1. Introduction
The term gastro-oesophageal reflux disease (GERD)
refers to a set of syndromes that include the signs and symptoms
associated with any pathological process, with retrosternal burning
(heartburn) and reflux as the characteristic symptoms. The Montreal
definition of GERD states that GERD is a disease that develops when
the reflux of stomach contents causes troublesome symptoms or
complications. The word ‘trouble’ in the definition accurately
captures how negative the symptoms appear to patients. The symptoms
of GERD are classified by this international evidence-based
consensus into oesophageal symptom syndrome (with oesophageal
symptoms but no evidence of oesophageal injury, including
non-erosive reflux disease) and oesophageal injury syndrome
[mucosal injury is a recognised aspect, and the most common
manifestation is reflux oesophagitis (RE), including stenosis,
Barrett's oesophagus, and adenocarcinoma], while the recognised
extra-oesophageal symptoms include reflux cough syndrome, reflux
laryngitis syndrome, reflux asthma syndrome, reflux tooth erosion
syndrome, etc. (1). In addition,
this new definition acknowledges that the reflux causing symptoms
might be weakly acidic or gaseous. According to extensive
population-based studies, the prevalence of GERD in Western Europe
and North America is 10-20% (2).
Symptomatic GERD adversely affects the quality of life of patients
with chronic liver disease (as affects the mood and general health
perception) (3).
The pathophysiology of GERD comprises several
factors, such as low basal pressure of the lower oesophageal
sphincter (LES), prolonged LES relaxation time, delayed oesophageal
clearance, and delayed gastric emptying (4). In addition, GERD is also closely
associated with unhealthy lifestyle habits, such as smoking,
obesity, strenuous exercise after meals, consumption of carbonated
drinks, poor eating habits, and excessive alcohol and coffee
consumption. Individuals with diabetes and metabolic syndrome are
more likely to experience GERD. RE is the most common manifestation
of oesophageal injury. RE is a risk factor for upper
gastrointestinal bleeding (UGIB) and greatly impacts the quality of
life. Patients with decompensated liver cirrhosis (LC) often
experience oesophageal variceal bleeding, and it appears that
effective anti-reflux treatment should be used. However, the
frequency and specific mechanism of RE in LC have not been
elucidated. Patients with LC might be prone to GERD, and some
studies indicate that the prevalence of GERD in patients with LC is
high (5-7).
What factors cause GERD in LC? Is it necessary to employ
anti-reflux measures? Some studies have reported that GERD might
promote the rupture of oesophageal varices (EV) in patients with
LC, resulting in an increased risk of UGIB (7-10).
In recent years, proton pump inhibitors (PPIs) have been widely
used empirically to lower the risk of oesophageal venous rupture
and bleeding in patients with LC. Herein, we attempt to explain the
possible pathogenesis of GERD in LC and discuss whether anti-reflux
measures should be employed.
2. Epidemiological investigation
GERD is very common worldwide, and the prevalence of
gastro-oesophageal reflux symptoms varies greatly among countries.
A recent survey indicates that the GERD prevalence in China is
approximately 2.5% (11).
According to a systematic review, the prevalence of GERD in East
Asia ranges from 2.5 to 6.7%; however, the data lacks quality
(12). A study comprehensively
analysed the epidemiological trend of GERD in 204 countries and
regions in the past 20 years and reported that the age-standardised
prevalence rate (ASPR) increased globally from 2015 to 2019. The
risk of GERD is closely associated with age, and the incidence of
GERD in women is marginally higher. In the past 20 years, the ASPRs
of Latin America, the Caribbean, South Asia, North Africa, and the
Middle East were the highest, while those of East Asia and China
were the lowest (below 5%) (13).
The increased risk of GERD in these areas is associated with
potential risk factors, such as obesity, alcohol consumption, and
smoking, which is consistent with the findings of previous studies
(11). The study conducted in 2018
also reported that despite the lack of clear evidence indicating
the high-risk factors of GERD, the observed prevalence rate of
individuals over 50 years of age, smokers, users of non-steroidal
anti-inflammatory drugs, and obese individuals is significantly
higher (14). Eating habits, such
as irregular eating patterns, large meals, and bedtime meals, might
be associated with GERD symptoms (15). In a prospective study in China,
1280 patients with chronic liver disease (879 patients with LC)
were endoscopically assessed, and a RE prevalence of 36.4% was
reported in patients with chronic liver disease (6). A recent retrospective study reported
that the 10-year incidence of RE in patients with LC was similar to
that of the general population (4.79%) (16). Currently, there are a few studies
on the prevalence and associated factors of GERD in patients with
LC, and a more representative research population and standardised
methods are warranted for further epidemiological study. There is a
lack of evidence proving the positive correlation between LC and
GERD to date because patients with LC are often accompanied by
complications, such as EV and portal hypertension (PHT), several
high-risk factors, such as previous endoscopic injection
sclerotherapy and portal vein thrombosis (PVT) exist, which might
result in severe RE in patients with LC. As most patients with RE
lack GERD symptoms and do not take acid inhibitors before the onset
of severe UGIB, the prevalence of severe RE that causes UGIB has
increased significantly in the past three decades (8). Ethanol can damage the mucosal
barrier, cause oesophageal mucosal inflammation, increase the risk
of oesophageal acid damage, and increase the prevalence of GERD or
RE among alcoholics (17,18). Alcohol is a risk factor for LC,
which might be one of the reasons why some patients with LC are
likely to develop GERD.
3. Pathophysiological changes of GERD
Under normal conditions, the intra-abdominal
pressure (IAP) is positive, and the intra-thoracic pressure is
negative, which is the physical basis of promoting the reflux of
stomach contents into the oesophagus. A small amount of reflux
occurs in all individuals throughout the day, and the primary
mechanism resulting in most physiological reflux events is referred
to as transient LES relaxation (TLESR). However, the normal anatomy
and physiology of the oesophagus, LES, diaphragm at hiatus, and
stomach can prevent pathological GERD. The most common causes of
pathological reflux are the destruction of the LES normal reflux
barrier and the pressure gradient change in the thoracic and
abdominal cavities (19,20). Several studies have reported that
ascites can increase intragastric and IAP (21-23).
Patients with LC have a high incidence of acid reflux (23) and oesophagitis (24). In patients with LC, a decreased LES
pressure is observed with increased ascitic fluid (23,25).
Therefore, ascites in patients with LC is a potentially important
factor for GERD development (7,21,23,26).
Fluctuations are observed in the normal oesophageal
mucosal environment between destruction and repair; therefore,
physiological TLESR occurs (20).
The LES function is closely associated with the incidence and
severity of GERD. The LES is the annular muscle layer at the distal
end of the oesophagus, which generates resting pressure higher than
the IAP, and the resting pressure generated by the LES is
sufficient to prevent the back-flow of gastric contents to the
oesophagus (27). The LES,
diaphragm, and the normal anatomy of the oesophagus and stomach are
involved in the anti-reflux mechanism.
