Introduction
Angiogenesis refers to the sprouting of capillaries
from existing blood vessels (1).
During angiogenesis, endothelial cells adopt different phenotypes,
which allow them to proliferate and migrate, and to form tube-like
structures that eventually result in the generation of new blood
vessels (2). Physiological
angiogenesis is essential for reproduction and placentation,
whereas persistent abnormal angiogenesis drives tumor metastasis,
blindness caused by ocular neovascularization and atherosclerotic
plaque formation (3,4). The process of angiogenesis is tightly
regulated by pro-angiogenic and anti-angiogenic molecules, and is
orchestrated by the extracellular microenvironment (5). Multiple intracellular signaling
pathways mediate the angiogenic response of endothelial cells,
including vascular endothelial growth factor (VEGF)/VEGF receptor
(VEGFR) signaling, TGF-β signaling and STAT signaling (4,6,7). In
addition, all of the three major subfamilies of MAPK signaling,
ERK, JNK and p38 MAPK, are involved in endothelial cell activation
(8).
Complement factor H (CFH) is an abundant serum
glycoprotein that is constitutively expressed in the liver
(9). CFH controls the alternative
pathway of complement activation in plasma (10). Mutations in the human CFH gene are
associated with age-related macular degeneration characterized by
disruption of the retina by excessive angiogenesis (11). Intravitreal injection of human CFH
has been shown to suppress the laser-induced choroidal
neovascularization in a rat model (12). In addition, Cfh-/- mice
have been shown to exhibit a pro-angiogenic phenotype in a Matrigel
plug assay and loss of CFH can increase endothelial cell migration
(13). An in vitro Matrigel
tube formation assay showed that an increased number of tubes were
formed by endothelial cells co-cultured with ARPE-19 cells
transfected with CFH-specific small interfering RNA (14). To date, the molecular mechanisms
underlying the anti-angiogenic effect of CFH remain unclear.
STAT3 belongs to the STAT family of transcription
factors (15). Upon activation,
STATs are phosphorylated on a tyrosine residue, leading to the
formation of homo- or heterodimers, and translocation to the
nucleus, where they activate the transcription of target genes
(16,17). STAT3 can be phosphorylated on the
Y705 site by the activation of JAK (6). Multiple studies have shown that STAT3
signaling mediates tumor angiogenesis (18-20).
Membrane progesterone receptor α has been reported to promote
endothelial cell migration and tube formation in lung
adenocarcinoma through STAT3 signaling (21). Furthermore, STATTIC, a specific
inhibitor of STAT3, was shown to reduce radiation-induced migration
and invasion in hepatocellular carcinoma cells (22). Knockdown of SETD7, a tumor
suppressor, can activate the STAT3 signaling pathway and enhance
lung cancer cell migration, whereas STATTIC abrogates the effect of
SETD7 on cell migration (23). In
addition, STAT3 has been shown to upregulate the transcription of
MMP-2, whereas blockade of STAT3 through the expression of a
dominant-negative STAT3 significantly suppresses MMP-2 expression
in metastatic tumor cells (24).
VEGF refers to a family of growth factors that have
potent pro-angiogenic activity (25-27).
VEGF family members bind to their tyrosine kinase cell receptors
(VEGFRs) on endothelial cells (28). VEGFR2 is considered to have the
strongest pro-angiogenic activity (29-31),
and has been proven to be a downstream effector of STAT3 signaling
(32,33). STAT3 has also been shown to
directly bind the promoter of the VEGFR2 gene and activate its
transcription (34).
The aim of this study was to evaluate the effect of
CFH on HUVEC viability and migration. In addition, the effects of
CFH on STAT3, MAPK and TGF-β signaling pathways in HUVECs were
measured, as well as the effects of CFH on VEGFR2 expression. This
study elucidated the molecular mechanisms underlying CFH-inhibited
angiogenesis.
Materials and methods
Cell culture and transduction
HUVECs (cat. no. 8000) were purchased from ScienCell
Research Laboratories, Inc. HUVECs were cultured in endothelial
cell medium (ECM; ScienCell Research Laboratories, Inc.)
supplemented with 5% (v/v) fetal bovine serum (FBS; Thermo Fisher
Scientific, Inc.), 1% endothelial cell growth supplements (ECGS;
ScienCell Research Laboratories, Inc.) and 1%
penicillin/streptomycin solution. The liver cancer cell line HepG2
(cat. no. CRL-10741) was purchased from American Type Culture
Collection. HepG2 cells were cultured in Dulbecco's modified
Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% (v/v) FBS and antibiotics (100 IU/ml penicillin and 100
mg/ml streptomycin). Cells underwent STR genotyping. The CFH
adenovirus (plasmid GV314 harboring CFH) and control virus (empty
GV314 plasmid) expressing enhanced green fluorescent protein were
obtained from Shanghai Jikai Gene Chemical Technology Co., Ltd.
Adenovirus infection of HepG2 cells was carried out according to
the manufacturer's protocol, as previously described (35). After 48 h, the culture medium
supernatant and HepG2 cells were harvested and expression of CFH
was tested by western blotting and reverse
transcription-quantitative PCR (RT-qPCR). The supernatant of HepG2
cells was collected to perform subsequent experiments.
Inhibition and stimulation of STAT3
signaling
The culture medium for CFH adenovirus-infected HepG2
cells (CFH-containing conditioned medium) or non-infected HepG2
cells (control medium) was collected. HUVECs were seeded in 6-well
plates at a density of 5x105 cells/well and cultured in
CFH-containing conditioned medium or control medium for 24 h at
37˚C. For STATTIC treatment, when cells reached 100% confluence,
STATTIC (Apexbio Technology LLC) was added to the media and
incubated with cells at different concentrations (0, 2.5, 5, 7.5
and 10 µM). Dimethyl sulfoxide (DMSO; Beijing Solarbio Science
& Technology Co., Ltd.) was used as a control. Cells were
harvested after 4 h of incubation at 37˚C and western blot analysis
was used to detect the effects of STATTIC on phosphorylated
(p)-STAT3 and the optimal dose. The optimal dose (7.5 µM) was used
to perform the subsequent experiments.
GV230 is a eukaryotic expression plasmid. The
full-length (NM_139276) gene of the Y705D-mutant STAT3 was cloned
into the GV230 expression plasmid (Shanghai Genechem Co., Ltd.) by
using XhoI and KpnI restriction sites. The Y705D
mutation is a phosphorylation mimicking form of STAT3(6). For STAT3 (Y705D) transfection, when
cells reached 40-90% confluence, Lipofectamine® 3000
(cat. no. L3000015; Invitrogen; Thermo Fisher Scientific, Inc.) and
2.5 µg STAT3 (Y705D) vector were incubated at room temperature for
15 min and then added to each well. Empty GV230 plasmid was used as
the control. Transfection was performed according to the
Lipofectamine 3000 manufacturer's protocol. The culture medium was
changed 6 h after transfection. After 24 h of transfection,
subsequent experiments were performed.
