerative colitis (UC). In UC, mucosal inflammation can
affect the colon and rectum, which comprises the mucosa and
submucosa of the large intestine (1). Although the pathogenesis of IBD
remains unclear, studies have shown that an imbalance in the
intestinal microenvironment, immune dysfunction, environmental
changes, and genetic factors may lead to UC. Under normal
conditions, the process of the inflammatory response can be
self-limiting, allowing for the complete remission of inflammation
and internal homeostasis (2). At
present, however, the excessive and persistent inflammation that
occurs in UC suggests an immunological pathogenesis (3).
More recently, an increasing number of studies have
shown that macrophages play crucial roles in preventing an
excessive immune response. Intestinal macrophages, as innate immune
cells, play a significant role in modulating the inflammatory
response in the gut mucosa (4).
Intestinal macrophages, composed of newly recruited circulating
monocytes and resident macrophages, can regulate tissue repair and
promote wound healing during infection and autoimmunity (5). Concurrently, intestinal macrophages
have also been reported to produce various cytokines and
inflammatory mediators that participate in gut homeostasis
(6-8).
For example, macrophages induce the production of IL-1β in the
microbiota and this promotes the release of colony-stimulating
factor-2 (CSF2) by type 3 innate lymphoid cells, and this
subsequently stimulates macrophages to secrete IL-10, thereby
maintaining intestinal homeostasis via antigen-specific CD4+CD25+
regulatory T(Treg) cell expansion (6). In addition, macrophages can polarize
into M1-like macrophages that produce various proinflammatory
cytokines, including TNF-a, IL-6, and iNOS, thus increasing
inflammatory damage to the intestinal mucosa. Conversely, M2-like
macrophages express mannose receptors (MRC1/CD206) and produced
several anti-inflammatory factors, such as IL-10, TGF-β, and
Fizz-1, further giving rise to the abatement of intestinal
inflammation (7,9). Thus, when macrophages are
dysregulated, it contributes to a cascade of inflammatory immune
responses in IBD, indicating that modulating macrophage
differentiation could be a promising therapeutic strategy for
treating patients with UC.
CD73 is an extracellular-5'-nucleotidase encoded by
the ecto-5'-nucleotidase CD73(NT5E) gene (10). The upstream CD39 catalyzes ATP to
produce adenosine monophosphate (AMP), which is converted into
adenosine by CD73, and adenosine binds to the downstream adenosine
receptor (A2AR) (11). It has been
shown that CD73-positive extracellular vesicles (EVs) contribute to
suppressing antitumor immune responses in the tumor
microenvironment, and that EVs derived from murine regulatory T
cells display AMP activity. Moreover, APCP can specifically block
the activity of AMP (12).
Previous studies have also demonstrated that APCP can block the
expression of CD73 (13,14). Additionally, APCP promotes tumor
immune escape, development, and metastasis by regulating the cell
cycle, apoptosis, and several signaling pathways, including EGFR,
NF-kB, VEGF, and AKT/ERK, which are key mediators of inflammatory
responses in the regulation of innate and adaptive immune responses
(15). Nevertheless, the
understanding of the effect of CD73 in UC remains to be
elucidated.
In the present study, the expression of CD73 in
colonic mucosal tissues from patients with UC and its correlation
with the endoscopic index was assessed. CD73 activity was also
blocked in macrophages to assess its effect on the production of
proinflammatory and anti-inflammatory cytokines, and to determine
the role of CD73 in the differentiation of macrophages. It was
found that CD73 inhibited M2 macrophage polarization in both mouse
and human-derived macrophages. In addition, blockade of CD73 in
mice reduced dextran sulfate sodium salt (DSS)-induced colitis.
Moreover, the results showed that the function of CD73 in the
modulation of inflammation induced by lipopolysaccharide (LPS)
involved regulation of the NF-kB and ERK signaling pathways.
Together, these findings show that CD73 inhibits M2 macrophage
polarization and that it may regulate inflammation via the NF-kB
and ERK signaling pathways to reduce UC.
Materials and methods
Patients
All colonic biopsy samples were collected from the
Department of Gastroenterology of the Affiliated Hospital of Jining
Medical College (UC: 8 men and 7 women; healthy controls: 9 men and
8 women). The median age of the patients with UC was 56 years and
the age range was 42-73 years. The median age of the healthy
controls was 41 years and the age range was 26-50 years. The
diagnostic criteria of UC were based on the clinical examination,
radiological examination, endoscopic examination, and histological
criteria (16). Written informed
consent was also obtained from all participants prior to the study.
Additionally, this study was approved by the Institutional Review
Board for Clinical Research. The severity of the disease and
intestinal mucosal lesions were graded using the Ulcerative Colitis
Endoscopic Index of Severity (UCEIS) criteria (17).
Mice
C57BL/6J mice were purchased from the Peng Yue
Experimental Animal Breeding Company and kept under specific
pathogen-free conditions in the animal house of Jining Medical
University. Mice were used for experiments at 8-10 weeks of age,
weighing 20-25 g. The number of mice used for the experiment was
14. All animal studies were approved by and conducted according to
the permission of the Ethical Committee of Jining Medical
University.
DSS-induced colitis model
Age- and sex-matched mice were divided into four
groups: The negative control-DSS group (intraperitoneal injection
of PBS), and the CD73 inhibitor-DSS group (intraperitoneal
injection of APCP) (4 mg/kg). There were four mice in each of these
two groups. Mice exposed to DSS received 2.5% DSS (MP Biomedicals,
M.W. 36,000-50,000 kDa) for 7 days, and then received sterile water
for a further 3 days from day 8. The other two groups of mice were
given sterile water for 9 continuous days as the negative controls.
There were three mice in each of these two groups. Subsequent
disease progression in the development of colitis was assessed by
monitoring indicators, including daily weight changes, fecal blood,
and diarrhea, which are common clinical indicators of colitis
(18). Colitis severity was
calculated using the disease activity index (DAI) based on a
composite score of weight loss, stool consistency, and bleeding,
ranging from 0 to 12, as described previously (19). All parameters were scored from days
0-9, and mice were sacrificed on day 9. The mice were executed by
asphyxiation with a 60% CO2 volume displacement rate
followed by cervical dislocation when the mice did not respond to
painful stimuli. Mice were euthanized when DSS mice reached ~85%
body weight loss. In the DSS group, 2 mice died due to colitis, and
the remaining 12 mice were euthanized as above. After ensuring that
the animals were free of vital signs (cardiac arrest, dilated
pupils, whitening of the eyelids, and no visual response), the dead
animals were placed in yellow waste bags and left for 30 min before
reconfirming whether there were any surviving animals.
Cell culture
A human monocyte leukemia cell line (THP-1) was
provided by the Medical Research Center of the Affiliated Hospital
of Jinan Medical University. THP1 cells were grown to a density of
2.5x105 cells/ml in 10% FBS-RPMI (Gibco; Thermo Fisher
Scientific, Inc.) medium at 37˚C in a humidified incubator supplied
with 5% CO2. In all experiments, THP-1 cells were
transformed into adherent macrophages in complete medium treated
with 200 ng/ml phorbol12-myristate 13-acetate (PMA)
(MilliporeSigma) for 24 h.
Isolation of peritoneal macrophages
(PMs)
PMs were harvested from mice injected
intraperitoneally with 3% Brewer's thioglycollate medium (Kingmore
Biotech Co., Ltd.). After 3 days, mice were sacrificed and their
abdominal cavities were flushed 2-3 times with DMEM (Gibco; Thermo
Fisher Scientific, Inc.) to obtain primary murine PMs, which were
cultured in complete DMEM supplemented with 10% FBS and 1%
penicillin/streptomycin (100 U/ml).
Isolation of bone marrow-derived
macrophages (BMDMs)
BMDMs were obtained from the tibia and femur of
C57BL/6 mice and cultured in complete DMEM supplemented with
granulocyte-macrophage (GM)-CSF (10 ng/ml; PeproTech, Inc.). On day
4, half of the medium was replaced with supplemented DMEM
containing GM-CSF (10 ng/ml). On day 7, BMDMs were harvested and
plated at a density of 2x106 cells/ml, and when the
cells had adhered and grown confluent, they were used for
subsequent experiments.
Cell stimulation
For inflammatory response analysis, cells were
stimulated with LPS (500 ng/ml) to induce an inflammatory state
with or without APCP (20 nm/ml) treatment after 6 h. For macrophage
polarization, cells were induced with LPS (200 ng/ml; PeproTech,
Inc.) and IFN-γ (10 ng/ml; PeproTech, Inc.) treatment to induce M1
polarization or with IL-13 (20 ng/ml; PeproTech, Inc.) and IL-4 (20
ng/ml; PeproTech, Inc.) treatment to induce M2 polarization in the
medium for the indicated lengths of times.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from cells or tissues using
TRIzol® reagent, according to the manufacturer's
protocol (Ambion; Thermo Fisher Scientific, Inc.). RNA quantity and
quality were assessed using a NanoVue spectrophotometer (GE
Healthcare Life Sciences), with a 260/280 ratio of 1.8 to 2.0
considered suitable. All primers were synthesized by ShengGong
BioTeck, and the sequences are shown in Table I. Total RNA was reverse transcribed
into cDNA using a 5x All-In-One RT MasterMix kit (ABM) according to
the manufacturer's protocol. mRNA expression was measured using a
fluorescence qPCR kit (UltraSYBR Mixture; CW Biosciences),
according to the manufacturer's instructions, on an iQ SYBR Green
on a CFX96 Real-Time System (Bio-Rad). The following cycling
conditions were used: 95˚C for 10 min, 95˚C for sec, and 60˚C for
30 sec, at which point, the plate was read. This was followed by 40
cycles of 95˚C for 1 min, 55˚C for 1 min, 55.0 to 95.0˚C in
increments of 0.5˚C for 10 sec which included a plate read to
obtain the melt curve. Data were analyzed using the
2-ΔΔCq method (20).
