Introduction
Septic cardiomyopathy (SC), with mortality rates of
up to 70%, is considered the primary cause of death in sepsis
(1). Since clinicians have
recognized that the myocardium is functionally and structurally
damaged in patients with SC (2),
its prevention and treatment measures have become a popular
research topic. There is currently no consensus on the mechanism of
SC (3). Cytokines,
pathogen-associated molecular patterns, endogenous
damage-associated molecular patterns, oxidative stress, changes in
nitric oxide metabolism and mitochondrial dysfunction have been
individually proposed to lead to SC through different pathways
(4-6).
However, the majority of treatments targeting specific cytokines
have failed in clinical trials (7). Further investigation of novel
molecular targets and effective therapeutic approaches involved in
SC continues to be of great interest.
Iron is essential to life because it is involved in
numerous biological processes, such as energy metabolism and
nucleotide synthesis and repair (8). However, iron overload can affect
normal cellular functions through increasing oxidative stress or
directly inducing ferroptosis by catalyzing the oxidation of
phospholipids in the cell membrane (9). Ferroptosis is a form of programmed
cell death, which is usually accompanied by a large amount of iron
accumulation and lipid peroxidation (10). A previous study reported that iron
chelators could suppress the generation of radicals and protect
against damage in experimental sepsis (11). Moreover, a recent study reported
that ferroptosis is an important process, which mediates the
pathogenesis and progression of sepsis-induced cardiomyopathy
(12). Similarly, it has been
previously reported that ferroptosis could be suppressed by
dexmedetomidine to alleviate septic heart injury (13). These results suggest that negative
regulation of sepsis-induced iron concentration and ferroptosis
could be beneficial for treatment of sepsis-induced cardiac
injury.
Cyclovirobuxine D (CVB-D) is a triterpenoid alkaloid
extracted from the traditional Chinese medicinal plant Buxus
microphylla, which has been widely used to treat a number of
cardiovascular diseases in China (14). Previous studies have suggested that
that CVB-D may play an important protective role in the
cardiovascular system by activating the Nrf2 signaling pathway to
inhibit oxidative stress, or by suppressing the accumulation of
reactive oxygen species and inflammation levels (15-17).
However, it is unclear whether CVB-D has a protective effect
against myocardial injury in sepsis. In the present study, whether
CVB-D had a cardioprotective effect during sepsis was assessed with
a focus on revealing the underlying mechanisms of action.
Materials and methods
Animal treatment
Experiments were performed on 8-week-old male
Sprague Dawley (SD) rats (weight, 180-220 g; Rong Heng Biological
Co., Ltd). A total of 30 SD rats were randomly assigned to five rat
cages (n=6/cage) after one week of acclimation, feeding in an
indoor animal room with a 12 h light/dark cycle, a temperature of
22±2˚C and 40-60% relative humidity with free access to food and
water. All procedures were performed with the approval of the
Animal Care and Use Committee of Hebei University of Chinese
Medicine (approval no. DWLL202206006).
Animal experiments were performed based on
scientific rationale according to the 3R principles of animal
experimentation. To reduce pain in rats, pentobarbital sodium was
used as an anesthetic during surgery. The rats were randomized into
five groups (n=6/group) as follows: Control group, CLP group, CLP +
CVB-D group, CVB-D group and CLP + Ferr-1 (Ferrostatin-1,
ferroptosis inhibitor) group. CLP surgery was performed to induce
septic cardiac dysfunction. CVB-D was dissolved in methanol (final
concentration, 10 mM) and stored at 4˚C as a stock solution. CVB-D
(cumulative dose of 4 mg/kg; Meilunbio Co., Ltd.) (11) or saline was administered by gavage
in rats for four consecutive days before CLP surgery. The rats
received an intraperitoneal injection of Ferr-1 (2 µmol/kg; Xcess
Biosciences) or saline once daily for four days before surgery.
The CLP surgery was performed as described
previously (18). Briefly, rats
were anesthetized by intraperitoneal injection of pentobarbital
sodium (40 mg/kg) then fixed on an operating table and incised 1.5
cm along the midline of the abdomen under aseptic conditions. After
the colon of the rat was exposed and ligated at 1/2 to the root,
the distal cecum was then punctured twice with a 9-gauge needle. A
small volume of fecal contents from the punctured cecum was spilled
into the abdominal cavity. The control group underwent the same
surgical procedure, but without cecal puncture. All animals were
placed on the operating table and immobilized with ketamine (40
mg/kg; intramuscular injection in the front of the thigh) and
xylazine (10 mg/kg; intraperitoneally administered) anesthesia,
after which 5 ml blood was obtained from the abdominal aorta. After
rats were sacrificed by exsanguination from the carotid artery,
blood, heart and liver tissues were obtained, rats were sacrificed
by exsanguination from the carotid artery. Blood (centrifuged at
3,000 x g for 15 min at 4˚C) was collected to obtain serum for
measuring cardiac function biomarker levels. After the hearts were
isolated, tissue samples from each heart were used for hematoxylin
and eosin (H&E) staining, transmission electron microscopy
(TEM), reverse transcription-quantitative PCR (RT-qPCR) and
biochemistry testing. Liver samples were also collected for
RT-qPCR.
Histopathological examination
Heart tissues were soaked in 4% paraformaldehyde at
4˚C for two days, then the tissue samples were gradually dehydrated
in an ascending gradient of ethanol 30 min each at room
temperature, embedded in paraffin and cut into 4 µm sections.
Slides were stained with H&E 5 min each at room temperature and
observed under a light microscope (Leica Microsystems GmbH).
Detection of cardiac injury
markers
Biochemical indicators of myocardial injury and
lipid peroxidation were detected by creatine kinase-muscle and
brain (CK-MB; cat. no. H197-1-1; Nanjing Jiancheng Institute of
Bioengineering), lactate dehydrogenase (LDH; cat. no. A020-2-2;
Nanjing Jiancheng Institute of Bioengineering) and cardiac troponin
I (cTnI; cat. no. H149-2; Nanjing Jiancheng Institute of
Bioengineering) content in serum, as well as malondialdehyde (MDA;
cat. no. A003-1-1; Nanjing Jiancheng Institute of Bioengineering),
superoxide dismutase (SOD; cat. no. A001-3-2; Nanjing Jiancheng
Institute of Bioengineering) and reduced glutathione (GSH; cat. no.
A006-2-1; Nanjing Jiancheng Institute of Bioengineering) content in
tissue. The content and activity of each sample indicator was
measured spectrophotometrically using biochemical detection kits
(Nanjing Jiancheng Bioengineering Institute).
Echocardiography
Cardiac function was examined in rats 24 h after
CLP. Rats were anesthetized by inhalation 2.5% isoflurane in 5%
carbon dioxide and 95% oxygen and their chest fur removed with
depilation cream (cat. no. VT-200; Veet; Reckitt Benckiser). A
coupling agent (Kofu Medical Technology Development Co., Ltd) was
applied evenly to the skin then M-mode echocardiograms were
recorded and analyzed using high-frequency echocardiography (Vevo
2100, VisualSonics, Inc.). Left ventricular ejection fraction (EF),
fractional shortening (FS), left ventricular end-systolic volume
(LVESV) and left ventricular end-diastolic volume (LVEDV) were
measured.
Transmission electron microscopy
Cardiac tissue samples were cut into 1 mm cubes and
fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer for at least
24 h at room temperature. All samples were postfixed in 1% osmium
tetroxide for 2 h at room temperature, dehydrated in an ascending
acetone gradient, embedded in Epon 812 for 72 h at 60˚C, cut into
70 nm-thick sections, mounted and double-stained with uranyl
acetate and lead citrate for 20 min at room temperature. The
samples were then imaged using a transmission electron microscope
(H-7600; Hitachi, Ltd.).
RT-qPCR
H9C2 cells were collected and pooled together to
achieve sufficient RNA. TRIzol (cat. no. RR047A; Takara Bio, Inc.)
total RNA extraction reagent was used for RNA extraction and a
PrimeScript™ RT reagent kit (cat. no. RR047A; Takara Bio, Inc.) was
used to synthesize cDNA according to the manufacture's protocol.
Detection of the mRNA expression levels of the hepcidin gene (hamp)
was performed against GAPDH (19).
The following thermocycling conditions were used for PCR: R67
Initial denaturation stage, 95˚C for 5 min. Cycling stages were as
follows: Denaturation, 95˚C for 30 sec; annealing, 60˚C for 30 sec;
and extension, 72˚C for 30 sec, for a total of 40 cycles. Final
extension stage: 72˚C for 5 min. Relative mRNA expression levels
were determined using the 2-ΔΔCq method (20). The primers sequences used were as
follows: Hamp forward (F), 5'-CCTGAGCAGCGGTGCCTATC-3' and reverse
(R), 5'-TGGTGTCTCGCTTCCTTCGC-3'; Prostaglandin-endoperoxide
synthase 2 (Ptgs2) F, 5'-ATGTTCGCATTCTTTGCCCAG-3' and R,
5'-TACACCTCTCCACCGATGAC-3'; and GAPDH F, 5'-GAGTCAACGGATTTGGTCGT-3'
and R, 5'-GACAAGCTTCCCGTTCTCAG-3'.
Tissue iron measurements
Non-heme levels of iron in the heart were measured.
Samples of heart tissue weighing ~50 mg were obtained then
incubated with 10 times the volume of tissue digestive liquid (3M
hydrochloric acid + 0.61M trichloroacetic acid) for 60 h at 65˚C.
After centrifugation (3,000 x g for 15 min at 4˚C), the supernatant
was collected for spectrophotometric quantification of the
formation of the Fe(II)-bathophenanthroline disulfonate complex.
Supernatant (0.5 ml) was added to 1.5 ml of iron chromogen and
boiled in a water bath for 5 min. Iron developing color working
solution (cat. no. A039-1-1; Nanjing Jiancheng Bioengineering
Institute) was freshly prepared and the absorbance was measured at
535 nm. A standard curve was generated and the non-heme iron
content of samples was calculated according to a formula provided
by the manufacturer.
Cell culture
H9C2 cardiomyoblasts purchased from Procell Life
Science & Technology Co., Ltd. were cultured in DMEM (cat. no.
C11995500BT; Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum (cat. no. 10270106; Gibco; Thermo
Fisher Scientific, Inc.) and 1% penicillin-streptomycin in an
incubator with a 5% CO2 atmosphere at 37˚C. Cell culture
medium was changed every other day. A stock solution of CVB-D
(final concentration, 10 mM) (cat. no. 860-79-7; Meilunbio Co.,
Ltd.) was prepared in methanol. Cells were pretreated with CVB-D at
doses of 0.1, 1, 10 or 100 µmol/l at 37˚C for 24 h, while the
control groups were treated with an equivalent volume of PBS. To
establish a model of septic myocardial injury in vitro, H9C2
cells were incubated at 37˚C for 12 h with LPS at doses of 0.1, 1,
10 or 100 µg/ml (Shanghai Aladdin Biochemical Technology Co., Ltd.)
(21). Cells were then harvested
for analysis.
Cell Counting Kit-8 (CCK-8) assay
The proliferation capacity of the cells was assessed
in a 96-well plate (2x105 cells/well) using the CCK-8
assay (22). The cells were
incubated in 96-well plates overnight before the addition of CCK-8.
After stimulation with CVB-D or LPS (cat. no. 93572-42-0;
MilliporeSigma) for 24 h, 10 µl of CCK-8 reagent was added and
incubated for 2 h at 37˚C under dark conditions. The absorbance
value at 450 nm of each well was recorded with a microplate
reader.