Severe RE and UGIB are often observed in patients
with LC owing to EV, PVT, PHT, etc., thereby deserving the
attention of hepatologists and gastroenterologists. However, the
existing research on GERD-related LC is scarce. Some studies
employed endoscopy and oesophageal manometry to demonstrate that
patients with LC have lower oesophageal motility disorder, abnormal
changes in the potential of hydrogen (pH) in the lower oesophageal
segment, and varying degrees of oesophageal mucosal injury
(Table I) (5-7,26,28).
 | Table IRelated research on oesophageal
monitoring in patients with LC. |
Table I
Related research on oesophageal
monitoring in patients with LC.
| First author/s,
year | Country | Research
objective | Observation methods
and outcome indicators | Related prevalence
rate | (Refs.) |
|---|
| Hassanin et
al, 2021 | Egypt | A total of 100
patients with HCV-associated LC were enrolled in the study and
underwent clinical examination, imaging examination and upper
gastrointestinal endoscopy. | Oesophageal mucosal
lesions were classified according to the Los Angeles classification
system as follows: Grade A, mucosal erosion <5 mm, not extending
between the upper limit of two mucosal folds; grade B, mucosal
erosion >5 mm, not extending between the upper limit of two
mucosal folds; grade C, confluent erosion that is continuous
between the upper limit of two or more mucosal folds but involves
<75% of the oesophageal circumference; and grade D, confluent
and circumferential erosion that involves ≥75% of the oesophageal
circumference. | 83 patients (83%)
were diagnosed with GERD by endoscopy, and the prevalence rates of
grades B and C were the highest. Among 62 patients with ascites, 56
patients (90.3%) were diagnosed with GERD under endoscopy. This
study reported that ascites are the only risk factor for GERD in
patients with LC. | (7) |
| Zhang et al,
2011 | China | A total of 78
patients with LC and 30 healthy controls confirmed to be EV-free by
endoscopy were included. | LESP, PA, PD, PV,
dynamic 24-h oesophageal pH and bilirubin monitoring were measured.
i) In the LC group, the LESP was 15.32±2.91 mmHg, the PA was
61.41±10.52 mmHg, the PD was 5.32±1.22 s, and the PV was 5.22±1.11
cm/s. ii) 24-h oesophageal pH and bilirubin monitoring results:
Compared with those in the control group, pathological oesophageal
pH and bilirubin measurements increased in the LC group, while bile
reflux events also increased. | The incidences of
RE and pathological reflux were 37.18 and 55.13%,
respectively. | (5) |
| Li et al,
2010 | China | Experimental group:
1,280 patients with chronic liver disease (879 patients with LC and
401 patients with chronic hepatitis). Control group: 29 patients
with acute hepatitis A or E. | Referring to the
Los Angeles classification scheme, the degree and scope of
oesophageal mucosal injury were observed by endoscopy, and RE was
classified into grades A, B, C and D. In the experimental group,
43% of the patients with LC (378/879) had different degrees of
oesophageal mucosal damage compared with 22% of the patients with
chronic hepatitis (91/401). | In this clinical
study, the prevalence of RE in patients with chronic liver disease
was 36.4% (469/1280), while it was 10.3% (3/29) in the control
group. | (6) |
| Schechter et
al, 2007 | Brazil | A total of 51
patients with LC with EV confirmed by endoscopy were included in
the study. | By manometry, pH
recording was performed by placing the probe 5 cm above the upper
limit of the LES. Reflux episodes and abnormal reflux were defined
according to the duration of oesophageal pH <4 in different
positions. Abnormal reflux of pH records was observed in 19
patients with LC with EV (37%), and 1 of the patients developed
erosive oesophagitis during endoscopy. | 27 patients (53%)
had typical GERD symptoms. An association was observed between
typical GERD symptoms and abnormal reflux. | (28) |
| Navarro-Rodriguez
et al, 2003 | Brazil | LESP and 24-h
oesophageal pH of 16 patients with ascites were monitored before
and after the puncture. | According to the
degree of intra-abdominal pressure reduction achieved, the patients
were divided into group A (decreased by >70%) and group B
(decreased by <70%). Before and after the reduction of
intra-abdominal pressure, the LESP in the two groups was as
follows: Group A, before puncture, 15.60 mmHg, after puncture,
18.09 mmHg; and group B, before puncture, 23.09 mmHg, after
puncture, 20.40 mmHg. However, 24-h pH monitoring showed
pathological reflux in patients with ascites that was reduced with
the paracentesis. In 16 patients, the mean total number of reflux
episodes before puncture was 520.26, and that after was 136.26. In
group A, the mean total number of reflux episodes was as follows:
Before puncture, 661.27; after puncture, 99.45. | The results
revealed that a reduction in intra-abdominal pressure of >70%
decreased gastro-oesophageal reflux. | (26) |
4. Mechanism
Relevant biological findings and clinical mechanism
analysis have been reported concerning GERD's abnormal mechanism.
GERD can be associated with bile (alkaline) reflux, gastric or
oesophageal distension, and dyskinesia. The LES plays a crucial
role in the incidence and severity of GERD. Currently, the
mechanism of RE in patients with LC has not been fully elucidated.
However, the incidence of RE in patients with LC is associated with
the following factors: i) Patients with LC often experience delayed
gastric emptying and might develop corresponding gastrointestinal
symptoms (29). ii) Ascites result
in an increased IAP, compressing the stomach and causing reflux of
the stomach contents (26). iii)
The existence of tense ascites results in decreased LES pressure.
During swallowing or coughing, the intragastric pressure
instantaneously increases, which might cause gastro-oesophageal
reflux (23,25). iv) With pathological changes in the
livers of patients with LC, the inducible nitric oxide (NO)
synthase (NOS) (iNOS) expression increases, and the endothelial NOS
(eNOS) activity decreases, resulting in a large number of harmful
pro-inflammatory NO mediators. On the one hand, the visceral artery
vessels dilate, and the plasma osmotic pressure decreases,
resulting in ascites (30-33).
On the other hand, excessive NO, promoting an increase in the TLESR
frequency, results in an increase in the total number of reflux
episodes (34,35).
Increased IAP and GERD
LC causes increased intrahepatic resistance, a
gradual increase in portal pressure, systemic visceral arterial
dilatation, and effective circulating blood volume insufficiency.
Through the antidiuretic process of the
renin-angiotensin-aldosterone pathway, the sympathetic nervous
system, and renal vasoconstriction, the human body increases the
total plasma volume through water-sodium retention to maintain
sufficient effective arterial blood volume. However, other factors,
such as hypoalbuminaemia and changes in intestinal capillary
pressure and permeability, can cause an accumulation of free fluid
in the abdominal cavity (36). A
study measuring the LES pressure (LESP) in patients with LC
reported that 10 control participants had a LESP of 21±1 mmHg,
whereas, among 15 patients with LC, 10 had a LESP of 22±1 mmHg,
while five LC patients with massive tension ascites had a LESP of
16±2 mmHg. After the resolution of ascites through diuresis, the
LESP increased to 25±3 mmHg in all LC patients with massive tension
ascites. Additionally, the remission of ascites not only increased
the LESP but also decreased the gastric pressure significantly,
with a significant linear correlation between the mean increase in
LESP and the mean decrease in gastric pressure (25). The results of another experimental
study revealed that intragastric pressure in patients with LC and
ascites is proportional to the volume of ascites, particularly in
patients with tension ascites; a sudden, transient increase in the
intragastric pressure during swallowing or vigorous coughing might
cause gastric oesophageal reflux (23). When abdominal compression is
applied, the LES relaxes, and gastro-oesophageal reflux prolongs
after swallowing (37).
Twenty-four-hour dynamic monitoring of oesophageal pH in patients
with ascites revealed that gastro-oesophageal reflux significantly
decreased when the IAP decreased by more than 70% of the
pre-puncture baseline, implying that a significant reduction in the
IAP significantly decreased gastro-oesophageal reflux (26).
