1. Introduction
Cutaneous melanin pigment production is a unique
characteristic of melanocytes, which has a critical role in the
protection against the harmful effects of sun exposure and
oxidative stress (1). It is
produced in melanosomes by melanocytes in a complex process: An
enzymatic transformation of L-tyrosine to dopaquinone and
subsequent chemical and biochemical reactions resulting in the
production of various 5,6-dihydroxyindole (DHI)-2-carboxylic acid
and DHI oligomers-main constituents of eumelanin, and the
benzothiazine and benzothiazole units of pheomelanin (2).
Increasing evidence indicates that numerous factors,
including ultraviolet radiation (UVR), as well as hormones at the
tissue, cellular and subcellular levels, may regulate the
biosynthesis of melanin, and UVR is known to be the most
significant factor (3). UVR,
mainly UVB, results in DNA thymidine breaks and generates genotoxic
cyclobutane pyrimidine dimers and 6-4 photoproducts in the skin.
The intermediates during this process also include reactive oxygen
species, which are highly cytotoxic and interact with multiple
cellular components and lead to oxidative DNA damage (4). Furthermore, DNA damage also increases
the expression of p53 in keratinocytes and activates the
transcription of pro-opiomelanocortin, which is cleaved to form
α-melanocyte-stimulating hormone and adrenocorticotropic hormone
(5), which then bind to their
receptor melanocortin 1 receptor on the cell membrane of
melanocytes to stimulate cyclic adenosine monophosphate
(AMP)-responsive production and activate the transcription of
microphthalmic-associated transcription factor (MITF)-induced
pigmentation enzymes, including tyrosinase (TYR) and
tyrosine-related protein 2(6). The
formation of the multi-enzyme complex in melanocytes control the
quantity and quality of melanin pigment production (7). In addition, UVR can also evoke a
transient increase in the cellular levels of diacylglycerol, a
component of the melanocyte membrane that activates protein kinase
C and regulates melanogenesis via TYR phosphorylation (8).
Recently, a ‘Yin and Yang’ action of melanogenesis
was proposed by Slominski et al (1). It means that under physiologic
conditions, melanogenesis is highly regulated because it takes
place within the boundaries of melanosomes, which has a protective
role against UVR-induced melanogenesis and is beneficial to the
skin (7); however, the presence of
melanin may be necessary for the initiation of malignant
transformation of melanocytes: Under pathological conditions, this
process is destructive through highly reactive intermediates of
melanogenesis leaking out of melanosomes, which affects the
behavior of melanoma cells and the outcomes of different types of
therapeutic approach. To be specific, the induction of
melanogenesis leads not only to stimulated expression of
hypoxia-inducible factor 1α (HIF-1α) protein in melanoma cells but
also to the robust upregulation of classical HIF-1-dependent target
genes involved in angiogenesis and cellular metabolism, including
glucose metabolism; furthermore, a highly oxidative environment
results in an immunosuppressive effect within the tumor environment
and/or systemically (2,9-11),
which inhibits the host responses and promotes melanoma
progression, and leads to therapeutic resistance. It may therefore
be suggested that inhibition of melanogenesis in advanced melanoma
may represent a realistic adjuvant strategy to attenuate melanoma
growth, as well as improve immuno-, radio- and chemotherapy.
Melanoma is the most aggressive and deadly type of
skin cancer (12). In total,
324,635 new cases and 57,043 deaths from melanoma were registered
in the GLOBOCAN 2020 database (13). Although early-stage melanoma is
considered to be curable with wide local excision (14), due to its potential to invade the
dermis within only a few months, melanoma is fatal when metastasis
occurs. Alarmingly, approximately one-third of patients with
advanced melanoma have already developed lung, liver or brain
metastasis by the time they receive a diagnosis (15). Overall, the 5-year survival rate
reaches 99% for patients with localized melanoma but decreases to
27.3% for those with distant metastasis (16). Thus, metastatic melanoma is usually
associated with poor prognosis. Recently, despite a steady rise in
the worldwide incidence of melanoma, novel therapeutic
interventions, such as targeted therapy and immunotherapy, have
resulted in rapid and extensive changes in mortality rates
(13-16).
Targeted agents mainly include mitogen-activated protein kinase
(MAPK) pathway inhibitors (15,17,18).
Immunotherapy includes immune checkpoint inhibitors, tumor
vaccinations and adoptive cell therapies (19-21).
For targeted therapy, classic v-Raf murine sarcoma viral oncogene
homologue B1 (BRAF) and MAPK kinase (MEK) inhibitors are applied
specifically for BRAF V600E/K mutation-positive melanoma. Targeted
drugs show high efficacy and increase the overall survival (OS) and
objective response rate (ORR) of most patients with metastatic
melanoma, though these patients may easily acquire drug resistance
(22). Immunotherapy, particularly
immune checkpoint inhibitors, may improve a patient's duration of
response (DOR), despite a slower onset of action (23). Providing insight into the
complementary advantages of these two regimens, the present article
reviewed clinical trials of current targeted therapies and
immunotherapy for the treatment of metastatic melanoma. The current
benefits and limitations of monotherapy or combination therapy may
encourage researchers to design strategies to allow for the use of
these treatments in more patients with metastatic melanoma.
2. Targeted therapy
BRAF and MEK inhibitors
MAPK cascades involve RAF, MEK and ERK kinases.
Approximately 50% of patients with metastatic melanoma harbor a
BRAF mutation (with >90% being the BRAF V600E mutation), which
mediates overactivation of the MAPK signaling pathway and the
survival, differentiation and proliferation of melanocytes
(24,25). This oncogenic signaling may be
blocked by BRAF (vemurafenib, dabrafenib and encorafenib) or MEK
(cobimetinib, trametinib and binimetinib) inhibitors (Fig. 1). Several combination therapies
comprising BRAF/MEK inhibitors with approved indications are
described in detail below and summarized in Table I.
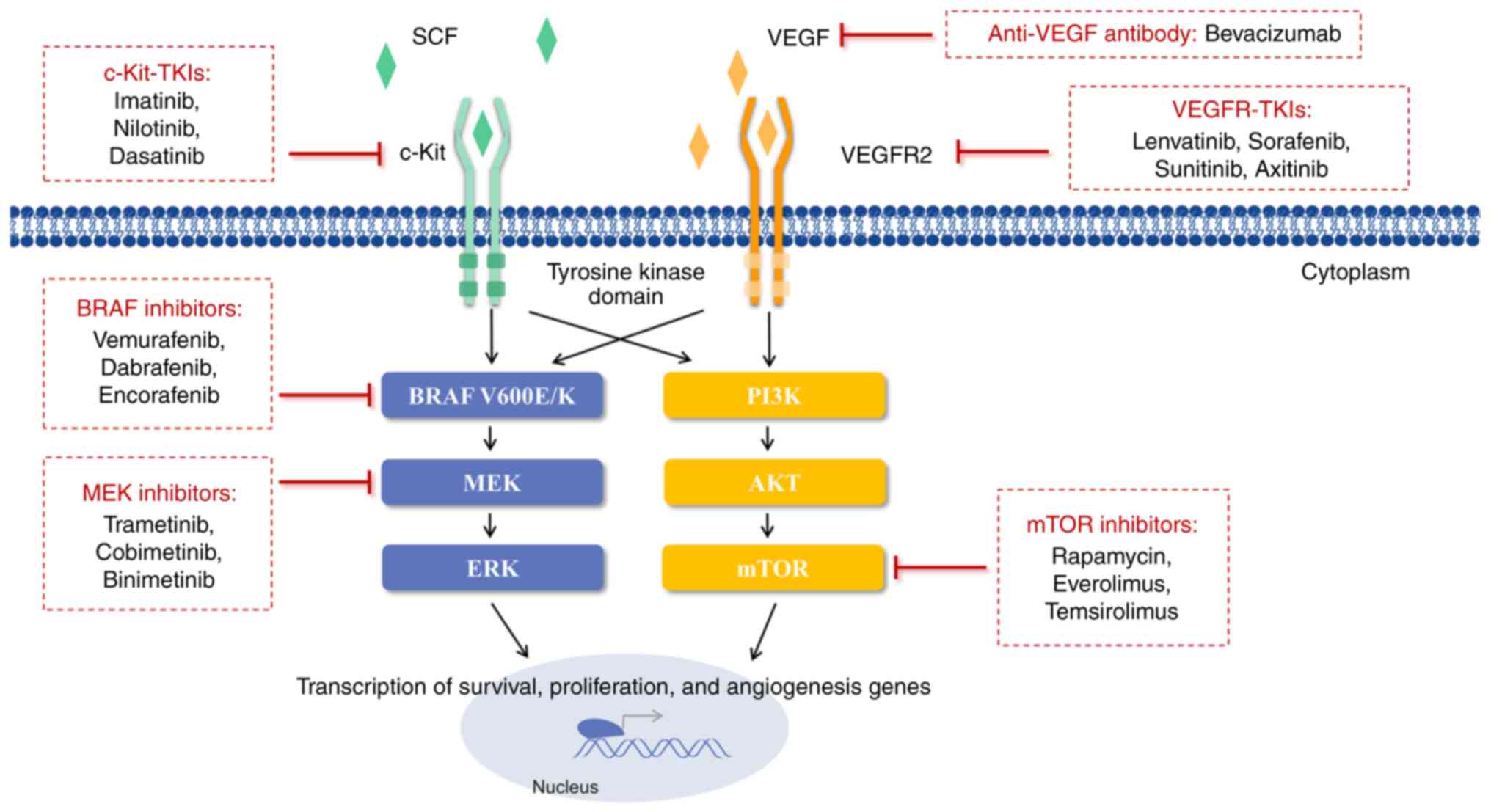 | Figure 1Signal transduction inhibitors for
metastatic melanoma. BRAF inhibitors (vemurafenib, dabrafenib and
encorafenib) and MEK inhibitors (trametinib, cobimetinib and
binimetinib) suppress abnormal MAPK signaling. mTOR inhibitors
(rapamycin and rapamycin analogues, everolimus and temsirolimus)
downregulate activation of the PI3K/AKT/mTOR pathway, which is
related to tumor cell proliferation and angiogenesis. VEGFR-TKIs
(lenvatinib, sorafenib, sunitinib and axitinib) inhibit
interactions between VEGF and VEGFR2. c-Kit-TKIs (imatinib,
nilotinib and dasatinib) inhibit interactions between SCF and
c-Kit. Furthermore, bevacizumab directly binds to VEGF, preventing
tumor angiogenesis and growth. SCF, stem cell factor; c-Kit,
receptor tyrosine kinase; TKI, tyrosine kinase inhibitor; VEGFR,
vascular endothelial growth factor receptor; MEK, mitogen-activated
protein kinase kinase; ERK, extracellular regulated protein kinase;
PI3K, phosphatidylinositol 3 kinase; AKT, protein kinase B; mTOR,
mammalian target of rapamycin; BRAF, v-Raf murine sarcoma viral
oncogene homologue B1. |
 | Table ICombined BRAF and MEK inhibition with
approved indications. Dabrafenib plus trametinib, vemurafenib plus
cobimetinib, and encorafenib and binimetinib are specifically
intended for a majority of patients with melanoma with the BRAF
V600 mutation. |
Table I
Combined BRAF and MEK inhibition with
approved indications. Dabrafenib plus trametinib, vemurafenib plus
cobimetinib, and encorafenib and binimetinib are specifically
intended for a majority of patients with melanoma with the BRAF
V600 mutation.
| Monotherapy or
combined therapy | Indication (year of
FDA approval) |
|---|
| Dabrafenib and
Trametinib | Metastatic,
unresectable melanoma with BRAF V600E/K+ (2014);
Adjuvant: Resected, stage III melanoma with BRAF
V600E/K+ (2018) |
| Vemurafenib and
Cobimetinib | Metastatic,
unresectable melanoma with BRAF V600E/K+ (2015) |
| Encorafenib and
Binimetinib | Metastatic,
unresectable melanoma with BRAF V600E/K+ (2018) |
Vemurafenib and Cobimetinib. In 2011,
vemurafenib became the first oral inhibitor for BRAF V600E-mutated
melanoma. Cobimetinib, a potent MEK inhibitor, was evaluated in
combination with vemurafenib in the Phase Ib BRIM 7 study on
patients with advanced BRAF V600E-mutated melanoma who had never
received a BRAF inhibitor. A confirmed ORR of 87%, including 10%
who had a complete response (CR), with a median PFS of 13.7 months,
was reported (26). Further
evidence of the efficacy of combined vemurafenib and cobimetinib
was reported in the international, multicentre, randomized phase
III CoBRIM study (27). In this
trial, 495 eligible participants were enrolled and randomized 1:1
to receive either dual cobimetinib plus vemurafenib therapy or
vemurafenib alone. With at least 5 years of follow-up, the median
OS was significantly increased, with 22.5 months in patients on
cobimetinib plus vemurafenib treatment compared with 17.4 months in
those on vemurafenib alone; OS rates were continuously improved
with this dual therapy compared with vemurafenib alone, with 38 vs.
31% at 3 years, 34 vs. 29% at 4 years, and 31 vs. 26% at 5 years.
Similar to the OS results, the median progression-free survival
(PFS) was 12.6 vs. 7.2 months, and PFS rates were 14 vs. 10% at 5
years (27). Identifying subgroups
of patients likely to have a beneficial long-term treatment outcome
is of great importance to informing treatment decisions when
managing patients with metastatic melanoma. Conventional prognostic
factors for survival outcomes in patients with metastatic melanoma
include disease stage, baseline lactate dehydrogenase (LDH) serum
level, baseline sum of the longest diameters of the target lesion
(SLD), baseline Eastern Cooperative Oncology Group performance
status (ECOG PS) and presence/absence of liver metastasis (28,29).
In this trial, the long-term survival outcomes with dual
cobimetinib and vemurafenib were most favourable in patients with
normal baseline LDH levels and a low tumor burden (defined as
either SLD ≤45 mm or <3 organ sites with metastasis), with
5-year OS rates of 52% in the subgroup defined by normal baseline
LDH and SLD ≤45 mm, and 68% in the subgroup defined by normal
baseline LDH and <3 organ sites (27). While the safety profile remained
consistent with previous data and there existed no new safety
signals (30), several
protocol-defined adverse events (AEs) of special interest were more
common in the cobimetinib plus vemurafenib group compared with the
vemurafenib monotherapy group, including retinal detachment or
central serous retinopathy, grade ≥3 photosensitivity, grade ≥3
liver laboratory abnormalities, grade ≥2 ejection fraction
reduction and grade ≥3 creatine phosphokinase elevation (27). In addition, the long-term OS
outcomes were least favourable in those with elevated baseline LDH
>2x upper limit of normal (ULN), and almost all patients had
died by 3 years. Therefore, it indicated an urgent need to design
different treatment strategies to improve long-term survival
outcomes for patients in poor prognosis subgroups, particularly
those with elevated LDH levels at baseline.