Detection of H9C2 reactive oxygen
species (ROS) generation
The level of fluorescence produced by the
fluorescent probe 2',7'-dichlorofluorescein diacetate (DCFH-DA;
cat. no. S0033S; Beyotime Institute of Biotechnology) was used to
quantify ROS generation. Groups of H9C2 cells were loaded with
1:1,000 DCFH-DA probes diluted with serum-free medium and incubated
for 20 min in a 37˚C cell incubator after 24 h of administration.
DCFH-DA is hydrolyzed into dichlorodihydrofluorescein (DCFH) after
entering the membrane of the H9C2 cell. DCFH, which is not
fluorescent, can be oxidized by intracellular ROS to fluorescent
DCF. The level of ROS was detected according to the fluorescence
intensity. The fluorescence intensity was imaged using a
fluorescence microscope (IX73; Olympus Corporation) and quantified
using ImageJ (version 1.5.1) software (National Institutes of
Health).
Quenching of calcein fluorescence by
the intracellular labile iron pool
Intracellular labile iron was quantified using
calcein-acetoxymethyl ester (Calcein-AM) (23). Cellular cytoplasmic esterase
converts a nonfluorescent acetoxy ester fraction, Calcein-AM, into
fluorescent calcein, which is impenetrable to other cells. When
iron binds to calcein, its fluorescence is quenched and then
restored when the iron loses its stronger chelating agent. Cells
were incubated in 24-well plates at 37˚C for 24 h. Then CVB-D or
LPS was given to incubate for 24 h. Calcein-AM (cat. no. 40719ES50;
Shanghai Yeasen Biotechnology Co., Ltd.) was added to cells in a
24-well plate at a final concentration of 0.25 µM and incubated for
30 min at 37˚C. The fluorescence level was then measured using an
inverted fluorescence microscope at the excitation and emission
wavelengths of 488 and 525 nm, respectively (IX73; Olympus
Corporation).
Western blot analysis
As previously described (24), total protein in myocardial tissue
and H9C2 cells were precipitated in RIPA lysis buffer solution
(cat. no. SL1020; Coolaber, Ltd.) containing protease inhibitors.
Tissue and cells were centrifuged at 4˚C for 20 min at 12,000 x g
to remove the insoluble residue. The supernatant was removed and
the concentration of total protein was determined using a BCA kit
(cat. no. CW0014; Beijing Kangwei Century Biotechnology Co., Ltd.).
Equal amounts of total protein were separated by electrophoresis
using 10 or 12% gel. Separated proteins were subsequently
transferred to polyvinylidene fluoride (EMD Millipore) membranes
and blocked overnight at 37˚C with TBS-0.1% Tween 20 in 5% skimmed
milk. Membranes were incubated with GPX4 (1:2,000; cat. no.
ET1706-45; HUABIO), SLC7A11 (1:1,000; cat. no. DF12509; Affinity
Biosciences), transferrin receptor 1 (TfR1; 1:2,000; cat. no.
abs131442; Absin), ferroportin 1 (FPN1; 1:3,000; cat. no.
GTX100573; Absin), divalent metal transporter 1 (DMT1; 1:2,000;
cat. no. abs112967; Absin), ferritin heavy chain (FtH; 1:1,000;
cat. no. AB183781; Abcam), IL-6 (1:1,000; cat. no. GB11117; Wuhan
Servicebio Technology Co., Ltd.), IL-1β (1:1,000; cat. no. AF5103;
Affinity Biosciences), tumor necrosis factor-α (TNF-α; 1:1,000;
cat. no. AF7014; Affinity Biosciences), phosphorylated-STAT3
(p-STAT3; 1:1,000; cat. no. AF3294; Affinity Biosciences), STAT3
(1:1,000; cat. no. AF6293; Affinity Biosciences), Nrf2 (1:1,000;
cat. no. M200-3; MBL International Co.), Lamin B1 (1:1,500; cat.
no. SI17-06; HUABIO) and β-tubulin (1:2,000; cat. no. A01857-1;
Wuhan Boster Biological Technology, Ltd.) antibodies primary
antibodies at 4˚C overnight. Goat anti-rabbit IgG (H+L)-HRP
(1:10,000; cat. no. S0001) or goat anti-mouse IgG(H+L)-HRP
(1:1,000; cat. no. S0002; both from Affinity Biosciences) were then
placed at room temperature for 2 h. The cell membranes were then
washed again and Visioncapt (V16.12; Vilber Lourmat) was used for
quantification.
Measurement of Ca2+
concentration by fluo-4 AM staining
The concentration of Ca2+ was assessed
using a Fluo-4 AM kit (cat. no. F8501; Beijing Solarbio Science
& Technology Co., Ltd) according to manufacturer's
instructions. 1x105 H9C2 cells were seeded into a
24-well plate and cultured at 37˚C for 24 h. Fluo-4 AM (1 ml, 4 µM)
was added to each well and incubated in darkness at 37˚C for 20
min. Hank's Balanced Salt Solution (HBSS; cat. no. F8501; Beijing
Solarbio Science & Technology Co.) containing 1% fetal bovine
serum was added and incubated at 37˚C for 40 min. After being
washed three times with HBSS and FBS (1 ml per well) was used each
time, cells were imaged to detect fluorescent calcium ions using
excitation and emission wavelengths of 494 and 516 nm,
respectively, with an inverted fluorescence microscope (IX73;
Olympus Corporation).
Measurement of ICa-L and
cell shortening
To further analyze whether CVB-D inhibits calcium
inwards flow through LTCCs, a diaphragm clamp technique was
utilized to detect L-type calcium current changes. With the use of
L-type calcium channel inhibitors to block calcium channels, the
inwards flow of calcium ions is reduced, resulting in protection
against myocardial injury. Verapamil (VER) is a typical calcium
channel blocker. The expression of the L-type calcium channel
(LTCC) in ventricular myocytes was analyzed utilizing the
whole-cell patch-clamp method. The intracellular solution consisted
of tetraethylammonium chloride (cat. no. 56-34-8; TCL),
MgCl2 (cat. no. 10012818; Sinopharm Group Chemical
Reagent Co., Ltd), CaCl2 (cat. no. 10005861; Sinopharm
Group Chemical Reagent Co., Ltd), Glucose (cat. no. 56-86-0;
Sigma-Aldrich; Merck KGaA) and
N-2-hydroxyethylpiperazine-N-ethane-sulphonicacid (cat. no.
7365-45-9, Beijing Bailingwei Technology Co., Ltd), and the
extracellular solution consisted of CsCl (cat. no. 7647-17-8;
Shanghai Yi En Chemical Technology Co., Ltd), tetraethylammonium
chloride, ATP-Mg (cat. no. 7480-12-9; Sigma-Aldrich; Merck KGaA),
N-2-hydroxyethylpiperazine-N-ethane-sulphonicacid and ethylene
glycol tetraacetic acid (cat. no. 67-42-5; Sigma-Aldrich; Merck
KGaA). Currents under filtered at 2 kHz were recorded with Axon
patch 200 B amplifier after the cells were connected with glass
electrodes (Sutter Instrument). The Axon Patch 200B amplifier
analyzed the current and pCLAMP™ software (version 10.0) was used
for data analysis (Molecular Devices, LLC). The contraction of
myocardial cells was measured. Cells were placed on an inverted
microscope (Ion Optix) platform and perfused with the external
solution to induce cardiac myocyte shortening 2 msec each time of
0.5 Hz. Cardiomyocytes with clear transverse lines and in good
condition were selected to measure contraction.
Statistical analysis
Experimental data are presented as the mean ±
standard deviation. Data were processed using GraphPad Prism
software (version 8.0; Dotmatics) using one-way ANOVA followed by
Tukey's test for comparisons among multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
CVB-D exerts a protective effect on
LPS-treated H9C2 cells
LPS is commonly used to generate pathophysiological
conditions of septic cardiomyopathy in investigation studies. CCK-8
assay results demonstrated that H9C2 cell viability was
significantly reduced by LPS in a dose-dependent manner (Fig. 1A). A dose of 10 µg/ml LPS at 37˚C
for 12 h was selected to induce a cellular sepsis model for further
use in the present study, as previously described (21,25,26).
Additionally, a CCK-8 assay was performed to assess the effects of
CVB-D on H9C2 cell viability. The results demonstrated that CVB-D
had no cytotoxic effect on H9C2 cells at 0.1 or 1 µM, but
significantly reduced H9C2 survival at 10 and 100 µM (Fig. 1A). Therefore, a dose of 1 µM CVB-D
was selected for use in the present study. Moreover, pretreatment
of H9C2 cells with 1 µM CVB-D significantly enhanced LPS-damaged
cell viability (Fig. 1B). As
expected, LPS distinctly weakened CK-MB, LDH activity and cTnI
levels (P<0.01), whereas the inhibitory effect was blocked by
CVB-D pretreatment (Fig. 1C).
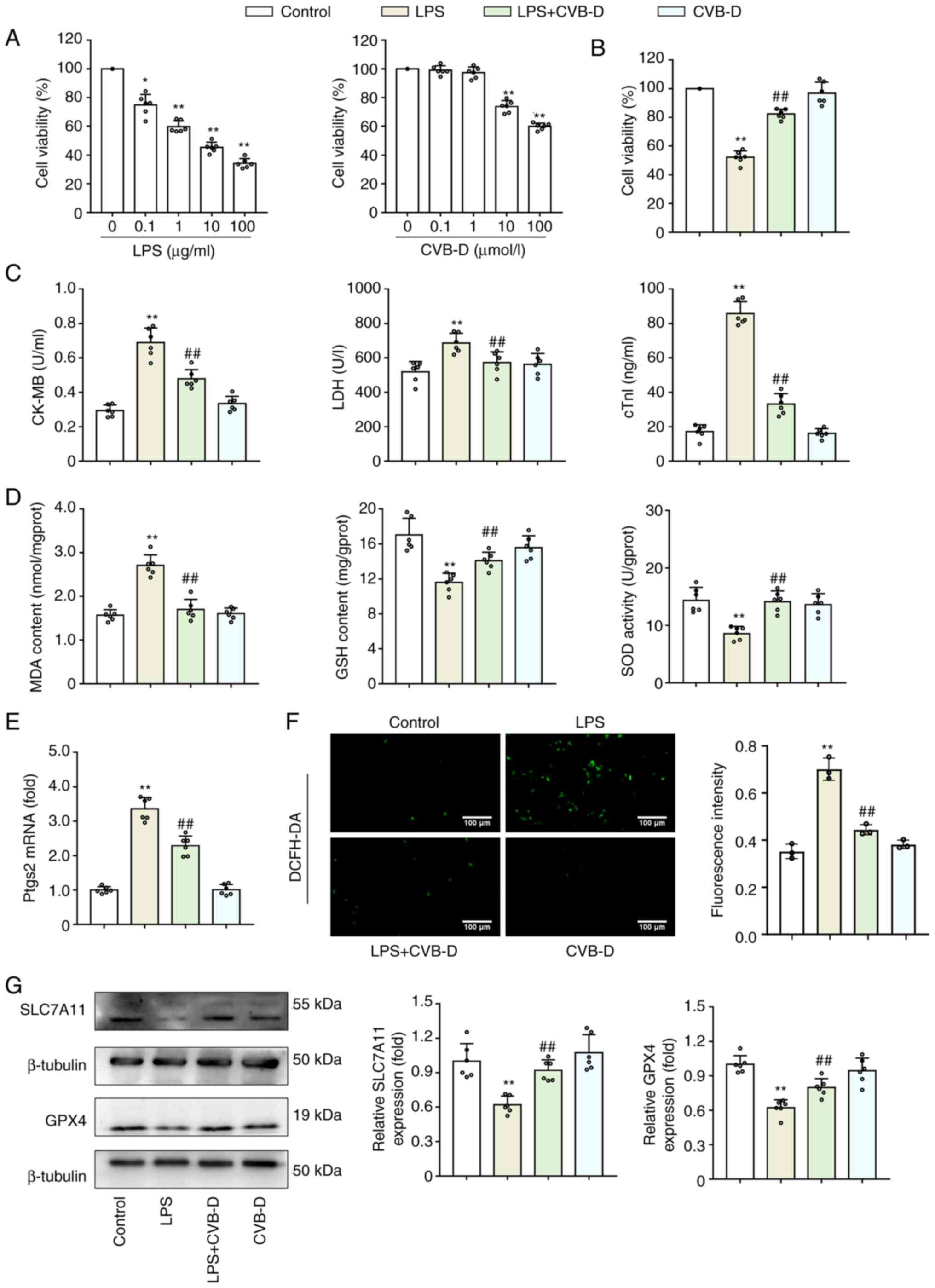 | Figure 1CVB-D exerts a protective effect on
LPS-treated H9C2 cells by inhibiting ferroptosis. (A) CCK-8 assay
was used to determine the dose of CVB-D and LPS for further study.