Western blot analysis and Coomassie
blue staining
The lysates of HepG2 or HUVECs were prepared with
RIPA buffer containing 1 mM PMSF (Beyotime Institute of
Biotechnology). Protein concentrations were quantified with a BCA
protein assay kit (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. Protein samples (20 µg) were loaded and
separated by SDS-PAGE on a 10% SDS-polyacrylamide gel. The gels
were incubated with Coomassie blue staining solution (Beijing
Solarbio Science & Technology Co., Ltd.) and then completely
destained in a 5% ethanol/10% acetic acid solution until clear
bands appeared. For western blotting, proteins were transferred
onto PVDF membranes (MilliporeSigma) and blocked with 5% nonfat dry
milk buffer for 2 h at room temperature, followed by incubation
with primary antibodies against CFH (1:1,000; cat. no. ab118820;
Abcam), p-ERK1/2 (1:2,000; cat. no. 4370; Cell Signaling
Technology, Inc.), ERK1/2 (1:1,000; cat. no. 4695; Cell Signaling
Technology, Inc.), p-JNK (1:1,000; cat. no. 4668; Cell Signaling
Technology, Inc.), JNK (1:1,000; cat. no. 9252; Cell Signaling
Technology, Inc.), p-p38 MAPK (1:1,000; cat. no. 4511; Cell
Signaling Technology, Inc.), p38 MAPK (1:1,000; cat. no. 8690; Cell
Signaling Technology, Inc.), TGFβ receptor 1 (TGFβR1; 1:1,000; cat.
no. ab235578; Abcam), p-Smad2 (1:1,000; cat. no. ab188334; Abcam),
Smad2 (1:2,000; cat. no. ab40855; Abcam), p-Smad3 (1:1,000; cat.
no. 9520; Cell Signaling Technology, Inc.), Smad3 (1:2,000; cat.
no. 9145; Cell Signaling Technology, Inc.), p-STAT3 (1:1,000; cat.
no. 9520; Cell Signaling Technology, Inc.), STAT3 (1:1,000; cat.
no. 9139; Cell Signaling Technology, Inc.), VEGFR2 (1:1,000; cat.
no. 2479S; Cell Signaling Technology, Inc.) and GAPDH (1:10,000;
cat. no. 10494-1-AP; Proteintech Group, Inc.) at 4˚C overnight.
Subsequently, the membranes were cultivated with HRP-conjugated
secondary antibody (Goat Anti-Rabbit IgG; 1:10,000; cat. no.
111-035-003; and HRP-Goat anti-Mouse IgG; 1:10,000; cat. no.
115-035-003; both from Jackson ImmunoResearch Laboratories, Inc.)
at room temperature for 2 h. Immobilon western chemiluminescent HRP
substrate (MilliporeSigma) was used to detect chemiluminescence.
The blots were visualized using an ECL imaging system and relative
protein expression levels were calculated using ImageJ 1.8.0
(National Institutes of Health).
RNA extraction and RT-qPCR
Total RNA was isolated from the HUVECs and HepG2
cells using the E.Z.N.A. Total RNA Kit (Omega Bio-Tek, Inc.). cDNA
synthesis and amplification were subsequently performed using
HiScript®III RT Super Mix (cat no. R323-01; Vazyme
Biotech Co., Ltd.) according to the manufacturer's instructions.
The RT procedure was: 2 min at 42˚C, 15 min at 37˚C, 5 sec at 85˚C
and then 4˚C for 30 min. ChamQ Universal SYBR qPCR Master Mix (cat
no. Q711-02; Vazyme Biotech Co., Ltd.) was used for qPCR analysis
with the following thermocycling conditions: 95˚C for 30 sec,
followed by 40 cycles at 95˚C for 10 sec and 60˚C for 30 sec. GAPDH
was used as an internal control and the relative expression levels
of the target gene were calculated using the 2-∆∆Cq
method (36). The primers were
designed as follows: hCFH, forward 5'-GTGAAGTGTTTACCAGTGACAGC-3',
reverse 5'-AACCGTACTGCTTGTCCAAA-3'; hVEGFR2, forward
5'-TTAGTGACCAACATGGAGTCGTG-3', reverse
5'-TAGTAAAGCCCTTCTTGCTTTCC-3'; and hGAPDH, forward
5'-TGATGACATCAAGAAGGTGGTGAAG-3', reverse
5'-TCCTTGGAGGCCATGTGGGCCAT-3'.
Cell viability assays
Cell viability was evaluated using the MTT assay kit
Beijing Solarbio Science & Technology Co., Ltd.) and the Cell
Counting Kit-8 (CCK-8; Apexbio Technology LLC). For the MTT assay,
HUVECs were seeded into a 96-well plate at a density of
5x104 cells/well and cultured in CFH-containing
conditioned medium (the supernatant of HepG2 cells) for 24 h at
37˚C, after which, 10 µl serum-free medium containing 5 mg/ml MTT
solution was added to each well. After 4 h incubation at 37˚C, the
supernatant was discarded, and 110 µl DMSO was added. The crystals
were sufficiently dissolved and the optical absorbance value was
measured at 490 nm using a microplate reader (Multiskan GO; Thermo
Fisher Scientific, Inc.). For the CCK-8 assay, HUVECs were
incubated with CFH-containing conditioned medium for 24 h at 37˚C,
followed by the addition of 10 µl CCK-8 solution to each well and
incubation of the plates for 4 h at 37˚C in a humidified incubator.
Absorbance was measured at 450 nm using a microplate reader
(Multiskan GO; Thermo Fisher Scientific, Inc.).
Wound healing assay
The HUVECs were seeded in 6-well plates at a cell
density of 5x105/well and at 37˚C where they reached
95-100% confluence. A 10-µl pipette tip was used to vertically
scratch the 6-well plate to create a line across the surface, and
the suspended cells were cleaned and removed with PBS. Cells were
cultured in CFH-containing conditioned medium in a humidified 5%
CO2 incubator at 37˚C for 24 h. Images were captured at
0 and 24 h under a light microscope (Olympus Corporation). The
migrated area was calculated using ImageJ software.
Transwell migration assay
A Transwell chamber (pore size, 8 µm; Corning, Inc.)
was used. HUVECs were diluted to 10x104/ml with
CFH-containing conditioned medium or control medium, and a 200-µl
cell suspension was added to the upper chamber. ECM (600 µl)
supplemented with 5% FBS, 1% ECGS and 1% antibiotics was added to
the lower chamber. The cells were allowed to migrate for 12 h at
37˚C. Subsequently, they were fixed with 4% formaldehyde solution
(1 ml/well) for 10 min at room temperature and washed three times
with PBS to remove the formaldehyde solution. They were then
stained with 0.1% crystal violet (1 ml/well) for 30 min at room
temperature and washed three times with PBS to remove the stain.
Finally, migrated HUVECs were counted and images were captured from
six random fields under an inverted light microscope.
Statistical analysis
Date analysis was performed using GraphPad Prism
8.0.2 software (Dotmatics). Data are presented as the mean ±
standard deviation and all experiments were repeated at least three
times. Statistical significance between two groups was assessed
using an unpaired Student's t-test. Significance among multiple
groups was calculated using one-way ANOVA and Bonferroni's post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Overexpression of CFH in HepG2 cells
and collection of conditioned media
CFH is primarily expressed in hepatocytes. To
imitate the in vivo environment, the culture medium of HepG2
cells transduced with a control vector, which contains HepG2
cell-secreted CFH, was used as the control to mimic the condition
in normal blood. In addition, a recombinant adenovirus containing
the full-length cDNA sequence of human CFH was constructed and
transduced into HepG2 cells to induce overexpression of CFH, thus
exogenously increasing the amount of CFH in the conditioned medium.