Increased IAP, formation of hiatal
hernia, and GERD
When the IAP increases, the oesophagogastric
junction (EGJ)actively contracts, and the tonic contraction of the
diaphragm results in increased LESP (38). During swallowing, the oesophageal
body is shortened due to contraction of the longitudinal muscles of
the oesophagus, causing the LES to move proximally, and a small
part of the proximal stomach enters the thoracic cavity through the
diaphragmatic hiatus. After swallowing, due to the elasticity of
the phreno-oesophageal ligament, all structures return to their
original anatomical positions. However, due to factors such as
excessive contraction of the longitudinal oesophageal muscles,
increased IAP, and age-related degeneration, these ligaments might
lose their elasticity, resulting in a hiatal hernia (39). The existence of a hiatal hernia
alters the pressure topography of the EGJ, which might increase the
susceptibility of gastro-oesophageal reflux events. When the LES
relaxes during swallowing, the LESP decreases, and when the
pressure of the hiatal hernia exceeds the LESP, barium back-flow
occurs from the hiatal pouch to the oesophagus (40). It is well known that GERD is
associated with the formation of hiatal hernia caused by increased
IAP; however, few studies have confirmed that hiatal hernia is
directly associated with LC. More evidence based on standardised
methods is required.
Excessive NO production and GERD
Several studies have revealed that liver pathology
varies in patients with LC, resulting in high NO levels and
elevated exhaled NO (eNO) (32,34,35,41-43).
NO is a novel signalling molecule associated with inflammation and
tissue damage and is the most known effective vasodilator. It can
dilate visceral blood vessels, increase visceral blood flow, and
aggravate PHT (44,45). In view of factors such as decreased
hepatic metabolism, toxin accumulation, increased intestinal
permeability, impaired intestinal motility, and changes and
translocation of the intestinal flora, endotoxins, and other
intestinal-derived metabolites, the blood vessels could be directly
stimulated in vivo or cytokines could be stimulated to
produce NOS. During NOS catalysis, L-arginine interacts with oxygen
to increase NO synthesis and release in vivo (46,47).
Does a change in the microbial community in patients with LC affect
ammonia metabolism? Compared with normal individuals, the structure
of the duodenal mucosal microflora in LC changes, and with the
development of LC, the intestinal microflora exhibits an increase
in Enterobacteriaceae, Staphylococcus, Streptococcus, and other
microflora, and these microflorae produce endotoxins and ammonia
through their urease activities, respectively (48,49).
A study revealed that gut urease-containing bacteria Streptococcus
salivarius was observed in patients with LC, and it was revealed
that a change in the salivary bacteria number in patients with LC
was positively correlated with ammonia accumulation (50). However, aseptic animals can also
produce ammonia through intestinal glutaminase activity. Therefore,
it is unclear whether the change in the intestinal microecology in
LC has additional effects on ammonia accumulation and excessive NO
production.
The increase in NO concentration increases the risk
of hepatic encephalopathy. Hyperammonaemia could increase NO, and
the NO concentration in brain regions with acute ammonia toxicity
increases, resulting in common learning and memory disorders and
brain oedema (51,52).
NO is produced by three isoforms of NOS: neuronal
NOS, iNOS, and eNOS. eNOS is constitutively expressed in hepatic
sinusoidal endothelial cells (LSECs) and produces a small amount of
NO. A small amount of NO keeps hepatic stellate cells (HSCs) and
Kupffer cells still, which is essential for controlling vascular
tension and blood flow in hepatic sinuses, and plays a crucial role
in vascular homeostasis and inhibiting hepatic pathological
conditions. However, iNOS is not expressed under normal conditions,
but its expression is induced by bacterial endotoxin
lipopolysaccharide secondary to intestinal bacterial translocation
and pro-inflammatory cytokines associated with liver
ischaemia-reperfusion injury. iNOS is up-regulated in various
hepatic cells (including LSECs, Kupffer cells, HSCs, smooth muscle
cells, bile duct cells (cholangiocytes), and other immune cells),
which produce a large amount of NO and promote liver injury. Under
pathological conditions, eNOS activity decreases, iNOS is
up-regulated, and NO production in LSECs decreases, resulting in
capillarisation of endothelial cells and HSC activation,
accompanied by extracellular matrix deposition, HSC contraction and
proliferation, finally resulting in increased intrahepatic
resistance and sinusoidal blood flow disorder (33,53-58).
Hepatic microvascular dysfunction and excessive NO production
result in apoptosis, inflammation, deoxyribonucleic acid damage,
and hepatocellular carcinoma.
A small increase in portal vein pressure and the
increase of related blood flow is first sensed by the intestinal
microcirculation. This increases vascular endothelial growth factor
production, triggering eNOS activation and subsequent NO
overproduction (30-32,41).
Therefore, excessive NO production might be associated with
ascites. Multiple studies (42,43)
demonstrated that NO, eNO, and plasma NO levels in peripheral and
hepatic veins of patients with LC patients are significantly
elevated, and NO is closely associated with TLESR. Reportedly, NO
can decrease the peristaltic wave amplitude in the distal
oesophagus and the peristaltic contraction rate in the proximal
oesophagus. NO plays a crucial role in TLESR secondary to fundus
distension, which is secondary to reflux attack (34). A study on healthy volunteers
revealed (35) that the use of
substances that inhibit NO synthesis [NG-monomethyl-L-arginine
(L-NMMA)] can significantly lower the TLESR frequency after
ingestion of solid food, and reduce the total reflux attack. It can
then be confirmed laterally that excessive NO might result in an
increase in the transient relaxation frequency of LES, thereby
resulting in an increase in the total number of reflux attacks.
Another study reported that L-NMMA inhibited the
increase in the number of TLESRs caused by gastric distension by
inhibiting NO synthesis (34). The
findings of Boulant's research on dogs also demonstrated that
L-NMMA reduces the gastric distension controlled by pressure in
dogs and reduces the number of TLESRs caused by gastric distension
(59). NO is involved in the
maintenance of basal gastric fundic tension and human diet-induced
gastric fundal relaxation. Prolonged L-NMMA infusion inhibits NO
synthesis, causing fundic contraction, which results in a decrease
in the basal fundus volume (60).
Non-adrenergic nerve mechanism involving the NO nerve can regulate
gastrointestinal smooth muscles and then affect the gastric fundus
tension (61).
Bad lifestyle and eating habits
A study on the risk factors of GERD reported that
unhealthy lifestyles, such as obesity; smoking; after-dinner and
strenuous physical activity; consumption of high-fat, fried, and
spicy food; excessive coffee/tea consumption; and consumption of
carbonated drinks and alcohol, contribute to GERD (15). Being overweight and obese increases
the risk of various digestive system-related diseases, such as GERD
and erosive oesophagitis (62). In
particular, diabetes, metabolic syndrome, and obesity might also
increase the risk of GERD (63).
On the other hand, metabolic syndrome, hyperglycaemia, and obesity
are independent risk factors for L of chronic hepatitis B (64). Therefore, metabolic factors might
reveal the relationship between GERD and chronic liver disease.
Several studies have indicated that ethanol can damage the mucosal
barrier, cause oesophageal mucosal inflammation, and increase the
risk of oesophageal acid injury. Alcohol-induced acute oesophageal
necrosis might occur in patients with high alcohol intake,
particularly in patients with immunosuppressive alcoholic
hepatitis, which further reveals that excessive alcohol intake
might be the key factor for oesophageal lesions and cirrhosis
(17,18,65).
In addition, a prospective study reported that there is a
significant correlation between chronic hepatitis B virus (HBV)
infection and GERD, particularly in women and HBV carriers with a
high aspartate aminotransferase to platelet ratio, and the
incidence of erosive oesophagitis increases (66).
The potential interactions between LC, TLESR,
increased IAP, increased intragastric pressure, excessive NO
production, and unhealthy lifestyle and eating habits are presented
in Fig. 1.