(B) Effect of CVB-D on LPS-treated H9C2 cell viability. (C) Effect
of CVB-D on CK-MB levels, LDH activity and cTnI levels of
LPS-treated H9C2 cells. (D) Effect of MDA generation, GSH levels
and SOD activity in LPS-treated H9C2 cells. (E) Effect of CVB-D on
the expression of ptgs2 mRNA levels (n=3). (F)
Representative fluorescent microscopy images of ROS production by
H9C2 cells (n=3). (G) SLC7A11 and GPX4 protein expression levels of
LPS- and CVB-D-treated H9C2 cells. Western blot images were taken
from different gels but a single replicate. *P<0.05
vs. control group, **P<0.01 vs. control group,
##P<0.01 vs. LPS-treated group. Data are presented as
the mean ± SD (n=6). CVB-D, cyclovirobuxine D; LPS,
lipopolysaccharide; CK-MB, creatine kinase isoenzyme; LDH, lactate
dehydrogenase; cTnI, cardiac troponin I; SOD, superoxide dismutase;
GSH, glutathione; MDA, malondialdehyde; ptgs2,
prostaglandin-endoperoxide synthase 2; ROS, reactive oxygen
species; DCFH-DA, 2',7'-dichlorofluorescein diacetate; SLC7A11,
solute carrier family 7 member 11; GPX4, glutathione peroxidase 4;
CCK-8, Cell Counting Kit-8. |
CVB-D inhibits ferroptosis in
LPS-treated H9C2 cells
The effect of CVB-D on H9C2 lipid peroxidation
levels and production of first line defense antioxidant enzymes GSH
and SOD were also assessed. LPS treatment of H9C2 cells
significantly increased production of MDA, but significantly
reduced GSH levels and significantly increased SOD activity;
however, CVB-D significantly reversed these effects (Fig. 1D). The enhancement of ptgs2 mRNA is
used as a putative marker of ferroptosis (9). CVB-D pretreatment significantly
reduced the stimulatory effects of LPS on ptgs2 mRNA (P<0.01)
(Fig. 1E). ROS production was
analyzed using the ROS sensitive DCFH-DA probe. The images from
this experiment demonstrated that LPS-treated H9C2 cells generated
increased ROS compared with controls. CVB-D significantly decreased
the ROS generation induced by LPS (Fig. 1F). Since the SLC7A11-GSH-GPX4
antioxidant axis is important in ferroptosis, the protein
expression levels of SLC7A11 and GPX4 were analyzed in LPS-induced
H9C2 cells pretreated with CVB-D. The protein expression levels of
SLC7A11 and GPX4 were significantly reduced after LPS treatment but
were elevated by treatment with CVB-D (Fig. 1G).
CVB-D inhibits LPS-induced iron
overload
Excess cellular iron has been previously reported to
promote the generation of both soluble and lipid ROS via the Fenton
reaction, which favors the ferroptotic effect (27). Thus, it was hypothesized that CVB-D
inhibited oxidative stress, lipid peroxidation and ferroptosis by
modulating the abnormal iron metabolism induced by LPS. In the
LPS-treated group of H9C2 cells, the significant decrease in
calcein fluorescence intensity indicated a high level of
calcein-bound iron, while CVB-D pretreatment significantly reversed
this change (Fig. 2A). In
addition, the presence of iron transporter proteins, such as DMT1,
TfR1, FPN1 and FtH, was detected by western blotting (Fig. 2B). Compared with the LPS-treated
group, CVB-D treatment significantly reduced DMT1, TfR1 and FtH
protein expression levels but significantly increased FPN1 protein
expression levels, which suggested that CVB-D may suppress iron
uptake and storage while increasing iron export. As FPN1 is the
only known iron exporter in mammalian cells, the expression levels
of hamp mRNA. The stimulation of LPS in cells has been reported to
induce the release of many inflammatory cytokines. Indeed, compared
with the LPS group, CVB-D pretreatment significantly decreased the
levels of hamp mRNA in H9C2 cells (Fig. 2C). Compared with LPS-treated cells,
CVB-D pretreatment significantly decreased the protein expression
levels of pro-IL-1β, TNF-α and IL-6 and the p-STAT3/STAT3 ratio in
H9C2 cells (Fig. 2D). These
findings demonstrated that CVB-D treatment could significantly
inhibit iron-overload by modulating iron metabolism. In particular,
CVB-D could eliminate LPS-induced hepcidin by blocking of the
IL-6/STAT3 axis.
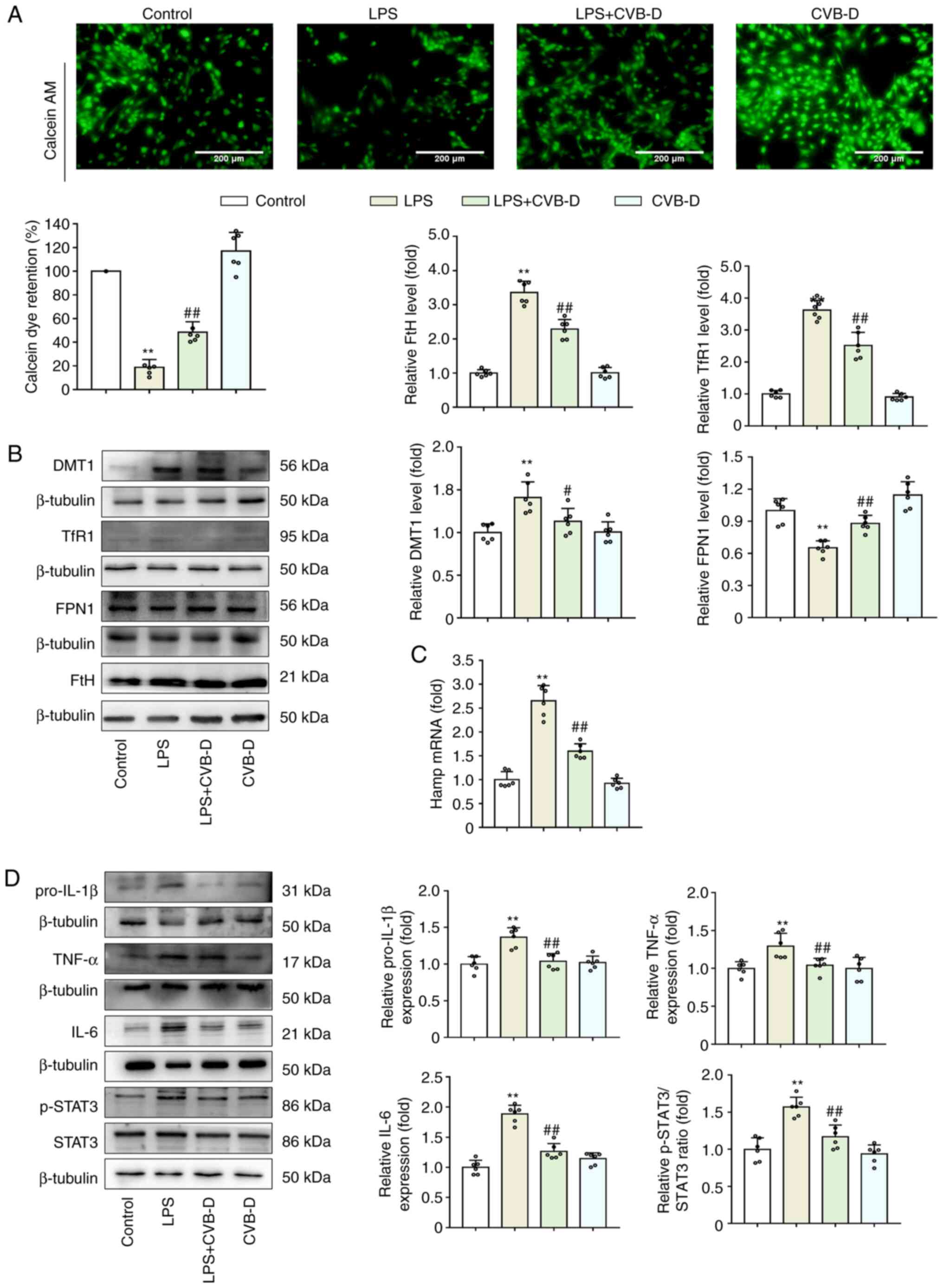 | Figure 2CVB-D inhibits cellular iron-overload
induced by LPS. (A) Calcein-AM imaging of intracellular iron levels
in H9C2 cells. (B) Western blot images and protein expression
levels of DMT1, TfR1, FPN1 and FtH in LPS- and CVB-D-treated H9C2
cells. (C) hamp mRNA expression levels in LPS- and
CVB-D-treated H9C2 cells. (D) Protein expression levels of
pro-IL-1β, TNF-α, IL-6, p-STAT3 and STAT3 in LPS- and CVB-D-treated
H9C2 cells. Data are presented as the mean ± SD (n=6).