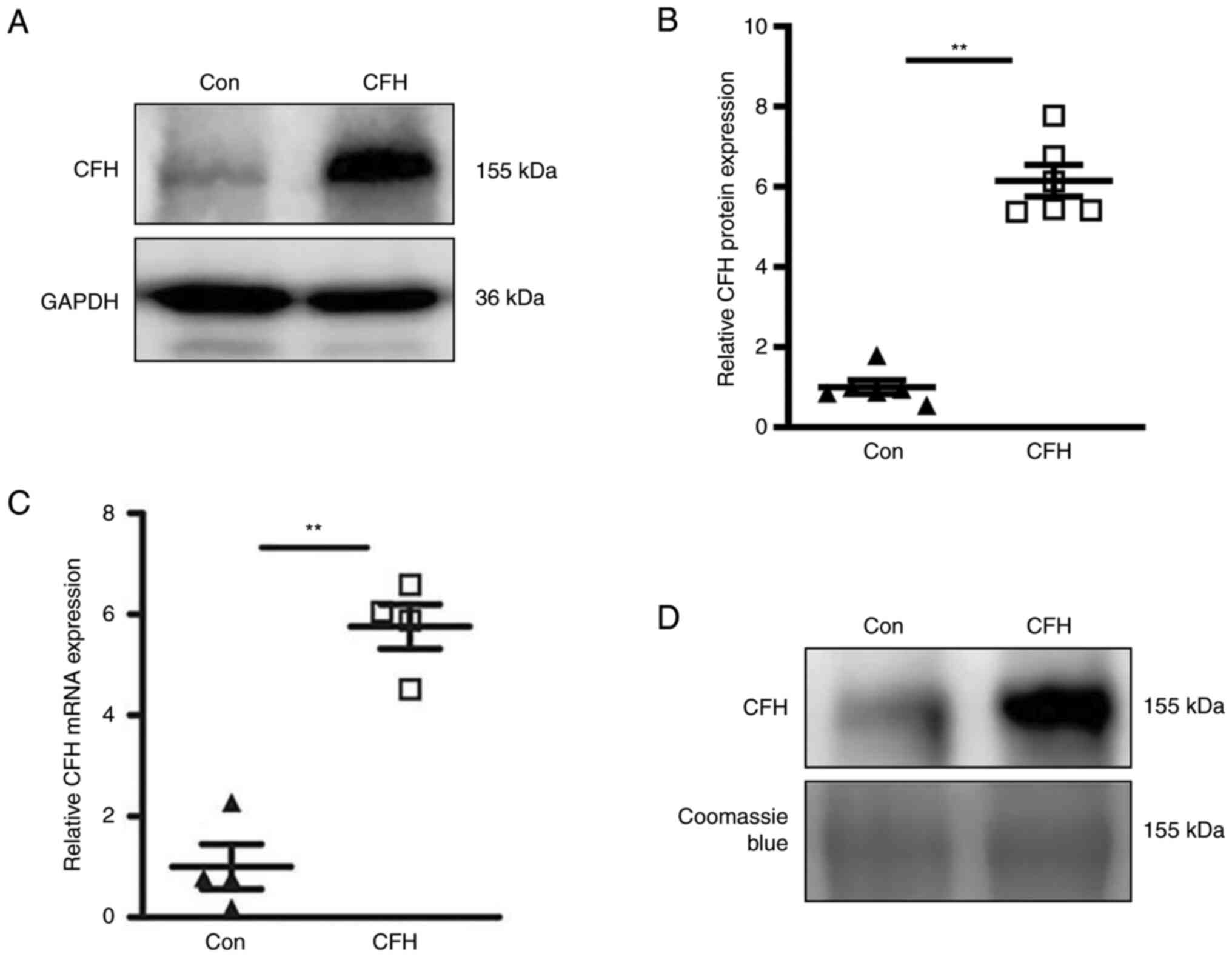
Western blotting was used to detect the protein expression levels
of CFH in HepG2 cells. The results revealed that the expression
levels of CFH in the CFH adenovirus infection group was
significantly higher than those in the control group (Fig. 1A and B). In addition, RT-qPCR results
demonstrated that the mRNA expression levels of CFH were
significantly elevated in HepG2 cells following CFH adenovirus
transduction (Fig. 1C). HepG2 cell
culture media were collected and subjected to western blot
analysis. As shown in Fig. 1D, CFH
protein expression in the conditioned medium of CFH-overexpressing
HepG2 cells was markedly elevated.
CFH does not alter the viability of
HUVECs but it does inhibit their migration
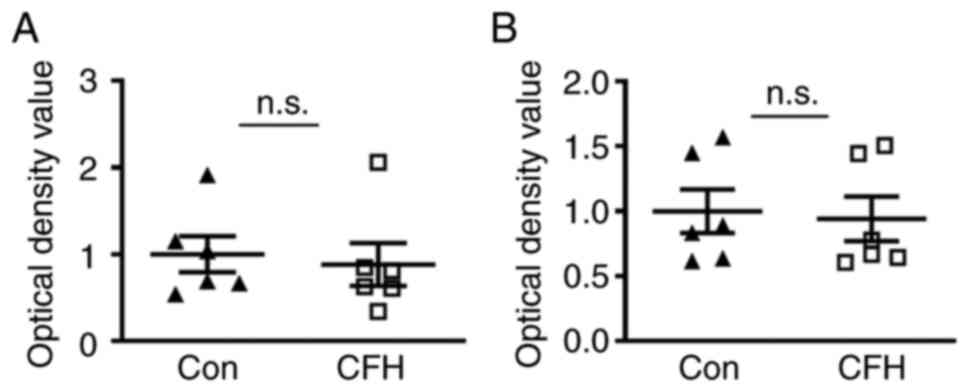
MTT and CCK-8 assays were applied to detect the
viability of HUVECs. Both assays showed no significant difference
in the viability of HUVECs between the CFH-containing conditioned
medium group and the control medium group (Fig. 2A and B). Therefore, CFH may not affect the
viability of HUVECs.
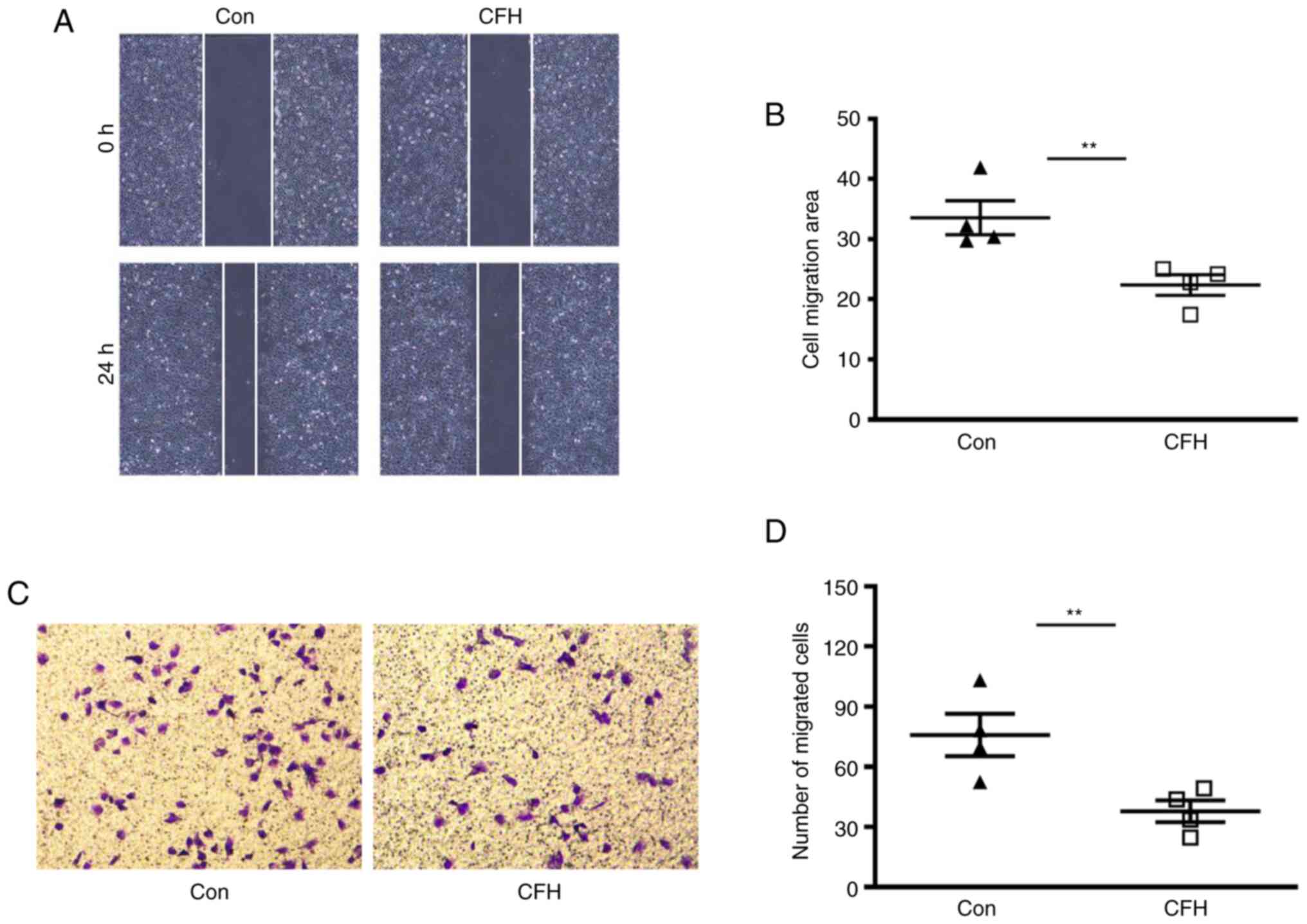
Wound healing assay and Transwell migration assay
were performed to determine the effect of CFH on the migration of
HUVECs. The wound healing assay indicated that the migration of
cells treated with the CFH-containing conditioned medium was
decreased compared with that in the control group (Fig. 3A and B). Transwell migration assays indicated
that the migration of HUVECs cultured in CFH-containing conditioned
medium was significantly reduced compared with that in the control
group (Fig. 3C and D). These results suggested that CFH
inhibits the migration of HUVECs.