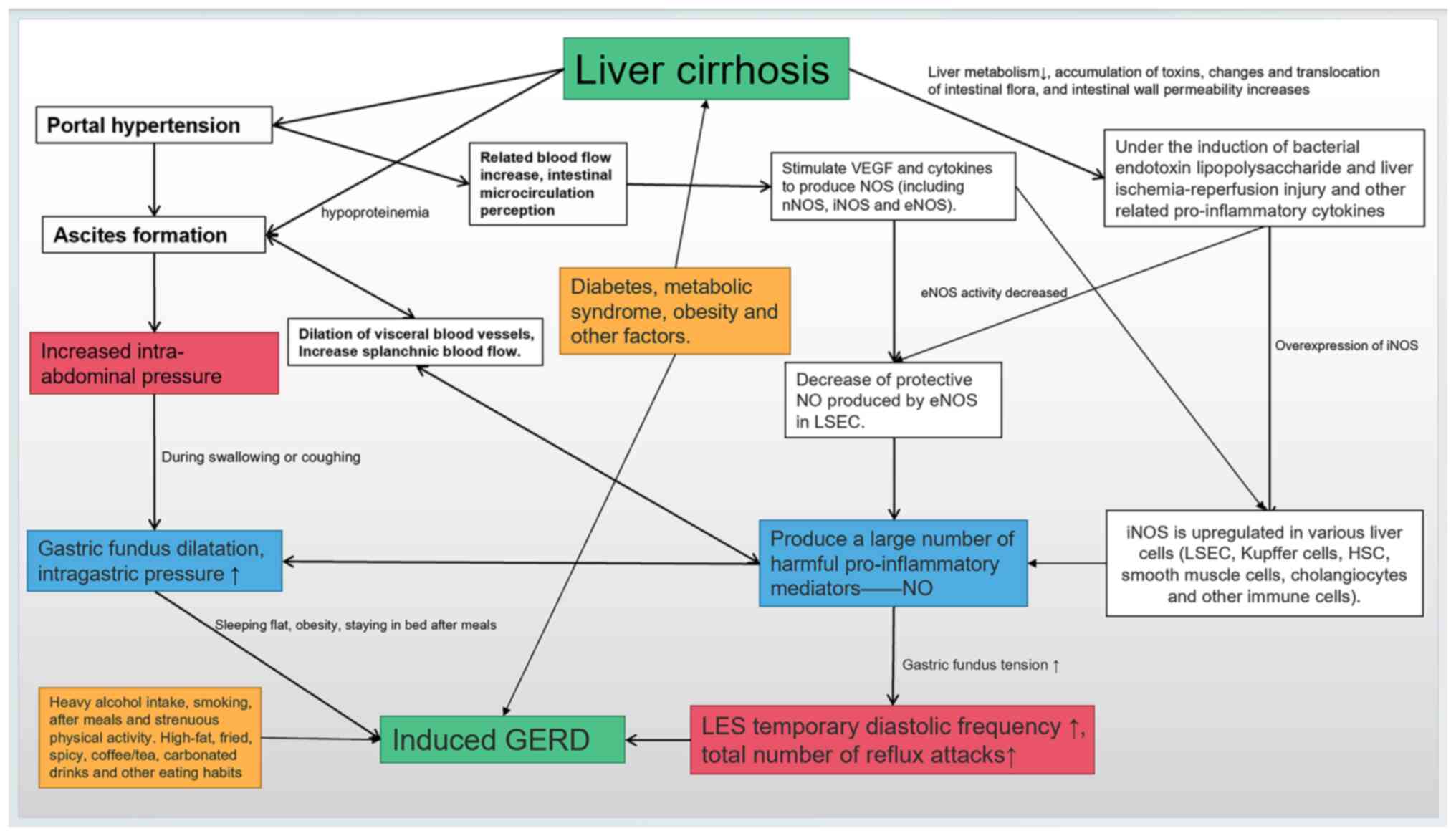 | Figure 1Potential interactions exist among
ascites, gastric fundus dilatation, excessive nitric oxide
production, increased LES relaxation frequency, metabolic diseases
and unhealthy lifestyle, among which the increase in transient LES
relaxation frequency is the key link, and ascites are a potentially
important factor affecting GERD development. Green indicates the
two diseases discussed, blue represents important factors that can
induce GERD in the progression of liver cirrhosis, red emphasizes
the key links in the pathogenesis, and yellow indicates the
adjustable lifestyle and metabolic factors. GERD,
gastro-oesophageal reflux disease; VEGF, vascular endothelial
growth factor; NOS, nitric oxide synthase; nNOS, neuron NOS; iNOS,
inducible NOS; eNOS, endothelial NOS; NO, nitric oxide; LSEC,
hepatic sinusoidal endothelial cells; LES, lower oesophageal
sphincter; HSC, hepatic stellate cells. |
5. Clinical intervention
RE might result in haemorrhagic oesophagitis and
variceal bleeding in patients with LC (8,67-69).
Although PPIs have been widely used in LC patients with EV in
recent years, it lacks strong evidence-based practice. On the one
hand, most patients with RE lack reflux symptoms; on the other
hand, almost two-thirds of the untreated patients with typical GERD
symptoms do not take acid inhibitors because of normal endoscopic
findings (70,71), thereby increasing the risk of
oesophageal bleeding. In a study on RE, only 36% of all RE patients
with UGIB were taking acid inhibitors before severe UGIB bleeding,
which might have resulted in an increase in UGIB incidence caused
by RE in recent years (8). The
question remains whether LC patients with reflux tendencies should
use acid inhibitors. A recent report on optimising GERD patient
management stated that if GERD diagnosis is clear after oesophageal
gastroscopy, PPIs can be used twice a day (before breakfast and
dinner). If necessary, another PPI should be administered. In the
case of nocturnal symptoms, any histamine-2 receptor antagonist
and/or alginate could be administered before bedtime (72). These drugs could alleviate the
symptoms of most patients with reflux, as they help cure
oesophageal injuries (such as oesophagitis and stenosis), improve
the quality of life, and improve sleep difficulties (73). However, long-term PPI use is
associated with spontaneous bacterial peritonitis (SBP) in patients
with LC. SBP is one of the most common complications in patients
with LC. At present, a popular dogma holds that frequent PPI use
could aggravate SBP occurrence. A recent meta-analysis reported a
weak correlation between SBP occurrence and PPI use; therefore,
this meta-analysis suggested that PPIs should be administered with
caution in patients with ascites in LC (74). Similar suggestions were made by
some other researchers. PPI should be administered to patients who
will benefit from its use. In elderly patients with severe liver
injury, particularly those with ascites in LC, PPI treatment should
be avoided or administered with caution to lower the risk of SBP
(75-77).
In the past decades, several studies have reported a
significant increase in the prevalence of RE that causes UGIB
(8-10).
Therefore, for LC patients with severe reflux tendency, surgical
treatment might be considered to reduce the risk of variceal
bleeding (78,79). The results of a recent study on
refractory GERD revealed that during the 12-month follow-up period,
patients who underwent laparoscopic fundoplication had the best
control of reflux symptoms compared with those who received
anti-reflux drugs (78). UGIB
following variceal rupture is the main cause of death in patients
with LC (80). Two types of
endoscopic treatments are considered the first choice to control
oesophageal variceal bleeding, namely endoscopic variceal
sclerotherapy (EVS) and endoscopic variceal ligation (EVL)
(81,82). However, several research findings
have reported that EVL is more effective and safer than EVS in
improving oesophageal motility disorder and eradicating EV
(83-85).