**P<0.01 vs. control group, #P<0.05,
##P<0.01 vs. LPS-treated group. Western blot images
were from different gels, but the same replicate. CVB-D,
cyclovirobuxine D; LPS, lipopolysaccharide; hamp, hepcidin; DMT1,
divalent metal transporter 1; TfR1, transferrin receptor 1; FPN 1,
ferroportin1; IL, interleukin; TNF-α, TNF tumor necrosis factor-α;
STAT3, signal transducer and activator of transcription 3; p,
phosphorylated. |
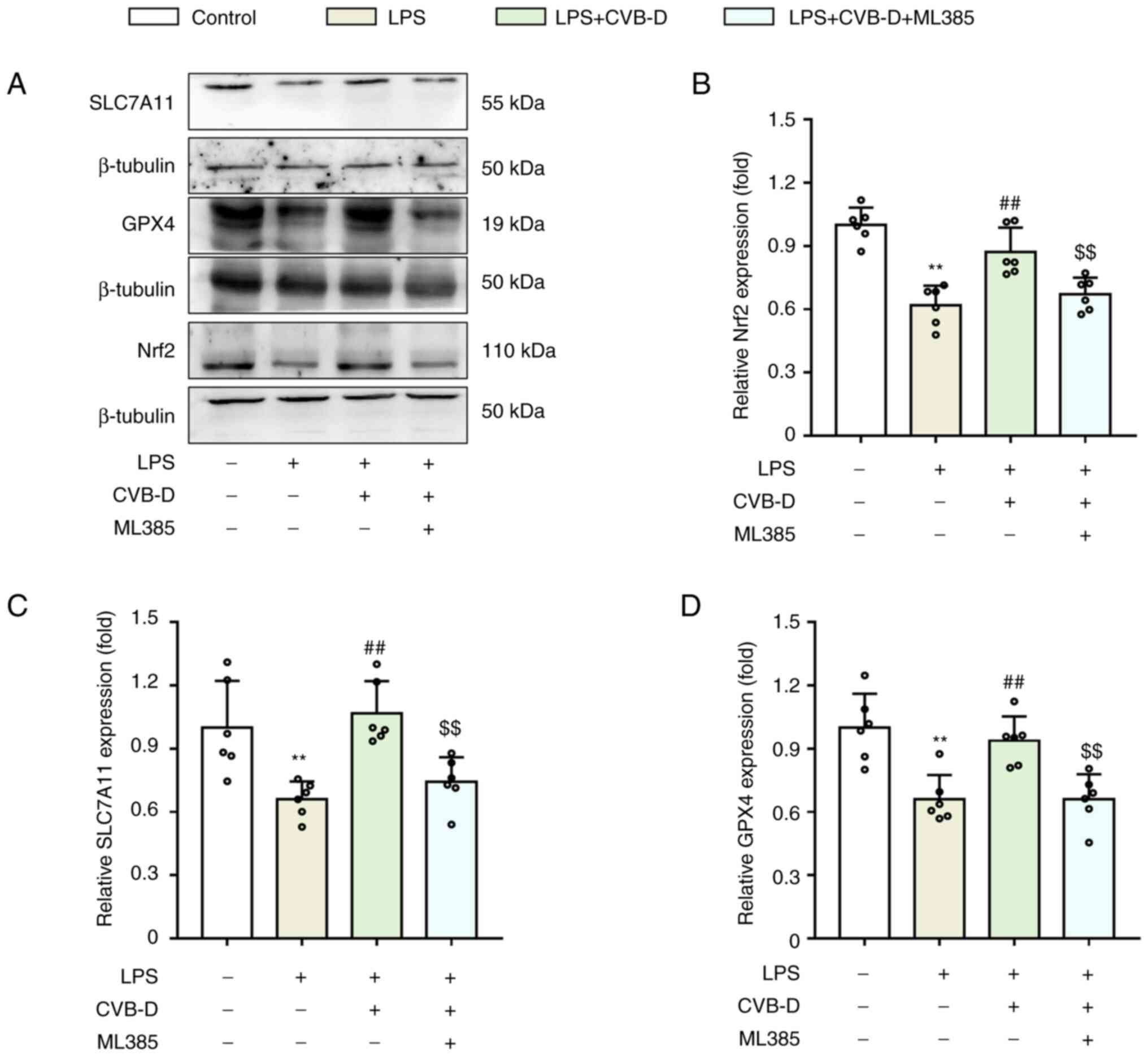
CVB-D protects against LPS-induced
ferroptosis in H9C2 cells by upregulating Nrf2
Nrf2 has previously been reported to promote
intracellular iron accumulation by downregulating FPN1(28). To confirm whether Nrf2 serves an
important role in protecting cells from septic harm, H9C2 cells
were treated with the Nrf2 inhibitor ML385 at a dose of 1 µM/l for
12 h. Nrf2, SLC7A11 and GPX4 protein expression levels in H9C2
cells were analyzed (Fig. 3A). LPS
significantly downregulated Nrf2 expression, and Nrf2 expression
was significantly increased when LPS was combined with CVB-D
treatment (Fig. 3B). Therefore,
the hypothesis that an enhancement of Nrf2 would be harmful to
cells by increasing iron by downregulating FPN1 was not supported.
SLC7A11 and GPX4 protein expression levels were significantly
upregulated in septic cells pretreated with CVB-D compared with
controls, while ML385 significantly reversed this change (Fig. 3C and D). This indicated that Nrf2 serves a role
in the protective effects of CVB-D against ferroptosis, but not via
FPN1.
CVB-D attenuates myocardial injury
induced by CLP surgery in rats
To evaluate whether CVB-D was beneficial for septic
heart injury, cardiac structure and function in rats were assessed.
H&E staining demonstrated that CVB-D treatment reduced the
myocardial tissue damage caused by CLP (Fig. 4A). The rats in the CLP surgery
group exhibited marked cardiomyocyte disorders, myocardial
abnormalities, inflammatory cell infiltration, visible fibrosis and
fiber breakage compared with the control surgery group. In
contrast, the rats in the CLP surgery group pretreated with CVB-D
demonstrated no obvious inflammatory cell infiltration or fibrosis
and the arrangement of myocardial fibers was neat. Serum levels of
CK-MB, LDH and cTnI, consistent with the degree of myocardial cell
injury, were significantly increased in the myocardium of CLP rats
compared with controls (Fig. 4B).
Furthermore, CVB-D pretreatment significantly inhibited CLP-induced
increases in CK-MB, LDH and cTnI (Fig.
4B). Type M-mode sonograms were performed to measure left
ventricular function (Fig. 1C).
The left ventricular ejection fraction shortening fraction was
significantly decreased in the CLP-treated group compared with the
control group (Fig. 4D). LVESV and
LVDEV were both significantly increased in the CLP-treated group
compared with the control. This change was significantly reversed
in the CLP + CVB-D treatment group, but no significant differences
were observed between the CVB-D group and the control. This
suggested that CVB-D demonstrated a significant ameliorative effect
on left ventricular dysfunction caused by CLP surgery.
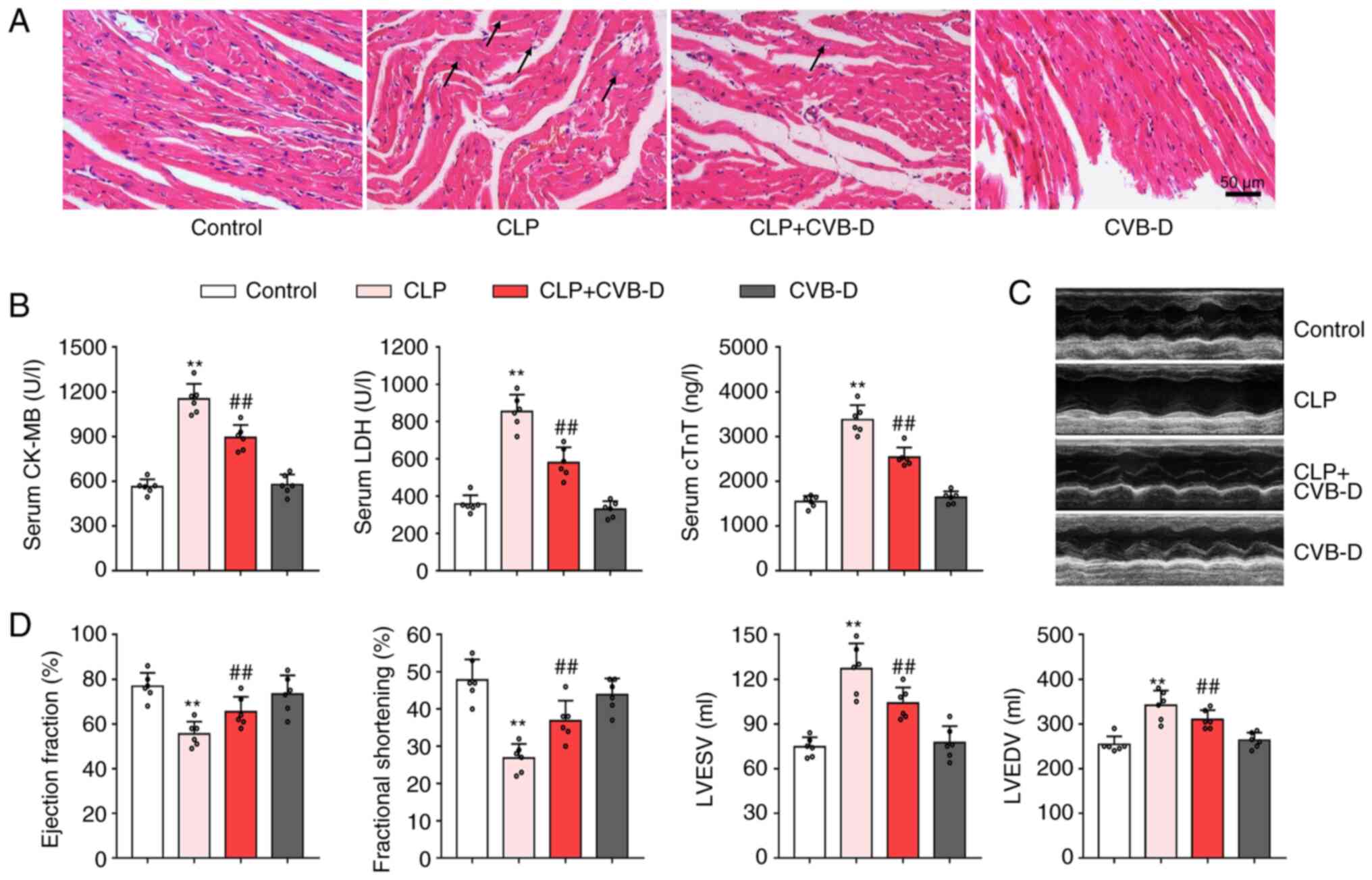 | Figure 4Protective effect of CVB-D on cardiac
injury after CLP. (A) Representative histopathological images
indicating myocardial tissue damage. Arrows indicate swollen
cardiomyocytes and inflammatory infiltrating cells. (B) Serum
levels of CK, LDH and cTnI in rats treated with CLP and CVB-D. (C)
Representative echocardiographic images of rats treated with CLP
and CVB-D. (D) Left ventricular ejection fraction, shortening
fraction, left ventricular end-systolic volume and left ventricular
end-diastolic volume of rats treated with CLP and CVB-D. Data are
presented as the mean ± SD (n=6). **P<0.01 vs.
control group, ##P<0.01 vs. CLP group. CVB-D,
cyclovirobuxine D; CLP, cecal ligation and puncture; CK-MB,
creatine kinase isoenzyme; LDH, lactate dehydrogenase; cTnI,
cardiac troponin I; LVESV, left ventricular end-systolic volume;
LVDEV, left ventricular end-diastolic volume. |
CVB-D reduces myocardial ferroptosis
induced by CLP surgery in rats
To evaluate whether ferroptosis was involved in the
cardioprotective effect of CVB-D on septic heart injury, levels of
ferroptosis related markers were examined in myocardial tissues.
TEM was used to image the ultrastructure of myocardial mitochondria
(Fig. 5A). In the control group,
the mitochondrial membrane was complete and the cristae were clear.
Cardiomyocytes in the CLP-treated rats demonstrated some shrunken
mitochondria with solidified mitochondrial membranes and decreased
mitochondrial cristae. Pretreatment with CVB-D prevented these
changes to mitochondrial ultrastructure, while CVB-D did not change
the mitochondrial ultrastructure in control rats (Fig. 5A). Prostaglandin-endoperoxide
synthase 2 (PTGS2) is considered a biomarker of ferroptosis
(29). The significant CLP-induced
increase in ptgs2 mRNA expression levels compared with
controls, was significantly reversed by CVB-D pretreatment
(Fig. 5B). Moreover, CVB-D
attenuated sepsis-induced lipid peroxidation, as demonstrated by
the significant decrease in MDA levels and the significant increase
in GSH levels (Fig. 5C and
D). Analysis of non-heme iron
levels demonstrated that CVB-D prevented ferroptosis by inhibiting
septic iron-overload (Fig. 5E).