CFH does not affect the MAPK and TGF-β
signaling pathways in HUVECs
To investigate the mechanisms underlying the effect
of CFH on migration, HUVECs were incubated with CFH-containing
conditioned medium for 24 h and proteins were extracted for western
blotting of the major components of the MAPK and TGF-β pathways,
which are involved in the regulation of cell migration (37,38).
As shown in Fig. 4A-C,
phosphorylation of ERK1/2, JNK and p38 MAPK, the three major
members of the MAPK pathway, were not markedly changed in HUVECs
treated with CFH-containing conditioned medium. The protein
expression levels of TGFβR1 and the phosphorylation of Smad2/3 were
also unchanged (Fig. 4D-F).
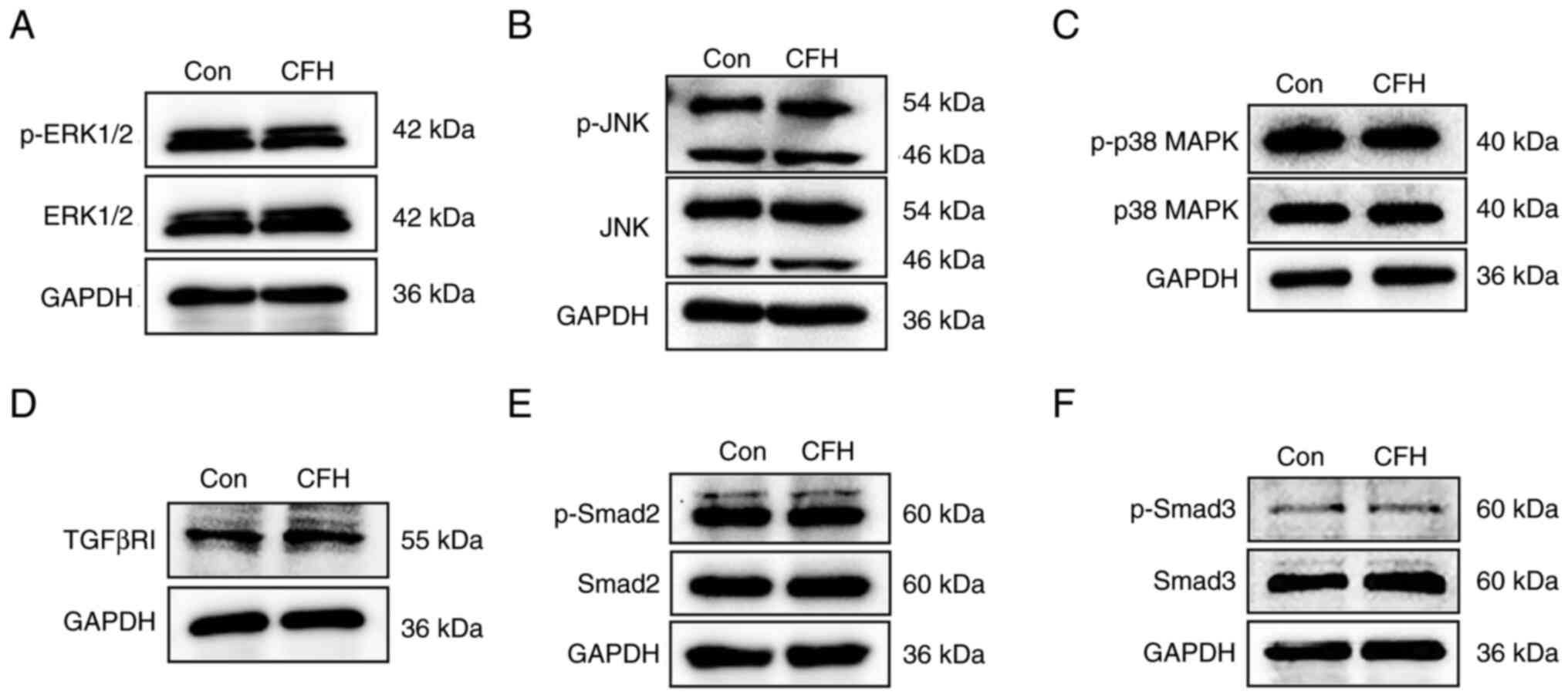 | Figure 4Effect of CFH on cell
migration-related signaling pathways in HUVECs. Western blot
analysis of proteins extracted from HUVECs incubated with
CFH-containing conditioned medium or control medium. (A) ERK1/2 and
p-ERK1/2, (B) JNK and p-JNK, (C) p38 MAPK and p-p38 MAPK, (D)
TGF-βRI, (E) Smad2 and p-Smad2, and (F) Smad3 and p-Smad3 were
detected. The p-ERK and ERK have bands at 42 and 44 kDa. For
p-Smad, the band for the small molecule is correct. The other band
is non-specific. GAPDH was used as a loading control. n=3. Con,
control; CFH, complement factor H; HUVECs, human umbilical vein
endothelial cells; p-, phosphorylated. |
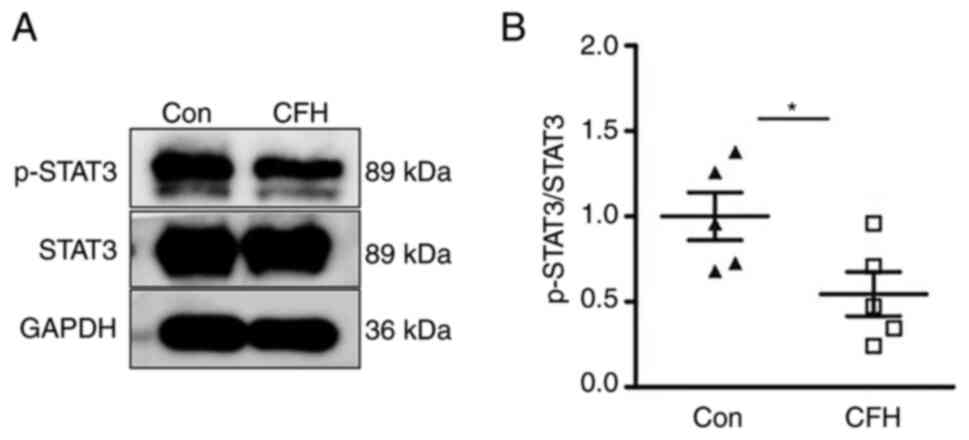
CFH inhibits the Y705 phosphorylation
of STAT3 in HUVECs
The STAT3 signaling pathway regulates endothelial
cell migration (23). After
treatment with CFH-containing conditioned medium, the
phosphorylation of STAT3 on Y705 was significantly decreased in
HUVECs, whereas the total STAT3 protein contents remained unchanged
compared with in the control group (Fig. 5A and B). These results suggested that CFH
inhibits the Y705 phosphorylation of STAT3 in HUVECs.