GAPDH was used as the housekeeping gene.
 | Table ISequences of the primers used for
quantitative PCR. |
Table I
Sequences of the primers used for
quantitative PCR.
| Gene | Species | Forward, 5'-3' | Reverse, 5'-3' |
|---|
| GAPDH | Human |
GGAGCCAAAAGGGTCATCATCT |
GAGGAGCCATCCACAGTCTTCT |
| CD73 | Human |
GCCTGGGAGCTTACGATTTTG |
TAGTGCCCTGGTACTGGTCG |
| GAPDH | Mouse |
AGGTCGGTGTGAACGGATTTG |
TGTAGACCATGTAGTTGAGGTCA |
| TGF-β | Mouse |
CTCCCGTGGCTTCTAGTGC |
GCCTTAGTTTGGACAGGATCTG |
| IL-10 | Mouse |
CTTACTGACTGGCATGAGGATCA |
GCAGCTCTAGGAGCATGTGG |
| CD206 | Mouse |
CTCTGTTCAGCTATTGGACGC |
CGGAATTTCTGGGATTCAGCTTC |
| Fizz-1 | Mouse |
CCAATCCAGCTAACTATCCCTCC |
CCAGTCAACGAGTAAGCACAG |
| IL-6 | Mouse |
TAGTCCTTCCTACCCCAATTTCC |
TTGGTCCTTAGCCACTCCTTC |
| IL-1β | Mouse |
GCAACTGTTCCTGAACTCAACT |
ATCTTTTGGGGTCCGTCAACT |
| INOS | Mouse |
GTTCTCAGCCCAACAATACAAGA |
GTGGACGGGTCGATGTCAC |
| TNF-α | Mouse |
CCCTCACACTCAGATCATCTTCT |
GCTACGACGTGGGCTACAG |
Immunohistochemistry (IHC)
Colonic biopsy samples from patients with UC and
healthy donors were fixed and embedded to determine CD73
expression. The sections were incubated with Envision flex
peroxidase-blocking reagent for 10 min at room temperature,
followed by incubation with a rabbit anti-human CD73 antibody
(1:200; cat. no. ab133582; Abcam) at 4˚C overnight. After washing
with PBS, the sections were incubated with conjugated anti-rabbit
antibodies (1:150; cat. no. ab170902; Abcam) for 60 min at 4˚C. The
color was developed using 3,3'-diaminobenzidine, and the sections
were counterstained with hematoxylin (1:20) at room temperature.
Observation of CD73+ cells was performed using a light
microscope (magnification, x5 and x200).
Western blot analysis
Cells were lysed using RIPA lysis buffer containing
protease inhibitors (cat. no. P0013B; Beyotime Institute of
Biotechnology). Cell lysates were quantified using a BCA assay,
followed by separation using 10% SDS-PAGE. The amount of protein
loaded per lane was 10 µl. Subsequently, proteins were transferred
to PVDF membranes (MilliporeSigma) for 1 h at 100 V. To block
non-specific binding sites, the membranes were placed in 3% BSA for
at least 1 h at room temperature and then incubated with primary
antibodies against p65 (1:2,000; cat. no. 8242T; Cell Signaling
Technology, Inc.), p-p65 (1:2,000; cat. no. 3033S; Cell Signaling
Technology, Inc.), ERK (1:2,000; cat. no. 4695T; Cell Signaling
Technology, Inc.), p-ERK (1:2,000; cat. no. 4370T; Cell Signaling
Technology, Inc.) or GAPDH (1:5,000; cat. no. D16H11; Cell
Signaling Technology, Inc.) at 4˚C overnight. After the incubation
with HRP-labeled goat anti-rabbit/mouse secondary antibodies
(1:3,000; cat. no. AS014/ZB-2305; Beyotime Institute of
Biotechnology) for at least 1 h at 4˚C, blots were analyzed using
an ECL kit (Thermo Fisher Scientific, Inc.) to detect the target
proteins. GAPDH was used as the internal control. Densitometry
analysis was performed using ImageJ (1.x; National Institutes of
Health).
Statistical Analyses
Data are presented as the mean ± SEM and were
analyzed using an unpaired Student's t-test or one-way ANOVA with a
Tukey's post hoc test. Spearman's correlation analysis was
performed to determine the correlation between CD73 expression and
UCEIS. Statistical analyses were performed using SPSS software
v21.0 (IBM Corp.). P<0.05 was considered to indicate a
statistically significant difference.
Results
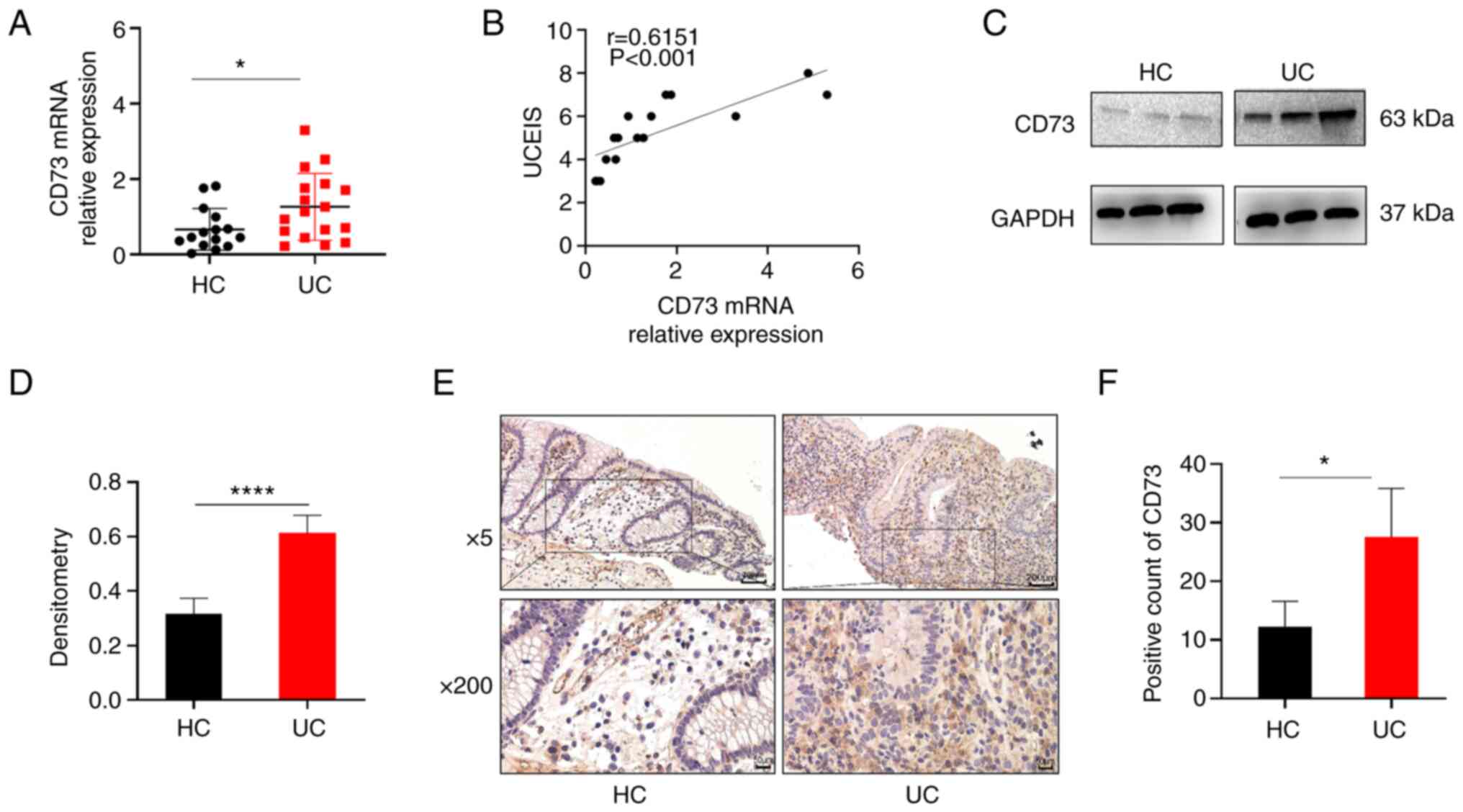
CD73 expression is increased in the
inflamed mucosa of patients with UC
Based on previous studies, CD73 has been reported to
participate in the pathogenesis of several immune diseases
(10,15). However, the role of CD73 in UC
remains unclear. Hence, inflamed mucosa samples from patients with
UC and healthy controls were obtained, and it was found that CD73
expression was notably increased in the inflamed mucosa of UC
patients compared with that in the healthy controls using RT-qPCR
(Fig. 1A). Furthermore, it was
hypothesized that CD73 expression may be associated with UC disease
activity. Thus, the correlation between CD73 expression and UCEIS,
the international standard criteria for assessing endoscopic
disease activity in patients with UC, was determined. As shown in
Fig. 1B, CD73 expression was
positively correlated with UCEIS in UC (r=0.6151, P<0.001).
Western blot analysis further indicated that CD73
protein expression was significantly increased in the mucosa of
patients with UC compared with that in healthy controls and this
increase was statistically significant (Fig. 1C and D). In addition, IHC staining showed that
the inflamed mucosa of patients with UC contained a significantly
higher number of CD73-positive cells than that in the healthy
controls (Fig. 1E and F). Together, these data indicate that
CD73 expression is upregulated in the inflamed tissues of patients
with UC and may therefore play a crucial role in UC
pathogenesis.