CVB-D intervention downregulated the liver hamp mRNA
expression levels in CLP-treated rats (Fig. 5F). These changes indicated that
CVB-D pretreatment effectively inhibited cardiac ferroptosis in
septic rats. To determine whether ferroptosis was closely related
to the pathophysiological processes of septic heart injury,
pretreatment with Ferr-1 was applied before CLP surgery. It was
subsequently demonstrated that inhibition of ferroptosis could
significantly elevate both ejection fraction and fractional
shortening compared with the CLP-treated group (Fig. 5G).
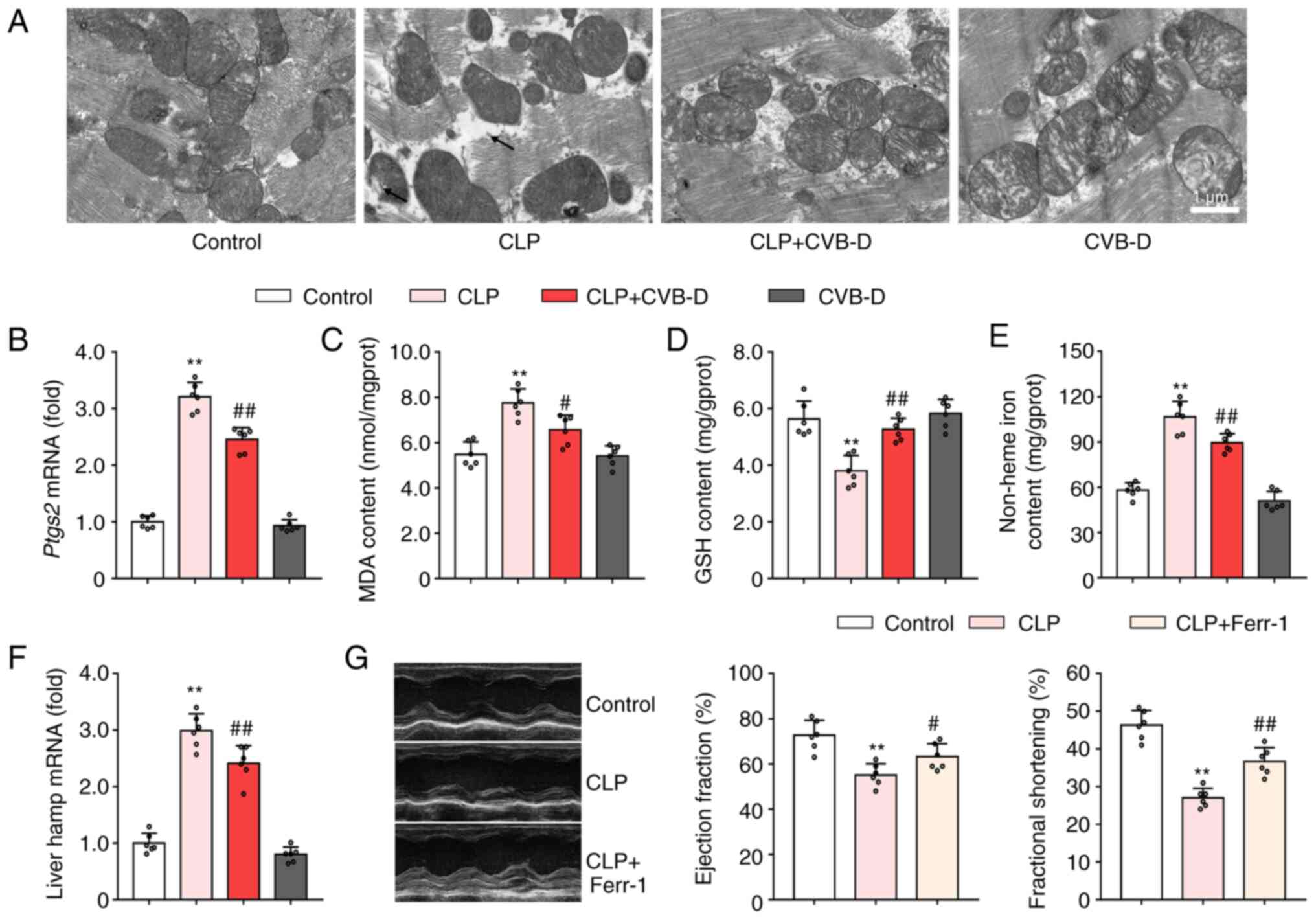 | Figure 5CVB-D reduces myocardial ferroptosis
induced by CLP surgery in rats. (A) Representative TEM images
showing myocardial mitochondria ultrastructure. (B) mRNA expression
levels of ptgs2 in hearts of rats treated with CLP and CVB-D. (C)
MDA levels in hearts of rats treated with CLP and CVB-D. (D) GSH
levels in hearts of rats treated with CLP and CVB-D. (E) Levels of
cardiac non-heme iron content in hearts of rats treated with CLP
and CVB-D. (F) mRNA expression levels of hamp in livers of
rats treated with CLP and CVB-D. (G) Representative
echocardiographic images, and left ventricular ejection fraction
and shortening fraction of CLP- and Ferr-1-treated rats. Data are
presented as the mean ± SD (n=6). **P<0.01 vs.
control group, #P<0.05, ##P<0.01 vs.
CLP-treated group. CVB-D, cyclovirobuxine D; CLP, cecal ligation
and puncture; PTGS2, prostaglandin-endoperoxide synthase 2;
hamp, hepcidin; SOD, superoxide dismutase; GSH, glutathione;
MDA, malondialdehyde. |
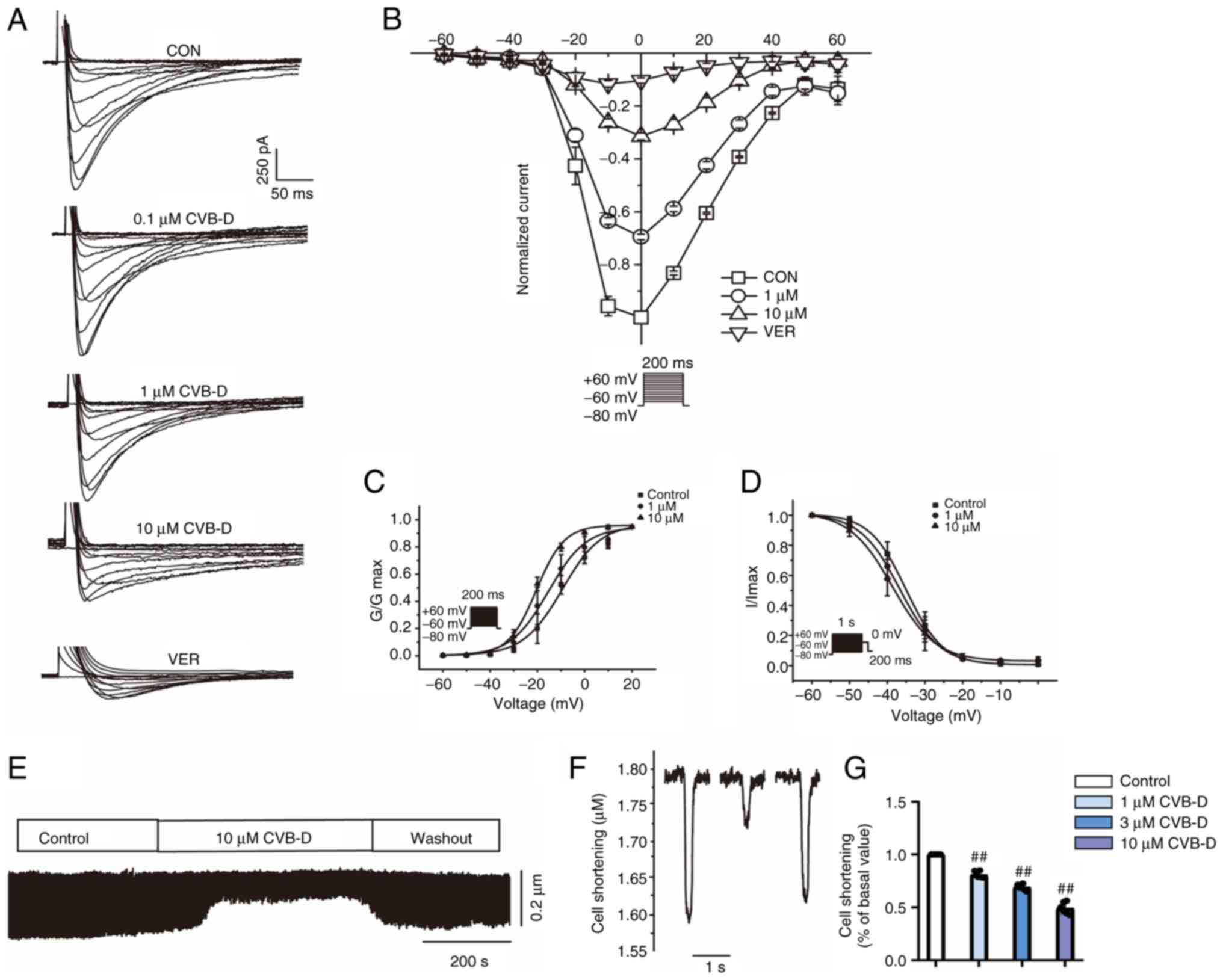
CVB-D alleviates the calcium overload
caused by LPS in H9C2 cells
LTCCs and T-type calcium channels also serve a role
in iron uptake into the heart (30). Therefore, the present study further
analyzed the effect of CVB-D on calcium channels in septic cells.
The influx of calcium ions was analyzed by detecting the
fluorescence intensity of Ca2+ using Fluo-4 AM staining.
The intracellular Ca2+ concentration was markedly
increased after LPS stimulation compared with the control (Fig. 6A). Intracellular calcium levels
were markedly reduced by CVB-D treatment.