A novel combined treatment strategy of adding one
immune checkpoint inhibitor to BRAF/MEK inhibitor combination
therapy emerged at an opportune time. The phase III IMspire150
study, examining how effective approved vemurafenib plus
cobimetinib combined with or without atezolizumab (A+V+C or P+V+C)
is for patients with BRAF V600 mutation-positive melanoma with
elevated LDH levels at baseline, is ongoing (31). Of note, patients with
anti-programmed cell death ligand 1 (PD-L1)- melanoma,
who have fewer PD-L1-expressing tumor-infiltrating cells and
generally benefit less from immunotherapy alone, appeared to derive
a clinical benefit from A+V+C similarly to those with
PD-L1+ tumors. Specifically, with a follow-up of 18
months, the median PFS for A+V+C in the PD-L1- and
PD-L1+ subgroups was 15.2 and 14.8 months, respectively.
In addition, the median PFS at 18 months follow-up for A+V+C in the
PD-L1- and high-LDH subgroup was higher than that in the
PD-L1+ and high-LDH subgroup, at 9.8 and 6.3 months,
respectively (32). However,
long-term benefits have not yet been reported and it is necessary
to identify other subgroups that may benefit from triplet A+C+V
therapy. TRICOTEL is another multicentre, single-arm, phase 2
clinical trial evaluating the efficacy and safety of this triplet
therapy (atezolizumab combined with vemurafenib plus cobimetinib)
in patients with BRAF V600 mutation-positive melanoma who were
receiving corticosteroids and had symptomatic central nervous
system (CNS) metastasis (33). The
results indicated that, at the 9.7-month median follow-up duration,
the intracranial ORR was 42%, which is comparable to that reported
with other available systemic treatments, with 46% of intracranial
ORR with the combination of nivolumab plus ipilimumab (34), and with 55% of extracranial ORR
with the combination of dabrafenib plus trametinib (35). Although the incidence of
treatment-related grade 3 or worse AEs was 68%, this was similar to
the incidences reported with A+V+C (79%) and P+V+C (73%) in
patients without CNS metastases in the IMspire150 trial (31,33).
Furthermore, the occurrence of serous retinopathy was generally
consistent with that observed with vemurafenib plus cobimetinib
combination in the coBRIM trial (36). Thus, the triplet combination
appears to be recommendable for patients with BRAF V600
mutation-positive melanoma, even with CNS metastases.
Dabrafenib and Trametinib. Dabrafenib is
another oral BRAF inhibitor that was approved for use in 2013. In
the next year, it was approved in combination with trametinib, an
oral MEK inhibitor, for the treatment of unresectable, metastatic
BRAF V600E/V600K-mutated melanoma. In the randomized,
double-blinded phase 3 study (COMBI d) with a median follow-up of
20 months for the combined therapy arm and 16 months for the
dabrafenib monotherapy arm, the combination of dabrafenib and
trametinib led to an improved median PFS (11 vs. 8.8 months) and
median OS (25.1 vs. 18.7 months), with a 2-year OS of 51 vs. 42%
for the dabrafenib monotherapy group (37). These findings were consistent with
those reported in another trial (COMBI v), which aimed to compare
the combination of dabrafenib and trametinib with vemurafenib
monotherapy (38). COMBI I is a
phase III trial to evaluate the efficacy of the anti-programmed
cell death protein 1 (PD-1) antibody spartalizumab combined with
dabrafenib and trametinib (sparta-DabTram) vs. placebo combined
with dabrafenib and trametinib (placebo-DabTram), with questionable
efficacy. After a minimum follow-up period of 24 months, only 20
and 18% of patients in the sparta-DabTram and placebo-DabTram arms,
respectively, achieved complete response (39). In addition, PD-1 blockade seemed to
add little to combined BRAF and MEK inhibition when treating BRAF
V600-mutated melanoma: The median investigator-assessed PFS was
16.2 and 12.0 months in the sparta-DabTram arm and placebo-DabTram
arm, respectively, but with no statistically significant difference
(39).
A prospective study on patients with advanced BRAF
V600-mutated cutaneous melanoma treated with dabrafenib plus
trametinib found that LDH, ECOG PS and a large number of metastatic
tumor sites are associated with disease progression. Relevant to
real-world practice, the study reported brain metastases as a major
prognostic factor (28).
Similarly, in the COMBI-MB trial, patients were recruited and
divided into 4 cohorts based on LDH levels, ECOG PS, type of
mutation and presence/absence of brain metastasis. The results
indicated that dabrafenib plus trametinib was efficient, with a
manageable safety profile, particularly in the BRAF V600-mutated
melanoma subgroup without brain metastasis, with a median PFS
ranging from 4.2 to 7.2 months and median DOR from 4.5 to 8.3
months (35). Therefore, it
appears necessary to explore a better treatment regimen for
patients with melanoma with brain metastasis to improve their
survival outcomes.
Efforts have been made to evaluate the role of
dabrafenib plus trametinib as an adjuvant treatment for high-risk
resected disease. In an adjuvant setting, according to the COMBI-AD
trial, patients with completely resected stage III melanoma with
BRAF V600 mutations treated with dabrafenib plus trametinib had a
significantly lower rate of recurrence (40,41).
During the follow-up period of 5 years, 52 and 65% of patients who
received adjuvant dabrafenib plus trametinib achieved relapse-free
and distant metastasis-free survival (DMFS) vs. 36 and 54% of those
receiving adjuvant placebo, respectively. Furthermore, the toxicity
was consistent with previous data regarding targeted therapy in the
metastatic setting and no new toxic effects were reported (42). Based on the positive results with
adjuvant dabrafenib and trametinib in this trial, this combination
has become a standard treatment option for adjuvant therapy in
patients with surgically resected stage III V600E/K BRAF-mutated
melanoma. Of note, in COMBI-AD, pyrexia was the most common adverse
event experienced by patients treated with dabrafenib plus
trametinib and 9% of all patients discontinued treatment due to
pyrexia. The subsequent COMBI-APlus trial, which aims to reduce the
burden of serious pyrexia-related events associated with this
treatment strategy, is currently being conducted (43). The investigators proposed an
adapted pyrexia management algorithm: Both drugs were interrupted
if patients developed signs and symptoms of possible
treatment-emergent pyrexia syndrome: Fever (body temperature
≥38˚C), chills, rigours, night sweats and influenza-like symptoms.
Treatment was restarted at the same dose once patients were
symptom-free for ≥24 h. The COMBI-APlus trial has now met its
primary endpoint of significant reduction in the incidence of
composite pyrexia events compared with a historical control from
the COMBI-AD trial (43), with
lower rates of grade 3/4 pyrexia (3.8%), hospitalization due to
pyrexia (4.3%) and discontinuation due to pyrexia (2.4%), compared
with COMBI-AD. It seems helpful for patients to manage pyrexia at
home, which may be beneficial for patients' quality of life.
Encorafenib and Binimetinib. In 2018,
encorafenib emerged as a second-generation BRAF inhibitor and was
approved by the food and drug administration (FDA) in combination
with another MEK inhibitor, binimetinib. In the randomized,
open-label phase III COLUMBUS trial, comparison of PFS by a blinded
independent central review revealed a median PFS of 14.9 months in
the encorafenib plus binimetinib arm and 9.6 months in the
encorafenib monotherapy arm; furthermore, the ORR was 63% for
patients who received the combination therapy, while it was 51% for
those who received the encorafenib monotherapy. In general, the
modified pharmacological properties of encorafenib are considered
crucial to its favourable efficacy and increased tolerability
profile for melanoma patients carrying the BRAF V600E mutation, as
a result of its enhanced on-target effects and less paradoxical
MAPK pathway activation (44).
This may be why the rate of grade 3 or 4 AEs was slightly reduced
with combined therapy as compared with BRAF inhibitors alone (47
vs. 63%) (45).
Combined BRAF and MEK inhibition has become the
standard-of-care treatment for BRAF V600E-mutated melanoma. Other
clinical trials to further evaluate the best dose and side effects
of the combination regimens are ongoing and may provide more
information on targeted therapy for melanoma over time
(NCT01909453, NCT03543969, NCT01989585, NCT05026983, NCT04741997,
NCT04221438, NCT01902173 and NCT02231775).
DNA damage response (DDR)
inhibitors
Different forms of DNA damage, including
single-strand breaks (SSBs) and double-strand DNA breaks (DSBs),
are repaired by different repair mechanisms. SSBs evoke responses
by the base excision repair (BER) pathway, whereas DSBs evoke
responses by the homologous recombination repair (HRR) and
non-homologous end-joining (NHEJ) pathways (46). It is worth noting that the HRR
process is based on template-directed DNA repair synthesis to
obtain error-free effective repair of DSBs but that NHEJ signaling
is an error-prone repair process that causes DNA rearrangements
(47,48).
Poly(ADP-ribose) polymerase (PARP)
inhibitors. PARP1 is a major factor in the BER process and it
is also critical for HRR and NHEJ. PARP inhibitors act through the
following two different mechanisms: Inhibition of canonical PARP
function and PARP ‘trapping’.
On the one hand, PARP inhibitors inhibit the
catalytic activity of PARP1, which leads to failure of SSB repair
and stalls and/or collapse of replication fork progression; hence,
deleterious DSBs may be generated and accumulate. In replicating
cells, these DSBs are normally repaired by HRR signaling; in
melanoma tumor cells with HRR deficiency, such as BRCA1, BRCA2 and
partner and localizer of BRCA2 (PALB2) mutations, NHEJ signaling
may be activated, which may result in modulation of DNA replication
dynamics, altered gene expression and tumor cell death (49,50).
This is called the ‘synthetic lethality’ effect (Fig. 2). On the other hand, PARP
inhibitors are reported to be more cytotoxic than PARP depletion
because the former block activated PARP1 on damaged DNA through a
poisonous allosteric effect (51).
Furthermore, the potency in trapping PARP varies markedly among
different inhibitors, in the order of talazoparib » niraparib >
olaparib/rucaparib » veliparib (51).
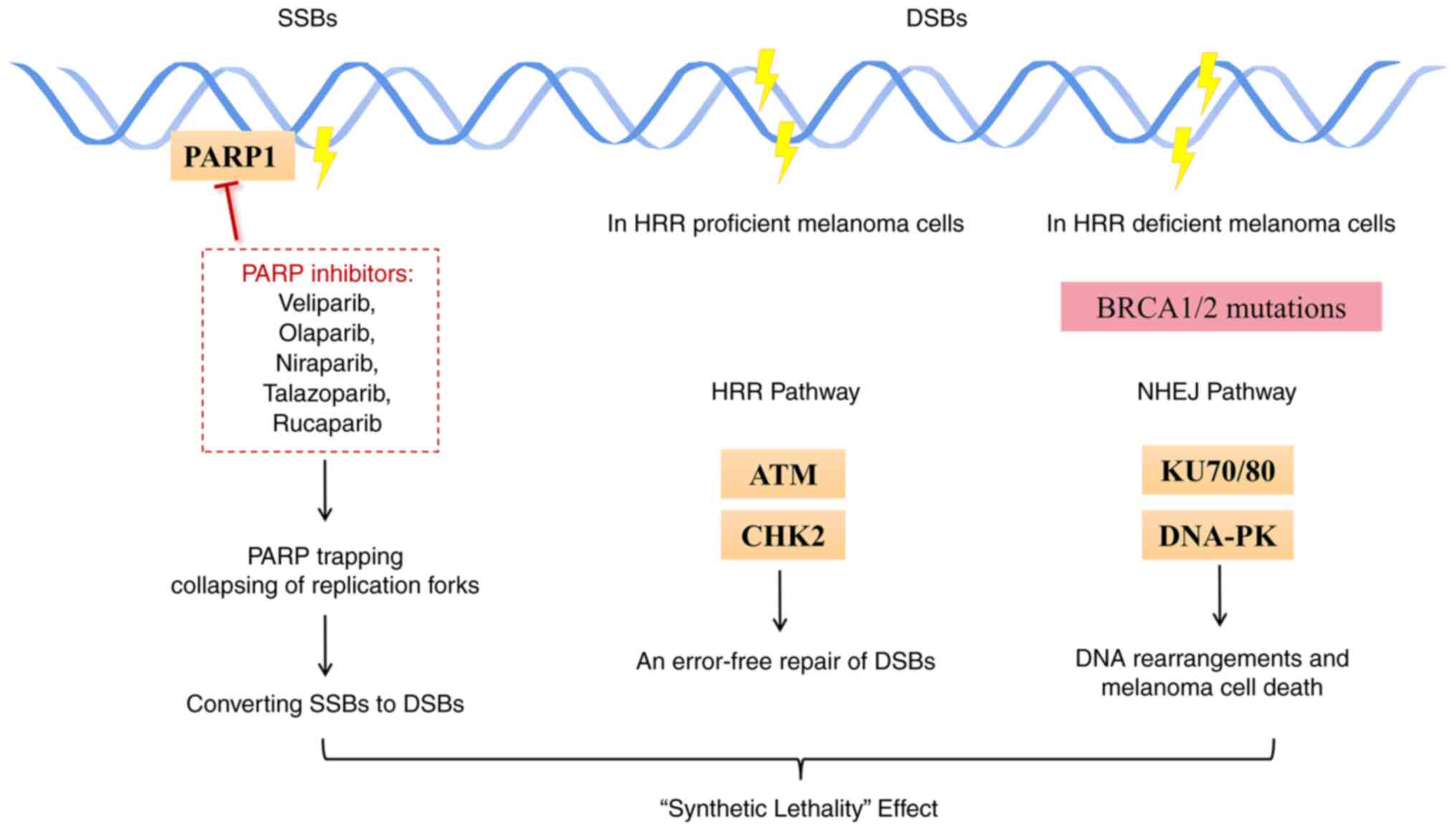 | Figure 2‘Synthetic lethality’ effect induced
by PARP inhibitors. PARP inhibitors impair the catalytic activity
of PARP1, which leads to failure of SSB repair and collapses the
progression of replication forks. Deleterious DSBs may accumulate.
In melanoma tumor cells with HRR deficiency, such as BRCA1/2
mutations, NHEJ signaling may be activated, which eventually
results in tumor cell death. ATM and CHK2, and KU70/80 and DNA-PK
are the main components of the HRR and NHEJ pathways, respectively.