CD73 blockade reduces proinflammatory
cytokines levels and increases anti-inflammatory cytokine levels in
mouse macrophages
The differentiation of inflammatory monocytes into
pro-degradative macrophages during chronic intestinal inflammation
induces UC and the secretion of large amounts of pro-inflammatory
factors (21). To determine
whether CD73 affects colitis by promoting the pro-inflammatory
response induced by LPS in macrophages, mouse PMs were isolated and
cultured from wild-type mice (Fig.
2A).
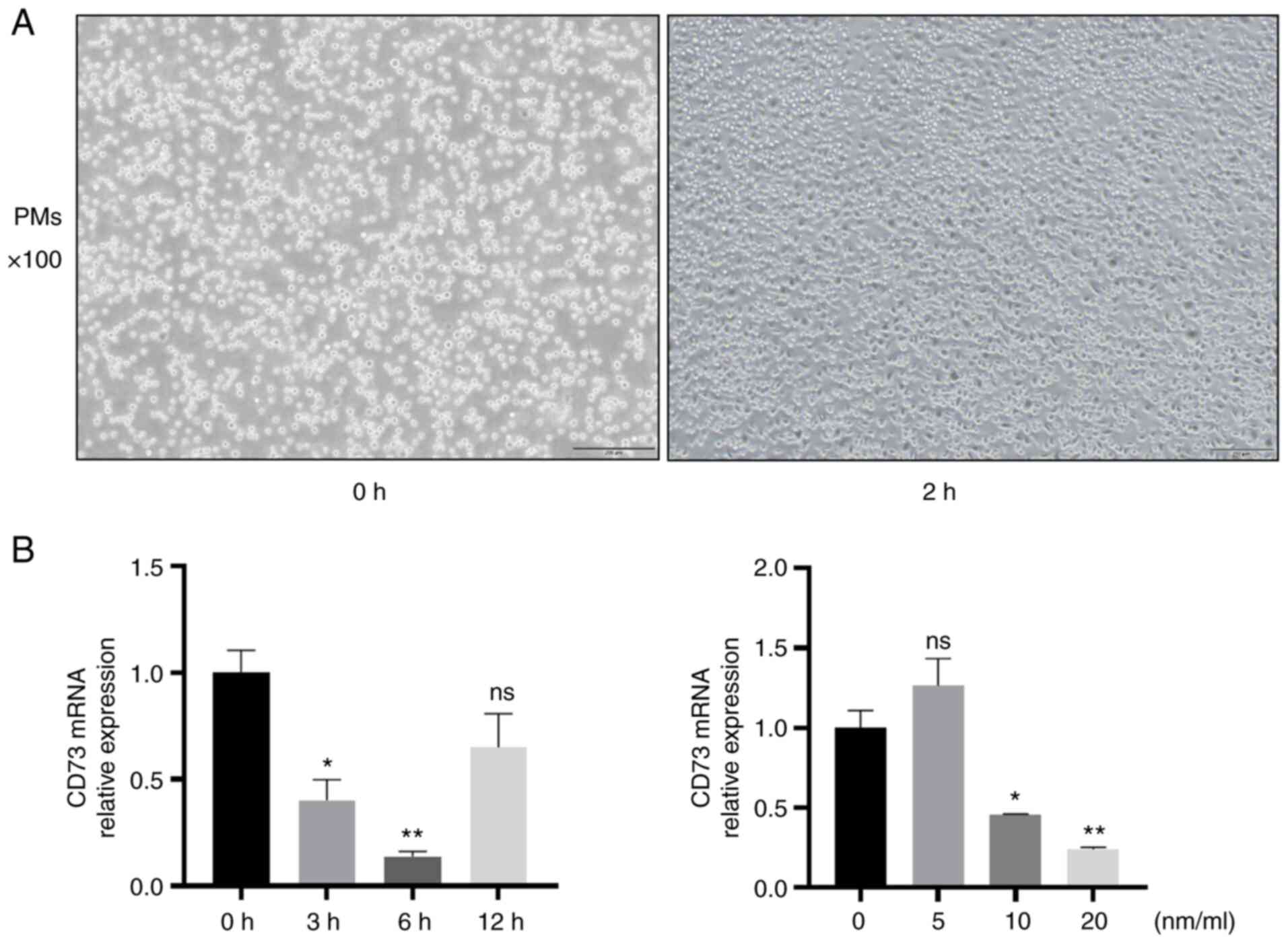 | Figure 2Determination of the optimum
concentration and time of APCP treatment. (A) Images of PMs.
Magnification, x100; scale bar, 200 µm. (B) PMs were treated with
APCP for 0, 3, 6, or 12 h with 0, 5, 10, or 20 nm/ml. The
expression of mRNA CD73 was assessed (n=3). *P<0.05
and **P<0.01. ns, not significant; PM, peritoneal
macrophage; APCP, adenosine 5'-(α, β-methylene) diphosphate. |
Firstly, the expression of CD73 was determined in M0
macrophages stimulated with different concentrations of APCP for
different lengths of time to confirm the optimum concentration and
stimulation time of APCP to inhibit the expression of CD73. The
expression of CD73 in PMs was significantly inhibited by APCP
(Fig. 2B). As shown in the
results, the optimal time was 6 h and the optimal concentration was
20 nm/ml. Next, PMs were treated with LPS to induce an inflammatory
state in the cells. The mRNA expression levels of
macrophage-related inflammatory cytokines obtained from PMs treated
with or without APCP after LPS administration were determined. As
shown in Fig. 3A, RT-qPCR analysis
showed that APCP significantly reduced IL-6, iNOS, and IL-1β levels
in LPS-stimulated PMs, but increased the expression of CD206, a
characteristic anti-inflammatory cytokine in M2 macrophages
(Fig. 3B). Thus, CD73 appears to
affect the expression of the inflammatory mediators related with
macrophages to promote an inflammatory reaction.
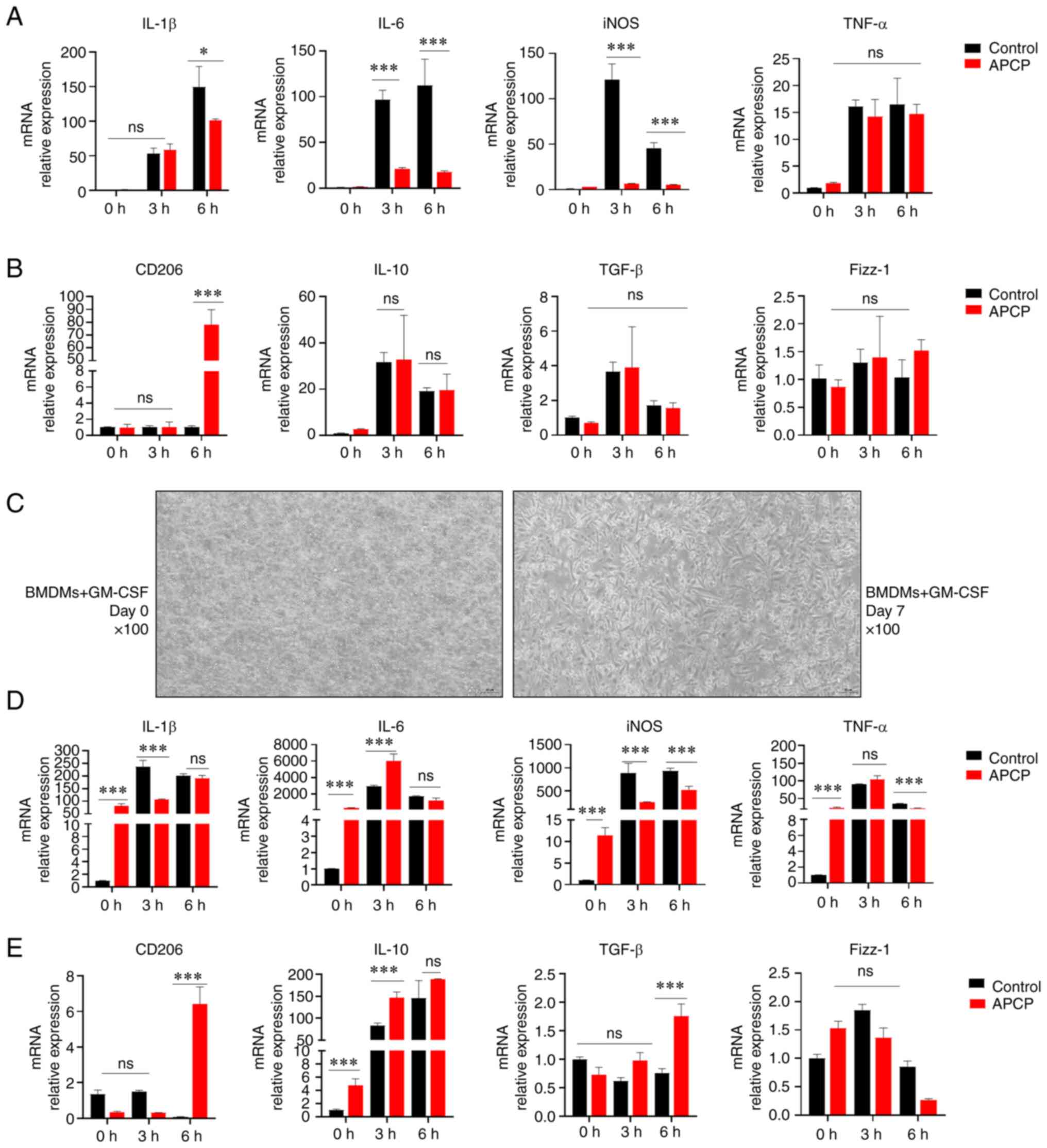 | Figure 3Blocking of CD73 suppresses the
production of proinflammatory cytokines and promotes the production
of anti-inflammatory cytokines. (A) mRNA expression of IL-1β, IL-6,
iNOS, and TNF-α in PMs treated with or without APCP after LPS (500
ng/ml) stimulation after 0, 3, and 6 h (n=3). (B) Gene expression
of CD206, IL-10, TGF-β, and Fizz-1 in PMs treated with or without
APCP after LPS (500 ng/ml) stimulation after 0, 3, and 6 h (n=3).