Once varicose veins are treated, patients should undergo an upper
gastrointestinal endoscopy every 3-6 months to evaluate the
recurrence of varicose veins and the need for repeated treatment
(86). However, although EVL is a
relatively safe surgical method, there is a risk of postoperative
bleeding. In rare cases, early spontaneous slippage of the rubber
band or rupture of the residual vein at the base of the oesophageal
ulcer could cause fatal bleeding (87,88).
The question remains whether long-term PPI use can
prevent varicose vein rupture and bleeding. Although there is
insufficient evidence currently, additional benefits might be
obtained from consistent PPI use after the endoscopic intervention
of varicose veins. Ulcer bleeding might rupture varicose veins
after EVL treatment, and the combination of PPIs and surgical
treatment might help lower the risk. The best evidence supports
that short-term (10 days) PPI use for patients with EV and
oesophageal ulcers after EVL treatment could decrease the
oesophageal ulcer size after selective oesophageal ligation and
even cure the ulcers after EVL (89,90).
Long-term PPI use (>1 month) could decrease the rebleeding rate
of patients with LC after endoscopic treatment while not affecting
bleeding-related mortality. Therefore, acid inhibition should also
be considered as a supplementary therapy after endoscopic treatment
(91-93).
Reportedly, chronic PPI use could increase the severity of hepatic
encephalopathy in patients with LC compared with patients who do
not use PPI (94). However, there
is a lack of higher-quality evidence proving the existence of an
association between them. Two isoenzymes (CYP2C19 and CYP3A4) are
involved in PPI metabolism in the liver, of which CYP2C19 is the
main metabolic pathway (95,96).
Older PPIs (including omeprazole, lansoprazole, and pantoprazole)
are primarily metabolised by CYP2C19, while rabeprazole is
primarily metabolised via the non-enzymatic pathway, which has
advantages over old PPIs. In the standard dose (20 mg once a day),
rabeprazole and esomeprazole provide better acid control than
omeprazole (97,98). Severe liver damage results in a 7-
to 9-fold increase in the area under the curve of all PPIs and an
extended half-life of 4 to 8 h (95). Therefore, when PPIs are used in
patients with LC, the increased half-life of these drugs in this
group of patients should be considered, and the dosage should be
reduced.
Brain toxicity caused by hyperammonaemia should also
be avoided in LC patients with excessive NO production. Several
studies have reported that the serum ammonia level can be decreased
by using lactulose, probiotics, and prebiotics. This is because
these agents regulate the intestinal flora and normalise the
intestinal flora. The reduction of blood ammonia level,
endotoxemia, and neurocognitive impairment lower the risk of
hepatic encephalopathy (99-101).
6. Lifestyle adjustment
Several patients with LC are accustomed to a
sedentary lifestyle owing to the decreased quality of life, which
might be associated with the occurrence of GERD. During drug
treatment, lifestyle changes such as weight loss, raising the
bedside, avoiding strenuous activities for a few hours after meals,
and refraining from consuming alcohol, coffee, carbonated drinks,
etc., should be incorporated before or during drug treatment. This
combination therapy helps relieve GERD symptoms (102). LC patients with varicose veins
should eat digestible soft food, stop alcohol consumption, avoid
staying up late, and be administered multivitamins. Patients with
hepatic encephalopathy should strictly limit their protein
intake.
7. Summary and comments
Herein, we outlined the possible relationship
between LC and GERD in terms of the pathological mechanisms. The
pathogenesis of GERD in LC is multifactorial. There are potential
interactions among TLESR, increased IAP, increased intragastric
pressure, and excessive NO production in patients with LC.
Increased intragastric pressure and an increased TLESR frequency
might be key factors for GERD development in LC. In view of the
evidence, we recommend that for LC patients with reflux tendency
(without ascites and EV), without other contraindications, acid
inhibitors are the appropriate choice for early protection of the
oesophageal mucosa, preferably with regular upper gastrointestinal
endoscopy for dynamically monitoring the oesophagus. For patients
with decompensated LC, such as portal hypertension, coagulation
dysfunction, and ascites (without EV), particularly those with
severe liver function damage, long-term PPI use should be avoided
to reduce the risk of SBP occurrence and further liver function
damage. In contrast, for decompensated patients with EV with a high
risk of bleeding, endoscopic oesophageal variceal eradication
should be considered, and acid inhibitors might be considered
postoperatively to reduce the rate of rebleeding after endoscopic
treatment in patients with LC. At the same time, LC patients with
unhealthy lifestyles should be given health education for lifestyle
modification. Finally, we eagerly anticipate more new evidence of
GERD in LC.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the research plan of
the National Natural Science Foundation of China (grant no.
325019038).
Availability of data and materials
All data generated and/or analyzed during this study
are included in this published article.
Authors' contributions
FY, XYH and YZ conceived the entire article, and
designed and drafted the manuscript. NL and HFF revised it
critically for important intellectual content. NL, HFF, ZJ and XYZ
investigated the present area of research and gathered relevant and
important information. LZ edited and modified the mechanism diagram
and table. FY and XYH approved the final version of the review.
Data authentication is not applicable. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vakil N, van Zanten SV, Kahrilas P, Dent J
and Jones R: Global Consensus Group. The Montreal definition and
classification of gastroesophageal reflux disease: A global
evidence-based consensus. Am J Gastroenterol. 101:1900–1920, 1943.
2006.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Dent J, El-Serag HB, Wallander MA and
Johansson S: Epidemiology of gastro-oesophageal reflux disease: A
systematic review. Gut. 54:710–717. 2005.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Suzuki K, Suzuki K, Koizumi K, Takada H,
Nishiki R, Ichimura H, Oka S and Kuwayama H: Effect of symptomatic
gastroesophageal reflux disease on quality of life of patients with
chronic liver disease. Hepatol Res. 38:335–339. 2008.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Cappell MS: Clinical presentation,
diagnosis, and management of gastroesophageal reflux disease. Med
Clin North Am. 89:243–291. 2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhang J, Cui PL, Lv D, Yao SW, Xu YQ and
Yang ZX: Gastroesophageal reflux in cirrhotic patients without
esophageal varices. World J Gastroenterol. 17:1753–1758.
2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Li B, Zhang B, Ma JW, Li P, Li L, Song YM
and Ding HG: High prevalence of reflux esophagitis among upper
endoscopies in Chinese patients with chronic liver diseases. BMC
Gastroenterol. 10(54)2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hassanin TM, Foaud Y, Mohamed H, Saad Z,
Elsayed A, Refaei S and Soliman W: Prevalence and risk factors of
endoscopically confirmed gastroesophageal reflux disease (GERD) in
patients with liver cirrhosis. Egypt Liver J. 11(27)2021.
|
|
8
|
Wangrattanapranee P, Khrucharoen U, Jensen
DM, Wongpongsalee T and Jensen ME: Severe Upper Gastrointestinal
Hemorrhage Caused by Reflux Esophagitis. Dig Dis Sci. 67:159–169.
2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wuerth BA and Rockey DC: Changing
epidemiology of upper gastrointestinal hemorrhage in the last
decade: A Nationwide Analysis. Dig Dis Sci. 63:1286–1293.
2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kim JJ, Sheibani S, Park S, Buxbaum J and
Laine L: Causes of bleeding and outcomes in patients hospitalized
with upper gastrointestinal bleeding. J Clin Gastroenterol.
48:113–118. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Eusebi LH, Ratnakumaran R, Yuan Y,
Solaymani-Dodaran M, Bazzoli F and Ford AC: Global prevalence of,
and risk factors for, gastro-oesophageal reflux symptoms: A
meta-analysis. Gut. 67:430–440. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wong BCY and Kinoshita Y: Systematic
review on epidemiology of gastroesophageal reflux disease in Asia.