PARP1, poly(ADP-ribose) polymerase 1; SSBs, single-strand breaks;
DSBs, double-strand DNA breaks; HRR, homologous recombination
repair; NHEJ, non-homologous end-joining; ATM, ataxia
telangiectasia-mutated gene; CHK2, checkpoint kinase 2; DNA-P2,
DNA-dependent protein kinase. |
The PARP inhibitors mentioned above have been
approved by the FDA for patients with familial breast or ovarian
cancer harboring germline BRCA1/2 mutations (52). This rapid translation from a
preclinical model to promising clinical data has driven the
development of several PARP inhibitors for different types of
tumor, including melanoma. Emerging evidence suggested that the
efficacy of PARP inhibitors in delaying the PARP-mediated repair of
DNA damage was potentiated by the combination of chemo- and/or
radiotherapeutic agents (53). A
phase I trial assessing the effect and best dose of veliparib when
given together with paclitaxel and carboplatin in patients with
metastatic melanoma is ongoing (NCT01366144). Another two phase II
trials focusing on the therapeutic outcome of temozolomide plus
veliparib or rucaparib in patients with metastatic melanoma
demonstrated a trend towards improvement in PFS, yet without
reaching statistical significance (54,55).
These disappointing data may be because the patients were not
stratified based on HR status. Indeed, one case report described
the effect of olaparib monotherapy in a patient with advanced
metastatic melanoma carrying a PALB2 mutation who had previously
progressed on ipilimumab plus nivolumab combined immunotherapy,
showing a partial response to the PARP inhibitor olaparib (56). As described by Khaddour et
al (57) in another recent
case report, a patient with unresectable stage IIIC melanoma with a
high HRR deficiency score, whose disease progressed on prior
nivolumab monotherapy, achieved a near-complete response to
olaparib plus nivolumab. Currently, the use of niraparib or
olaparib in patients with melanoma known to have mutations in
BRCA1/2, PALB2, BRCA1-Associated Protein 1 (BAP1) or other
components of the DDR pathway through synthetic lethality is under
clinical validation (NCT05169437, NCT03925350, NCT03207347 and
NCT05482074), highlighting the importance of testing for genetic HR
mutations in patients with melanoma and determining the mutations'
impact on treatment decisions.
Inhibitors of other DDR factors are also under
development, including those targeting ataxia-telangiectasia
mutated (ATM) and ataxia-telangiectasia mutated and Rad3-related
(ATR) kinases, cell cycle checkpoint kinase 1/2 (CHK1/2), and WEE1
checkpoint kinase. The DDR-DDR inhibitor combination appears to be
an emergent approach to treating melanoma, which is able to target,
at the same time, modulators involved in the different pathways
melanoma relies on (e.g. pro-survival pathways). Preclinical
studies demonstrated that the PARP inhibitor veliparib, as well as
the PARP inhibitor Olaparib combined with ATR inhibitor
ceralasertib, could reduce the viability of melanoma cells
sensitive and resistant to BRAF/MEK inhibitors, respectively, in
vitro (58,59); however, there are no further
related clinical trials of them in patients with melanoma
refractory to standard targeted therapy or no trials assessing the
efficacy of the combination of DDR inhibitor plus standard targeted
therapy in patients with melanoma.
Certain evidence pointed to the emerging role of
PARP inhibitors in the tumor immune microenvironment and suggested
that the addition of PARP inhibitors may potentiate the therapeutic
response to checkpoint inhibitors. The immune role of PARP
inhibitors is further discussed below and clinical trials of this
combined regimen are summarized in section 4 and Table II.
| Table IIOngoing clinical trials combining
targeted therapy and immunotherapy. Clinical trials of combined
therapy involving BRAF/MEK inhibitors plus immune checkpoint
inhibitors or involving angiogenesis inhibitors plus immune
checkpoint inhibitors are listed. |
Table II
Ongoing clinical trials combining
targeted therapy and immunotherapy. Clinical trials of combined
therapy involving BRAF/MEK inhibitors plus immune checkpoint
inhibitors or involving angiogenesis inhibitors plus immune
checkpoint inhibitors are listed.
| NCT number | Trial phase | Targeted
therapy | Immunotherapy | Status |
|---|
| NCT04511013 | II | Encorafenib,
Binimetinib | Ipilimumab,
Nivolumab | Recruiting |
| NCT03235245 | II | Encorafenib,
Binimetinib | Ipilimumab,
Nivolumab | Recruiting |
| NCT02631447 | II | Encorafenib,
Binimetinib | Ipilimumab,
Nivolumab | Active, not
recruiting |
| NCT02224781 | III | Dabrafenib,
Trametinib | Ipilimumab,
Nivolumab | Active, not
recruiting |
| NCT01940809 | I | Dabrafenib,
Trametinib | Ipilimumab,
Nivolumab | Active, not
recruiting |
| NCT04741997 | I | Encorafenib,
Binimetinib | Nivolumab | Recruiting |
| NCT02910700 | II | Encorafenib,
Binimetinib | Nivolumab | Recruiting |
| | | Dabrafenib,
Trametinib | | |
| NCT04375527 | II | Binimetinib | Nivolumab | Recruiting |
| NCT04310397 | II | Dabrafenib,
Trametinib | Spartalizumab | Active, not
recruiting |
| NCT02967692 | III | Dabrafenib,
Trametinib | Spartalizumab | Active, not
recruiting |
| NCT01950390 | II | Bevacizumab | Ipilimumab | Active, not
recruiting |
| NCT04996823 | II | Axitinib | Ipilimumab | Recruiting |
| NCT03086174 | Ib | Axitinib | Toripalimab | Active, not
recruiting |
| NCT03175432 | II | Bevacizumab,
Cobimetinib | Atezolizumab | Recruiting |
| NCT03554083 | II | Vemurafenib,
Cobimetinib | Atezolizumab | Recruiting |
| NCT04356729 | II | Bevacizumab | Atezolizumab | Recruiting |
| NCT02902042 | I/II | Encorafenib,
Binimetinib | Pembrolizumab | Completed |
| NCT04657991 | III | Encorafenib,
Binimetinib | Pembrolizumab | Recruiting |
| NCT02858921 | II | Dabrafenib,
Trametinib | Pembrolizumab | Active, not
recruiting |
| NCT02298959 | I | Aflibercept | Pembrolizumab | Recruiting |
| NCT04633902 | II | Olaparib | Pembrolizumab | Recruiting |
| NCT04187833 | II | Talazoparib | Pembrolizumab | Recruiting |
| NCT03820986 | III | Lenvatinib | Pembrolizumab | Active, not
recruiting |
Angiogenesis inhibitors
Angiogenesis is closely associated with the growth
and metastasis of solid tumors, including melanoma (60). As the tumor grows, its cells
consume a large amount of oxygen and nutrients, leading to hypoxia
in the tumor microenvironment, which subsequently induces
upregulation of proangiogenic factors, mainly vascular endothelial
growth factor (VEGF). The interaction between VEGF and its main
receptor, VEGF receptor 2 (VEGFR2), induces several downstream
angiogenic signaling pathways, such as Src/vascular endothelial
cadherin (VE-cadherin) signaling and phosphatidylinositol 3 kinase
(PI3K)/protein kinase B (AKT)/endothelial nitric oxide (NO)
synthase/NO signaling, to become aberrantly activated (61,62).
Therefore, VEGF and VEGFR have become predominant targets for the
development of anti-angiogenic agents (Fig. 1).
Anti-VEGF antibody. Bevacizumab, a humanized
immunoglobulin G1 (IgG1) monoclonal antibody, has been the first
approved anti-angiogenic agent since 2004. It is designed to
selectively bind to VEGF and block interactions between VEGF and
its receptors, thereby preventing tumor angiogenesis and growth.
Based on promising previous clinical data, bevacizumab leads to
particularly favourable outcomes and has an acceptable safety
profile when used in combination with chemotherapy for the
treatment of malignant melanoma (63-66).
In the most recent randomized controlled phase II trial of
bevacizumab in combination with carboplatin plus paclitaxel (a
first-line chemotherapeutic regimen) for advanced mucosal melanoma,
the addition of bevacizumab markedly increased the median PFS from
3.0 to 4.8 months and the median OS from 9.0 to 13.6 months
(66). A phase III study further
evaluating the benefits of this new therapeutic protocol
carboplatin-paclitaxel-bevacizumab (CPB) is underway (NCT02023710).
Overall, the CPB protocol may become an alternative for the
treatment of mucosal melanoma in the first-line setting. In
addition, Hodi et al (19)
found that bevacizumab showed significant synergistic efficacy when
used in combination with ipilimumab for metastatic melanoma. These
authors recently showed that patients with a long-term or delayed
increase in soluble PD-L1 had clinically beneficial outcomes,
highlighting the cross-talk between tumor immunity and angiogenesis
within the tumor microenvironment (67).
VEGFR tyrosine kinase inhibitors (TKIs).
Several VEGFR-TKIs have been approved for use as targeted therapy,
mainly in metastatic lung cancer, including sorafenib, lenvatinib,
sunitinib and axitinib. Recently, the clinical efficacy of
VEGFR-TKIs in metastatic melanoma, as a part of combination
medications or a single agent, has been evaluated and showed
limited but promising outcomes (68-72).
High expression of VEGF in was observed in patients with metastatic
melanoma, indicating that high vascularity may be a prognostic
factor for tumor progression and metastasis. Following the novel
idea of dual VEGF and VEGFR signaling blockade, a phase II trial of
combined therapy of bevacizumab plus sorafenib evaluated efficacy
and safety for advanced melanoma. The results suggested that of 14
patients with malignant melanoma who received treatment, 57%
achieved stable disease (SD) lasting ≥16 weeks, including 3 with SD
lasting ≥1 year. The median time to progression (TTP) was 32 weeks.
Of note, patients with low serum VEGF tended to achieve longer TTP
than those with high serum VEGF (50 vs. 15 weeks) (73). In terms of drug safety,
hypertension, fatigue and foot syndrome were the most frequently
reported drug-related AEs.
To date, the therapeutic strategy of dual VEGF and
VEGFR inhibition has not yet been validated, mainly because dual
inhibition is likely to generate significant toxicities compared
with monotherapy. For instance, despite improved anti-tumor
efficacy, the clinical trial of bevacizumab plus sunitinib in
melanoma and renal cell carcinoma was suspended due to
microangiopathy (74).
Other targeted therapies. Receptor
tyrosine kinase (c-Kit)-TKIs
c-Kit, also named cluster of differentiation
(CD)117, is a class III transmembrane receptor tyrosine kinase
(75). Melanomas harbouring c-Kit
alteration or amplification are mainly found in mucosal, acral and
chronically sun-damaged skin (76). c-Kit becomes activated via
phosphorylation after stem cell factor binding and is primarily
responsible for cell growth, survival and migration. Thus, the
c-Kit gene is regarded as an oncogene and small molecule inhibitors
targeting c-Kit in metastatic melanoma have been developed and
investigated in clinical trials (76). The relevant oncogenic cascades and
several targeted agents for melanoma are depicted in Fig. 1.
Guo et al (77) first conducted a phase II trial of
the application of imatinib for the treatment of patients with
metastatic melanoma harbouring c-Kit mutation and/or of c-Kit gene
copy number amplification. The results of this trial indicated that
the use of imatinib was associated with a median PFS of 3.5 months
and tumor regression in 41.9% of patients, with an overall 6-month
PFS rate of 36.6%, an overall 1-year survival rate of 51.0% and an
overall disease control rate of 55% in the cohort. It was also
reported that imatinib may be preferred for patients with c-Kit
genetic aberrations (77).
Recently, Jung et al (78)
performed a pooled analysis (n=130) based on retrospective,
‘real-world’ experience of imatinib for melanoma. In this study,
patients with mucosal melanoma appeared to have a higher response
rate (38%) than those with acral melanoma (25%). Patients harboring
L576P (exon 11) or K642E (exon 13) mutations displayed the greatest
response rates (52 and 42%) and disease control rates (65 and 92%);
of note, no patients with mutations in exon 17 had response or
disease control. In addition, seemingly longer PFS (median, 4.3 and
4.5 vs. 1.1 months) and OS (median, 19.7 and 15.4 vs. 12.1 months)
were observed in patients with exon 11 and 13 vs. exon 17
alterations, but there was no statistical significance. This result
indicates that more refined genetic selection strategies for
imatinib as a treatment of c-Kit-altered melanoma are needed in
subsequent trials.
A new meta-analysis revealed the highest ORR of 20%
for nilotinib, another promising c-Kit selective inhibitor, when
compared with dasatinib and sunitinib (8 and 8%), though very
similar to imatinib, with an ORR of 19% (17). Furthermore, five clinical trials of
this small molecule inhibitor have been conducted, in which all
eligible participants received nilotinib 400 mg twice daily
(79-83).
Among these trials, an ORR of 26.2% was observed in the single-arm,
phase II trial of nilotinib in 42 patients with c-Kit-mutated
advanced or inoperable melanoma (80). The most recent results from the
phase II multicentre trial performed by the French Skin Cancer
Network reported a durable tumor response in 16% of patients at 6
months after nilotinib initiation. The best ORR was 20% and the
disease control rate was 56%; however, these results were limited
to those harbouring exon 11 or 13 mutation (81). These nilotinib studies showed
similar ORRs, similar to historical data for imatinib treatment
(77,84), indicating that nilotinib may serve
as an active agent for patients with disease progression after
receiving imatinib.
Mammalian target of rapamycin (mTOR)
inhibitors. mTOR is a key kinase downstream of PI3K/AKT
signaling and is considered a regulator of cell proliferation,
survival, differentiation, apoptosis, angiogenesis and metabolism
(85). Aberrant activation of the
mTOR pathway is strongly linked to the pathogenesis of melanoma and
relevant clinical agents targeting mTOR itself have been developed.
In vitro, rapamycin and the rapamycin analogues everolimus
and temsirolimus show a significant inhibitory effect on melanoma
cell proliferation (86).
Temsirolimus was reported to be safe when combined with
bevacizumab, with enhanced systemic immune function for patients
with BRAF-wild-type melanoma (87). In addition, everolimus plus
bevacizumab was demonstrated to be well tolerated, despite frequent
grade 1 or 2 toxicities, for patients with metastatic melanoma who
had previously received chemotherapy and/or immunotherapy (88). Although mTOR inhibitors have an
essential role in anti-angiogenesis, a recent clinical trial
(NCT02023710) suggested that the addition of everolimus to the CPB
therapeutic protocol may not improve PFS among patients with
unresectable stage IV melanoma. Overall, the efficacy, safety and
scientific validity in metastatic melanoma produced by different
treatment options with mTOR inhibition remain unclear and more
preclinical and clinical studies are needed to evaluate
mTOR-targeted therapeutic approaches.