(C) Images of treated BMDMs (magnification, x100; scale bar, 50
µm). (D) Gene expression of IL-1β, IL-6, iNOS, and TNF-α in BMDMs
treated with or without APCP after LPS (500 ng/ml) stimulation
after 0, 3, and 6 h (n=3). (E) Gene expression of CD206, IL-10,
TGF-β, and Fizz-1 in BMDMs treated with or without APCP after LPS
(500 ng/ml) stimulation after 0, 3, and 6 h (n=3). Gene expression
was normalized to GAPDH in each group. *P<0.05 and
***P<0.001. ns, not significant; PM, peritoneal
macrophage; APCP, adenosine 5'-(α, β-methylene) diphosphate; BMDM,
bone marrow-derived macrophage; iNOS, inducible nitric oxide
synthase; GM-CSF, granulocyte-macrophage-colony stimulating
factor. |
It has been suggested that PMs have distinct
heterogeneities, and yet BMDMs are the most homogeneous due to
macrophage CSF (M-CSF) induction (22). To avoid the variation in
polarization caused by the induction of macrophages from different
sources, our experimental hypothesis was verified using BMDMs
(Fig. 3C). The expression of
related inflammatory mediators in BMDMs was consistent with that in
PMs. The mRNA levels of proinflammatory factors, such as IL-6,
IL-1β, and iNOS, were lower in the APCP-treated groups than those
in the control groups; however, the expression of anti-inflammatory
factors such as CD206, IL-10, and TGF-β was higher in the
APCP-treated groups (Fig. 3D-E).
Interestingly, the effect of CD73 on BMDMs towards M2 macrophage
polarization was greater than that on PMs in an inflammatory state.
Taken together, these data suggested that CD73 blockade by APCP
significantly inhibited the pro-inflammatory responses induced by
LPS in macrophages.
CD73 blockade promotes M2 macrophage
polarization
To explore the effect of CD73 on the polarization of
intestinal macrophages, the expression of several genes related to
M1 and M2 macrophages was detected by RT-qPCR. M0 macrophages were
induced towards M1 polarization with LPS and IFN-γ treatment, or
towards M2 polarization using IL-4 and IL-13 treatment. The
expression of M1 macrophage markers, such as IL-1β and iNOS, was
decreased in the groups treated with LPS and IFN-γ. However, this
was not statistically significant (Fig. 4A). In contrast, the inhibition of
CD73 measurably increased the percentage of CD206, TGF-β, Fizz-1,
and IL-10 in mouse PMs treated with IL-4 and IL-13 (Fig. 4B), which are the characteristic
markers of M2 macrophages. Furthermore, these results additionally
suggested that the blockade of CD73 could enhance polarization
towards M2 macrophages in regulating inflammatory responses.
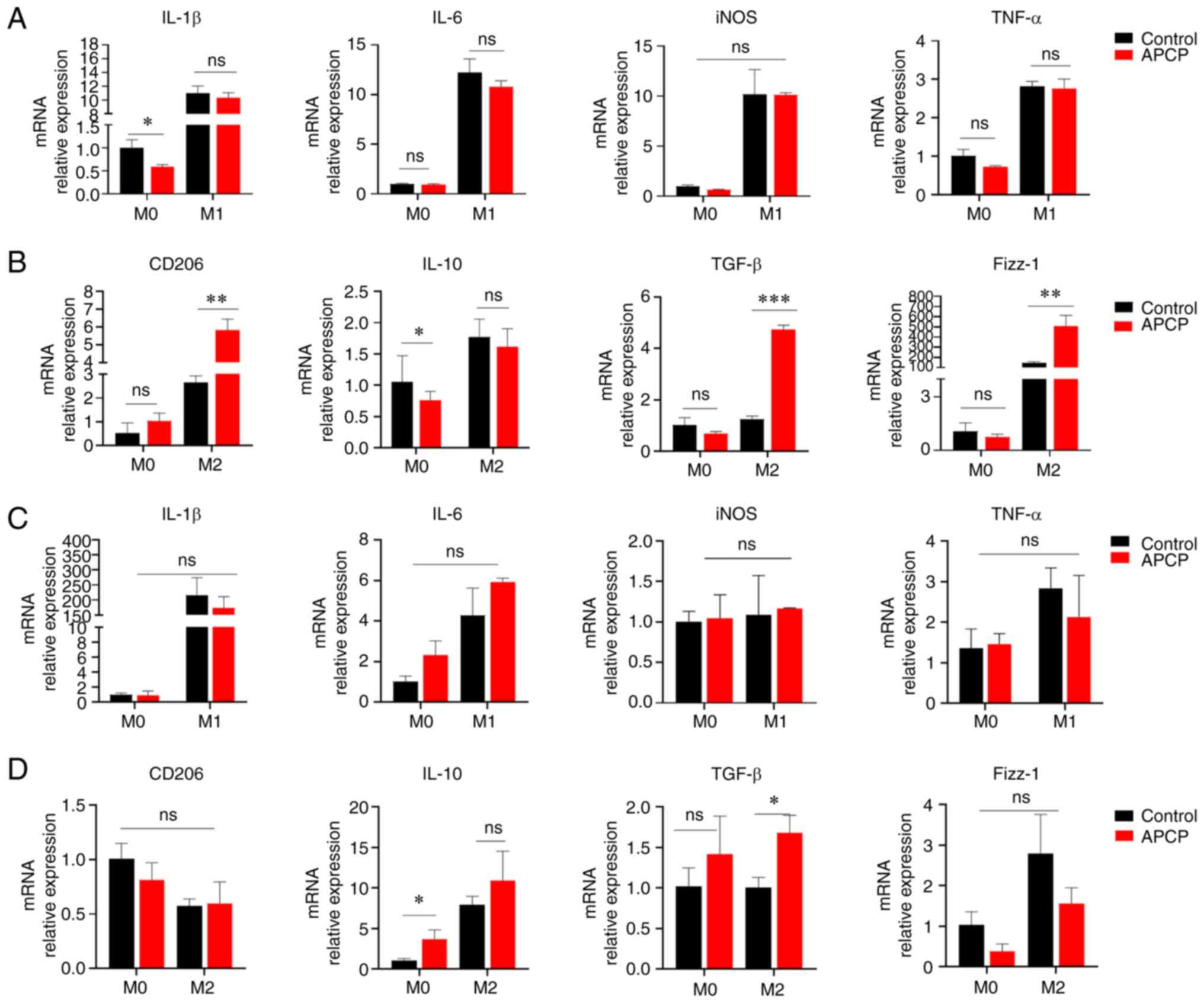 | Figure 4Blocking of CD73 increases M2
macrophage polarization. (A) The mRNA expression levels of IL-6,
TNF-α, IL-1β, and iNOS in PMs were determined after inducing
differentiation into M0, M1, or M2 macrophages for 72 h (n=3). (B)
The mRNA levels of TGF-β, Fizz-1, IL-10, and CD206 in PMs were
determined (n=3). (C) The mRNA levels of IL-6, TNF-α, IL-1β, and
iNOS in BMDMs were determined (n=3). (D) The mRNA levels of TGF-β,
Fizz-1, IL-10, and CD206 in BMDMs were determined (n=3).
*P<0.05, **P<0.01 and
***P<0.001. ns, not significant; BMDM, bone
marrow-derived macrophage; PM, peritoneal macrophage. |
To further confirm the findings, analogous
polarization experiments were conducted using BMDMs. Inhibition of
CD73 increased the expression of the characteristic markers of M2
macrophages and decreased the expression of M1 macrophage genes
(Fig. 4C and D). Interestingly, the suppression of M2
macrophage polarization induced by CD73 on BMDMs was not
significant compared to that in PMs. Therefore, these data
demonstrate that CD73 regulates LPS-induced pro-inflammatory
responses in macrophages, while suppressing M2-like macrophage
polarization.
CD73 blockade promotes
anti-inflammatory cytokine production and enhances M2 macrophage
polarization in THP1 cells
Next, whether CD73 had a similar effect on M2
macrophage polarization on human THP1 cells. As shown in Fig. 5A, THP1 cells were pulsed with PMA
to induce macrophage differentiation. Consistently, THP1
macrophages expressed high levels of the M2 markers such as CD206
upon LPS treatment (Fig. 5B).
Moreover, THP1 macrophages expressed upregulated levels of the M2
marker Fizz-1 upon culture with IL-4 and IL-13 (Fig. 5C). Collectively, these results
further indicated that the blockade of CD73 increased M2 macrophage
polarization in human macrophage cells, thereby exerting
anti-inflammatory effects.