Clin Gastroenterol Hepatol. 4:398–407. 2006.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zhang D, Liu S, Li Z and Wang R: Global,
regional and national burden of gastroesophageal reflux disease,
1990-2019: Update from the GBD 2019 study. Ann Med. 54:1372–1384.
2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Corley DA and Kubo A: Body mass index and
gastroesophageal reflux disease: A systematic review and
meta-analysis. Am J Gastroenterol. 101:2619–2628. 2006.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Taraszewska A: Risk factors for
gastroesophageal reflux disease symptoms related to lifestyle and
diet. Rocz Panstw Zakl Hig. 72:21–28. 2021.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Liu Z, Wei L and Ding H: Clinical
characteristics of reflux esophagitis among patients with liver
cirrhosis: A case-control study. Scand J Gastroenterol. 57:384–391.
2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Minatsuki C, Yamamichi N, Shimamoto T,
Kakimoto H, Takahashi Y, Fujishiro M, Sakaguchi Y, Nakayama C,
Konno-Shimizu M, Matsuda R, et al: Background factors of reflux
esophagitis and non-erosive reflux disease: A cross-sectional study
of 10,837 subjects in Japan. PLoS One. 8(e69891)2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Bor S, Bor-Caymaz C, Tobey NA,
Abdulnour-Nakhoul S and Orlando RC: Esophageal exposure to ethanol
increases risk of acid damage in rabbit esophagus. Dig Dis Sci.
44:290–300. 1999.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Mikami DJ and Murayama KM: Physiology and
pathogenesis of gastroesophageal reflux disease. Surg Clin North
Am. 95:515–525. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Iwakiri K, Hayashi Y, Kotoyori M, Tanaka
Y, Kawakami A, Sakamoto C and Holloway RH: Transient lower
esophageal sphincter relaxations (TLESRs) are the major mechanism
of gastroesophageal reflux but are not the cause of reflux disease.
Dig Dis Sci. 50:1072–1077. 2005.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Bhatia SJ, Narawane NM, Shalia KK, Mistry
FP, Sheth MD, Abraham P and Dherai AJ: Effect of tense ascites on
esophageal body motility and lower esophageal sphincter pressure.
Indian J Gastroenterol. 18:63–65. 1999.PubMed/NCBI
|
|
22
|
Mittal RK and McCallum RW: Characteristics
and frequency of transient relaxations of the lower esophageal
sphincter in patients with reflux esophagitis. Gastroenterology.
95:593–599. 1988.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Simpson JA and Conn HO: Role of ascites in
gastroesophageal reflux with comments on the pathogenesis of
bleeding esophageal varices. Gastroenterology. 55:17–25.
1968.PubMed/NCBI
|
|
24
|
Polish E and Sullivan BH: Esophagitis
associated with hemorrhage from esophageal varices. Ann Intern Med.
54:908–911. 1961.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Nebel OT: Lower esophageal sphincter
function in cirrhosis. Am J Dig Dis. 22:1101–1105. 1977.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Navarro-Rodriguez T, Hashimoto CL,
Carrilho FJ, Strauss E, Laudanna AA and Moraes-Filho JPP: Reduction
of abdominal pressure in patients with ascites reduces
gastroesophageal reflux. Dis Esophagus. 16:77–82. 2003.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Mittal RK and Balaban DH: The
esophagogastric junction. N Engl J Med. 336:924–932.
1997.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Schechter RB, Lemme EMO and Coelho HSM:
Gastroesophageal reflux in cirrhotic patients with esophageal
varices without endoscopic treatment. Arq Gastroenterol.
44:145–150. 2007.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Souza RC and Lima JHC: Helicobacter pylori
and gastroesophageal reflux disease: A review of this intriguing
relationship. Dis Esophagus. 22:256–263. 2009.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Abraldes JG, Iwakiri Y, Loureiro-Silva M,
Haq O, Sessa WC and Groszmann RJ: Mild increases in portal pressure
upregulate vascular endothelial growth factor and endothelial
nitric oxide synthase in the intestinal microcirculatory bed,
leading to a hyperdynamic state. Am J Physiol Gastrointest Liver
Physiol. 290:G980–G987. 2006.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Fernandez M, Mejias M, Angermayr B,
Garcia-Pagan JC, Rodés J and Bosch J: Inhibition of VEGF receptor-2
decreases the development of hyperdynamic splanchnic circulation
and portal-systemic collateral vessels in portal hypertensive rats.
J Hepatol. 43:98–103. 2005.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Tsai MH, Iwakiri Y, Cadelina G, Sessa WC
and Groszmann RJ: Mesenteric vasoconstriction triggers nitric oxide
overproduction in the superior mesenteric artery of portal
hypertensive rats. Gastroenterology. 125:1452–1461. 2003.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Iwakiri Y and Kim MY: Nitric oxide in
liver diseases. Trends Pharmacol Sci. 36:524–536. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Hirsch DP, Holloway RH, Tytgat GN and
Boeckxstaens GE: Involvement of nitric oxide in human transient
lower esophageal sphincter relaxations and esophageal primary
peristalsis. Gastroenterology. 115:1374–1380. 1998.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Hirsch DP, Tiel-Van Buul MM, Tytgat GN and
Boeckxstaens GE: Effect of L-NMMA on postprandial transient lower
esophageal sphincter relaxations in healthy volunteers. Dig Dis
Sci. 45:2069–2075. 2000.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Fortune B and Cardenas A: Ascites,
refractory ascites and hyponatremia in cirrhosis. Gastroenterol Rep
(Oxf). 5:104–112. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Vanderstappen G and Texter EC Jr: Response
of the physiologic gastroesophageal sphincter to increased
intra-abdominal pressure. J Clin Invest. 43:1856–1868.
1964.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Mittal RK, Fisher M, McCallum RW,
Rochester DF, Dent J and Sluss J: Human lower esophageal sphincter
pressure response to increased intra-abdominal pressure. Am J
Physiol. 258:G624–G630. 1990.PubMed/NCBI View Article : Google Scholar
|
|
39
|
van Herwaarden MA, Samsom M and Smout
AJPM: The role of hiatus hernia in gastro-oesophageal reflux
disease. Eur J Gastroenterol Hepatol. 16:831–835. 2004.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Sloan S and Kahrilas PJ: Impairment of
esophageal emptying with hiatal hernia. Gastroenterology.
100:596–605. 1991.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Iwakiri Y, Tsai MH, McCabe TJ, Gratton JP,
Fulton D, Groszmann RJ and Sessa WC: Phosphorylation of eNOS
initiates excessive NO production in early phases of portal
hypertension. Am J Physiol Heart Circ Physiol. 282:H2084–H2090.
2002.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Battista S, Bar F, Mengozzi G, Zanon E,
Grosso M and Molino G: Hyperdynamic circulation in patients with
cirrhosis: Direct measurement of nitric oxide levels in hepatic and
portal veins. J Hepatol. 26:75–80. 1997.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Delclaux C, Mahut B, Zerah-Lancner F,
Delacourt C, Laoud S, Cherqui D, Duvoux C, Mallat A and Harf A:
Increased nitric oxide output from alveolar origin during liver
cirrhosis versus bronchial source during asthma. Am J Respir Crit
Care Med. 165:332–337. 2002.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Asano K, Chee CB, Gaston B, Lilly CM,
Gerard C, Drazen JM and Stamler JS: Constitutive and inducible
nitric oxide synthase gene expression, regulation, and activity in
human lung epithelial cells. Proc Natl Acad Sci USA.