3. Immunotherapy
Immunotherapy has the ability to prevent tumor
growth and recurrence long-term by sensitizing the host's immune
system or strengthening the anti-tumor response. Based on decades
of research on solid tumor immunology, immunotherapy is considered
a promising new modality not only for metastatic melanoma but also
for other solid cancers, including kidney cancer, non-small cell
lung cancer and head and neck cancer. This approach has definitely
led to marked improvements in survival in these patients. To date,
several immunotherapy drugs, particularly blockade of negative
immune regulatory checkpoints, have been approved by the FDA for
metastatic melanoma (Fig. 3).
Immune checkpoint blockade
CD28, which is present on the surface of T cells,
acts as a co-stimulatory molecule to induce full activation of T
cells by binding to B7-1 (CD80) and B7-2 (CD86) on dendritic cells.
Cytotoxic T lymphocyte-associated protein 4 (CTLA-4) is a member of
the CD28 family. CTLA-4 is regarded to have a higher affinity to
compete with CD28 for binding to its ligands and thereby to mediate
T-cell exhaustion, perhaps via inhibition of interleukin 2 (IL-2)
accumulation and cell cycle progression (89). PD-1, which is also called PDCD1 or
CD279, is another inhibitory molecule on the surface of various
immune cells. Upon binding to the ligands PD-L1 (CD274) and PD-L2
(CD273) on tumor cells or antigen-presenting cells (APCs), PD-1
inhibits the T-cell receptor (TCR) signaling pathway and thus
blocks the immune response (90).
The 2018 Nobel Prize in Physiology or Medicine was
awarded to Professor James Allison and Professor Tasuku Honjo for
their work on CTLA-4 and PD-1, respectively (91,92).
From theory to reality, the idea of unleashing the host's immune
system to kill cancer cells using CTLA-4 and PD-1/PD-L1 blockade
has led to impressive results in cancer therapy.
CTLA-4 blockade. In 2011, ipilimumab, a fully
humanized IgG1 monoclonal antibody designed to block the
CTLA-4-CD80/86 interaction, was first approved for the treatment of
advanced unresectable stage III or IV melanoma, ushering in a new
era of immune-based therapies for cancer. In a phase III study
among patients with previously treated metastatic melanoma, rates
of OS were significantly higher with ipilimumab monotherapy (3
mg/kg) or ipilimumab plus glycoprotein 100 (gp100), a peptide
vaccine, than with gp100 monotherapy (45.6 and 43.6 vs. 25.3% at 12
months, 33.2 and 30.0 vs. 16.3% at 18 months, and 23.5 and 21.6 vs.
13.7% at 24 months). However, the removal of self-tolerance induces
autoimmune toxicities, which is termed immune-related adverse
events (irAEs). The incidence of irAEs was reported to be higher
with ipilimumab alone or combined with gp100 (including
dermatologic, pruritus, rash, vitiligo, gastrointestinal and
diarrhea) compared with gp100 alone (14.5 and 10.2 vs. 3.0%)
(93). In an adjuvant setting,
approval from the FDA was granted in 2015 based on the results of
the EORTC 18071 phase III trial. The trial aimed to evaluate the
efficacy of adjuvant ipilimumab at 10 mg/kg in patients with stage
III cutaneous melanoma who had undergone a complete regional lymph
node dissection. At a mean follow-up of 5.3 years, 5-year rates of
recurrence-free survival (RFS), OS, and DMFS were 40.8, 65.4 and
48.3% in patients on adjuvant ipilimumab treatment compared with
30.3, 54.4 and 38.9% in the placebo group, respectively (94). The frequency of grade 3 or 4 irAEs
in the ipilimumab group was higher than that in the placebo group
(41.6 vs. 2.7%), with the most common ones being gastrointestinal
disorders, hepatitis and endocrinopathy. At the cost of substantial
toxic effects, this updated analysis was consistent with the
previously observed prolongation of RFS in patients on adjuvant
ipilimumab treatment (95). While
ipilimumab achieved high efficacy and durability of the
anti-melanoma response, the clinical use of this drug in tumor
immunotherapy is limited because of severe irAEs. Indeed, these
irAEs may vary in severity and are occasionally life-threatening.
Pancreatitis, nephritis, severe skin reactions including Steven
Johnson syndrome, neurologic conditions like inflammatory myopathy,
aseptic meningitis, posterior reversible encephalopathy syndrome
and myasthenia gravis are also described in the literature,
underscoring a wide spectrum of effects of immune activation
(96,97).
Tremelimumab is another anti-CTLA-4 monoclonal
antibody. At the beginning of phase I and II testing in patients
with solid malignancies, promising preliminary anti-tumor activity
and safety with single-agent tremelimumab (15 mg/kg) were
demonstrated (98,99). The most common AEs were diarrhea,
colitis, dermatologic events and fatigue, which were similar to
those of ipilimumab treatment. However, in the phase III study, the
incidence of AE-related dose discontinuation was 12.5%, compared
with 5% in the phase II study. The rates of grade 5 AEs were 2 and
0.8%, respectively. Therefore, the side effect profile of
tremelimumab appears comparable to that of ipilimumab (100).
PD-1/PD-L1 blockade. In 2014, the FDA granted
accelerated approval for two fully humanized anti-PD-1 IgG4 isotype
antibodies, nivolumab and pembrolizumab, as first-line treatment of
unresectable or metastatic melanoma, thereby markedly advancing
treatment. As demonstrated in the CheckMate 037 and KEYNOTE 002
trials, both nivolumab and pembrolizumab had superior efficacy and
an improved safety profile compared with investigator-choice
chemotherapy for the treatment of ipilimumab-refractory advanced
melanoma (101,102). Of note, compared with the
ipilimumab trial, a lower rate of high-grade AEs (10-15%) and
discontinuations due to AEs (5-7%) was detected in nivolumab and
pembrolizumab clinical trials (101,102). In general, anti-PD-1 therapy
appears to have fewer severe side effects than ipilimumab. This is
probably due to the activation and expansion of a wider variety of
T-cell subpopulations from CTLA-4 inhibition (103). In a phase II trial of patients
with advanced melanoma, known BRAF V600 mutation status and an ECOG
PS of 0 or 1 (CheckMate 069), at a median follow-up of 24 months,
the OS rates were 63.8% in the nivolumab plus ipilimumab group and
53.6% in the ipilimumab group. The incidence of grade 3 or 4 AEs in
the combination group was 54 vs. 20% in the ipilimumab monotherapy
group (104). While this dual
therapy has shown impressive efficacy relative to other available
therapies, toxicity seems a major barrier. Furthermore, it has
demonstrated consistent results in a phase III trial (CheckMate
067). In patients with BRAF-mutated tumors, the patient follow-up
data indicated 6.5-year OS rates of 57% in the nivolumab plus
ipilimumab group vs. 43% in the nivolumab monotherapy group and 25%
in the ipilimumab monotherapy group; in those patients with
BRAF-wild-type tumors, the 6.5-year OS rates were 46, 42 and 22%,
respectively (105). In terms of
the safety profile, almost all patients (82.1-95.5%) experienced a
treatment-related AEs (TRAEs), and more than half of the patients
in the combination group (55.0%) experienced a grade 3/4 AE,
compared with 16.3 and 27.3% in the nivolumab and ipilimumab
groups, respectively, with diarrhea, colitis, increased alanine
aminotransferase levels, and increased aspartate aminotransferase
levels being the most common ones. With the use of
immunosuppressive or immunomodulatory agents, most select AEs were
also manageable with established treatment guidelines, with no
treatment-related death reported in the combination arm (106). Therefore, due to the durable,
sustained survival benefits with an acceptable safety profile,
nivolumab plus ipilimumab combined therapy was approved as the
first dual checkpoint inhibition regimen in patients not only with
BRAF-wild type melanoma but also with BRAF-mutated melanoma
(21). In addition, nivolumab
monotherapy or in combination with ipilimumab was also reported to
be effective in immunotherapy-naïve patients with melanoma brain
metastases. A high proportion of patients achieved an intracranial
response with the combination (107).
Atezolizumab, a monoclonal anti-PD-L1 antibody, was
approved for clinical use in combination with vemurafenib and
cobimetinib as a first-line treatment in patients with BRAF
V600-mutated advanced melanoma. The ongoing phase III IMspire150
study is examining how efficacious the approved BRAF/MEK inhibitor
combinations (vemurafenib and cobimetinib) with or without
atezolizumab are for patients with unresectable, advanced or
metastatic BRAF V600 mutation-positive melanoma (31). Given that the adjuvant roles of
nivolumab and pembrolizumab have been verified, a study of
neoadjuvant atezolizumab in cutaneous melanoma is currently
underway (NCT04020809). In addition, with an increasing
understanding of the interactions between the immune system and
melanocytes, the efficacy and safety of atezolizumab in combination
with other major immune checkpoint blockades may be promising; this
is currently under clinical validation (NCT03554083 and
NCT03829501).
Furthermore, the adjuvant role of PD-1/PD-L1
blockade in patients with high-risk melanoma is gradually being
revealed and data continue to mature. The phase III CheckMate 238
randomized study compared adjuvant nivolumab to ipilimumab in
patients with resected stage IIIB/C and stage IV melanoma without
evidence of disease and demonstrated a 12-month RFS rate of 70.5%
and 18-month RFS rate of 66.4% in the nivolumab group compared to
60.8 and 52.7% in the ipilimumab group, respectively, along with
significantly decreased toxicity (108). In addition, longer DMFS was
observed in the nivolumab group as compared with the ipilimumab
group (108). An update reported
a 5-year RFS of 50% with nivolumab vs. 39% with ipilimumab and a
5-year OS of 76 vs. 72%, respectively (109). Furthermore, a recent clinical
trial (NCT04099251) of adjuvant nivolumab (CheckMate 76K) has
indicated a great reduction in the risk of recurrence or death of
58% in those with completely resected stage IIB/C melanoma. Based
on these results, in 2017, nivolumab was approved in the adjuvant
setting for resected melanoma. In a phase III trial for resected
stage III melanoma (KEYNOTE 054), adjuvant pembrolizumab resulted
in a clinically relevant 20% improvement in 3-year RFS compared
with placebo (63.7 vs. 41%) (110). At a 42.3-month median follow-up,
pembrolizumab improved DMFS (65.3 vs. 45.4%) and recurrence-free
survival (59.8 vs. 41.4%) compared with the placebo group, leading
to its approval in the USA and Europe for patients with stage IIB
or IIC melanoma following complete resection (111). A trial by the Southwest Oncology
Group (SWOG 1404) also demonstrated a significant improvement in
RFS with adjuvant pembrolizumab in patients with resected stage III
melanoma with a high risk of recurrence, compared with high-dose
interferon α2b (IFNα2b) or ipilimumab (112). As expected, the incidence of
grade 3 or higher toxicity was greater with IFNα2b and ipilimumab
compared with pembrolizumab (66 and 43 vs. 17%) (112). Similarly, in the KEYNOTE 716
trial in patients with stage IIB/C melanoma, after a median
follow-up of 27 months, adjuvant pembrolizumab markedly improved
both 24-month RFS (81.2 vs. 72.8%) and DMFS (88.1 vs. 82.2%)
compared with the placebo group (113). Based on these results, the FDA
extended its indications to adult and paediatric (12 years of age
and older) patients with surgically resected high-risk stage IIB,
IIC or III melanoma in December 2021. To date, however, these
studies haven't demonstrated any improvement in OS, the reason for
which is unclear but it is probably affected by post-relapse
treatment, insufficient follow-up or biologic and immune issues not
yet fully understood (114). It
is also unclear whether it is more efficacious to treat when there
is a residual microscopic disease or to wait until patients have
disease recurrence to avoid treating those who may have been cured
by surgery alone. Integrating biomarkers into adjuvant trials may
allow for better selection of those truly benefiting from adjuvant
therapy (114). Disease
recurrence was still observed in >30% of those patients with
high-risk melanoma receiving adjuvant immunotherapies within 2
years after surgery (94,108,114). Administering immunotherapies in a
neoadjuvant setting before surgery is a promising strategy. Due to
the presence of the tumor at the beginning of the therapy,
neoadjuvant therapies may induce a deeper immune response, thereby
reducing the tumor burden and facilitating cancer surgery.
Recently, a phase II SWOG S1801 trial indicated a 42% reduction in
2-year event-free survival risk with neoadjuvant compared to
adjuvant pembrolizumab in patients with resectable stage IIIB-D/IV
melanoma (72 vs. 49%), indicating that neoadjuvant single-agent
immunotherapy may serve as a new standard of care. In the phase Ib
OpACIN trial, 20 participants with macroscopic stage III melanoma
were randomized either to receive four cycles of adjuvant
ipilimumab plus nivolumab every 3 weeks following therapeutic lymph
node dissection or to receive two cycles of neoadjuvant ipilimumab
plus nivolumab every 3 weeks before surgery, subsequently followed
by total lymph node dissection at week 6 and another two cycles of
treatment at week 12. The results in the neoadjuvant arm were
promising, with an unexpectedly high pathologic partial response
(PPR) of 78% at a median follow-up of 25.6 months, and these
responding patients remained free of relapse at 2 years (115). However, this regimen induced
similarly high toxicity in both arms, with ~90% of patients
experiencing one or more grade 3 or 4 irAEs, resulting in early
treatment discontinuation in 18/20 patients (115). To further identify a dosing
schedule of the neoadjuvant application of ipilimumab plus
nivolumab to make it less toxic but equally effective, the
subsequent multicentre phase II OpACIN-neo trial was launched. A
total of 86 patients were enrolled and randomly assigned to one of
three different dosing groups, and then therapeutic lymph node
dissection was planned at week 6 without additional adjuvant
therapy. Finally, this trial identified a tolerable neoadjuvant
treatment regimen consisting of two cycles of ipilimumab 1 mg/kg
plus nivolumab 3 mg/kg every 3 weeks, which may be suitable for
broader clinical use, with a high PPR of 77 and 20% grade 3 or 4
irAEs (116). After a median
follow-up of 4 years, the OpACIN and OpACIN-neo trials showed that
this treatment strategy induced durable RFS in >80% of patients.
The investigators also found that those with a high IFN-γ score and
high tumor mutational burden (TMB) had a PPR of 100%; For those
with a low IFN-γ score/low TMB, the PPR was only 39%, while for
those with only a high IFN-γ score or only high TMB, the PPR was 91
and 88%, respectively (117).
This indicated that the presence of a pathologic response was
possibly a surrogate marker for long-term benefits and showed the
predictive potential of TMB and IFN-γ score, which may help
discriminate responders from non-responders (117). Recently updated data from the
OpACIN and OpACIN-neo trials showed that at a median follow-up of
69 months for OpACIN, only 14.3% of patients with a pathologic
response to neoadjuvant application of combined checkpoint
inhibitions had disease recurrence; at a median follow-up of 47
months for OpACIN-neo, the estimated 3-year RFS rate was 95% for
those with a pathologic response vs. 37% for those without
pathologic response (118).