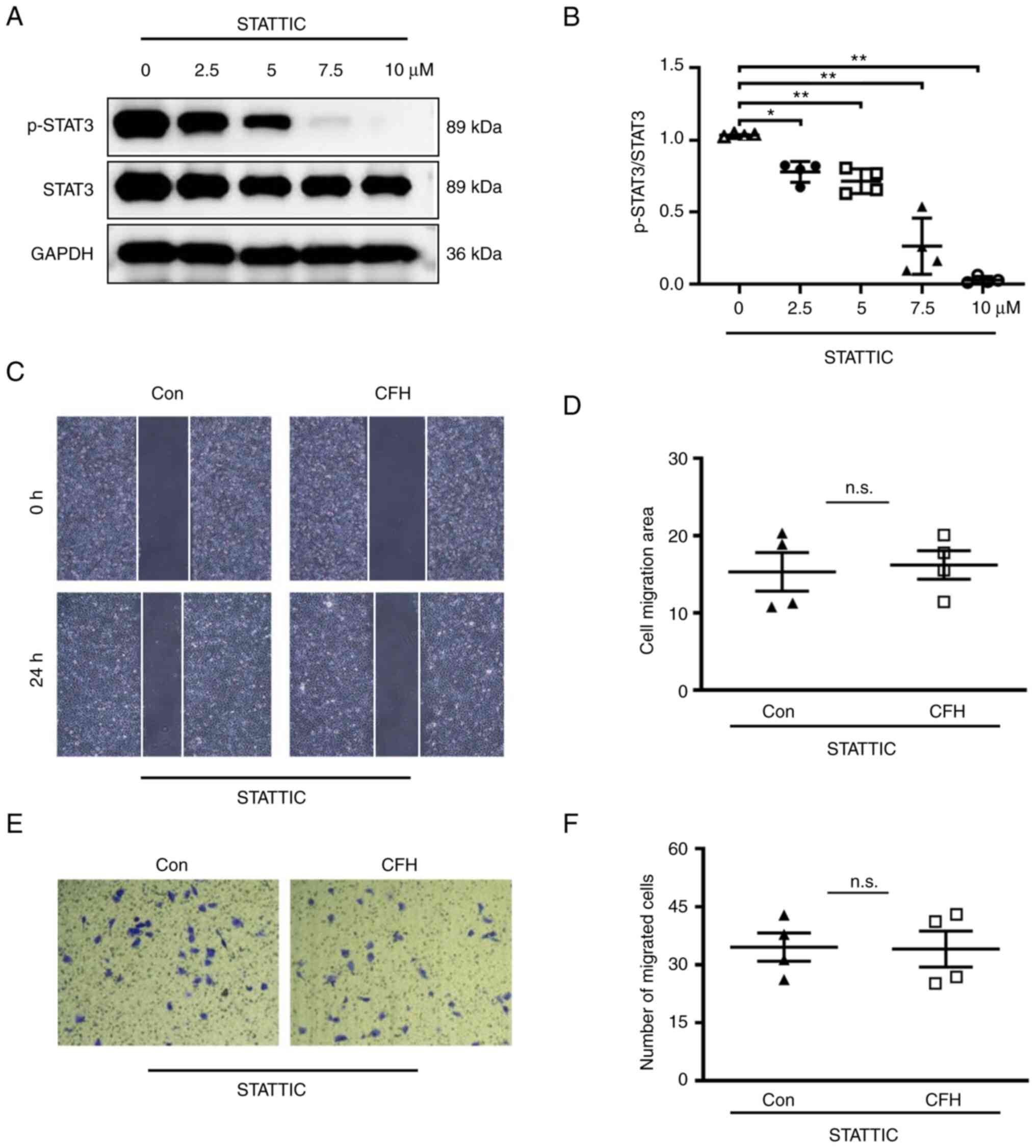
Inhibition of STAT3 signaling
compromises CFH-inhibited HUVEC migration
STATTIC is a small molecule that suppresses the
physiological binding of tyrosine-phosphorylated peptides to the
STAT3 SH2 domain, and reduces STAT3 dimerization and DNA binding
(39). To confirm that the
migration of HUVECs is dependent on STAT3, STATTIC was used to
inhibit STAT3 signaling in HUVECs. As shown in Fig. 6A and B, 7.5 and 10 µM STATTIC completely
inhibited STAT3 phosphorylation. After incubation with 7.5 µM
STATTIC, the migration of HUVECs cultured in CFH-containing
conditioned medium and control medium displayed no significant
difference, as determined by wound healing and Transwell migration
assays (Fig. 6C-F). These results
indicated that CFH does not induce additional inhibition of the
migration of HUVECs treated with STATTIC. If CFH inhibited HUVEC
migration via other pathways, it would induce additional inhibition
of the migration of STATTIC-treated HUVECs. Therefore, these
results suggested that CFH decreases HUVEC migration through
downregulation of STAT3 signaling.
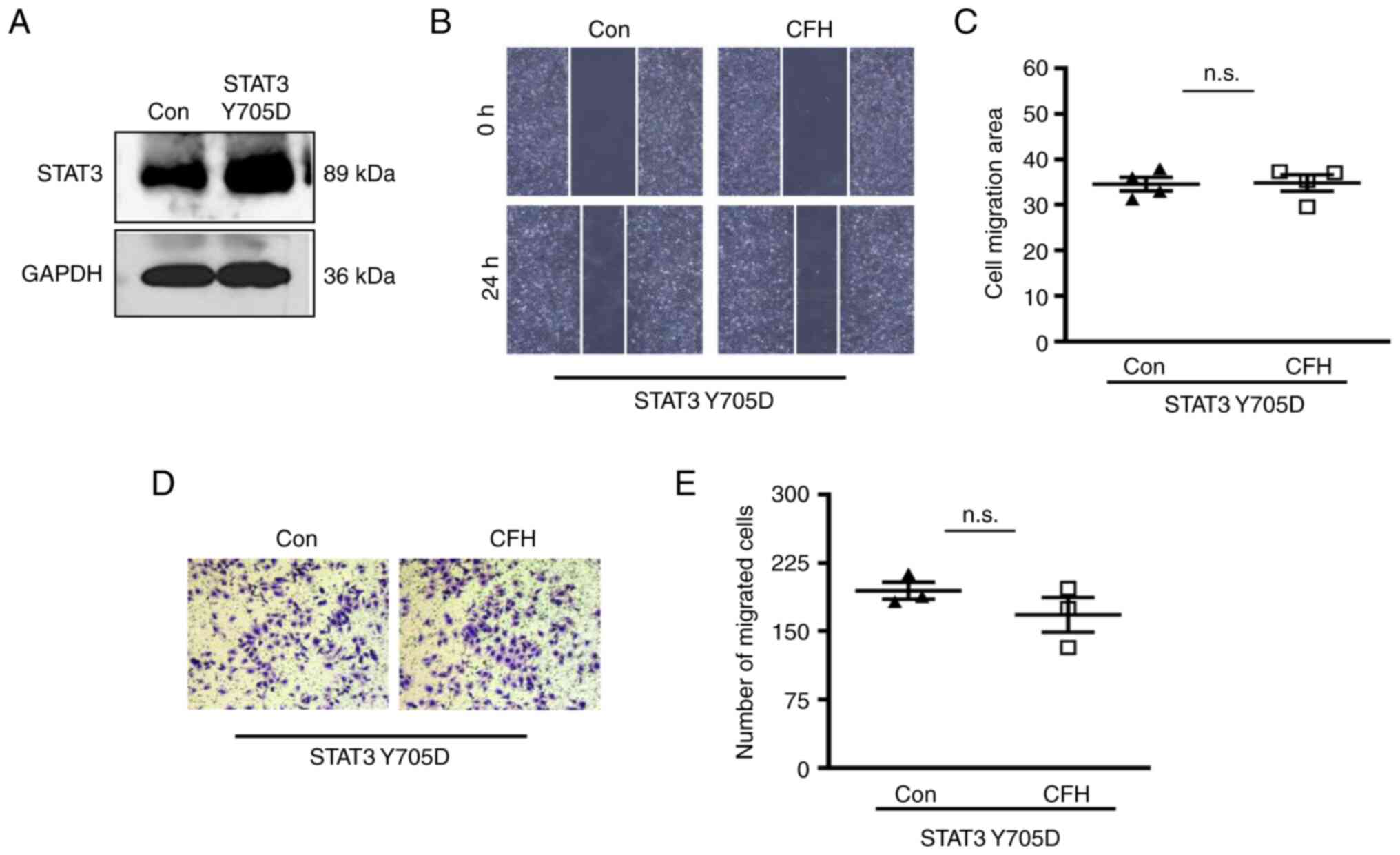
Activation of STAT3 signaling
compromises CFH-inhibited HUVEC migration
Phosphorylation-mimetic (constitutively active)
STAT3 (Y705D) has a substitution of tyrosine to aspartate at
position 705. The plasmid expressing STAT3 (Y705D) was transfected
into HUVECs to mimic activation of STAT3 signaling (Fig. 7A). After transfection, wound
healing and Transwell migration assays revealed no significant
difference in migration between the CFH-containing conditioned
medium group and the control group (Fig. 7B-E). Therefore, activation of STAT3
signaling also eliminated CFH-induced inhibition of HUVEC
migration. These findings indicated that CFH inhibits migration of
HUVECs in a STAT3-dependent manner.