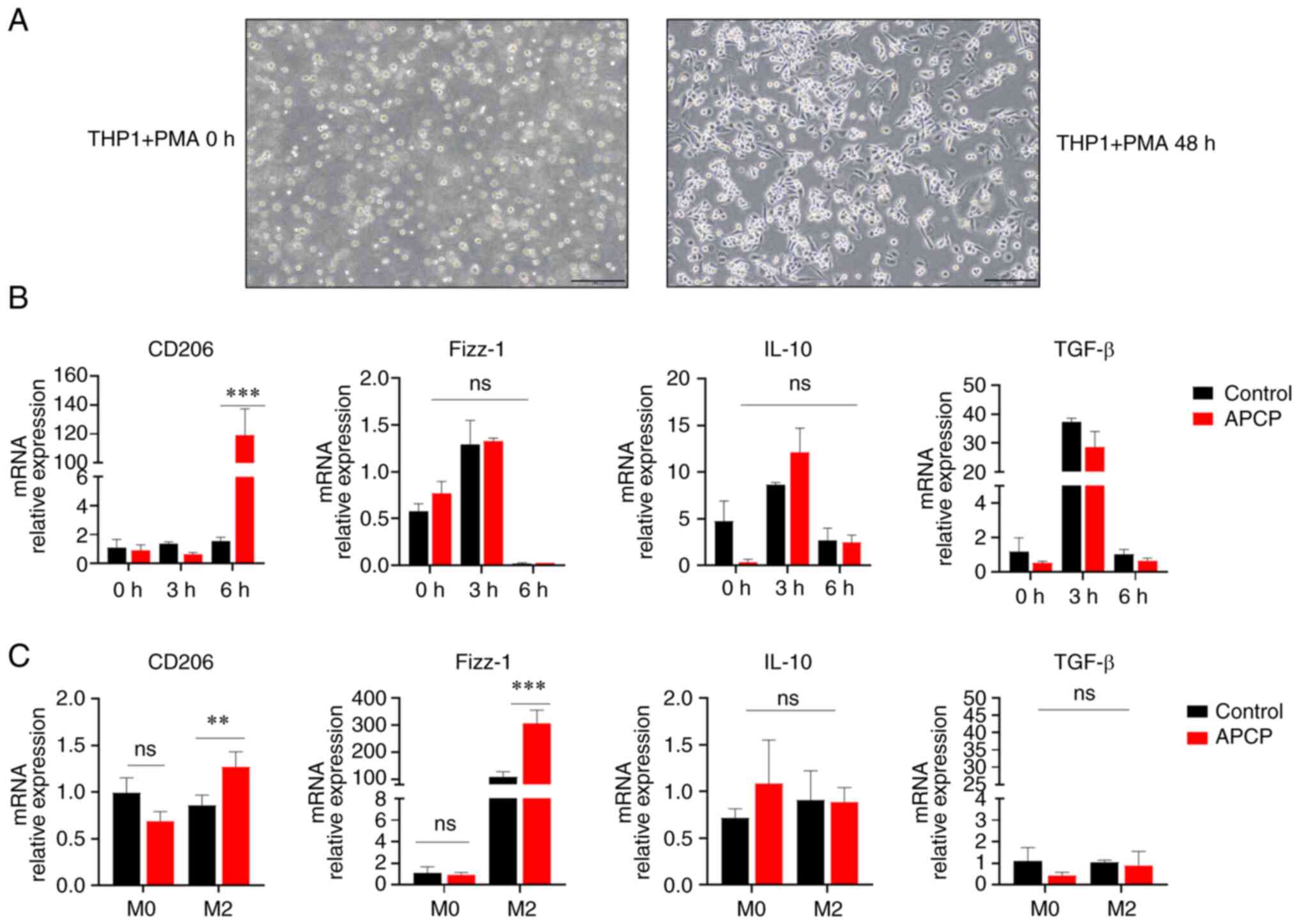 | Figure 5Blocking of CD73 promotes the
production of anti-inflammatory cytokines and enhances M2
macrophage polarization in THP1 cells. (A) Images of THP1 cells
treated with PMA. Magnification, x100; scale bar, 200 µm. (B) mRNA
expression of CD206, IL-10, TGF-β, and Fizz-1 in THP1 following
stimulation with LPS (500 ng/ml) for 0, 3, or 6 h following
treatment with APCP (20 nmol/ml) (n=3). (C) The mRNA levels of
TGF-β, Fizz-1, IL-10, and CD206 in BMDMs were determined after IL-4
and IL-13-induced differentiation into M2 macrophages for 72 h
(n=3). *P<0.01 and ***P<0.001. ns, not
significant; BMDM, bone marrow-derived macrophage; PMA,
phorbol12-myristate 13-acetate. |
CD73 blockade alleviates DSS-induced
colitis in mice
To verify the effect of CD73 on mucosal
inflammation, the changes in intestinal inflammation after blocking
of CD73 in an animal model of DSS-induced colitis was assessed
(Fig. 6A). DSS-treated and
non-DSS-treated mice were injected intraperitoneally with APCP
daily; PBS was additionally used as a negative control. Clinical
disease severity was scored based on weight loss, diarrhea, and
bleeding. Mice that were treated with APCP exhibited a significant
increase in body weight compared with the negative control-DSS
group (Fig. 6B). Treatment with
DSS resulted in severe diarrhea and bleeding, and mice in the
negative control DSS group had a shortened colonic length, severe
diarrhea, and more bleeding than those treated with APCP (Fig. 6C). It has been previously that
histological inflammation in UC patients is correlated with
endoscopic inflammation of the ileal pouch, reflecting the severity
of colonic inflammation to some extent (23). Following DSS treatment, the
intestine produced remarkable inflammatory responses, primarily
manifesting as inflammatory cell infiltration, loss of cup cells,
crypt loss, crypt abscess formation, and reactive epithelial
hyperplasia. Importantly, mice treated with APCP exhibited a
significantly alleviated inflammatory reaction, compared with those
treated with PBS, as demonstrated by hematoxylin and eosin
histopathology analysis (Fig. 6D).
These data demonstrate that the blocking of CD73 reduces
inflammation in the DSS-induced colitis model.
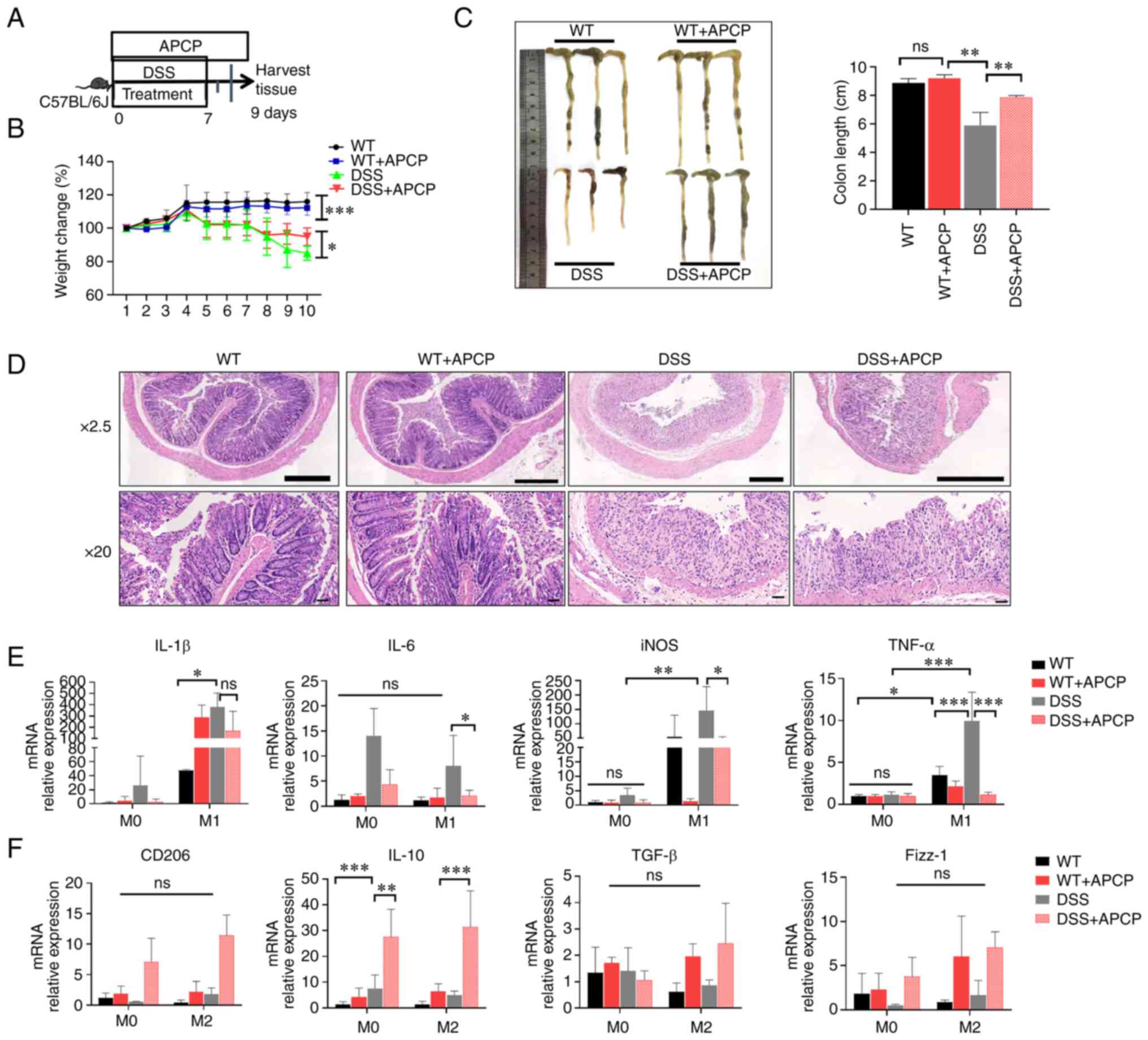 | Figure 6Blocking of CD73 alleviates
DSS-induced colitis in mice. (A) Flow diagram of the establishment
of the mouse model. (B) Changes in body weight were observed and
are presented as a percentage of the initial body weight at the
start of experiments. (C) Extracted colons and statistical analysis
of the length of colons 9 days after DSS induction. (D) H&E
biopsies of colon tissues in the different groups. Magnification,
x2.5 and x20; scale bar, 50 µm. (E) The mRNA levels of IL-6, TNF-α,
IL-1β, and iNOS in BMDMs from DSS-induced colitis mice. (F) The
mRNA levels of TGF-β, Fizz-1, IL-10, and CD206 in BMDMs from
DSS-induced colitis mice. *P<0.05,
**P<0.01, ***P<0.001. ns, not
significant; H&E, hematoxylin and eosin; DSS, dextran sulfate
sodium; BMDM, bone marrow-derived macrophage. |
Furthermore, the production of macrophage-associated
inflammatory cytokines was also examined in the BMDMs using
RT-qPCR. The levels of IL-1β, TNF-a, iNOS, and IL-6 were
significantly decreased in M1 macrophages of mice that received
APCP treatment compared with the mice that did not receive APCP.