In order to increase RFS in the non-responders among
patients with melanoma, the investigators raised the concept of a
pathologic response-driven treatment strategy (119,120). In a multicentre phase 2 PRADO
extension cohort of OpACIN-neo, 99 patients with IIIB-D nodal
melanoma received 6 weeks of neoadjuvant ipilimumab plus nivolumab.
Subsequently, for those who achieved major pathologic response
(MPR, ≤10% viable tumor) in their index lymph node (the largest
lymph node metastasis at baseline), therapeutic lymph node
dissection (TLND) and adjuvant therapy were omitted; those with a
PPR (>10 to ≤50% viable tumor) underwent TLND only, while those
with a pathologic non-response (PNR, >50% viable tumor)
underwent TLND and subsequent adjuvant therapy (nivolumab in BRAF
V600E/K wild-type and dabrafenib plus trametinib in BRAF
V600E/K-mutant patients). Surprisingly, in contrast to the 4-year
RFS rate of 100% for patients with PPR in the OpACIN-neo cohort,
these same patients with PPR in the PRADO study had a 2-year RFS
rate of only 64%, with a rate of 93 and 71% in patients with MPR
and PNR, respectively (119).
Thus, the investigators questioned whether MPR, instead of
pathologic response, would be a better predictor of outcome.
According to the PRADO data, the currently recruiting phase III
NADINA trial comparing neoadjuvant to adjuvant therapies in
macroscopic stage III melanoma was amended: In arm A, patients with
MPR will receive two cycles of ipilimumab plus nivolumab and will
undergo TLND at week 6. For those with a PPR or PNR, surgery will
be followed by adjuvant nivolumab or BRAF/MEK inhibition (in case
of BRAF V600E/K mutation-positivity). Meanwhile, in arm B, patients
will undergo upfront TLND followed by adjuvant nivolumab (121). Taken together, the outcomes of
the S1801, OpACIN, OpACIN-neo, PRADO, and the awaited results of
the NADINA trial not only encouraged neoadjuvant checkpoint
immunotherapy to become a new standard of care in patients with
high-risk melanoma but also indicated the importance of the concept
of personalized treatment strategies based on pathologic response
after neoadjuvant therapy.
Novel immune checkpoint blockades
Lymphocyte activation gene 3 (LAG-3) is expressed on
T cells and is the third immune checkpoint co-inhibitor receptor to
be exploited in cancer immunotherapy (122). With its higher affinity for major
histocompatibility complex class II than CD4, it mediates the
downregulation of T-cell activation and proliferation (122). Preclinical studies have
demonstrated enhanced tumor-specific immunity and disruption to
melanoma tumor growth not only in dual anti-LAG-3 and anti-PD-1
antibody-treated mice but also in
Lag3-/-Pdcd1-/- (Pdcd1 encodes PD-1) mice,
suggesting a potentially beneficial combinatorial strategy of dual
LAG-3/PD-1 blockade for melanoma (123). In a phase I/IIa cohort expansion
study of the fully human LAG-3-specific antibody relatlimab
administered alone and in combination with nivolumab in
participants with melanoma who progressed during prior
anti-PD-1/PD-L1 therapy, escalation to nivolumab plus relatlimab
resulted in an ORR of 11.5% in all patients and of 18% in patients
with LAG-3 expression ≥1% (124).
The randomized, double-blind phase II/III RELATIVITY-047 study of
nivolumab with or without relatlimab for treating unresectable
melanoma or melanoma that has spread has been underway. Results
thus far reported a 12-month PFS rate of 47.7% with a fixed-dose
combination of relatlimab and nivolumab compared with 36.0% with
single-agent nivolumab. In terms of drug safety, the combination
was well tolerated with 21.1% of patients experiencing grade 3 or 4
TRAEs (125). Given its efficacy
and favourable toxicity profile in RELATIVITY-047, in March 2022,
the FDA approved the use of fixed-dose relatlimab/nivolumab
combination in patients with unresectable or metastatic melanoma.
Of note, the currently recruiting phase III RELATIVITY-098
(NCT05002569) trial aimed to test this combination vs. nivolumab
alone after complete resection of stage III-IV melanoma in the
adjuvant setting. In another randomized trial evaluating how well
the relatlimab/nivolumab combination worked in treating resectable
clinical stage III or oligometastatic stage IV melanoma in the
neoadjuvant and adjuvant setting, the eligible 30 participants
first received 2 neoadjuvant doses of relatlimab 160 mg and
nivolumab 480 mg every 4 weeks followed by surgery and then 10
doses of adjuvant relatlimab/nivolumab combination therapy. These
neoadjuvant and adjuvant checkpoint blockades resulted in a high
MPR rate of 57% and an improvement in the 1- and 2-year RFS rate
(100 and 92%, respectively) in patients achieving any pathologic
response compared to those without a pathologic response. Safety
during neoadjuvant therapy is favourable, with no grade 3 or 4
irAEs experienced, while 26% grade 3 or 4 toxicities were observed
in the adjuvant setting (126).
Though the study was limited by its small sample size, these
initial results were encouraging and similar to the individual arms
in the OpACIN-neo trial, providing further confirmation of the
efficacy and safety of this new immunotherapy regimen. Additional
follow-up may be necessary to fully assess the clinical impact and
the durability of responses.
Adoptive cell therapy
Adoptive cell therapy uses either natural host cells
that exhibit anti-tumor reactivity or host cells that have been
genetically modified with anti-tumor TCRs or chimeric antigen
receptors (CARs). In the mid-1980s, the demonstration from
Rosenberg et al (127)
that adoptive transfer of IL-2-stimulated lymphokine-activated
killer (LAK) cells resulted in complete durable tumor regressions
provided a stimulus to identify the tumor-specific T cells involved
in cancer immunotherapy. Their subsequent in-vivo studies
suggested that autologous tumor-infiltrating lymphocytes (TILs), a
form of T cells originating from tumor tissues with broad-spectrum
heterogeneity, in conjunction with IL-2 provide 50 to 100 times
more effective anti-tumor activity than LAK cells (128). Currently, TIL therapy has emerged
as a promising option to treat patients with solid tumors who were
refractory to checkpoint inhibitors and targeted therapies
(129-133).
Lifileucel, a one-time autologous TIL product, achieved an
investigator-assessed ORR of 36% in 66 patients with metastatic
melanoma who had progressed on standard checkpoint inhibitors and
BRAF ± MEK targeted agents (if BRAF V600 mutation-positive), while
only 4-10% of these patients had objective responses to cytotoxic
chemotherapy (132). In a recent
large multicentre phase II trial (C-144-01) that included 153
patients with advanced metastatic melanoma, combining the 66
previously reported patients, the cryopreserved TIL product
lifileucel provided flexibility in treatment scheduling in the
real-world clinical setting and demonstrated an ORR of 31.4%, with
8 complete responses and 40 partial responses; the median DOR was
not reached at a median follow-up of 27.6 months and nearly half of
the patients had responses maintained for ≥18 months. Based on
these encouraging results, the investigators supported the use of
lifileucel as a novel treatment option for patients with advanced
melanoma to address a highly unmet need: The patients with advanced
melanoma following failure of checkpoint inhibitors, and targeted
agents where appropriate, irrespective of baseline tumor
characteristics, should be considered for lifileucel as second-line
therapy (third-line if BRAF V600 mutation-positive) if they have an
ECOG PS and organ functions adequate for receiving a
nonmyeloablative lymphodepletion regimen and a shortened course of
IL-2. Of note, the FDA has granted a regenerative medicine advanced
therapy designation for lifileucel in advanced melanoma.
Furthermore, in two ongoing phase II multicentre, multicohort,
prospective, open-label studies (IOV-COM-202 and C-145-04), in PD-1
inhibitor-naïve patients, lifileucel combined with pembrolizumab
also produced a high ORR (60%), supporting the potential for
improved response rates with earlier TIL cell therapy (134). The recently performed phase III
trial of lifileucel plus pembrolizumab in frontline advanced
melanoma was expected to be well underway at the time of a
potential approval.
Talimogene laherparepvec (T-VEC), an attenuated
herpes simplex virus 1 encoding granulocyte-macrophage
colony-stimulating factor, has been approved for use as the first
oncolytic virus therapy for the local treatment of metastatic
melanoma that cannot be surgically removed. Previous findings
indicated that T-VEC plus ipilimumab or pembrolizumab combination
therapy has great efficacy, with a tolerable safety profile, in
treating advanced melanoma (135,136). In a phase II trial, the clinical
response rate was doubled from 18% in patients with ipilimumab
alone to 38% with T-VEC plus ipilimumab (137). In addition, T-VEC plus
pembrolizumab led to a high CRR of 43% in the MASTERKEY-265 phase
Ib study (136). According to
recent results from the MASTERKEY-265 phase III study, there was no
statistically significant difference in median PFS or OS between
the combined therapy and pembrolizumab monotherapy groups (138); however, there existed a numerical
difference of 5.8 months favouring the T-VEC plus pembrolizumab
group (14.3 vs. 8.5 months) and the researchers found that among
patients with baseline LDH ≤ ULN, patients with baseline SLD ≤
median and patients enrolled in the United States, the
T-VEC-pembrolizumab combination was beneficial for PFS (138). This combination therapy is still
under active investigation in those who were refractory to
anti-PD-1-based therapy in advanced melanoma (NCT04068181).
Immune-mobilizing monoclonal T-cell
receptors against cancer (ImmTACs)
ImmTACs are a novel class of T-cell redirector
molecules as well as anti-tumor reagents that utilize
affinity-enhanced soluble TCRs stabilized by a disulfide bond and
fused to an anti-CD3 single-chain variable fragment that engages T
cells. This ImmTAC platform allows for highly specific access to
the vast pool of intracellular targets (139). The most advanced ImmTAC molecule,
tebentafusp (IMCgp100), redirects T cells towards human leukocyte
antigen (HLA)-A*02:01-positive melanoma cells expressing a gp100
peptide (HLA-A*02:01 is an important restriction element for
peptide presentation to T cells in disease and cancer), inducing
the formation of an immune synapse to kill targeted tumor cells
(140). In August 2021,
tebentafusp was given approval by the FDA to treat
HLA-A*02:01-positive uveal melanoma. To date, three clinical trials
of tebentafusp have shown promising results in patients with
metastatic uveal melanoma: IMCGp-100-01 (NCT01211262),
IMCGp-100-102 (NCT02570308) and IMCGp-100-202 (NCT03070392).
The IMCGp-100-01 trial reported that a nearly 40%
increased dose of tebentafusp through a 3-week step-up dosing
regimen (20-30-68 µg) compared with a fixed weekly dose of 50 µg
had a manageable side-effect profile and a signal of efficacy in
HLA-A*02- or HLA-A*02:01-positive uveal melanoma (141). This novel treatment regimen of
tebentafusp was subsequently used in the IMCGp-100-202 phase III
trial, in which patients with metastatic uveal melanoma were
assigned in a 2:1 ratio to a tebentafusp monotherapy group or
control group (dacarbazine, ipilimumab or pembrolizumab
monotherapy). Surprisingly, the data revealed a 1-year OS of 73%
with tebentafusp compared with 59% with systemic treatments
(142). Indeed, this promising OS
result was higher than that recently reported for ipilimumab in
combination with nivolumab. Conversely, two single-arm, phase II
trials comprising patients with metastatic uveal melanoma treated
with ipilimumab plus nivolumab only achieved a 1-year OS rate of
51.9% (143) and 56% (144).
4. Combination of targeted therapy and
immunotherapy
MAPK inhibitors plus immune checkpoint
inhibitors
For the patient population with highly symptomatic
disease and an ECOG PS score of at least 2, and disease affecting
crucial anatomical sites, such as symptomatic brain metastasis
requiring corticosteroid therapy, which are not amenable to local
therapy, BRAF and MEK inhibitors seem to be preferred over immune
checkpoint inhibitors. Their duration of response to the first-line
targeted agents is often short. However, one limitation of immune
checkpoint inhibitors is their slower onset of action. Instead of
targeting the tumor cells directly, CTLA-4, PD-1 and PD-L1
blockades first activate the immune system and then let a high
number of active immune cells become tumor killers. This same
patient population therefore responds poorly to them, although a
small percentage may have long-term durable control (145).
Numerous efforts have been made to combine MAPK
inhibitors with immune checkpoint inhibitors. In general,
understanding the effect of BRAF and MEK inhibition on melanoma
tumors and their microenvironment may prove critical in supporting
such a combination approach. The scientific rationale for the
combination is based on ‘immunosensitization’, whereby
pharmacologic modulation with specific inhibitors of oncogenic
events in cancer cells sensitizes cancer cells to immune attack
(146). For instance, Koya et
al (147) showed that the
BRAF V600E-specific inhibitor vemurafenib improved the anti-tumor
effects of TCR-engineered ACT in a BRAF V600E-driven murine model
of melanoma, with higher immune-stimulating cytokine IFN-γ
secretion and better gp100-specific lytic activity. In addition,
Wilmott et al (148)
examined melanoma tumor biopsies and observed an increase in the
density of CD8+ and CD4+ TILs following
treatment with a BRAF inhibitor. Frederick et al (149) showed that BRAF blockade is
associated with increased expression of melanoma-associated
antigens [gp100, MART-1 (a melanocyte lineage-specific protein;
melanoma antigen recognized by T cells 1), and TYRP-1/2
(tyrosinase-related protein-1/2)], increased markers of T-cell
cytotoxicity (perforin and granzyme B) and decreased expression of
immunosuppressive cytokines (IL-6 and IL-8), all enhancing the
tumor microenvironment. Paradoxically, an increase in the T-cell
exhaustion markers T-cell immunoglobulin domain and mucin domain 3
(TIM-3), PD-1 and the immunosuppressive ligand PD-L1 was also noted
during BRAF inhibition treatment, which may be one reason for the
initiation of immune evasion (149). These results support the
hypothesis that combining BRAF-targeted therapy with immunotherapy
may have superior anti-tumor efficacy in patients with advanced
melanoma. With regard to immune evasion, oncogenic BRAF induces
decreased expression of melanocyte differentiation antigens (MDAs),
indicating defective recognition of melanoma cells by
antigen-specific T lymphocytes. Surprisingly, the study by Boni
et al (150) corroborated
that impaired melanocyte antigen expression is reversed by a
selective BRAF V600E inhibitor, without compromising T lymphocyte
function. As recognition of MDAs is central to immunotherapy for
melanoma, it may also provide support for potential synergistic
effects of BRAF-targeted therapy plus immunotherapy.