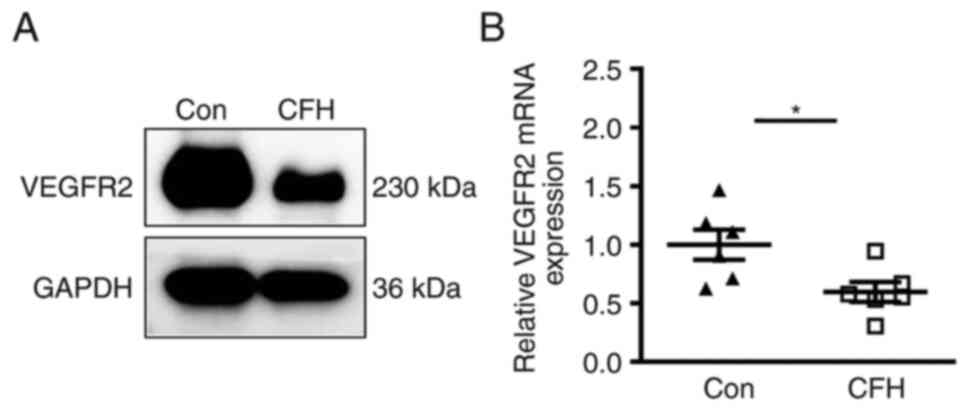
CFH decreases the expression of
VEGFR2
As a downstream target gene of STAT3, the expression
of VEGFR2 is upregulated by phosphorylation of STAT3(34). The present study detected the
expression levels of VEGFR2 in HUVECs after incubation with
CFH-containing conditioned medium. Western blotting and RT-qPCR
demonstrated that the protein and mRNA expression levels of VEGFR2
in HUVECs cultured in CFH-containing conditioned medium were
markedly reduced (Fig. 8A and
B). Therefore, CFH may decrease
the expression of VEGFR2 in HUVECs.
Discussion
Complement factor H has been reported to inhibit
angiogenesis (40-42);
however, the underlying molecular mechanisms remain unknown. The
present study collected CFH-containing conditioned medium from
HepG2 cells transduced with CFH-expressing adenovirus, and used
this to culture HUVECs. Exogenous CFH did not affect viability, but
it did significantly inhibit the migration of HUVECs. Western
blotting results suggested that CFH had no significant effect on
the MAPK and TGF-β signaling pathways, but it did inhibit the Y705
phosphorylation of STAT3 in HUVECs. It was further confirmed that
the inhibitory effect of CFH on HUVECs migration was
STAT3-dependent, which was verified by STATTIC treatment and STAT3
(Y705D) overexpression. In addition, CFH decreased the mRNA and
protein expression levels of VEGFR2.
In the alternative complement pathway, CFH
accelerates the decay of C3 and C5 convertases, and is also a
cofactor for factor I-mediated inactivation of C3b, resulting in
reduction of the generation of C3a and C5a, and the formation of
the membrane attack complex (C5b-9) (43). CFH is a plasma protein that
functions directly on endothelial cells in the circulatory system.
The present findings suggested that exogenous CFH could inhibit
HUVEC migration, but not viability. In Cfh-/- mice, no
alteration was previously found in the aortic ring assay, but a
significant increase in angiogenic vessels was shown in the
Matrigel plug assay (13). In
addition, plasma from Cfh-/- mice have been shown to
significantly increase endothelial cell migration (13). Thus, CFH may directly interact with
endothelial cells and inhibit their migration.
Endothelial cell migration is essential for
angiogenesis. This dynamic and multistep process is directionally
regulated by chemotactic, haptotactic and mechanotactic stimuli,
and requires the activation of various signaling pathways (44). It has previously been shown that
endothelial cell migration is regulated by a variety of
intracellular signaling pathways. Reconstituted HDL-apoE3 can
promote endothelial cell migration through ERK1/2 and p38 MAPK
(45). In mice, downregulation of
CFH expression can lead to increases in the expression levels of
VEGF and TGF-β (46). Furthermore,
signaling of TGF-β through ALK5 and subsequent Smad2/3
phosphorylation has been reported to lead to inhibition of
endothelial cell proliferation and migration (47). The present study demonstrated that
CFH-containing conditioned medium did not affect the MAPK and TGF-β
signaling pathways; however, it inhibited the STAT3 signaling
pathway. To further confirm whether CFH inhibited endothelial cell
migration via STAT3 signaling, HUVECs were treated with a STAT3
inhibitor, STATTIC, or were transfected with phospho-mimetic STAT3
(Y705D) to reduce or activate the STAT3 signaling. STATTIC is a
nonpeptidic small molecule shown to selectively inhibit the
function of the STAT3 SH2 domain regardless of the STAT3 activation
state in vitro (39). The
Y705D mutation is a phospho-mimetic form of STAT3(48). STAT3 is phosphorylated at Y705
within its SH2 domain, dimerizes and translocates into the nucleus
to regulate target gene expression; dysfunction of the SH2 domain
inhibits STAT3 activity (49). The
results revealed that both inhibiting and increasing the
phosphorylation of STAT3 abolished CFH-inhibited HUVEC migration.
These results indicated that CFH inhibits the migration of
endothelial cells in a STAT3-dependent manner. Yahata et al
(16) also indicated that
translocation of STAT3 into the nuclei is essential for triggering
human dermal microvascular endothelial cell migration. Thus, the
present study confirmed an important mechanism underlying
CFH-inhibited endothelial cell migration.
VEGFR is expressed by endothelial cells and mediates
angiogenic signaling (29-31).
Upon binding with VEGF, VEGFR2 is phosphorylated, which then
induces various downstream angiogenic signaling pathways, including
the MEK-ERK pathways, resulting in cell proliferation, survival,
migration and increased vascular permeability (34,50).