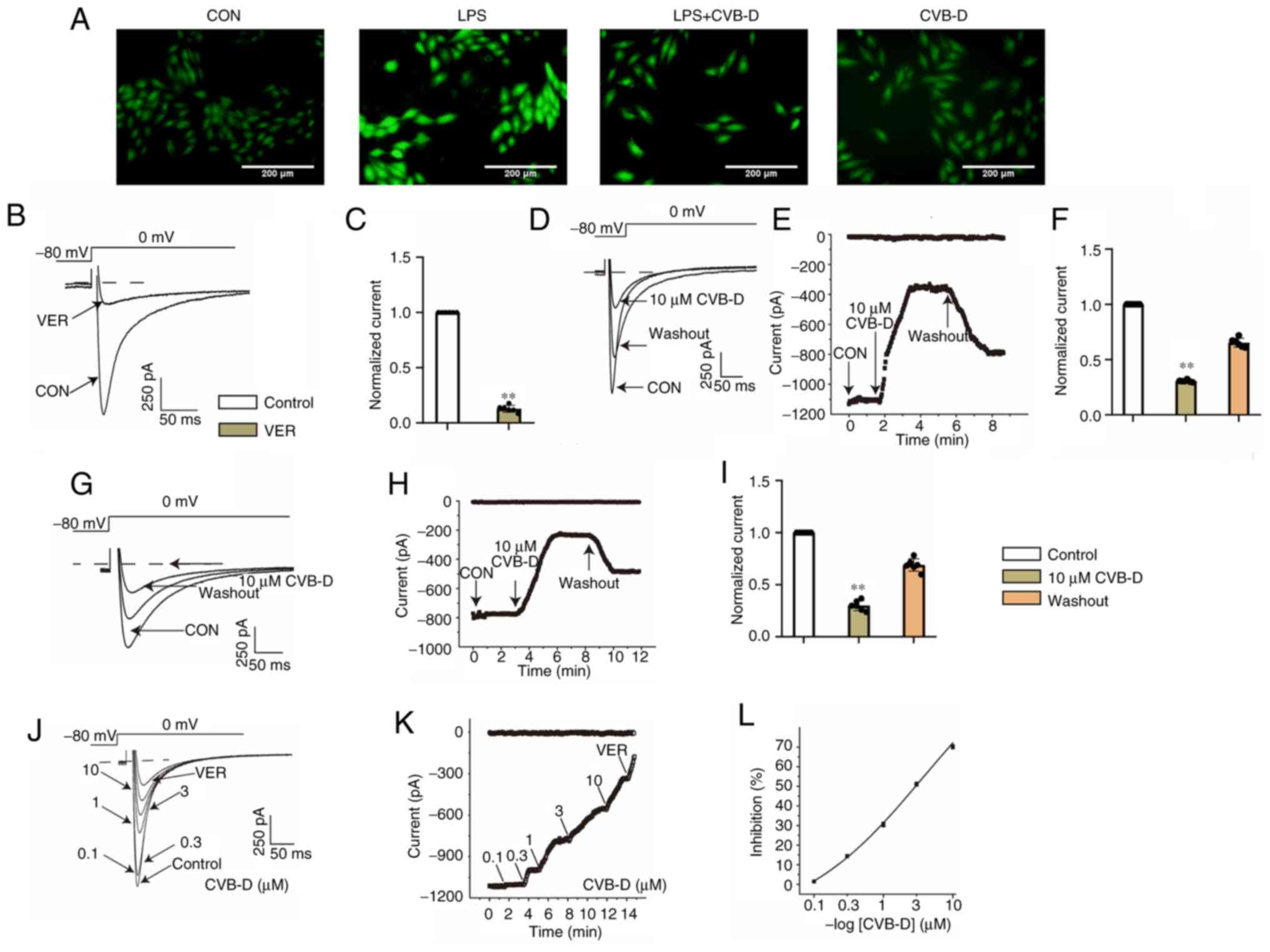 | Figure 6CVB-D attenuates calcium overload
caused by LPS. (A) Fluorescence microscopy image of different
treatment groups of H9C2 cells: CON, LPS, LPS + CVB-D and CVB-D.
(B) Exemplary trace and (C) summary data of ICa-L recordings of
VER. (D) Typical traces of normal cells. (E) Time course of
ICa-L in normal cells. (F) Pooled data recorded normal
cells under CON, 10 µM CVB-D and washout conditions. (G) Typical
traces of the CLP group cells. (H) Time course of ICa-L
in the CLP group cells. (I) Pooled data recorded the CLP group
cells under CON, 10 µM CVB-D and washout conditions. (J) Exemplary
traces and (K) time course of ICa-L exposure to 0.1,
0.3, 1, 3 and 10 µM CVB-D and 1 µM VER were recorded. (L)
Dose-response curves show that CVB inhibited ICa-L in
ventricular myocytes. **P<0.01 vs. control group.
Data are presented as the mean ± SD (n=6). CON, control; CVB-D,
cyclovirobuxine D; LPS, lipopolysaccharide; VER, verapamil; ICa-L,
L-type Ca2+ current. |
CVB-D reduces L-type Ca2+
currents (ICa-L) and myocyte contraction
Calcium channels are ion channels which selectively
allow for the passage of calcium ions. Voltage-gated calcium
channels and ligand-gated calcium channels are two categories of
calcium channels. Voltage-gated channels include L-type, T-type and
N-type calcium channels. LTCCs are the main source of inward
calcium flow in cardiac myocytes (31).
Measurement of ICa-L. VER (1 µM),
a specific L-type calcium current blocker, almost completely
blocked LTCC, which indicated that the flowing current was
Ca2+ current. Partial recovery of LTCC was observed
after external fluid administration (Fig. 6B and C).
CVB-D inhibits ICa-L currents in
normal and ischemic cardiomyocytes. During CVB-D (10 µM)
exposure, ICa-L decreased significantly, by 69.43±0.42%.
The partial recovery of ICa-L after topical solution
washing indicated that the effect of CVB-D on ICa-L was
reversible (Fig. 6D-F). The peak
amplitude of ICa-L in CLP group ventricular myocytes was
reduced by 70.22±1.98% at 10 µM CVB-D. The L-type Ca2+
current was partially recovered after the current was stabilized
using an external solution, which indicated that CVB-D inhibited
ICa-L in normal and post-infected rat ventricular
myocytes with a partially reversible effect (Fig. 6G-I).
CVB-D concentration-dependent inhibition of
ICa-L. The current trajectories from the test of
potential depolarization from -80 mV to 0 mV under the action of
various concentrations of CVB-D were analyzed (Fig. 6J). With increasing CVB-D
concentrations (0.1-10 µmol/l), ICa-L was gradually
inhibited. CVB-D (10 µM) significantly decreased ICa-L
in normal cells and LPS-treated ventricular myocytes by increasing
the concentration of CVB-D, and the time course of ICa-L
was gradually decreased (Fig. 6K).
The time dependence of CVB-D on ICa-L was measured
(Fig. 6L). The inhibition rates of
CVB-D at 0.1, 0.3, 1, 3 and 10 µM were 1.48±0.12, 14.41±0.76,
30.51±1.15, 51.09±0.96 and 72.04±1.31%, respectively.
Effect of CVB-D on the I-V relationship of
ICa-L. The current-voltage curves of the control and
0.1, 0.3, 1, 3 and 10 µM CVB-D and VER (1 µM) treatment groups
recorded during the steady-state activation procedure were measured
(Fig. 7A). Potentials from -60-60
mV were measured at different treatment concentrations and a
representative trace was generated. In addition, CVB-D demonstrated
a marked dose-dependent effect on the current-voltage (I-V) curve
(Fig. 7B).
Effects of CVB-D on the homeostatic activation
and inactivation of ICa-L. The effect of CVB-D
concentrations (1 and 10 µmol/l) on the steady-state activation
caused a significant left shift and inactivation curve did not
change significantly. In the ICa-L activation and
inactivation curves, CVB-D caused a marked leftward movement. The
V2/1 value/slope factor (k) of activated CON and CVB-D (1 and 10
µmol/l) were -9.685±1.774/8.880±1.631, -15.609±1.565/8.541±1.486
and -20.646±0.7345/6.1189±0.668, respectively (Fig. 7C). The V2/1/slope factor (k) values
of the inactivation curves in CON and CVB-D (from 1 and 10 µmol/l)
were -35.037±0.066/4.831±0.054, -36.649±0.091/5.333±0.080 and
-38.624±0.262/5.712±0.241, respectively (Fig. 7D).
CVB-D inhibits ventricular myocyte
contractility
Recordings of the process of cardiomyocyte
shortening were performed (Fig.
7E) and the effect of CVB-D treatment (10 µmol/l) on myocyte
shortening was analyzed (Fig. 7F).
CVB-D significantly inhibited myocyte shortening by 48.87±5.44%.
After washing out, the contractility partially returned (Fig. 7G).
Discussion
Sepsis-derived lesions can involve numerous organs,
including the heart and kidneys (32). SC is an important link in the
process of multiple organ dysfunction syndrome. In previous years,
numerous active ingredients have improved myocardial injury caused
by sepsis through various mechanisms (33). For instance, luteolin increases
autophagy through adenosine 5'-monophosphate-activated protein
kinase (AMPK) signaling, thereby ameliorating LPS-induced
myocardial injury (34), Ferr-1
alleviates sepsis-induced cardiac dysfunction through the toll-like
receptor 4//NF-κB signaling pathway (35) and puerarin prevents iron deposition
by acting on the AMPK signaling pathway and protecting the
myocardium from LPS-induced injury (36). Proinflammatory cytokines act
synergistically to reduce myocardial contractility. And ROS-induced
oxidative stress-mediated mitochondrial damage accelerates
mitochondrial malfunction, improves cardiac function and reduces
mortality (37). Although much
progress has been made in understanding the mechanisms underlying
this disease, clinically available treatments are still limited,
and thus, finding new drugs for the treatment of SC is
important.
CVB-D is utilized in China in the prevention and
therapy of a variety of cardiovascular dysfunction-related
disorders including angina pectoris, coronary heart disease,
arrhythmia and heart failure (38). CVB-D has been reported to protect
against diabetic cardiomyopathy by triggering Nrf2-mediated
antioxidant responses (16). It
has also been reported to improve the anti-inflammatory response in
LPS-stimulated mouse macrophages (39). In the present study, it was
hypothesized that CVB-D may protect the myocardium from LPS-induced
damage by inhibiting oxidative stress. Inflammation is the initial
biological process associated with SC and is a hallmark for the
development of SC (40). TNF-α,
IL-6 and IL-1β are major inflammatory mediators, which are
overproduced in sepsis (41). The
present study demonstrated that CVB-D reduced the transcription
levels of inflammatory mediators TNF-α, IL-6 and IL-1β in
LPS-stimulated cardiomyocytes. In addition, the overproduction of
ROS caused lipid oxidation, further compromising cellular
integrity. CVB-D has been reported to play an anti-oxidative stress
role in in cardiomyocytes (42).
Therefore, the present study measured the protein expression levels
of the cysteine/glutamate transporter SLC7A11, which is the main
source of cysteine used for synthesizing glutathione, and of the
lipid hydroperoxidase GPX4, which serves an important role in
eliminating lipid peroxidation (43). Therefore, CVB-D may alleviate
myocardial injury by regulating ferroptosis in H9C2 cells.
Ferroptosis is a highly iron-dependent and lipid
peroxidation-driven form of cell death (44). Ferroptosis is essential for the
physiological function of biological systems through the
involvement of iron in various pathways such as iron metabolism,
oxidative stress and lipid peroxidation (45). In a state of oxidative stress,
superoxide is produced over a short period of time, reducing ferric
iron stored in ferritin (composed of FtH and FtL) to ferrous iron
and leading to its release from the cell (45). DMT1 and TFR1 act as an ‘on’ switch
for cellular iron homeostasis to take up extracellular iron into
the cell, and FPN1 acts as an ‘off’ switch in the system
responsible for exporting excess iron from inside to outside of the
cells (44). The results of the
present study demonstrated that CVB-D treatment significantly
reduced intracellular iron content. Hepcidin is a hormone that
serves a crucial role in iron homeostasis management (46). STAT3 has previously been identified
as a critical transcription factor involved in the stimulation of
iron-regulated hormone expression of genes in the liver by
IL-6(47). The findings of the
present study support that CVB-D reduces intracellular iron by
suppressing inflammation and decreasing hepcidin transcription
produced by the IL-6/STAT3 pathway, enhancing iron release.
To further investigate the results observed in the
present study, production of ferroptosis-related proteins was
measured. GPX4, a member of the GPX protein family, serves a key
role as a critical regulator in ferroptosis, mainly through the
inhibition of lipid peroxide formation (48). System xc- is located in
phospholipid duplexes and serves as an important part of the
cellular antioxidant system, comprising SLC7A11 and SLC3A2(49). Inhibition of system xc-
activities influences GSH synthesis by inhibiting cystine uptake,
which leads to decreased GPX activity, reduced cellular antioxidant
capacity and lipid peroxidation accumulation, resulting in
oxidative damage and ferroptosis (50). As demonstrated in the present
study, the group of LPS-treated H9C2 cells expressed lower levels
of GPX4 and SLC7A11 and CVB-D prevented this impact, which
indicated that ferroptosis is implicated in the development of SC
and CVB-D suppresses ferroptosis.