Several clinical trials of such a combinatorial
approach that resulted in promising efficacies in patients with
advanced melanoma are listed in Table
II (NCT02130466, NCT02858921, NCT02967692, NCT02908672,
NCT02224781, and NCT03235245).
Dabrafenib and trametinib plus pembrolizumab is a
triplet combination therapy assessed as a feasible treatment
approach in the KEYNOTE 022 phase I, dose-identification trial
(NCT02130466). Ribas et al (151) reported that this approach
increases the frequency of long-lasting responses in patients with
BRAF V600-mutated melanoma and is most suitable for treating those
with a poor prognosis on monotherapy. In terms of safety and
toxicity, 20% of patients had dose-limiting toxicities and 73%
experienced one or more grade 3 or 4 TRAE. Among the AEs, two
events (pneumonitis and autoimmune hepatitis) prompted treatment
discontinuation. In a parallel phase II study (NCT02130466),
Ascierto et al (152)
found that at a median follow-up of 9.6 months, the primary
endpoint of PFS did not show statistically significant improvement
in the triplet arm (dabrafenib and trametinib plus pembrolizumab)
compared with the doublet arm (dabrafenib and trametinib plus
placebo) (16.0 and 10.3 months, respectively). With a longer median
follow-up of 36.6 months, the changes of PFS (16.9 and 10.7 months,
respectively), DOR (25.1 and 12.1 months, respectively) and OS (not
reached and 26.3 months, respectively) were more notable between
the two arms (153). Despite
clinical advantages, considerable toxicity was the major limitation
of this strategy. The incidence of grade 3 through 5 TRAEs was 58%
with the triplet regimen (including most commonly fever, increased
transaminase levels and rash) but only 25% with the doublet
therapy. Of the patients on triplet therapy, 40% discontinued at
least one of the agents due to TRAEs, compared with 20% on doublet
therapy. There was one death from treatment-related pneumonitis
(153). Recently, Maio et
al (154) identified the
maximum tolerated doses (MTDs) for concurrent and intermittent
dosing strategies of pembrolizumab plus trametinib, indicating that
both were feasible with manageable toxicity and safety profile
among participants with BRAF-wild-type melanoma or advanced solid
tumors, irrespective of BRAF mutation. A phase II, randomized study
is ongoing, with the aim of addressing whether different
intermittent or dose-sequencing regimens may be able to reduce the
size of tumors prior to surgery in advanced melanoma and prevent
recurrence of melanoma after surgery while reducing toxicity
(NCT02858921).
COMBI I (NCT02967692) is a global, randomized, phase
III study of spartalizumab plus dabrafenib and trametinib
(sparta-DabTram) for patients with BRAF V600-mutated metastatic
melanoma, though it did not meet its primary endpoint of PFS
(155). In that study, at the
data cut-off, median PFS was 16.2 months in the sparta-DabTram arm
compared with 12.0 months in the placebo-DabTram arm and the median
OS was not reached in either arm, with ongoing analyses. Similar to
the KEYNOTE 022 study, this triple combination was associated with
greater toxicity than the doublet regimen. The incidence of grade 3
through 5 TRAEs was 55% with the triplet regimen and 33% with the
doublet therapy.
Positive results were also reported from the phase
III IMspire150 trial (NCT02908672): Adding atezolizumab to
vemurafenib and cobimetinib prolonged the primary endpoint of
investigator-assessed PFS from 10.6 to 15.1 months in previously
untreated patients with BRAF mutation-positive advanced melanoma.
From this, a delayed separation of PFS curves (started after 7
months) was evident, maintaining a benefit in favour of the triplet
arm. The DOR was also improved with the addition of atezolizumab
(21.0 vs. 12.6 months) with, importantly, little change in the
grade 3/4 toxicity rate (the main grade 3/4 TRAEs were presented
similarly in the atezolizumab and control groups, including
increased creatine phosphokinase, increased liver
enzymes/lipase/amylase and rash). It is also notable that the OS
curves in the KEYNOTE 022 trial separated later. Taken together,
the addition of PD-1/PD-L1 checkpoint inhibitors to BRAF/MEK
inhibitors may provide response rates that are similar to those
observed with BRAF/MEK-targeted therapy alone but with prolonged
durable responses, which may be a critical factor in the design of
immunotherapy combination evaluation.
Overall, according to the data from KEYNOTE 022,
COMBI I and IMspire150, the role of BRAF and MEK targeted agents
plus immune checkpoint inhibitors was highlighted in patients with
treatment-naïve BRAF V600-mutated melanoma. However, combination
therapies appear to increase toxicity. On the other hand, the
differences among these three published studies in terms of study
design, patient population and agents investigated preclude direct
comparison (153). A full
evaluation of the long-term benefit of these treatments is needed
from ongoing clinical trials to determine their feasibility. In
particular, these triplet combinations may be applicable for
treating urgent cases in which the patient has limited time to wait
for a response to a checkpoint inhibitor.
Although the results do not support the routine use
of first-line immunotherapy plus targeted therapy, certain
prognostic biomarkers may be helpful for informing treatment
selection in patient subpopulations (156). In the COMBI I study, a baseline
intra-tumoral and blood CD4+/CD8+ T-cell
ratio above the median was a useful non-invasive biomarker and
associated with the response to sparta-DabTram therapy. In
addition, patients with a clinically higher tumor burden and
detectable baseline circulating tumor DNA shedding appeared to
derive a PFS benefit with sparta-DabTram vs. placebo-DabTram.
Similarly, a small proportion of this subgroup of patients carrying
the BRAF V600K mutation tended to have greater survival benefits
with sparta-DabTram treatment. Thus, stringent patient selection
based on biomarkers is of utmost importance for delineating
patients who require this triplet therapy.
However, there is still no definitive answer
regarding which treatment should be given first or which treatment
sequence is preferable. Recently, a retrospective study of 114
patients with melanoma showed that those who had progressed on
anti-PD-1 agents experienced inferior survival outcomes after
starting subsequent BRAF-targeted therapy compared with those who
were not previously treated with anti-PD-1 agents. Similarly,
pre-treatment with BRAF-targeted therapy was seemingly associated
with worse outcomes after receiving anti-PD-1(157). Other data also suggested that
anti-PD-1 exposure may influence the frequency and magnitude of
AEs, including rapid onset of pyrexia or a sepsis-like syndrome
with or without hypotension, which was associated with subsequent
BRAF and MEK inhibition (158).
Currently, initial treatment with
ipilimumab/nivolumab followed by dabrafenib/trametinib compared to
the same sequence in reverse is being evaluated in the randomized
phase III DREAMseq trial (NCT02224781) (159). In this trial, 265 participants
with treatment-naïve BRAF V600-mutated metastatic melanoma were
randomly assigned (1:1) to receive ipilimumab/nivolumab (arm A) or
dabrafenib/trametinib (arm B). Upon disease progression, patients
were enrolled to receive the alternative combination therapy:
Dabrafenib/trametinib (arm C) or ipilimumab/nivolumab (arm D). The
data indicated that after a median follow-up duration of 27.7
months, the sequence starting with dual immune checkpoint blockade
followed by dual BRAF/MEK inhibitor resulted in a 20% absolute
improvement in 2-year OS compared to the inverse sequence (71.8 vs.
51.5%). Other efficacy endpoints, such as median PFS duration (11.8
months in arm A vs. 8.5 months in arm B) and median DOR (not
reached in arm A vs. 12.7 months in arm B), also indicated a
benefit of prior first-line immunotherapy. Of note, the ORR was
similar between arms A and B, at 46.0 and 43.0%, respectively, but
rates were inferior in arm D vs. arm C (29.6 vs. 47.8%). In terms
of safety, grade ≥3 toxicities occurred with similar frequency
between arms, with more grade 4 toxicities seen in arm A. TRAEs in
arms A and D were primarily immune-related and for arms B and C,
they were primarily fevers, leukopenia and hyponatremia.
Furthermore, prior ipilimumab plus nivolumab was associated with
higher mortality, with 24 patients (18%) dying within 10 months,
with notable rapid disease progression and poor prognostic features
(159). Further relevant clinical
trials are needed to identify whether this subgroup with the
aggressive disease may benefit from at least a brief preceding
course of BRAF/MEK-targeted therapy. Ascierto et al
(160) reported the results of
the randomized phase II SECOMBIT study (NCT02631447). In this
trial, 209 patients were randomly assigned to three treatment arms
(arm A: Immunotherapy with ipilimumab plus nivolumab followed by
targeted therapy with encorafenib plus binimetinib at disease
progression; arm B: The reverse order of arm A; arm C: A sandwich
arm with a short course of targeted therapy followed by a switch to
immunotherapy followed by targeted therapy at disease progression).
Similar to the phase III DREAMseq trial, the survival results of
2-year OS rates for patients receiving initial immunotherapy were
65% in arm A, 73% in arm B and 69% in arm C, and no new safety
signals emerged (160). The phase
II EBIN EORTC trial (NCT03235245) is further exploring the effect
of ipilimumab plus nivolumab preceded or not by encorafenib plus
binimetinib in patients with BRAF V600 mutation-positive
unresectable or metastatic melanoma. A key difference between the
DREAMseq and SECOMBIT trials is inclusion of the sandwich approach,
which also showed clinical benefits. This warrants further
investigation, as there is no one-size-fits-all strategy for
treating patients with BRAF V600-mutated melanoma.
Such a sequencing trial is clinically critical to
defining optimal sequential approaches and the sequencing trials
are already underway in patients with melanoma to identify
efficacious treatment approaches in PD-(L)1 refractory disease in
the adjuvant setting (NCT03991130, NCT03385486, NCT05061134 and
NCT04250246) (161).
Anti-angiogenesis agents plus immune
checkpoint inhibitors
Using a murine model of melanoma, Schmittnaegel
et al (162) demonstrated
that dual VEGF and angiopoietin 2 blockade increase the proportion
and cross-presentation capability of intra-tumoral APCs. Normalized
tumor blood vessels and increased extravasation of IFNγ-expressing
CD8+ CTLs were also observed. Schoenfeld et al
(163) found that CTLA-4 blockade
elicited a humoral reaction that broadly targeted multiple
angiogenic cytokines. According to another study, anti-VEGF
targeted therapy has the ability to downregulate the expression of
exhaustion markers such as PD-1, LAG-3 and TIM-3 on tumor-educated
T cells upon resistance to immune checkpoint inhibitors (164). Such reduced VEGF levels were also
shown to be associated with improved response to PD-1 blockade in
patients with melanoma (165).
These results indicate that antiangiogenic treatments may be
effectively combined with immune checkpoint inhibitors to improve
patient outcomes.
Currently, several clinical trials of such a
combinatorial approach are ongoing (Table II) (NCT03820986, NCT01950390,
NCT04356729, NCT03175432, NCT04996823, and NCT02298959).
Lenvatinib can shift the tumor microenvironment to
an immune-stimulatory state by targeting VEGF and fibroblast growth
factor signaling (166), and the
combination of lenvatinib and pembrolizumab activates IFNγ-positive
CD8+ T cells through a reduction in tumor-associated
macrophages (166,167). Clinically, lenvatinib monotherapy
in previously treated participants yielded an ORR of only 9.7%
(168); however, the ORR of 48%,
median DOR of 12.5 months and median PFS of 5.5 months in a phase
IB/II trial of lenvatinib plus pembrolizumab indicated an
encouraging improvement (169).
The single-arm LEAP-004 trial was designed to identify whether this
combination therapy is able to improve survival in the population
of patients with unresectable stage III/IV melanoma experiencing
rapid clinical progression on anti-PD-1/PD-L1 monoclonal antibody
treatment alone, anti-PD-1/PD-L1 plus anti-CTLA-4 therapy and, if
applicable, BRAF/MEK-targeted therapy. The eligible 103 patients
received lenvatinib 20 mg orally once a day plus up to 35 cycles of
pembrolizumab 200 mg by intravenous infusion once every 3 weeks. At
the 15.3-month follow-up, an ORR of 21.4%, 12-month PFS rate of
18.5% and 12-month OS rate of 54.5% were observed in the total
population; an ORR of 33.3% was also observed in the subgroup (30
patients) refractory to prior anti-PD-1 plus anti-CTLA-4 therapy
(170). Of note, lenvatinib plus
pembrolizumab induced antitumor activity and tolerability
irrespective of baseline tumor characteristics (55.3% of patients
had elevated LDH) or number of previous therapies [58.3% of
patients were treated with ≥2 prior lines of therapy (170)], and it may thus be a treatment
option for this growing population with a highly unmet medical
need. Another phase III LEAP-003 trial assessing the safety and
efficacy of this combined therapy as a first-line intervention in
adults with previously untreated stage III/IV melanoma is ongoing
(NCT03820986).
In addition, a randomized phase II trial
(NCT01950390) to compare OS among patients with advanced melanoma
who receive ipilimumab plus bevacizumab or ipilimumab monotherapy
is currently underway. Another phase II study (NCT04356729) is
evaluating the safety and effectiveness of a combination of
atezolizumab and bevacizumab in unresectable, metastatic melanoma,
which is currently in the patient recruitment phase. In particular,
for patients with active melanoma brain metastasis, the safety,
tolerability and preliminary efficacy of atezolizumab plus
bevacizumab with or without cobimetinib is also being investigated
in a phase II trial (NCT03175432). Another phase II study of
axitinib plus ipilimumab combined therapy in patients with advanced
melanoma refractory to anti-PD-1 therapy is also ongoing
(NCT04996823). Aflibercept is a soluble decoy VEGF receptor with
anti-angiogenic effects, and given that it has been confirmed to
have potent preclinical anti-melanoma activity (171), a phase I study (NCT02298959)
testing whether aflibercept plus pembrolizumab is an appropriate
combinatorial approach in patients with metastatic melanoma and
other solid tumors is ongoing.
PARP inhibitors plus immune checkpoint
inhibitors
An emerging body of evidence supports that the
addition of PARP inhibitors can potentiate the therapeutic response
to checkpoint inhibitors. The rationale for PARP inhibitor plus
checkpoint inhibitor combined therapy mainly involves the following
three aspects: i) The accumulated DSBs induced by PARP inhibitors
that are not repaired may release tumor neo-antigens and eventually
induce a profound anti-tumor immune response. In addition, there is
evidence of a close association between melanoma with innate
deficiencies in DDR genes and a durable benefit from immunotherapy
(172). ii) PARP inhibition
promotes the accumulation of toxic DNA DSBs in tumor cells with DNA
repair deficiency, such as BRCA1/2 mutation, and PARP inhibition
also triggers the DNA-sensing cyclic GMP-AMP synthase-stimulator of
interferon genes pathway and upregulates type I interferons to
induce an anti-tumor immune response that is independent of DNA
repair deficiency (173). iii)
PARP inhibitor treatment can upregulate the expression of PD-L1 in
cancer cells and attenuate its efficacy due to immunosuppression,
which is reversible by blocking the PD-1/PD-L1 interaction
(174).