Lee et al (34)
demonstrated that VEGF induced VEGFR2 expression in brain
endothelial cells through STAT3 activation. Although p-STAT3 enters
the nucleus, the direct interaction of STAT3 and the human VEGFR2
promoter has not been studied (16,17).
The present study revealed that CFH decreases the expression of
VEGFR2 in HUVECs, but the lack of chromatin immunoprecipitation on
the VEGFR2 promoter is a limitation of this study. Collectively,
the present results indicated that CFH may reduce VEGFR2 expression
through inhibition of the phosphorylation of STAT3, and
subsequently decrease endothelial cell migration.
In conclusion, the present study determined that CFH
suppresses endothelial cell migration via inhibition of the STAT3
signaling pathway. As a plasma protein, CFH may be targeted
directly through the blood circulation, thus it could be considered
a new therapeutic target for anti-angiogenic therapy. Future work
shall elucidate the detailed molecular mechanisms underlying the
effects of CFH on endothelial cells.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the National Nature Science
Foundation of China (grant nos. 82171318 and 82241030), the Natural
Sciences Foundation of Shandong Province (grant no. ZR2021MH128),
the Academic Promotion Program of Shandong First Medical University
(grant no. 2019QL014) and the Shandong Taishan Scholarship.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JiL, HH, SX, MF, XW and JZ contributed to
experimental design, acquisition of data and data analysis. KW, SH
and AG contributed to the writing and editing of the manuscript,
and the analysis and interpretation of the data. JuL contributed to
the conception, experimental design, acquisition of data, data
analysis, and the writing and editing of the manuscript. JiL and
JuL confirm the authenticity of all the raw data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The use of the primary human cells in this study was
approved by the Ethics Committee of Shandong Provincial Qianfoshan
Hospital (approval no. 2017-106; Jinan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Grant ZL, Whitehead L, Wong VH, He Z, Yan
RY, Miles AR, Benest AV, Bates DO, Prahst C, Bentley K, et al:
Blocking endothelial apoptosis revascularizes the retina in a model
of ischemic retinopathy. J Clin Invest. 130:4235–4251.
2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Dahham SS, Tabana Y, Asif M, Ahmed M, Babu
D, Hassan LE, Ahamed MBK, Sandai D, Barakat K, Siraki A and Majid
AMSA: β-Caryophyllene induces apoptosis and inhibits angiogenesis
in colorectal cancer models. Int J Mol Sci.
22(10550)2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Unterleuthner D, Neuhold P, Schwarz K,
Janker L, Neuditschko B, Nivarthi H, Crncec I, Kramer N, Unger C,
Hengstschläger M, et al: Cancer-associated fibroblast-derived WNT2
increases tumor angiogenesis in colon cancer. Angiogenesis.
23:159–177. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wang X, Abraham S, McKenzie JAG, Jeffs N,
Swire M, Tripathi VB, Luhmann UFO, Lange CAK, Zhai Z, Arthur HM, et
al: LRG1 promotes angiogenesis by modulating endothelial TGF-β
signalling. Nature. 499:306–311. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bell CL, Vandenberghe LH, Bell P, Limberis
MP, Gao GP, Van Vliet K, Agbandje-McKenna M and Wilson JM: The AAV9
receptor and its modification to improve in vivo lung gene transfer
in mice. J Clin Invest. 121:2427–2435. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Gao P, Wang LL, Liu J, Dong F, Song W,
Liao L, Wang B, Zhang W, Zhou X, Xie Q, et al: Dihydroartemisinin
inhibits endothelial cell tube formation by suppression of the
STAT3 signaling pathway. Life Sci. 242(117221)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Li L, Dong F, Xu D, Du L, Yan S, Hu H,
Lobe CG, Yi F, Kapron CM and Liu J: Short-term, low-dose cadmium
exposure induces hyperpermeability in human renal glomerular
endothelial cells. J Appl Toxicol. 36:257–265. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Yang X, Song W, Zhang K, Wang Y, Chen F,
Chen Y, Huang T, Jiang Y, Wang X and Zhang C: p38 mediates T-2
toxin-induced Leydig cell testosterone synthesis disorder.
Ecotoxicol Environ Saf. 253(114695)2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Mao X, Zhou L, Tey SK, Ma APY, Yeung CLS,
Ng TH, Wong SWK, Liu BHM, Fung YME, Patz EF Jr, et al: Tumour
extracellular vesicle-derived complement factor H promotes
tumorigenesis and metastasis by inhibiting complement-dependent
cytotoxicity of tumour cells. J Extracell Vesicles.
10(e12031)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Esparza-Gordillo J, Soria JM, Buil A,
Almasy L, Blangero J, Fontcuberta J and Rodríguez de Córdoba S:
Genetic and environmental factors influencing the human factor H
plasma levels. Immunogenetics. 56:77–82. 2004.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Klein RJ, Zeiss C, Chew EY, Tsai JY,
Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne
ST, et al: Complement factor H polymorphism in age-related macular
degeneration. Science. 308:385–389. 2005.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kim SJ, Kim J, Lee J, Cho SY, Kang HJ, Kim
KY and Jin DK: Intravitreal human complement factor H in a rat
model of laser-induced choroidal neovascularisation. Br J
Ophthalmol. 97:367–370. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Liu J and Hoh J: Loss of complement factor
H in plasma increases endothelial cell migration. J Cancer.
8:2184–2190. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang Y, Huang Q, Tang M, Zhang J and Fan
W: Complement factor H expressed by retinal pigment epithelium
cells can suppress neovascularization of human umbilical vein
endothelial cells: An in vitro study. PLoS One.
10(e0129945)2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hu Z, Han Y, Liu Y, Zhao Z, Ma F, Cui A,
Zhang F, Liu Z, Xue Y, Bai J, et al: CREBZF as a key regulator of
STAT3 pathway in the control of liver regeneration in mice.
Hepatology. 71:1421–1436. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Yahata Y, Shirakata Y, Tokumaru S,
Yamasaki K, Sayama K, Hanakawa Y, Detmar M and Hashimoto K: Nuclear
translocation of phosphorylated STAT3 is essential for vascular
endothelial growth factor-induced human dermal microvascular
endothelial cell migration and tube formation. J Biol Chem.
278:40026–40031. 2003.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhang M, Zhou L, Xu Y, Yang M, Xu Y,
Komaniecki GP, Kosciuk T, Chen X, Lu X, Zou X, et al: A STAT3
palmitoylation cycle promotes TH17 differentiation and
colitis. Nature. 586:434–439. 2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Shen Y, Wang X, Liu Y, Singhal M,
Gürkaşlar C, Valls AF, Lei Y, Hu W, Schermann G, Adler H, et al:
STAT3-YAP/TAZ signaling in endothelial cells promotes tumor
angiogenesis. Sci Signal. 14(eabj8393)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Song D, Lan J, Chen Y, Liu A, Wu Q, Zhao
C, Feng Y, Wang J, Luo X, Cao Z, et al: NSD2 promotes tumor
angiogenesis through methylating and activating STAT3 protein.
Oncogene. 40:2952–2967. 2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Huang J, Tang L, Zhao Y and Ding W: TRIM11
promotes tumor angiogenesis via activation of STAT3/VEGFA signaling
in lung adenocarcinoma. Am J Cancer Res. 9:2019–2027.
2019.PubMed/NCBI
|
|
21
|
Xia Z, Xiao J, Dai Z and Chen Q: Membrane
progesterone receptor α (mPRα) enhances hypoxia-induced vascular
endothelial growth factor secretion and angiogenesis in lung
adenocarcinoma through STAT3 signaling. J Transl Med.