Importantly, there was no significant difference in M0 macrophages
that did undergo LPS-induced polarization (Fig. 6E). Interestingly, the expression of
M2-associated inflammatory factors was elevated in both M0 and M2
macrophages in the APCP-treated mice. However, only the elevated
levels of IL-10 were statistically significant, while CD206, TGF-β,
and Fizz-1 were not significantly differentially expressed. Thus,
CD73 affected the differentiation of macrophages in vivo,
and also promoted intestinal inflammation by promoting M1
macrophage polarization and inhibiting M2 polarization.
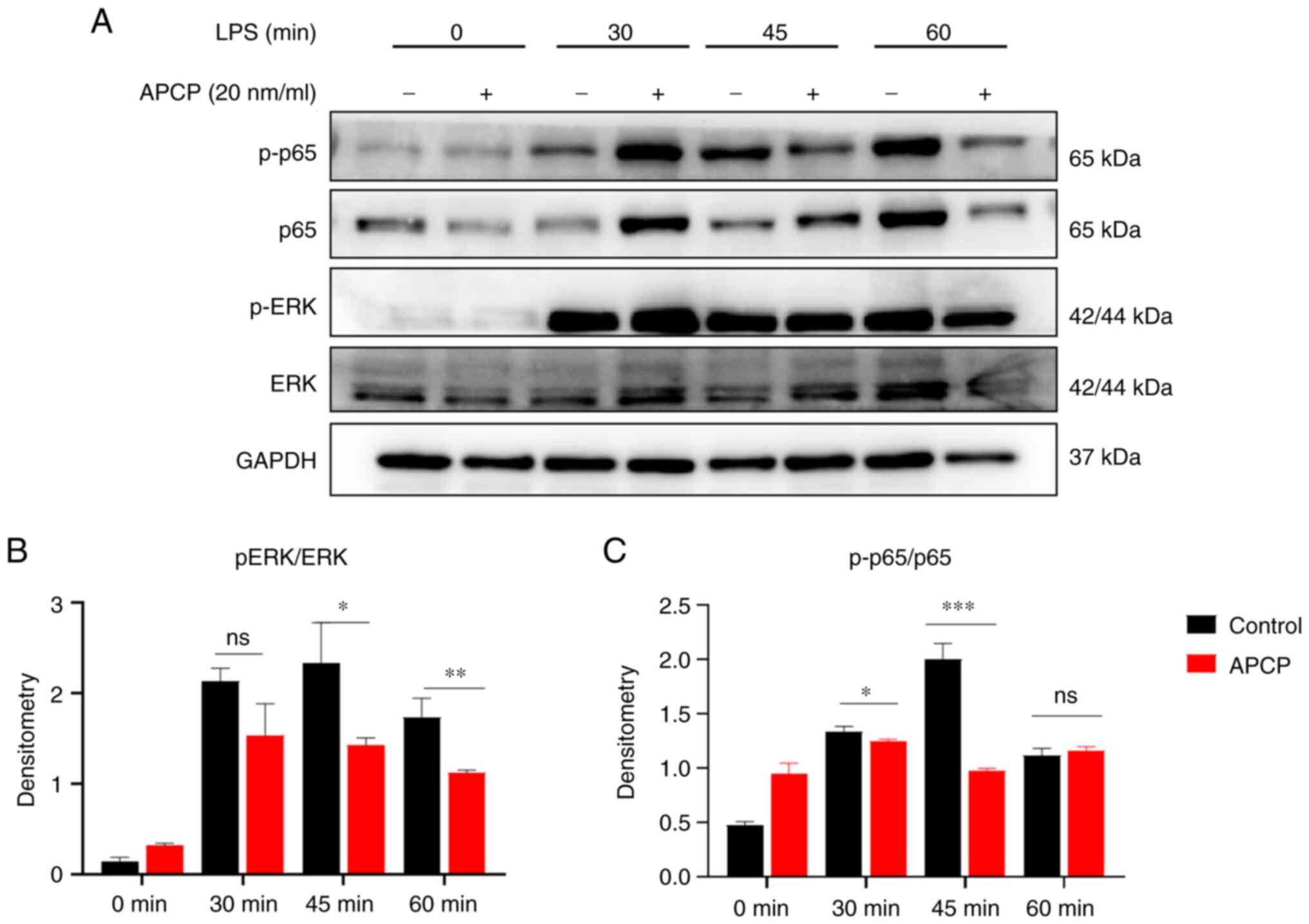
CD73 regulates macrophage polarization
via inhibition of p-p65 and ERK phosphorylation
To determine the mechanism underlying the effects of
CD73 in colitis, the signaling pathways closely related to
macrophage polarization related to CD73 were assessed.
Mechanistically, previous studies have reported that CD73 promotes
optimal TLR-mediated response in tumor cells (24). LPS activates TLR4 to activate the
NF-kB and ERK signaling pathways (25). Thus, PMs were treated with LPS for
lengths of time and the levels of p-p65 were assessed. The results
showed that there were significantly detectable differences between
the control and CD73-inhibited macrophages. Additionally,
inhibition of CD73 increased the expression levels of several
proteins in The ERK signaling pathway, including p-p65 and ERK
(Fig. 7A). Subsequently, the ratio
of phosphorylated proteins to the respective non-phosphorylated
proteins in these pathways was assessed to further confirm that the
p-p65 and ERK pathways were inhibited after CD73 blockade (Fig. 7B and C). Taken together, these data indicate
that CD73 regulates LPS-induced pro-inflammatory responses in
macrophages by modulating the activation of the NF-kB and ERK
signaling pathways.
Discussion
UC is a multifactorial chronic disease prone to
relapse that is characterized by abnormal systemic and local
dysregulation of the mucosal immune response (26). Immune modulation is necessary to
reduce the immune response and limit immunopathology following an
intestinal infection or inflammation, which would otherwise cause
the accumulation of large quantities of immune cells (12). Our understanding of the etiology
and complexity of immune regulation in UC is still relatively
limited (3,27). Although it has been shown that CD73
acts as an immunological factor that dampens the antitumor
T-cell-mediated immune response, the investigation of CD73 function
in UC is limited. The expression in CD73 on immune cells is
species-specific (12). It is
expressed on most immune cells including B cells, T cell subsets
such as Tregs, and NK cells (28,29).
Given the immunoregulatory abilities of CD73 in the immune
response, it was hypothesized that CD73 may play an important part
in IBD-associated macrophages by regulating pathways related to
inflammation and immunity. In the present study, it was found that
CD73 expression was upregulated in the inflamed mucosa of patients
with UC, and that increased CD73 expression was positively
correlated with disease activity and UCEIS. Importantly, colitis in
mice was significantly alleviated after inhibition of CD73 with
APCP. These data indicate that CD73 may play an important role in
UC pathogenesis by regulating the immune response.
Macrophages, the key innate immune system cells,
play important roles in several diseases, including IBD (2,30).
Previous studies have demonstrated that LPS enhances M1
polarization, whereas IL13 and IL4 are crucial for polarization
towards M2-associated genes (30).
However, the mechanism underlying M1/M2-switch regulation
associated with UC development remains to be elucidated.
Classically activated M1-like macrophages are involved in
initiating and maintaining inflammation, whereas alternatively
activated M2-like macrophages are associated with inflammation
regression (31). M1-like
macrophages produce a wide range of proinflammatory cytokines,
including TNF-a, IL-6, and iNOS (32), while M2-like macrophages produce
large quantities of anti-inflammatory cytokines, including IL-10,
TGF-β, and CD206, which can suppress intestinal inflammation
(33). Macrophages with increased
M1 polarization and decreased M2 polarization are typically
observed in the colon tissues of UC patients, and an M1/M2
imbalance is associated with the pathogenesis of UC (34). In the present study, the production
of M1 and M2-related cytokines during inflammation was assessed,
and the results showed that CD73 blockade reduced LPS-induced
proinflammatory cytokine expression and increased the levels of
markers of M2 macrophages, confirming that CD73 inhibited M2
macrophage activation while promoting M1 macrophage activation in
intestinal mucosal inflammation of UC.