At present, there are no FDA-approved standard PARP
inhibitors for the treatment of metastatic melanoma. However, given
the immunological role of PARP inhibitors, combination therapy with
immune checkpoint inhibitors may become an option. Two clinical
studies of this combined regimen for treating melanoma are
currently in the patient recruitment phase (Table II). NCT04633902 is an open-label
phase II trial aiming to explore the benefit of olaparib plus
pembrolizumab in patients with advanced melanoma with genetic HR
alterations, particularly BAP1, ATM, AT-rich interactive
domain-containing protein 1A/B, CHK2, and BRCA1/2, whose disease
progressed on prior immunotherapy and/or BRAF-targeting therapy.
Similarly, the efficacy of talazoparib plus nivolumab in patients
with BRCA1/2 or other DDR mutations is also being assessed
(NCT04187833).
5. New directions to overcome primary or
acquired resistance to targeted therapy and immunotherapy
In spite of the numerous targeted therapy- and
immunotherapy-based treatments that are now available for clinical
use in melanoma, a large proportion of patients with melanoma do
not show any durable benefit, with either no clinical response or
disease progression. There remains the question of how to identify
better treatment strategies to overcome resistance in order to
confer clinical benefits for these patients.
Under targeted therapy and/or immunotherapy,
certain tumor cells are still durable by showing resistance to cell
death and escaping from immunological surveillance in cancer
patients (including melanoma and other cancer cells), impairing the
immune system and enabling cancer recurrence. Thus, the vital
immune cells and the regulatory signaling networks involved in the
interaction between tumor cells and the immune system are main
factors to modulate the susceptibility of cancer cells to death
(175). Immunogenic cell death
(ICD) is a new key factor in the treatment of metastatic melanoma,
which is featured by the release or expression of molecules with
danger-associated molecular patterns, such as calreticulin, ATP,
high mobility group box 1, heat-shock proteins, Annexin A1 and type
1 IFN. By binding to their receptors, these molecules result in the
recruitment and stimulation of immune cells and finally cause
damage to tumor cells through the maturation of dendritic cells,
the activation of CTLs, the enhancement of the cytotoxic activity
of natural killer cells, as well as the production of
proinflammatory cytokines and chemokines (176). The ICD inducer SD-101 is a
synthetic CpG oligonucleotide that stimulates Toll-like receptor 9
(TLR9). Early data from a phase Ib study demonstrated that by
concomitantly releasing PD-1-mediated inhibition with
pembrolizumab, the injection of SD-101 into peripheral or visceral
lesions was able to induce broad immune activation in the tumor
microenvironment at that site. Among the 13 patients with melanoma
who had prior anti-PD-1 therapy, the ORR was 15%. Of note, one
patient refractory to prior anti-PD-1 therapy receiving 1 mg of
SD-101 had a partial response ongoing at 10.5 months of follow-up
(177). Similarly, intratumoral
vidutolimod, a virus-like particle-encapsulated CpG-A TLR9 agonist,
combined with pembrolizumab also improved cancer immunotherapy
outcomes in patients with advanced anti-PD-1-refractory melanoma
through triggering a strong IFN response to induce and attract
antitumor T cells. Durable responses were observed in 25% of
patients, with tumor regression in both vidutolimod-injected and
noninjected target lesions, including visceral metastases (178). Among the intralesional agents,
oncolytic viruses would also be anticipated to work by stimulating
host anti-tumor immunity through preferential replication in tumor
cells and production of cytokines and other immunomodulatory
molecules. However, according to the results from the MASTERKEY-265
phase III study of T-VEC plus pembrolizumab, there was no
statistically significant difference in median PFS or OS between
the combined therapy and pembrolizumab monotherapy groups (138). This combination therapy is still
under active investigation (NCT04068181) in those patients with
advanced melanoma who were refractory to anti-PD-1-based
therapy.
Microbiota and their metabolites have been
demonstrated to have a significant impact on potentiating immune
checkpoint inhibitor therapy (179). Fecal microbiota transplantation
(FMT), as an effective strategy for manipulating the gut
microbiota, may transfer the fecal material isolated from a healthy
donor to a recipient via colonoscopy, nasogastric tube or prepared
capsules (180). In preclinical
mouse models, reconstitution of germ-free mice with fecal material
from anti-PD-1-responding patients resulted in augmented T-cell
responses and greater efficacy of PD-1 blockade. On the contrary,
if the mice were reconstituted with non-responder fecal material,
the corresponding mice were also non-responders to PD-1 blockade
(181). Based on this strong
correlation between commensal microbial composition and clinical
response to immunotherapy, several clinical trials in patients with
metastatic melanoma are underway to test the potential for FMT to
enhance the efficacy of immune checkpoint blockade therapy. Whether
reprogramming the gut microbiota can overcome resistance to
anti-PD-1 in patients with advanced melanoma remains to be
evaluated. To address this question, Davar et al (182) performed a phase II study
(NCT03341143) of concurrent FMT together with pembrolizumab in
patients with PD-1-resistant melanoma. This combination was
reported to have favourable safety results and provided a clinical
benefit in 6 of 15 patients, along with a rapid and durable
microbiota perturbation. A response to this combination was
associated with an increased number of CD8+ T cells in
the tumor microenvironment. Among the responding patients, several
circulating cytokines and chemokines were also detected to be
decreased after FMT, including C-C motif chemokine ligand 2, IL-8
and IL-18, which have been reported to be associated with adverse
prognosis to PD-1 blockade in multiple cancer types, including
melanoma (183), indicating that,
beyond the immunostimulatory potential, the microbiota may also be
employed to decrease tumor-associated immunosuppression. The other
recent clinical trial (NCT03353402) also confirmed the safety and
feasibility of FMT and anti-PD-1 therapy in patients with
anti-PD-1-refractory metastatic melanoma, and treatment with FMT
was also associated with favourable changes in intra-tumoral
CD8+ T-cell infiltration, IFN-γ mediated signalling
pathway, dendritic cell differentiation and T helper type 1 immune
response in the tumor microenvironment (184).
Furthermore, tumor cell plasticity also contributes
to targeted therapy and immunotherapy resistance. An expression
profiling of melanoma cell lines has established the existence of
two major transcription programs. The two programmes are expressed
in distinct cell populations, defined as either of ‘proliferative
cellular phenotypes’ or ‘invasive cellular phenotypes’ (185). The former contributes to high
rates of proliferation and low motility, while the latter is
characterized by lower rates of proliferation and high metastatic
motility (186). As replicated
in vivo, melanoma cells may perform
proliferative-to-invasive phenotype switching in response to the
tumor microenvironment, which is thought to drive melanoma
progression and influence the ability to adjust to drug exposure
during treatment (186). MITF is
a marker of gene expression signatures linked to the proliferative
phenotype, and the tyrosine-protein kinase receptor, AXL, is linked
to the invasive phenotype. In the initial response phase, >78%
of patients with melanoma on BRAF and MEK inhibitors treatment
displayed high MITF expression in MITFhigh cells, which
may lead to a drug-tolerant state and overcome the cytotoxic
effects of drugs, as MITF-mediated survival signalling may
counteract BRAF or MEK inhibitor-induced melanoma cell death
(187). MITF knockdown may
enhance the overall cytotoxicity of BRAF inhibitors (188) and this molecule is therefore an
important therapeutic target for melanoma. The compound TT-012 has
been identified to destroy the growth of MITFhigh B16F10
and GAK melanoma cell lines, and potently suppress the tumor growth
and metastasis with tolerable toxicity to liver and immune cells in
animal models, indicating that the inhibition of MITF through
TT-012 may present a novel approach to benefit melanoma treatment
(188). Smith et al
(189) also identified that in
BRAF V600E-melanoma allografts and in patients with BRAF-mutant
melanoma, BRAF and MEK inhibitors increase the number of
tumor-associated macrophages and lead to high expression of MITF
and TNFα, a crucial melanoma growth factor. They indicated that
targeting the NF-κB pathway inhibited the MITF- and TNFα-mediated
resistance to the MAPK-targeted agent. Of note, Müller et al
(190) detected an inverse
correlation between the loss of MITF and gain of expression of AXL
in the context of acquired resistance. A current phase Ib/II
randomised open-label trial of the AXL inhibitor BGB324 is being
undertaken in patients with advanced non-resectable or metastatic
melanoma to assess the efficacy of BGB324 given together with
standard treatment, pembrolizumab or dabrafenib and trametinib,
compared to standard treatment alone (NCT02872259) (191). However, this study did not
include any participants with primary or acquired resistance to
targeted therapy or immunotherapy.
Overall, it may be considered that continuously
analyzing the role of ICD and gut microbiota in the tumor
microenvironment is necessary, as both factors influence immune
response and outcome of treatment with immune checkpoint
inhibitors. According to preclinical and clinical studies, two
strategies, including TLR9 agonist-mediated inhibition with
pembrolizumab as well as FMT together with pembrolizumab, appear
promising in patients with advanced anti-PD-1-refractory melanoma.
On the other hand, BRAF and MEK inhibitors induce TNFα production,
NF-κB pathway activation and higher MITF expression, and thus,
focusing on inhibiting TNFα/MITF-mediated survival signals that
protect melanoma cells from MAPK pathway-targeted therapies may be
helpful to overcome the resistance. As
MITFlow/AXLhigh aggressive phenotype in
melanoma cells also contributes to resistance to both MAPK pathway
and PD-1 inhibitors (192,193),
AXL inhibitors combined with pembrolizumab or dabrafenib/trametinib
may also provide a new direction expected to enhance the efficacy
of these therapies in metastatic melanoma.
6. Conclusion
Melanoma develops due to sun exposure-induced DNA
damage, which triggers the malignant transformation of melanocytes.
Since 2011, with several new drugs having been approved for
clinical use, the treatment of patients with metastatic melanoma
has seen drastic changes and therapeutic options are now available.
In the present review, several promising dual BRAF and MEK
inhibition and PARP inhibitors, VEGF/VEGFR inhibitors and other
molecularly targeted therapies, such as c-Kit inhibitors and mTOR
inhibitors, were extensively discussed. In the field of
immunotherapy, in addition to cancer vaccination and adoptive cell
therapy, monoclonal antibodies blocking CTLA-4 and PD-1/PD-L1 have
been successfully developed.
Currently, there is no one-size-fits-all strategy
for treating patients affected by metastatic melanoma. The
decision-making in the first-line setting for BRAF-mutant
metastatic melanoma is still guided by clinical parameters and the
biological aspects of melanoma, including LDH level, organs
involved, performance status, tumor burden and disease progression
kinetics. On one hand, considering patients with BRAF-mutant and
aggressive diseases and those needing an immediate benefit in the
reduction of tumor burden, targeted therapy remains the backbone of
any first-line approach with a rapid onset of action. On the other
hand, for patients with less extensive BRAF-mutant and non-rapidly
progressive disease and more favourable parameters, first-line
therapy with immunotherapy should generally be preferred.
BRAF and MEK-targeted therapies show high efficacy
and increase the OS and ORR of a relatively high proportion of
patients with melanoma with somatic activating BRAF mutations in a
very short time, although a main characteristic of acquired
treatment resistance remains. Meanwhile, immunotherapy,
particularly immune checkpoint inhibitors, demonstrates durable
antitumor activity in a subset of patients with both BRAF-mutant
and wild-type melanoma, despite a slower onset of action. Thus, the
identification of a novel treatment strategy of combining immune
checkpoint inhibitors with BRAF and MEK-targeted agents seems a
promising perspective for this setting. Several clinical and
real-life studies have confirmed the role of this combination in
improving the prognosis of patients with metastatic melanoma;
however, this benefit is at the cost of substantial toxic effects
compared with monotherapy, which also indicates a ‘Yin and Yang’
action of combination therapy. Making the treatment more effective
with fewer side effects remains a critical objective in patients on
combination therapy.
According to most but not all results from clinical
studies of different sequencing strategies, frontline ipilimumab
plus nivolumab followed by targeted therapy seems to produce more
excellent response rates in patients with melanoma containing BRAF
mutations, compared with the inverse sequence. More and larger
studies are still required in order to confirm whether
immunotherapies are less effective if given after targeted therapy,
and confirm whether patients exactly benefit more from prior immune
checkpoint blockade. The need to answer the above questions has led
investigators to design clinical trials of novel combinatory
strategies. Because the safety profile of encorafenib plus
binimetinib is advantageous compared with other combinations of
BRAF and MEK inhibitors, two promising clinical trials (NCT04657991
and NCT04655157) were designed to investigate the efficacy of
triple therapy, in particular encorafenib plus binimetinib plus
pembrolizumab and encorafenib with or without binimetinib plus
nivolumab and low-dose ipilimumab, respectively.
Of note, a large proportion of patients with
melanoma remains who do not experience any durable benefits under
targeted therapy- and immunotherapy-based treatment, with either no
clinical response or disease progression. More evidence is needed
to clarify the optimal combinations between recent treatments and
new therapeutics to manage patients with recurrent melanoma while
taking into account patient-related characteristics, such as
currently targetable BRAF mutations, frontline treatment regimens
(single-agent or combination), type of resistance pattern (primary
or acquired) and CNS metastasis (presence or absence).
Acknowledgements
The authors thank Professor Bo Gao (Department of
Immunology, Shanghai Medical College, Fudan University, Shanghai,
China) for helpful and constructive discussions and comments.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
ZQ and MZ both made a significant contribution to
the study, including establishing the background, analysis or
interpretation, or all of these areas. Both drafted, wrote,
substantially revised and critically reviewed the manuscript. All
authors read and approved the final version to be published, agreed
on the journal to which the article has been submitted and agreed
to be accountable for all aspects of the work. Data authentication
is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Slominski RM, Sarna T, Płonka PM, Raman C,
Brożyna AA and Slominski AT: Melanoma, melanin, and melanogenesis:
The yin and yang relationship. Front Oncol.
12(842496)2022.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Slominski A, Tobin DJ, Shibahara S and
Wortsman J: Melanin pigmentation in mammalian skin and its hormonal
regulation. Physiol Rev. 84:1155–1228. 2004.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Videira IF, Moura DF and Magina S:
Mechanisms regulating melanogenesis. An Bras Dermatol. 88:76–83.