20(72)2022.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Xu G, Zhu L, Wang Y, Shi Y, Gong A and Wu
C: Stattic enhances radiosensitivity and reduces radio-induced
migration and invasion in HCC cell lines through an apoptosis
pathway. Biomed Res Int. 2017(1832494)2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Cao L, Ren Y, Guo X, Wang L, Zhang Q, Li
X, Wu X, Meng Z and Xu K: Downregulation of SETD7 promotes
migration and invasion of lung cancer cells via JAK2/STAT3 pathway.
Int J Mol Med. 45:1616–1626. 2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Xie TX, Wei D, Liu M, Gao AC, Ali-Osman F,
Sawaya R and Huang S: Stat3 activation regulates the expression of
matrix metalloproteinase-2 and tumor invasion and metastasis.
Oncogene. 23:3550–3560. 2004.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chen J and Wang Y, Wang S, Zhao X, Zhao L
and Wang Y: Salvianolic acid B and ferulic acid synergistically
promote angiogenesis in HUVECs and zebrafish via regulating VEGF
signaling. J Ethnopharmacol. 283(114667)2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Porter AM, Klinge CM and Gobin AS:
Biomimetic hydrogels with VEGF induce angiogenic processes in both
hUVEC and hMEC. Biomacromolecules. 12:242–246. 2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Cerezo AB, Hornedo-Ortega R,
Alvarez-Fernandez MA, Troncoso AM and Garcia-Parrilla MC:
Inhibition of VEGF-induced VEGFR-2 activation and HUVEC migration
by melatonin and other bioactive indolic compounds. Nutrients.
9(248)2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mac Gabhann F and Popel AS: Dimerization
of VEGF receptors and implications for signal transduction: A
computational study. Biophys Chem. 128:125–139. 2007.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Rezzola S, Di Somma M, Corsini M, Leali D,
Ravelli C, Polli VAB, Grillo E, Presta M and Mitola S: VEGFR2
activation mediates the pro-angiogenic activity of BMP4.
Angiogenesis. 22:521–533. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Melincovici CS, Boşca AB, Şuşman S,
Mărginean M, Mihu C, Istrate M, Moldovan IM, Roman AL and Mihu CM:
Vascular endothelial growth factor (VEGF)-key factor in normal and
pathological angiogenesis. Rom J Morphol Embryol. 59:455–467.
2018.PubMed/NCBI
|
|
31
|
Nagarkoti S, Kim YM, Ash D, Das A, Vitriol
E, Read TA, Youn SW, Sudhahar V, McMenamin M, Hou Y, et al: Protein
disulfide isomerase A1 as a novel redox sensor in VEGFR2 signaling
and angiogenesis. Angiogenesis. 26:77–96. 2023.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zou S, Gao Y and Zhang S: lncRNA HCP5 acts
as a ceRNA to regulate EZH2 by sponging miR-138-5p in cutaneous
squamous cell carcinoma. Int J Oncol. 59(56)2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhao M, Hu X, Xu Y, Wu C, Chen J, Ren Y,
Kong L, Sun S, Zhang L, Jin R and Zhou X: Targeting of EZH2
inhibits epithelial-mesenchymal transition in head and neck
squamous cell carcinoma via regulating the STAT3/VEGFR2 axis. Int J
Oncol. 55:1165–1175. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lee HT, Xue J, Chou PC, Zhou A, Yang P,
Conrad CA, Aldape KD, Priebe W, Patterson C, Sawaya R, et al: Stat3
orchestrates interaction between endothelial and tumor cells and
inhibition of Stat3 suppresses brain metastasis of breast cancer
cells. Oncotarget. 6:10016–10029. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhang F, Hu G, Chen X, Zhang L, Guo L, Li
C, Zhao H, Cui Z, Guo X, Sun F, et al: Excessive branched-chain
amino acid accumulation restricts mesenchymal stem cell-based
therapy efficacy in myocardial infarction. Signal Transduct Target
Ther. 7(171)2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zhu T, Yao Q, Hu X, Chen C, Yao H and Chao
J: The role of MCPIP1 in ischemia/reperfusion injury-induced HUVEC
migration and apoptosis. Cell Physiol Biochem. 37:577–591.
2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Chen W, He S and Xiang D: Hypoxia-induced
retinal pigment epithelium cell-derived bFGF promotes the migration
and angiogenesis of HUVECs through regulating TGF-β1/smad2/3
pathway. Gene. 790(145695)2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Schust J, Sperl B, Hollis A, Mayer TU and
Berg T: Stattic: A small-molecule inhibitor of STAT3 activation and
dimerization. Chem Biol. 13:1235–1242. 2006.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Lyzogubov VV, Tytarenko RG, Jha P, Liu J,
Bora NS and Bora PS: Role of ocular complement factor H in a murine
model of choroidal neovascularization. Am J Pathol. 177:1870–1880.
2010.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Rohrer B, Long Q, Coughlin B, Wilson RB,
Huang Y, Qiao F, Tang PH, Kunchithapautham K, Gilkeson GS and
Tomlinson S: A targeted inhibitor of the alternative complement
pathway reduces angiogenesis in a mouse model of age-related
macular degeneration. Invest Ophthalmol Vis Sci. 50:3056–3064.
2009.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Borras C, Delaunay K, Slaoui Y, Abache T,
Jorieux S, Naud MC, Sanharawi ME, Gelize E, Lassiaz P, An N, et al:
Mechanisms of FH protection against neovascular AMD. Front Immunol.
11(443)2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Moore SR, Menon SS, Cortes C and Ferreira
VP: Hijacking factor H for complement immune evasion. Front
Immunol. 12(602277)2021.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Siamwala JH, Reddy SH, Majumder S, Kolluru
GK, Muley A, Sinha S and Chatterjee S: Simulated microgravity
perturbs actin polymerization to promote nitric oxide-associated
migration in human immortalized Eahy926 cells. Protoplasma.
242:3–12. 2010.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Valanti EK, Dalakoura-Karagkouni K,
Fotakis P, Vafiadaki E, Mantzoros CS, Chroni A, Zannis V, Kardassis
D and Sanoudou D: Reconstituted HDL-apoE3 promotes endothelial cell
migration through ID1 and its downstream kinases ERK1/2, AKT and
p38 MAPK. Metabolism. 127(154954)2022.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Bora NS, Kaliappan S, Jha P, Xu Q, Sohn
JH, Dhaulakhandi DB, Kaplan HJ and Bora PS: Complement activation
via alternative pathway is critical in the development of
laser-induced choroidal neovascularization: role of factor B and
facto H. J Immunol. 177:1872–1878. 2006.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Goumans MJ, Valdimarsdottir G, Itoh S,
Lebrin F, Larsson J, Mummery C, Karlsson S and ten Dijke P: Activin
receptor-like kinase (ALK)1 is an antagonistic mediator of lateral
TGFbeta/ALK5 signaling. Mol Cell. 12:817–828. 2003.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Min TR, Park HJ, Park MN, Kim B and Park
SH: The root bark of Morus alba L. Suppressed the migration of
human non-small-cell lung cancer cells through inhibition of
epithelial-mesenchymal transition mediated by STAT3 and Src. Int J
Mol Sci. 20(2244)2019.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Coleman DR IV, Ren Z, Mandal PK, Cameron
AG, Dyer GA, Muranjan S, Campbell M, Chen X and McMurray JS:
Investigation of the binding determinants of phosphopeptides
targeted to the SRC homology 2 domain of the signal transducer and
activator of transcription 3. Development of a high-affinity
peptide inhibitor. J Med Chem. 48:6661–6670. 2005.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Jin J, Yuan F, Shen MQ, Feng YF and He QL:
Vascular endothelial growth factor regulates primate
choroid-retinal endothelial cell proliferation and tube formation
through PI3K/Akt and MEK/ERK dependent signaling. Mol Cell Biochem.
381:267–272. 2013.PubMed/NCBI View Article : Google Scholar
|