Nrf2 is considered to be a key regulator of
resistance to oxidative stress and a number of its downstream
targets serve critical roles in redox homeostasis (51), during which it impacts the
expression of SLC7A11 and GPX4(52). The present study demonstrated that
CVB-D increased Nrf2 in LPS-treated H9C2 cells, which indicated
that CVB-D triggered the translocation of Nrf2; however, the Nrf2
inhibitor ML385 decreased this effect of CVB-D on GPX4 and SLC7A11
levels in LPS-treated H9C2 cells. The results from the present
study suggest that CVB-D activated the Nrf2/SLC7A11/GPX4 signaling
axis in LPS-treated H9C2 cells.
Previous studies have reported that calcium serves a
critical role in regulating ferroptosis through several pathways
(53). It has been reported that
excess Ca2+ levels increase calpain protease activity,
which is responsible for cleaving BH3-interacting domain death
agonist and apoptosis-inducing factor, two proteins that contribute
to ferroptosis (54). Moreover,
elevated intracellular Ca2+ contributes to activated
calcium-dependent phospholipase A2, which promotes arachidonic acid
release from cell membranes and initiates ferroptosis through the
accumulation of lipid peroxides after the esterification of
arachidonic acid into phosphatidylethanolamines (55). In line with this notion, a recent
study reported that luminal Ca2+ stores alter
sensitivity to ferroptosis by inducing lipid remodeling,
specifically by altering lipid elongation and saturation state
(56). Additionally, calcium can
also regulate ferroptosis by modulating the activation of the
endoplasmic reticulum stress response (57). Notably, calcium can lead to an
increase in ROS by affecting the electron transport chain and
regulating oxidative phosphorylation (58). Thus, in turn, lipid peroxidation is
promoted and ferroptosis is triggered (59). Understanding how calcium regulates
ferroptosis could provide potential therapeutic targets for
diseases associated with dysregulated cell death.
The present study demonstrated the beneficial effect
of CVB-D on sepsis-induced cardiac dysfunction by decreasing
ferroptosis. CVB-D may decrease sepsis-induced cardiomyocyte
ferroptosis by inhibiting oxidative stress, iron overload, lipid
peroxidation and the inward flow of calcium ions. Mechanistically,
CVB-D inhibited LPS-induced iron overload by promoting
ferroportin-mediated cellular iron release via the
IL-6/STAT3/hepcidin axis. Furthermore, CVB-D was demonstrated to
inhibit ferroptosis by activating the Nrf2/SLC7A11/GPX4 axis in
cardiomyocytes, which may contribute to its protective effects
against myocardial injury. Additionally, CVB-D may inhibit LTCC,
thus suppressing elevated cellular calcium and iron levels. These
findings emphasize the potential therapeutic value of CVB-D in the
management of sepsis-induced cardiac dysfunction (Fig. 8).
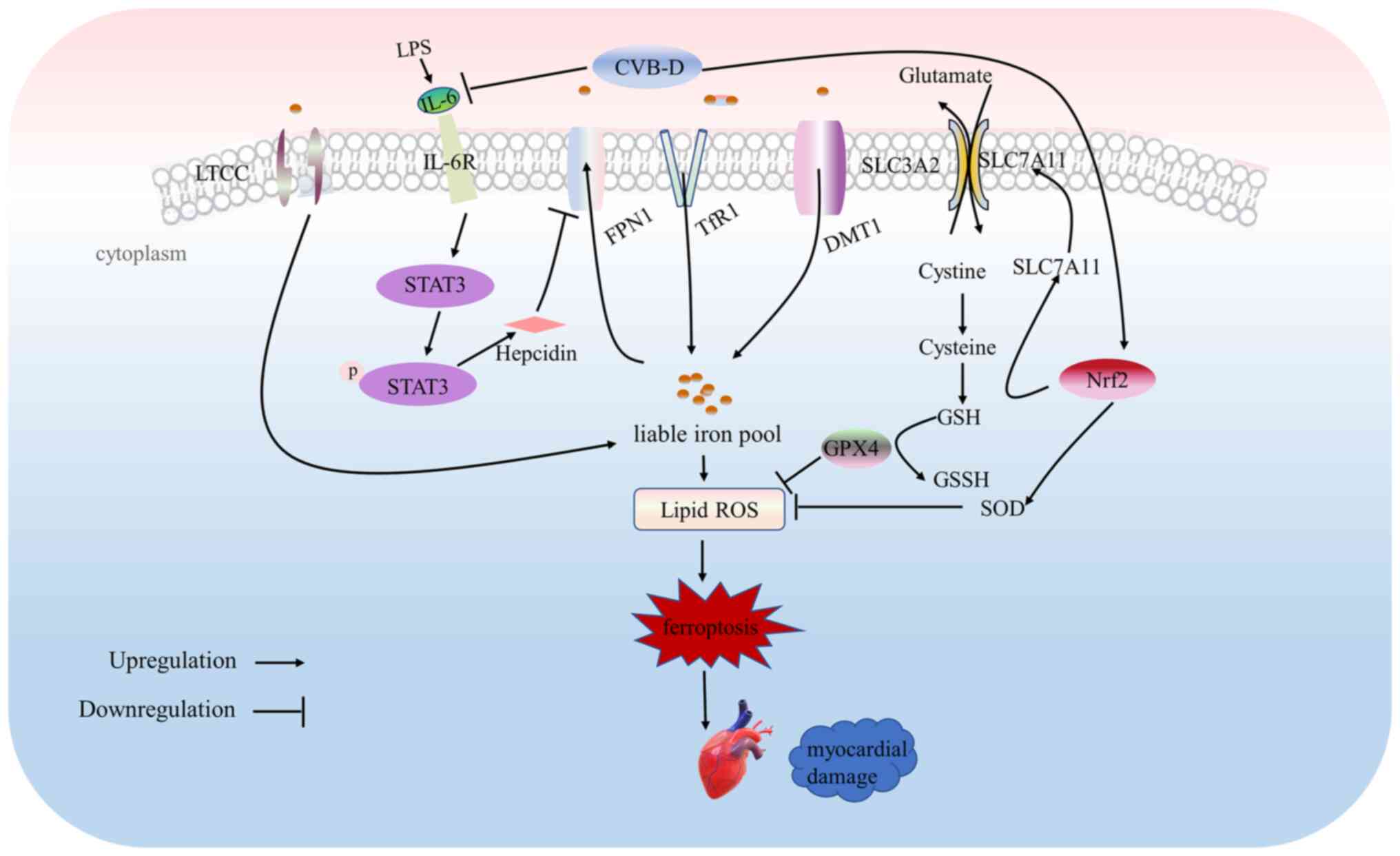 | Figure 8Proposed signaling pathways involved
in the protective effect of CVB-D in septic cardiomyopathy.
Cellular injury, ferroptosis and oxidative stress induced by LPS
are attenuated by CVB-D. CVB-D downregulated cellular iron
accumulation by alleviating cellular inflammatory responses.
Furthermore, CVB-D also upregulated the Nrf2/SLC7A11/GPX4 pathway.
CVB-D may reduce the influx of calcium ions by inhibiting LTCCs,
thus reducing myocardial injury. CVB-D, cyclovirobuxine D; LPS,
lipopolysaccharide; LTCCs, L-type calcium channels; SLC7A11, solute
carrier family 7 member 11; Nrf2, nuclear factor erythroid
2-related factor 2; GPX4, glutathione peroxidase 4; ROS, reactive
oxygen species; p, phosphorylated; IL, interleukin; R, receptor;
SLC3A2, solute carrier family 3 member 2; STAT3, signal transducer
and activator of transcription 3; FPN1, ferroportin 1; TfR1,
transferrin receptor 1; DMT1, divalent metal transporter 1; GSH,
glutathione; GSSH, glutathione disulfide, SOD, superoxide
dismutase. |
CVB-D ameliorated the impairment of cardiac function
induced by CLP surgery in rats. In addition, CVB-D regulated the
expression of iron metabolism-related proteins in LPS-stimulated
H9C2 cells and ameliorated the inflammatory response by inhibiting
the IL-6/STAT3 signaling pathway and reducing the expression of
hepcidin. Furthermore, in LPS-stimulated H9C2 cells, CVB-D exerted
a protective effect through the Nrf2/SLC7A11/GPX4 pathway. The
results of the present study are the first to report that CVB-D
ameliorates ferroptosis by increasing cell survival and thus
effectively ameliorates LPS-induced SC in association with
activation of Nrf2/SLC7A11/GPX4 pathway targets. It was also
demonstrated that CVB-D significantly reduced calcium
contractility, which could be associated with inhibition of LTCCs.
The present study demonstrated that CVB-D could reduce myocardial
injury by inhibiting LTCCs to reduce calcium ion influx. Further
in vivo and clinical studies are required to assess the
safety and efficacy of CVB-D in SC, and to fully elucidate the
mechanistic links between CVB-D treatment, ferroptosis and
cardioprotection.
Acknowledgements
Not applicable.
Funding
Funding: This project was supported by the Science Foundation of
Hebei Normal University (grant no. L2021B47).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ESJ and NW conceived and planned the study, provided
supervision and obtained funding. JXW and PG collected data and
drafted the manuscript. YC and MX analyzed data and reviewed and
edited the manuscript. JXW and PG confirm the authenticity of all
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All procedures were performed with the approval of
the Animal Care and Use Committee of Hebei University of Chinese
Medicine (approval no. DWLL202206006).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Beesley SJ, Weber G, Sarge T, Nikravan S,
Grissom CK, Lanspa MJ, Shahul S and Brown SM: Septic
cardiomyopathy. Crit Care Med. 46:625–634. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tullo G, Candelli M, Gasparrini I, Micci S
and Franceschi F: Ultrasound in Sepsis and septic shock-from
diagnosis to treatment. J Clin Med. 12(1185)2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Flierl MA, Rittirsch D, Huber-Lang MS,
Sarma JV and Ward PA: Molecular events in the cardiomyopathy of
sepsis. Mol Med. 14:327–336. 2008.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Shang X, Zhang Y, Xu J, Li M, Wang X and
Yu R: SRV2 promotes mitochondrial fission and Mst1-Drp1 signaling
in LPS-induced septic cardiomyopathy. Aging. 12:1417–1432.
2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zheng Z, Ma H, Zhang X, Tu F, Wang X, Ha
T, Fan M, Liu L, Xu J, Yu K, et al: Enhanced glycolytic metabolism
contributes to cardiac dysfunction in polymicrobial sepsis. J
Infectious Dis. 215:1396–1406. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhu X, Sun M, Guo H, Lu G, Gu J, Zhang L,
Shi L, Gao J, Zhang D, Wang W, et al: Verbascoside protects from
LPS-induced septic cardiomyopathy via alleviating cardiac
inflammation, oxidative stress and regulating mitochondrial
dynamics. Ecotoxicol Environ Saf. 233(113327)2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yang Y, Lei W, Qian L, Zhang S, Yang W, Lu
C, Song Y, Liang Z, Deng C, Chen Y, et al: Activation of NR1H3
signaling pathways by psoralidin attenuates septic myocardial
injury. Free Radical Biol Med. 204:8–19. 2023.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Venkataramani V: Iron homeostasis and
metabolism: Two sides of a coin. Adv Exp Med Biol. 1301:25–40.