91:10089–10093. 1994.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Iwakiri Y: Pathophysiology of portal
hypertension. Clin Liver Dis. 18:281–291. 2014.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Huang X, Thansamay S, Yang K, Luo T and
Chen S: Measurement of exhaled nitric oxide in cirrhotic patients
with esophageal and gastric varices. Biomed Res Int.
2019(9673162)2019.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Matsumoto A, Ogura K, Hirata Y, Kakoki M,
Watanabe F, Takenaka K, Shiratori Y, Momomura S and Omata M:
Increased nitric oxide in the exhaled air of patients with
decompensated liver cirrhosis. Ann Intern Med. 123:110–113.
1995.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Chen Y, Ji F, Guo J, Shi D, Fang D and Li
L: Dysbiosis of small intestinal microbiota in liver cirrhosis and
its association with etiology. Sci Rep. 6(34055)2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Kang DJ, Betrapally NS, Ghosh SA, Sartor
RB, Hylemon PB, Gillevet PM, Sanyal AJ, Heuman DM, Carl D, Zhou H,
et al: Gut microbiota drive the development of neuroinflammatory
response in cirrhosis in mice. Hepatology. 64:1232–1248.
2016.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Zhang Z, Zhai H, Geng J, Yu R, Ren H, Fan
H and Shi P: Large-scale survey of gut microbiota associated with
MHE via 16S rRNA-based pyrosequencing. Am J Gastroenterol.
108:1601–1611. 2013.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Rao VLR: Nitric oxide in hepatic
encephalopathy and hyperammonemia. Neurochem Int. 41:161–170.
2002.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Swamy M, Zakaria AZ, Govindasamy C,
Sirajudeen KNS and Nadiger HA: Effects of acute ammonia toxicity on
nitric oxide (NO), citrulline-NO cycle enzymes, arginase and
related metabolites in different regions of rat brain. Neurosci
Res. 53:116–122. 2005.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Abu-Amara M, Yang SY, Seifalian A,
Davidson B and Fuller B: The nitric oxide pathway-evidence and
mechanisms for protection against liver ischaemia reperfusion
injury. Liver Int. 32:531–543. 2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
La Mura V, Pasarín M, Rodriguez-Vilarrupla
A, García-Pagán JC, Bosch J and Abraldes JG: Liver sinusoidal
endothelial dysfunction after LPS administration: A role for
inducible-nitric oxide synthase. J Hepatol. 61:1321–1327.
2014.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Carnovale CE and Ronco MT: Role of nitric
oxide in liver regeneration. Ann Hepatol. 11:636–647.
2012.PubMed/NCBI
|
|
56
|
Fujita K, Nozaki Y, Yoneda M, Wada K,
Takahashi H, Kirikoshi H, Inamori M, Saito S, Iwasaki T, Terauchi
Y, et al: Nitric oxide plays a crucial role in the
development/progression of nonalcoholic steatohepatitis in the
choline-deficient, l-amino acid-defined diet-fed rat model. Alcohol
Clin Exp Res. 34 (Suppl 1):S18–S24. 2010.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Langer DA, Das A, Semela D, Kang-Decker N,
Hendrickson H, Bronk SF, Katusic ZS, Gores GJ and Shah VH: Nitric
oxide promotes caspase-independent hepatic stellate cell apoptosis
through the generation of reactive oxygen species. Hepatology.
47:1983–1993. 2008.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Xie G, Wang X, Wang L, Wang L, Atkinson
RD, Kanel GC, Gaarde WA and Deleve LD: Role of differentiation of
liver sinusoidal endothelial cells in progression and regression of
hepatic fibrosis in rats. Gastroenterology. 142:918–927.e6.
2012.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Boulant J, Fioramonti J, Dapoigny M,
Bommelaer G and Bueno L: Cholecystokinin and nitric oxide in
transient lower esophageal sphincter relaxation to gastric
distention in dogs. Gastroenterology. 107:1059–1066.
1994.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Kuiken SD, Vergeer M, Heisterkamp SH,
Tytgat GNJ and Boeckxstaens GEE: Role of nitric oxide in gastric
motor and sensory functions in healthy subjects. Gut. 51:212–218.
2002.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Russo A, Fraser R, Adachi K, Horowitz M
and Boeckxstaens G: Evidence that nitric oxide mechanisms regulate
small intestinal motility in humans. Gut. 44:72–76. 1999.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Nam SY: Obesity-Related digestive diseases
and their pathophysiology. Gut Liver. 11:323–334. 2017.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Lee YC, Yen AM, Tai JJ, Chang SH, Lin JT,
Chiu HM, Wang HP, Wu MS and Chen TH: The effect of metabolic risk
factors on the natural course of gastro-oesophageal reflux disease.
Gut. 58:174–181. 2009.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Wong GL, Wong VW, Choi PC, Chan AW, Chim
AM, Yiu KK, Chan HY, Chan FK, Sung JJ and Chan HL: Metabolic
syndrome increases the risk of liver cirrhosis in chronic hepatitis
B. Gut. 58:111–117. 2009.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Săftoiu A, Cazacu S, Kruse A, Georgescu C,
Comănescu V and Ciurea T: Acute esophageal necrosis associated with
alcoholic hepatitis: Is it black or is it white? Endoscopy.
37:268–271. 2005.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Hsu CS, Wang CC, Wang PC, Lin HH, Tseng
TC, Chen CH, Su WC, Liu CJ, Chen CL, Lai MY, et al: Increased
incidence of gastroesophageal reflux disease in patients with
chronic hepatitis B virus infection. Hepatol Int. 4:585–593.
2010.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Ahmed AM, al Karawi MA, Shariq S and
Mohamed AE: Frequency of gastroesophageal reflux in patients with
liver cirrhosis. Hepatogastroenterology. 40:478–480.
1993.PubMed/NCBI
|
|
68
|
Sakaguchi M, Manabe N, Ueki N, Miwa J,
Inaba T, Yoshida N, Sakurai K, Nakagawa M, Yamada H, Saito M, et
al: Factors associated with complicated erosive esophagitis: A
Japanese multicenter, prospective, cross-sectional study. World J
Gastroenterol. 23:318–327. 2017.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Costa ND, Cadiot G, Merle C, Jolly D,
Bouche O, Thiéfin G and Zeitoun P: Bleeding reflux esophagitis: A
prospective 1-year study in a university hospital. Am J
Gastroenterol. 96:47–51. 2001.PubMed/NCBI View Article : Google Scholar
|
|
70
|
El-Serag HB, Sweet S, Winchester CC and
Dent J: Update on the epidemiology of gastro-oesophageal reflux
disease: A systematic review. Gut. 63:871–880. 2014.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Vakil N, van Zanten SV, Kahrilas P, Dent J
and Jones R: Global Consensus Group. The Montreal definition and
classification of gastroesophageal reflux disease: A global
evidence-based consensus. Am J Gastroenterol. 101:1900–1920.
2006.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Törnblom H, Tsoposidis A, Wallenius V,
Kostic S, Axelsson H, Lundell L, Håkanson B, Simrén M and Thorell
A: Management of patients with gastroesophageal reflux disease can
be optimized. Lakartidningen. 119(21177)2022.PubMed/NCBI(In Swedish).
|
|
73
|
Spechler SJ: Proton Pump Inhibitors: What
the Internist Needs to Know. Med Clin North Am. 103:1–14.