It has been shown that CD73 expression on tumor
cells sufficiently facilitates tumor growth and metastasis by
generating extracellular adenosine to mediate immune evasion and
promote colitis-associated tumorigenesis in mice (15). The activity of CD73 regulates
macrophage function by converting M2 to M1 phenotypes. A growing
body of clinical evidence and research data supports the
involvement of purinergic signaling in the pathogenesis of IBD
through inflammatory responses and modulation of bacterial
alterations (35,36). Thus, how CD73 regulated intestinal
inflammation in vivo was assessed. It was found that CD73
blockade by APCP could markedly alleviate DSS-induced colitis in
mice. Moreover, CD73 blockade significantly reduced the quantities
of inflammatory cytokines in BMDMs from DSS-induced colitis mice,
which was consistent with the results in vitro. However,
M1-related pro-inflammatory factors were significantly suppressed
in mice injected intraperitoneally with APCP, while M2-related
anti-inflammatory factors were not significantly elevated. It is
hypothesized that the reason for this condition is the inflammatory
state developed in mice by DSS.
One limitation of the present study was that a
selective inhibitor rather than CD73 knockout mice was used.
CD73-knockout mice are being established to further validate the
results of the present study. Additionally, this study only
investigated the inflammatory effect of CD73 on DSS-induced colitis
in mice, and the study of other types of colitis should be
assessed. It is hypothesized that CD73 knockout mice could be used
to investigate the effects of CD73 on colonic inflammation and the
bacterial flora in the Trinitro-benzene-sulfonic acid colitis model
and the Citrobacter Rodentium colitis model (37,38).
A previous study showed that the efficacy of organoid
transplantation on the treatment of intestinal inflammation in
experimental colitis models is relatively mature (39); thus organoid transplantation
technology may be useful for further investigation of the mechanism
of CD73 action on the intestinal epithelium in UC at a later
stage.
Excessive NF-κB activation exacerbates the severity
of intestinal inflammation in UC patients (40). As such, NF-κB blockade has become a
promising therapeutic strategy for the management of UC (41). The phosphorylation of p65, which
initiates the activation of NF-κB, is a key regulator of the NF-κB
pathway (42), which regulates
multiple aspects of innate and adaptive immunity. Canonical NF-κB
induces the production of proinflammatory cytokines, such as TNF-α
and IL-1β in the innate immune system, resulting in an inflammatory
response (43). Interestingly, it
was shown that CD73 blockade significantly inhibited p-p65 and p65
expression, suggesting that CD73 may exert immune effects in
inflammation via NF-κB activation. ERK is a serine/threonine
protein kinase that mediates fundamental biological processes and
cellular responses to external stress signals, and also regulates
the synthesis of inflammation and apoptosis mediators (44). Accumulating evidence has revealed
that ERK also regulates the transcriptional activity of
NF-κB-p65(45). Thus, the protein
expression levels of p-ERK and ERK were assessed following CD73
inhibition. In agreement with the p-p65 results, the blockade of
CD73 also decreased the expression of the p-ERK and ERK. These data
suggested that CD73 exhibited pro-inflammatory effects via
activation of NF-κB p65 and ERK. Together, it was shown that CD73
expression regulates the intestinal inflammatory response through
the regulation of the clinical phenotype and inflammatory mediator
mechanisms of macrophages.
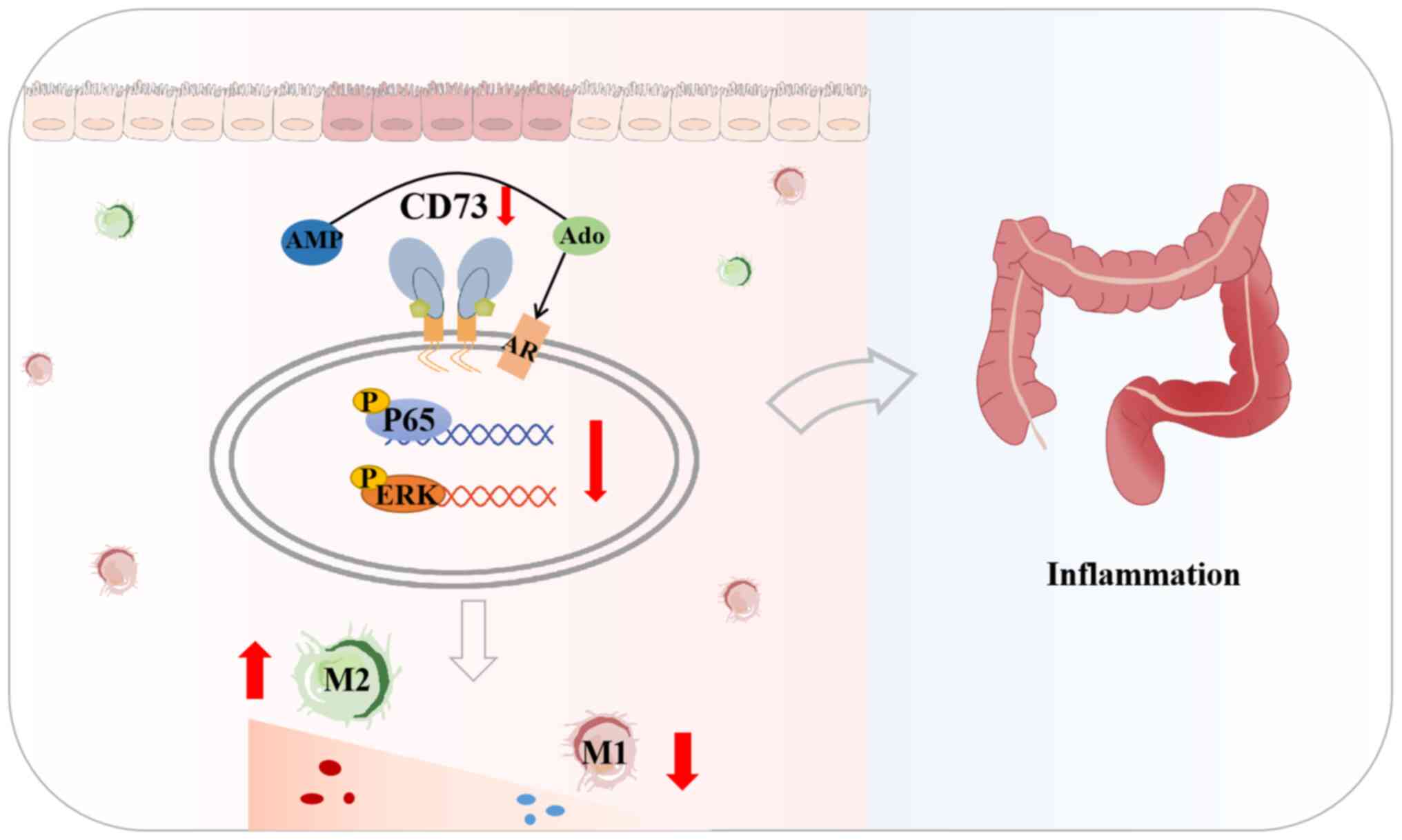
In conclusion, the results of the present study
showed that CD73 regulates the intestinal inflammatory immune
response by switching M1/M2 macrophage polarization as well as the
expression of related inflammatory factors via NF-κB and ERK
signaling, thereby exerting proinflammatory effects (Fig. 8). Therefore, CD73 may be considered
as a diagnostic biomarker and drug target for the treatment of
inflammation-related diseases. Targeting CD73 in the appropriate
cell types should be considered as a potential treatment of
inflammatory diseases such as UC.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by grants from the Tai Shan
Young Scholar Foundation of Shandong Province (grant no.
tsqn202103190), the National Natural Science Foundation of China
(grant nos. 82270562, 82200591, and 81901655), and the Key research
and development plan of Jining City (grant nos. 2021YXNS045, and
2021YXNS144).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
RW and GZ conceived and designed the study. CW and
RW performed the experiments. YiW, YaW contributed to acquisition
of the clinical data and specimens. FZ, RW, YaW and YY analyzed the
data. GJ, YY, YiW and RW helped to interpret the data, and write
and review the manuscript. GJ, GZ and YY confirm the authenticity
of all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was reviewed and approved by the
Ethics Committee of Jining Medical University (approval no.
2021-09-C002; Jining, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Larabi A, Barnich N and Nguyen HTT: New
insights into the interplay between autophagy, gut microbiota and
inflammatory responses in IBD. Autophagy. 16:38–51. 2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Na YR, Stakenborg M, Seok SH and Matteoli
G: Macrophages in intestinal inflammation and resolution: A
potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol.
16:531–543. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Schett G and Neurath MF: Resolution of
chronic inflammatory disease: Universal and tissue-specific
concepts. Nat Commun. 9(3261)2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Matheis F, Muller PA, Graves CL, Gabanyi
I, Kerner ZJ, Costa-Borges D, Ahrends T, Rosenstiel P and Mucida D:
Adrenergic signaling in muscularis macrophages limits
infection-induced neuronal loss. Cell. 180:64–78 e16.
2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lu H, Zhang C, Wu W, et al: MCPIP1
restrains mucosal inflammation by orchestrating the intestinal
monocyte to macrophage maturation via an ATF3-AP1S2 axis. Gut.
72:882–895. 2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mortha A, Chudnovskiy A, Hashimoto D,
Bogunovic M, Spencer SP, Belkaid Y and Merad M:
Microbiota-dependent crosstalk between macrophages and ILC3
promotes intestinal homeostasis. Science.