2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Rochette L, Dogon G, Rigal E, Zeller M,
Cottin Y and Vergely C: Lipid peroxidation and Iron metabolism: Two
corner stones in the homeostasis control of ferroptosis. Int J Mol
Sci. 24(449)2022.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Islam S, Jarosch S, Zhou J, Parquet Mdel
C, Toguri JT, Colp P, Holbein BE and Lehmann C: Anti-inflammatory
and anti-bacterial effects of iron chelation in experimental
sepsis. J Surg Res. 200:266–273. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C,
Wu H, Deng W, Shen D and Tang Q: Ferritinophagy-mediated
ferroptosis is involved in sepsis-induced cardiac injury. Free
Radical Biol Med. 160:303–318. 2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wang C, Yuan W, Hu A, Lin J, Xia Z, Yang
CF, Li Y and Zhang Z: Dexmedetomidine alleviated sepsis-induced
myocardial ferroptosis and septic heart injury. Mol Med Rep.
22:175–184. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
National Pharmacopoeia Committee, ed.
Pharmacopoeia of People's Republic of China. Beijing: Chemical
Industry Press, 2010.
|
|
15
|
Guo Q, Guo J, Yang R, Peng H, Zhao J, Li L
and Peng S: Cyclovirobuxine D attenuates doxorubicin-induced
cardiomyopathy by suppression of oxidative damage and mitochondrial
biogenesis impairment. Oxid Med Cell Longev.
2015(151972)2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Jiang Z, Fu L, Xu Y, Hu X, Yang H, Zhang
Y, Luo H, Gan S, Tao L, Liang G and Shen X: Cyclovirobuxine D
protects against diabetic cardiomyopathy by activating
Nrf2-mediated antioxidant responses. Sci Rep.
10(6427)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Xiang ZN, Su JC, Liu YH, Deng B, Zhao N,
Pan J, Hu ZF, Chen FH, Cheng BY, Chen JC and Wan LS: Structurally
diverse alkaloids from Buxus sempervirens with cardioprotective
activity. Bioorg Chem. 109(104753)2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Fang H, Gong C, Fu J, Liu X, Bi H, Cheng
Y, Liu Y, Tang Y and Wang D: Evaluation of 2 rat models for sepsis
developed by improved cecal ligation/puncture or feces
intraperitoneal-injection. Med Sci Monitor.
26(e919054)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Qin LY, Guan P, Wang JX, Chen Y, Zhao YS,
Yang SC, Guo YJ, Wang N and Ji ES: Therapeutic potential of
Astragaloside IV against adriamycin-induced renal damage in rats
via ferroptosis. Front Pharmacol. 13(812594)2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chen DD, Wang HW and Cai XJ: Transcription
factor Sp1 ameliorates sepsis-induced myocardial injury via
ZFAS1/Notch signaling in H9C2 cells. Cytokine.
140(155426)2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Xiong B, Chen L, Huang Y, Lu G, Chen C,
Nong J and Pan H: ZBTB16 eases lipopolysaccharide-elicited
inflammation, apoptosis and degradation of extracellular matrix in
chondrocytes during osteoarthritis by suppressing GRK2
transcription. Exp Ther Med. 25(276)2023.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Varghese J, James J, Vaulont S, McKie A
and Jacob M: Increased intracellular iron in mouse primary
hepatocytes in vitro causes activation of the Akt pathway but
decreases its response to insulin. Biochim Biophys Acta Gen Subj.
1862:1870–1882. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Guan P, Sun ZM, Wang N, Zhou J, Luo LF,
Zhao YS and Ji ES: Resveratrol prevents chronic intermittent
hypoxia-induced cardiac hypertrophy by targeting the PI3K/AKT/mTOR
pathway. Life Sci. 233(116748)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Liu L, Liu F, Sun Z, Peng Z, You T and Yu
Z: LncRNA NEAT1 promotes apoptosis and inflammation in LPS-induced
sepsis models by targeting miR-590-3p. Exp Ther Med. 20:3290–3300.
2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hu X and Miao H: MiR-539-5p inhibits the
inflammatory injury in septic H9c2 cells by regulating IRAK3. Mol
Biol Rep. 49:121–130. 2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Cheng J, Zhu Y, Xing X, Xiao J, Chen H,
Zhang H, Wang D, Zhang Y, Zhang G, Wu Z and Liu Y:
Manganese-deposited iron oxide promotes tumor-responsive
ferroptosis that synergizes the apoptosis of cisplatin.
Theranostics. 11:5418–5429. 2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kong Y, Hu L, Lu K, Wang Y, Xie Y, Gao L,
Yang G, Xie B, He W, Chen G, et al: Ferroportin downregulation
promotes cell proliferation by modulating the Nrf2-miR-17-5p axis
in multiple myeloma. Cell Death Dis. 10(624)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kumfu S, Chattipakorn SC, Fucharoen S and
Chattipakorn N: Dual T-type and L-type calcium channel blocker
exerts beneficial effects in attenuating cardiovascular dysfunction
in iron-overloaded thalassaemic mice. Exp Physiol. 101:521–539.
2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Hardy N, Viola HM, Johnstone VP, Clemons
TD, Cserne Szappanos H, Singh R, Smith NM, Iyer KS and Hool LC:
Nanoparticle-mediated dual delivery of an antioxidant and a peptide
against the L-Type Ca2+ channel enables simultaneous reduction of
cardiac. ACS Nano. 9:279–289. 2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Bai T, Wang S, Zhao Y, Zhu R, Wang W and
Sun Y: Haloperidol, a sigma receptor 1 antagonist, promotes
ferroptosis in hepatocellular carcinoma cells. Biochem Biophys Res
Commun. 491:919–925. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Xin T and Lu C: SirT3 activates
AMPK-related mitochondrial biogenesis and ameliorates
sepsis-induced myocardial injury. Aging. 12:16224–16237.
2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wu B, Song H, Fan M, You F, Zhang L, Luo
J, Li J, Wang L, Li C and Yuan M: Luteolin attenuates
sepsis-induced myocardial injury by enhancing autophagy in mice.
Int J Mol Med. 45:1477–1487. 2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xiao Z, Kong B, Fang J, Qin T, Dai C,
Shuai W and Huang H: Ferrostatin-1 alleviates
lipopolysaccharide-induced cardiac dysfunction. Bioengineered.
12:9367–9376. 2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhou B, Zhang J, Chen Y, Liu Y, Tang X,
Xia P, Yu P and Yu S: Puerarin protects against sepsis-induced
myocardial injury through AMPK-mediated ferroptosis signaling.
Aging. 14:3617–3632. 2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Liu YC, Yu MM, Shou ST and Chai YF:
Sepsis-induced cardiomyopathy: Mechanisms and treatments. Front
Immunol. 8(1021)2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Ke Z, Hou X and Jia XB: Design and
optimization of self-nanoemulsifying drug delivery systems for
improved bioavailability of cyclovirobuxine D. Drug Design Dev
Ther. 10:2049–2060. 2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Guo D, Li JR, Wang Y, Lei LS, Yu CL and
Chen NN: Cyclovirobuxinum D suppresses lipopolysaccharide-induced
inflammatory responses in murine macrophages in vitro by blocking
JAK-STAT signaling pathway. Acta Pharmacol Sinica. 35:770–778.
2014.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kakihana Y, Ito T, Nakahara M, Yamaguchi K
and Yasuda T: Sepsis-induced myocardial dysfunction:
Pathophysiology and management. J Intensive Care.
4(22)2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Baek HS, Min HJ, Hong VS, Kwon TK, Park
JW, Lee J and Kim S: Anti-inflammatory effects of the novel PIM
kinase inhibitor KMU-470 in RAW 264.7 cells through the
TLR4-NF-κB-NLRP3 pathway. Int J Mol Sci. 21(5138)2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Yu B, Fang TH, Lü GH, Xu HQ and Lu JF:
Beneficial effect of Cyclovirobuxine D on heart failure rats
following myocardial infarction. Fitoterapia. 82:868–877.
2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Koppula P, Zhuang L and Gan B: Cystine
transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient
dependency, and cancer therapy. Protein Cell. 12:599–620.
2021.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Fang X, Ardehali H, Min J and Wang F: The
molecular and metabolic landscape of iron and ferroptosis in
cardiovascular disease. Na Rev Cardiol. 20:7–23. 2023.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Broxmeyer HE, Cooper S, Levi S and Arosio
P: Mutated recombinant human heavy-chain ferritins and
myelosuppression in vitro and in vivo: A link between ferritin
ferroxidase activity and biological function. Proc Natl Acad Sci
USA. 88:770–774. 1991.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Zhang H, Ostrowski R, Jiang D, Zhao Q,
Liang Y, Che X, Zhao J, Xiang X, Qin W and He Z: Hepcidin promoted
ferroptosis through Iron metabolism which is associated with DMT1
signaling activation in early brain injury following subarachnoid
hemorrhage. Oxidative Med Cell Longevity.
2021(9800794)2021.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Pietrangelo A, Dierssen U, Valli L, Garuti
C, Rump A, Corradini E, Ernst M, Klein C and Trautwein C: STAT3 is
required for IL-6-gp130-dependent activation of hepcidin in vivo.
Gastroenterology. 132:294–300. 2007.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao
N, Sun B and Wang G: Ferroptosis: Past, present and future. Cell
Death Dis. 11(88)2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Fotiadis D, Kanai Y and Palacín M: The
SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med.
34:139–158. 2013.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Lei P, Bai T and Sun Y: Mechanisms of
ferroptosis and relations with regulated cell death: A review.
Front Physiol. 10(139)2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Dodson M, Castro-Portuguez R and Zhang DD:
NRF2 plays a critical role in mitigating lipid peroxidation and
ferroptosis. Redox Biol. 23(101107)2019.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Ma H, Wang X, Zhang W, Li H, Zhao W, Sun J
and Yang M: Melatonin suppresses ferroptosis induced by high
glucose via activation of the Nrf2/HO-1 signaling pathway in type 2
diabetic osteoporosis. Oxid Med Cell Longev.
2020(9067610)2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Dhaouadi N, Vitto VAM, Pinton P, Galluzzi
L and Marchi S: Ca(2+) signaling and cell death. Cell Calcium.
113(102759)2023.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Neitemeier S, Jelinek A, Laino V, Hoffmann
L, Eisenbach I, Eying R, Ganjam GK, Dolga AM, Oppermann S and
Culmsee C: BID links ferroptosis to mitochondrial cell death
pathways. Redox Biol. 12:558–570. 2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Li D and Li Y: The interaction between
ferroptosis and lipid metabolism in cancer. Signal Transduct Target
Ther. 5(108)2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Xin S, Mueller C, Pfeiffer S, Kraft VAN,
Merl-Pham J, Bao X, Feederle R, Jin X, Hauck SM, Schmitt-Kopplin P
and Schick JA: MS4A15 drives ferroptosis resistance through
calcium-restricted lipid remodeling. Cell Death Differ. 29:670–686.
2022.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Fu F, Wang W, Wu L, Wang W, Huang Z, Huang
Y, Wu C and Pan X: Inhalable biomineralized liposomes for cyclic
Ca(2+)-Burst-centered endoplasmic reticulum stress enhanced lung
cancer ferroptosis therapy. ACS Nano. 17:5486–5502. 2023.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Madreiter-Sokolowski CT, Thomas C and
Ristow M: Interrelation between ROS and Ca(2+) in aging and
age-related diseases. Redox Biol. 36(101678)2020.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Peng TI and Jou MJ: Oxidative stress
caused by mitochondrial calcium overload. Ann N Y Acad Sci.
1201:183–188. 2010.PubMed/NCBI View Article : Google Scholar
|