2019.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Alhumaid S, Al Mutair A, Al Alawi Z, Zaidi
ARZ, Rabaan AA, Elhazmi A and Al-Omari A: Proton Pump inhibitors
use and risk of developing spontaneous bacterial peritonitis in
cirrhotic patients: A systematic review and meta-analysis. Gut
Pathog. 13(17)2021.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Zhang M, Liu W, Xu X, Chen T and Qi JY:
Association between Proton pump inhibitor therapy and spontaneous
bacterial peritonitis occurrence in cirrhotic patients: A clinical
review. Curr Med Sci. 42:673–680. 2022.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Dahabra L, Kreidieh M, Abureesh M, Abou
Yassine A and Deeb L: Proton Pump inhibitors use and increased risk
of spontaneous bacterial peritonitis in cirrhotic patients: A
retrospective cohort analysis. Gastroenterology Res. 15:180–187.
2022.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Shaikh BA, Shaikh ZA, Shah AH and Kumar A:
Determining the Risk of Spontaneous Bacterial Peritonitis due to
increase use of Proton Pump Inhibitors among cirrhotic patients
with ascites. Pak J Med Sci. 37:1075–1079. 2021.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Spechler SJ, Hunter JG, Jones KM, Lee R,
Smith BR, Mashimo H, Sanchez VM, Dunbar KB, Pham TH, Murthy UK, et
al: Randomized trial of medical versus surgical treatment for
refractory heartburn. N Engl J Med. 381:1513–1523. 2019.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Roark R, Sydor M, Chatila AT, Umar S,
Guerra RDL, Bilal M and Guturu P: Management of gastroesophageal
reflux disease. Dis Mon. 66(100849)2020.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Holster IL, Tjwa ET, Moelker A, Wils A,
Hansen BE, Vermeijden JR, Scholten P, van Hoek B, Nicolai JJ,
Kuipers EJ, et al: Covered transjugular intrahepatic portosystemic
shunt versus endoscopic therapy + β-blocker for prevention of
variceal rebleeding. Hepatology. 63:581–589. 2016.PubMed/NCBI View Article : Google Scholar
|
|
81
|
D'Amico G, Pagliaro L and Bosch J: The
treatment of portal hypertension: A meta-analytic review.
Hepatology. 22:332–354. 1995.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Laine L and Cook D: Endoscopic ligation
compared with sclerotherapy for treatment of esophageal variceal
bleeding. A meta-analysis. Ann Intern Med. 123:280–287.
1995.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Viazis N, Armonis A, Vlachogiannakos J,
Rekoumis G, Stefanidis G, Papadimitriou N, Manolakopoulos S and
Avgerinos A: Effects of endoscopic variceal treatment on
oesophageal function: A prospective, randomized study. Eur J
Gastroenterol Hepatol. 14:263–269. 2002.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Berner JS, Gaing AA, Sharma R, Almenoff
PL, Muhlfelder T and Korsten MA: Sequelae after esophageal variceal
ligation and sclerotherapy: A prospective randomized study. Am J
Gastroenterol. 89:852–858. 1994.PubMed/NCBI
|
|
85
|
Hou MC, Yen TC, Lin HC, Kuo BI, Chen CH,
Lee FY, Liu RS, Chang FY and Lee SD: Sequential changes of
esophageal motility after endoscopic injection sclerotherapy or
variceal ligation for esophageal variceal bleeding: A scintigraphic
study. Am J Gastroenterol. 92:1875–1878. 1997.PubMed/NCBI
|
|
86
|
Cremers I and Ribeiro S: Management of
variceal and nonvariceal upper gastrointestinal bleeding in
patients with cirrhosis. Therap Adv Gastroenterol. 7:206–216.
2014.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Mishin I and Dolghii A: Early spontaneous
slippage of rubber bands with fatal bleeding: A rare complication
of endoscopic variceal ligation. Endoscopy. 37:275–276.
2005.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Toyoda H, Fukuda Y, Katano Y, Ebata M,
Nagano K, Morita K, Yokozaki S, Takeuchi M and Hayakawa T: Fatal
bleeding from a residual vein at the esophageal ulcer base after
successful endoscopic variceal ligation. J Clin Gastroenterol.
32:158–160. 2001.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Lo EA, Wilby KJ and Ensom MH: Use of
proton Pump inhibitors in the management of gastroesophageal
varices: A systematic review. Ann Pharmacother. 49:207–219.
2015.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Boo GB, Oh JC, Lee BJ, Lee DM, Kim YD,
Park CG and Kim MW: The effect of proton pump inhibitor on healing
of post-esophageal variceal ligation ulcers. Korean J
Gastroenterol. 51:232–240. 2008.PubMed/NCBI(In Korean).
|
|
91
|
Lin L, Cui B, Deng Y, Jiang X, Liu W and
Sun C: The efficacy of proton pump inhibitor in cirrhotics with
variceal bleeding: A systemic review and meta-analysis. Digestion.
102:117–127. 2021.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Kang SH, Yim HJ, Kim SY, Suh SJ, Hyun JJ,
Jung SW, Jung YK, Koo JS and Lee SW: Proton pump inhibitor therapy
is associated with reduction of early bleeding risk after
prophylactic endoscopic variceal band ligation: A retrospective
cohort study. Medicine (Baltimore). 95(e2903)2016.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Hidaka H, Nakazawa T, Wang G, Kokubu S,
Minamino T, Takada J, Tanaka Y, Okuwaki Y, Watanabe M, Tanabe S, et
al: Long-term administration of PPI reduces treatment failures
after esophageal variceal band ligation: A randomized, controlled
trial. J Gastroenterol. 47:118–126. 2012.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Fasullo M, Rau P, Liu DQ, Holzwanger E,
Mathew JP, Guilarte-Walker Y and Szabo G: Proton pump inhibitors
increase the severity of hepatic encephalopathy in cirrhotic
patients. World J Hepatol. 11:522–530. 2019.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Thjodleifsson B: Treatment of acid-related
diseases in the elderly with emphasis on the use of proton pump
inhibitors. Drugs Aging. 19:911–927. 2002.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Ishizaki T and Horai Y: Review article:
Cytochrome P450 and the metabolism of proton pump
inhibitors-emphasis on rabeprazole. Aliment Pharmacol Ther. 13
(Suppl 3):S27–S36. 1999.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Lind T, Rydberg L, Kylebäck A, Jonsson A,
Andersson T, Hasselgren G, Holmberg J and Röhss K: Esomeprazole
provides improved acid control vs. omeprazole in patients with
symptoms of gastro-oesophageal reflux disease. Aliment Pharmacol
Ther. 14:861–867. 2000.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Williams MP, Sercombe J, Hamilton MI and
Pounder RE: A placebo-controlled trial to assess the effects of 8
days of dosing with rabeprazole versus omeprazole on 24-h
intragastric acidity and plasma gastrin concentrations in young
healthy male subjects. Aliment Pharmacol Ther. 12:1079–1089.
1998.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Liu Q, Duan ZP, Ha DK, Bengmark S,
Kurtovic J and Riordan SM: Synbiotic modulation of gut flora:
Effect on minimal hepatic encephalopathy in patients with
cirrhosis. Hepatology. 39:1441–1449. 2004.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Prasad S, Dhiman RK, Duseja A, Chawla YK,
Sharma A and Agarwal R: Lactulose improves cognitive functions and
health-related quality of life in patients with cirrhosis who have
minimal hepatic encephalopathy. Hepatology. 45:549–559.
2007.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Shukla S, Shukla A, Mehboob S and Guha S:
Meta-analysis: The effects of gut flora modulation using
prebiotics, probiotics and synbiotics on minimal hepatic
encephalopathy. Aliment Pharmacol Ther. 33:662–671. 2011.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Chapelle N, Ben Ghezala I, Barkun A and
Bardou M: The pharmacotherapeutic management of gastroesophageal
reflux disease (GERD). Expert Opin Pharmacother. 22:219–227.
2021.PubMed/NCBI View Article : Google Scholar
|