343(1249288)2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chistiakov DA, Myasoedova VA, Revin VV,
Orekhov AN and Bobryshev YV: The impact of interferon-regulatory
factors to macrophage differentiation and polarization into M1 and
M2. Immunobiology. 223:101–111. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Lissner D, Schumann M, Batra A, Kredel LI,
Kühl AA, Erben U, May C, Schulzke JD and Siegmund B: Monocyte and
M1 macrophage-induced barrier defect contributes to chronic
intestinal inflammation in IBD. Inflamm Bowel Dis. 21:1297–1305.
2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Biswas A, Shouval DS, Griffith A, Goettel
JA, Field M, Kang YH, Konnikova L, Janssen E and Redhu NS:
WASP-mediated regulation of anti-inflammatory macrophages is IL-10
dependent and is critical for intestinal homeostasis. Nat Commun.
9(1779)2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Liu XH, Wu XR, Lan N, Zheng XB, Zhou C, Hu
T, Chen YF, Cai ZR, Chen ZX, Lan P and Wu XJ: CD73 promotes
colitis-associated tumorigenesis in mice. Oncol Lett. 20:1221–1230.
2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zarek PE, Huang CT, Lutz ER, Kowalski J,
Horton MR, Linden J, Drake CG and Powell JD: A2A receptor signaling
promotes peripheral tolerance by inducing T-cell anergy and the
generation of adaptive regulatory T cells. Blood. 111:251–259.
2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Schneider E, Rissiek A, Winzer R, Puig B,
Rissiek B, Haag F, Mittrücker HW, Magnus T and Tolosa E: Generation
and function of non-cell-bound CD73 in inflammation. Front Immunol.
10(1729)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Petruk N, Tuominen S, Akerfelt M, Mattsson
J, Sandholm J, Nees M, Yegutkin GG, Jukkola A, Tuomela J and
Selander KS: CD73 facilitates EMT progression and promotes lung
metastases in triple-negative breast cancer. Sci Rep.
11(6035)2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lopes DV, de Fraga Dias A, Silva LFL,
Scholl JN, Sévigny J, Battastini AMO and Figueiró F: Influence of
NSAIDs and methotrexate on CD73 expression and glioma cell growth.
Purinergic Signal. 17:273–284. 2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Roh M, Wainwright DA, Wu JD, Wan Y and
Zhang B: Targeting CD73 to augment cancer immunotherapy. Curr Opin
Pharmacol. 53:66–76. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Núñez FP, Krugliak Cleveland N, Quera R
and Rubin DT: Evolving role of endoscopy in inflammatory bowel
disease: Going beyond diagnosis. World J Gastroenterol.
27:2521–2530. 2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhang XF, Li P, Ding XL, Chen H, Wang SJ,
Jin SB, Guo J and Tian ZB: Comparing the clinical application
values of the Degree of Ulcerative Colitis Burden of Luminal
Inflammation (DUBLIN) score and Ulcerative Colitis Endoscopic Index
of Severity (UCEIS) in patients with ulcerative colitis.
Gastroenterol Rep (Oxf). 9:533–542. 2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chassaing B, Aitken JD, Malleshappa M and
Vijay-Kumar M: Dextran sulfate sodium (DSS)-induced colitis in
mice. Curr Protoc Immunol. 104:15.25.1–15.25.14. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yang R, Liao Y, Wang L, He P, Hu Y, Yuan
D, Wu Z and Sun X: Exosomes Derived From M2b Macrophages Attenuate
DSS-Induced Colitis. Front Immunol. 10(2346)2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Huang B, Chen Z, Geng L, Wang J, Liang H,
Cao Y, Chen H, Huang W, Su M, Wang H, et al: Mucosal profiling of
pediatric-onset colitis and IBD reveals common pathogenics and
therapeutic pathways. Cell. 179:1160–1176 e24. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhao YL, Tian PX, Han F, Zheng J, Xia XX,
Xue WJ, Ding XM and Ding CG: Comparison of the characteristics of
macrophages derived from murine spleen, peritoneal cavity, and bone
marrow. J Zhejiang Univ Sci B. 18:1055–1063. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Toritani K, Kimura H, Otani M, Fukuoka H,
Kunisaki R, Watanabe J, Ishibe A, Misumi T, Inayama Y and Endo I:
Inflammatory bowel disease-specific findings are common
morphological changes in the ileal pouch with ulcerative colitis.
Sci Rep. 12(20361)2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Liu B, Zheng Y, Yin F, Yu J, Silverman N
and Pan D: Toll receptor-mediated hippo signaling controls innate
immunity in Drosophila. Cell. 164:406–419. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ren Q, Guo F, Tao S, Huang R, Ma L and Fu
P: Flavonoid fisetin alleviates kidney inflammation and apoptosis
via inhibiting Src-mediated NF-κB p65 and MAPK signaling pathways
in septic AKI mice. Biomed Pharmacother. 122(109772)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Maloy KJ and Powrie F: Intestinal
homeostasis and its breakdown in inflammatory bowel disease.
Nature. 474:298–306. 2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Liu TC and Stappenbeck TS: Genetics and
pathogenesis of inflammatory bowel disease. Annu Rev Pathol.
11:127–148. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Raczkowski F, Rissiek A, Ricklefs I, Heiss
K, Schumacher V, Wundenberg K, Haag F, Koch-Nolte F, Tolosa E and
Mittrücker HW: CD39 is upregulated during activation of mouse and
human T cells and attenuates the immune response to Listeria
monocytogenes. PLoS One. 13(e0197151)2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Liang D, Zuo A, Zhao R, Shao H, Born WK,
O'Brien RL, Kaplan HJ and Sun D: CD73 Expressed on gammadelta T
cells shapes their regulatory effect in experimental autoimmune
uveitis. PLoS One. 11(e0150078)2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lawrence T and Natoli G: Transcriptional
regulation of macrophage polarization: Enabling diversity with
identity. Nat Rev Immunol. 11:750–761. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
31
|
Liu X, Ren X, Zhou L, Liu K, Deng L, Qing
Q, Li J, Zhi F and Li M: Tollip orchestrates macrophage
polarization to alleviate intestinal mucosal inflammation. J Crohns
Colitis. 16:1151–1167. 2022.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yao X, Dong G, Zhu Y, Yan F, Zhang H, Ma
Q, Fu X, Li X, Zhang Q, Zhang J, et al: Leukadherin-1-Mediated
Activation of CD11b Inhibits LPS-Induced pro-inflammatory response
in macrophages and protects mice against endotoxic shock by
blocking LPS-TLR4 interaction. Front Immunol.
10(215)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Shouval DS, Biswas A, Goettel JA, McCann
K, Conaway E, Redhu NS, Mascanfroni ID, Al Adham Z, Lavoie S,
Ibourk M, et al: Interleukin-10 receptor signaling in innate immune
cells regulates mucosal immune tolerance and anti-inflammatory
macrophage function. Immunity. 40:706–719. 2014.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Guilliams M, Thierry GR, Bonnardel J and
Bajenoff M: Establishment and maintenance of the macrophage niche.
Immunity. 52:434–451. 2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Longhi MS, Moss A, Jiang ZG and Robson SC:
Purinergic signaling during intestinal inflammation. J Mol Med
(Berl). 95:915–925. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Costales MG, Alam MS, Cavanaugh C and
Williams KM: Extracellular adenosine produced by
ecto-5'-nucleotidase (CD73) regulates macrophage pro-inflammatory
responses, nitric oxide production, and favors Salmonella
persistence. Nitric Oxide. 72:7–15. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wirtz S, Popp V, Kindermann M, Gerlach K,
Weigmann B, Fichtner-Feigl S and Neurath MF: Chemically induced
mouse models of acute and chronic intestinal inflammation. Nat
Protoc. 12:1295–1309. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Yang W, Liu H, Xu L, Yu T, Zhao X, Yao S,
Zhao Q, Barnes S, Cohn SM, Dann SM, et al: GPR120 inhibits colitis
through regulation of CD4(+) T Cell Interleukin 10.
Gastroenterology. 162:150–165. 2022.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Watanabe S, Kobayashi S, Ogasawara N,
Okamoto R, Nakamura T, Watanabe M, Jensen KB and Yui S:
Transplantation of intestinal organoids into a mouse model of
colitis. Nat Protoc. 17:649–671. 2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Andresen L, Jorgensen VL, Perner A, Hansen
A, Eugen-Olsen J and Rask-Madsen J: Activation of nuclear factor
kappaB in colonic mucosa from patients with collagenous and
ulcerative colitis. Gut. 54:503–509. 2005.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhu Y, Yang S, Zhao N, Liu C, Zhang F, Guo
Y and Liu H: CXCL8 chemokine in ulcerative colitis. Biomed
Pharmacother. 138(111427)2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Barnabei L, Laplantine E, Mbongo W,
Rieux-Laucat F and Weil R: NF-κB: At the borders of autoimmunity
and inflammation. Front Immunol. 12(716469)2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Yu H, Lin L, Zhang Z, Zhang H and Hu H:
Targeting NF-κB pathway for the therapy of diseases: Mechanism and
clinical study. Signal Transduct Target Ther. 5(209)2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zhang Y, Zhou S, Zhou J, Wang D and Zhou
T: Regulation of NF-κB/MAPK signaling pathway attenuates the acute
lung inflammation in Klebsiella pneumonia rats by mollugin
treatment. Microb Pathog. 132:369–373. 2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zhuo Y, Li D, Cui L, Li C, Zhang S, Zhang
Q, Zhang L, Wang X and Yang L: Treatment with
3,4-dihydroxyphenylethyl alcohol glycoside ameliorates
sepsis-induced ALI in mice by reducing inflammation and regulating
M1 polarization. Biomed Pharmacother. 116(109012)2019.PubMed/NCBI View Article : Google Scholar
|