2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Garmyn M, Young AR and Miller SA:
Mechanisms of and variables affecting UVR photoadaptation in human
skin. Photochem Photobiol Sci. 17:1932–1940. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Slominski RM, Raman C, Chen JY and
Slominski AT: How cancer hijacks the Body's homeostasis through the
neuroendocrine system. Trends Neurosci. 46:263–275. 2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wolf Horrell EM, Boulanger MC and D'Orazio
JA: Melanocortin 1 receptor: Structure, function, and regulation.
Front Genet. 7(95)2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Li C, Kuai L, Cui R and Miao X:
Melanogenesis and the targeted therapy of melanoma. Biomolecules.
12(1874)2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kawaguchi M, Valencia JC, Namiki T, Suzuki
T and Hearing VJ: Diacylglycerol kinase regulates tyrosinase
expression and function in human melanocytes. J Invest Dermatol.
132:2791–2799. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Slominski A, Wortsman J, Mazurkiewicz JE,
Dietrich J, Lawrence K, Gorbani A and Paus R: Detection of
proopiomelanocortin-derived antigens in normal and pathologic human
skin. J Lab Clin Med. 122:658–666. 1993.PubMed/NCBI
|
|
10
|
Slominski A, Paus R and Mihm MC:
Inhibition of melanogenesis as an adjuvant strategy in the
treatment of melanotic melanomas: Selective review and hypothesis.
Anticancer Res. 18:3709–3715. 1998.PubMed/NCBI
|
|
11
|
Slominski A, Zmijewski MA and Pawelek J:
L-tyrosine and L-dihydroxyphenylalanine as hormone-like regulators
of melanocyte functions. Pigment Cell Melanoma Res. 25:14–27.
2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Huang AC and Zappasodi R: A decade of
checkpoint blockade immunotherapy in melanoma: Understanding the
molecular basis for immune sensitivity and resistance. Nat Immunol.
23:660–670. 2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global Cancer Statistics 2020:
GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36
Cancers in 185 Countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kozovska Z, Gabrisova V and Kucerova L:
Malignant melanoma: Diagnosis, treatment and cancer stem cells.
Neoplasma. 63:510–517. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Luke JJ, Flaherty KT, Ribas A and Long GV:
Targeted agents and immunotherapies: Optimizing outcomes in
melanoma. Nat Rev Clin Oncol. 14:463–482. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Heistein JB and Acharya U: Malignant
Melanoma. StatPearls. Treasure Island (FL), StatPearls Publishing
Copyright ©. 2022, StatPearls Publishing LLC., 2022.
|
|
17
|
Steeb T, Wessely A, Petzold A, Kohl C,
Erdmann M, Berking C and Heppt MV: c-Kit inhibitors for
unresectable or metastatic mucosal, acral or chronically
sun-damaged melanoma: A systematic review and one-arm
meta-analysis. Eur J Cancer. 157:348–357. 2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
El Zaoui I, Bucher M, Rimoldi D, Nicolas
M, Kaya G, Pescini Gobert R, Bedoni N, Schalenbourg A, Sakina E,
Zografos L, et al: Conjunctival melanoma targeted therapy: MAPK and
PI3K/mTOR pathways inhibition. Invest Ophthalmol Vis Sci.
60:2764–2772. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou
J, Sasada T, Zeng W, Giobbie-Hurder A, Atkins MB, Ibrahim N, et al:
Bevacizumab plus ipilimumab in patients with metastatic melanoma.
Cancer Immunol Res. 2:632–642. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wu X, Giobbie-Hurder A, Liao X, Lawrence
D, McDermott D, Zhou J, Rodig S and Hodi FS: VEGF Neutralization
Plus CTLA-4 blockade alters soluble and cellular factors associated
with enhancing lymphocyte infiltration and humoral recognition in
melanoma. Cancer Immunol Res. 4:858–868. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Andtbacka RH, Kaufman HL, Collichio F,
Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I,
Agarwala SS, et al: Talimogene laherparepvec improves durable
response rate in patients with advanced melanoma. J Clin Oncol.
33:2780–2788. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
McArthur GA, Chapman PB, Robert C, Larkin
J, Haanen JB, Dummer R, Ribas A, Hogg D, Hamid O, Ascierto PA, et
al: Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K
mutation-positive melanoma (BRIM-3): Extended follow-up of a phase
3, randomised, open-label study. Lancet Oncol. 15:323–332.
2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Overman MJ, Lonardi S, Wong KYM, Lenz HJ,
Gelsomino F, Aglietta M, Morse MA, Van Cutsem E, McDermott R, Hill
A, et al: Durable clinical benefit with nivolumab plus ipilimumab
in DNA mismatch repair-deficient/microsatellite instability-high
metastatic colorectal cancer. J Clin Oncol. 36:773–779.
2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Gerosa L, Chidley C, Fröhlich F, Sanchez
G, Lim SK, Muhlich J, Chen JY, Vallabhaneni S, Baker GJ, Schapiro
D, et al: Receptor-Driven ERK pulses reconfigure MAPK signaling and
enable persistence of Drug-Adapted BRAF-Mutant melanoma cells. Cell
Syst. 11:478–494.e9. 2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Tang KT and Lee CH: BRAF mutation in
papillary thyroid carcinoma: Pathogenic role and clinical
implications. J Chin Med Assoc. 73:113–1128. 2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Long GV, Stroyakovskiy D, Gogas H,
Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A,
Grob JJ, et al: Dabrafenib and trametinib versus dabrafenib and
placebo for Val600 BRAF-mutant melanoma: A multicentre,
double-blind, phase 3 randomised controlled trial. Lancet.
386:444–451. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ascierto PA, Dréno B, Larkin J, Ribas A,
Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L,
et al: 5-Year outcomes with cobimetinib plus vemurafenib in
BRAFV600 Mutation-positive advanced melanoma: Extended Follow-up of
the coBRIM study. Clin Cancer Res. 27:5225–5235. 2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hauschild A, Larkin J, Ribas A, Dréno B,
Flaherty KT, Ascierto PA, Lewis KD, McKenna E, Zhu Q, Mun Y and
McArthur GA: Modeled prognostic subgroups for survival and
treatment outcomes in BRAF V600-mutated metastatic melanoma: Pooled
analysis of 4 randomized clinical trials. JAMA Oncol. 4:1382–1388.
2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Dickson PV and Gershenwald JE: Staging and
prognosis of cutaneous melanoma. Surg Oncol Clin N Am. 20:1–17.
2011.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Larkin J, Ascierto PA, Dréno B, Atkinson
V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas
L, et al: Combined vemurafenib and cobimetinib in BRAF-mutated
melanoma. N Engl J Med. 371:1867–1876. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gutzmer R, Stroyakovskiy D, Gogas H,
Robert C, Lewis K, Protsenko S, Pereira RP, Eigentler T, Rutkowski
P, Demidov L, et al: Atezolizumab, vemurafenib, and cobimetinib as
first-line treatment for unresectable advanced BRAFV600
mutation-positive melanoma (IMspire150): Primary analysis of the
randomised, double-blind, placebo-controlled, phase 3 trial.
Lancet. 395:1835–1844. 2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ascierto P, Robert C, Lewis K, Gutzmer R,
Stroyakovskiy D, Gogas HJ, Protsenko S, Pereira RP, Kurt Eigentler
T, Rutkowski P, et al: 1102P Clinical benefit in BRAFV600
mutation-positive melanoma defined by programmed death ligand 1
(PD-L1) and/or lactate dehydrogenase (LDH) status: Exploratory
analyses from the IMspire150 study. Ann Oncol. 31(S745)2020.
|
|
33
|
Dummer R, Queirolo P, Abajo Guijarro AM,
Hu Y, Wang D, de Azevedo SJ, Robert C, Ascierto PA, Chiarion-Sileni
V, Pronzato P, et al: Atezolizumab, vemurafenib, and cobimetinib in
patients with melanoma with CNS metastases (TRICOTEL): A
multicentre, open-label, single-arm, phase 2 study. Lancet Oncol.
23:1145–1155. 2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tawbi HA, Forsyth PA, Algazi A, Hamid O,
Hodi FS, Moschos SJ, Khushalani NI, Lewis K, Lao CD, Postow MA, et
al: Combined Nivolumab and Ipilimumab in melanoma metastatic to the
brain. N Engl J Med. 379:722–730. 2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Davies MA, Saiag P, Robert C, Grob JJ,
Flaherty KT, Arance A, Chiarion-Sileni V, Thomas L, Lesimple T,
Mortier L, et al: Dabrafenib plus trametinib in patients with
BRAFV600-mutant melanoma brain metastases (COMBI-MB): A
multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol.
18:863–873. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
de la Cruz-Merino L, Di Guardo L, Grob JJ,
Venosa A, Larkin J, McArthur GA, Ribas A, Ascierto PA, Evans JTR,
Gomez-Escobar A, et al: Clinical features of serous retinopathy
observed with cobimetinib in patients with BRAF-mutated melanoma
treated in the randomized coBRIM study. J Transl Med.
15(146)2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ribas A, Gonzalez R, Pavlick A, Hamid O,
Gajewski TF, Daud A, Flaherty L, Logan T, Chmielowski B, Lewis K,
et al: Combination of vemurafenib and cobimetinib in patients with
advanced BRAF(V600)-mutated melanoma: A phase 1b study. Lancet
Oncol. 15:954–965. 2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Grimaldi AM, Simeone E and Ascierto PA:
Vemurafenib plus cobimetinib in the treatment of mutated metastatic
melanoma: The CoBRIM trial. Melanoma Manag. 2:209–215.
2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Callahan MK and Chapman PB: PD-1 or PD-L1
blockade adds little to combination of BRAF and MEK inhibition in
the treatment of BRAF V600-mutated melanoma. J Clin Oncol.
40:1393–1395. 2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Long GV, Hauschild A, Santinami M,
Atkinson V, Mandalà M, Chiarion-Sileni V, Larkin J, Nyakas M,
Dutriaux C, Haydon A, et al: Adjuvant dabrafenib plus trametinib in
stage III BRAF-mutated melanoma. N Engl J Med. 377:1813–1823.
2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Schadendorf D, Hauschild A, Santinami M,
Atkinson V, Mandalà M, Chiarion-Sileni V, Larkin J, Nyakas M,
Dutriaux C, Haydon A, et al: Patient-reported outcomes in patients
with resected, high-risk melanoma with BRAFV600E or
BRAFV600K mutations treated with adjuvant dabrafenib
plus trametinib (COMBI-AD): A randomised, placebo-controlled, phase
3 trial. Lancet Oncol. 20:701–710. 2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Dummer R, Hauschild A, Santinami M,
Atkinson V, Mandalà M, Kirkwood JM, Chiarion Sileni V, Larkin J,
Nyakas M, Dutriaux C, et al: Five-year analysis of adjuvant
dabrafenib plus trametinib in stage III melanoma. N Engl J Med.
383:1139–1148. 2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Atkinson V, Robert C, Grob JJ, Gogas H,
Dutriaux C, Demidov L, Gupta A, Menzies AM, Ryll B, Miranda F, et
al: Improved pyrexia-related outcomes associated with an adapted
pyrexia adverse event management algorithm in patients treated with
adjuvant dabrafenib plus trametinib: Primary results of
COMBI-APlus. Eur J Cancer. 163:79–87. 2022.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Delord JP, Robert C, Nyakas M, McArthur
GA, Kudchakar R, Mahipal A, Yamada Y, Sullivan R, Arance A, Kefford
RF, et al: Phase I dose-escalation and -expansion study of the BRAF
inhibitor encorafenib (LGX818) in metastatic BRAF-mutant melanoma.
Clin Cancer Res. 23:5339–5348. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Dummer R, Ascierto PA, Gogas HJ, Arance A,
Mandala M, Liszkay G, Garbe C, Schadendorf D, Krajsova I, Gutzmer
R, et al: Encorafenib plus binimetinib versus vemurafenib or
encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A
multicentre, open-label, randomised phase 3 trial. Lancet Oncol.
19:603–615. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
O'Connor Mark J: Targeting the DNA damage
response in cancer. Mol Cell. 60:547–560. 2015.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Lieber MR: The mechanism of double-strand
DNA break repair by the nonhomologous DNA end-joining pathway. Annu
Rev Biochem. 79:181–211. 2010.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Moynahan ME and Jasin M: Mitotic
homologous recombination maintains genomic stability and suppresses
tumorigenesis. Nat Rev Mol Cell Biol. 11:196–207. 2010.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Khan AQ, Travers JB and Kemp MG: Roles of
UVA radiation and DNA damage responses in melanoma pathogenesis.
Environ Mol Mutagen. 59:438–460. 2018.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Schlacher K, Wu H and Jasin M: A distinct
replication fork protection pathway connects Fanconi anemia tumor
suppressors to RAD51-BRCA1/2. Cancer Cell. 22:106–116.
2012.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Murai J, Huang SY, Das BB, Renaud A, Zhang
Y, Doroshow JH, Ji J, Takeda S and Pommier Y: Trapping of PARP1 and
PARP2 by clinical PARP inhibitors. Cancer Res. 72:5588–5599.
2012.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Fong PC, Boss DS, Yap TA, Tutt A, Wu P,
Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et
al: Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA
mutation carriers. N Engl J Med. 361:123–134. 2009.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Palma JP, Rodriguez LE, Bontcheva-Diaz VD,
Bouska JJ, Bukofzer G, Colon-Lopez M, Guan R, Jarvis K, Johnson EF,
Klinghofer V, et al: The PARP inhibitor, ABT-888 potentiates
temozolomide: Correlation with drug levels and reduction in PARP
activity in vivo. Anticancer Res. 28:2625–2635. 2008.PubMed/NCBI
|
|
54
|
Plummer R, Lorigan P, Steven N, Scott L,
Middleton MR, Wilson RH, Mulligan E, Curtin N, Wang D, Dewji R, et
al: A phase II study of the potent PARP inhibitor, Rucaparib
(PF-01367338, AG014699), with temozolomide in patients with
metastatic melanoma demonstrating evidence of chemopotentiation.
Cancer Chemother Pharmacol. 71:1191–1199. 2013.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Middleton MR, Friedlander P, Hamid O, Daud
A, Plummer R, Falotico N, Chyla B, Jiang F, McKeegan E, Mostafa NM,
et al: Randomized phase II study evaluating veliparib (ABT-888)
with temozolomide in patients with metastatic melanoma. Ann Oncol.
26:2173–2179. 2015.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Lau B, Menzies AM and Joshua AM: Ongoing
partial response at 6 months to olaparib for metastatic melanoma
with somatic PALB2 mutation after failure of immunotherapy: A case
report. Ann Oncol. 32:280–282. 2021.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Khaddour K, Ansstas M and Ansstas G:
Clinical outcomes and longitudinal circulating tumor DNA changes
after treatment with nivolumab and olaparib in immunotherapy
relapsed melanoma with detected homologous recombination
deficiency. Cold Spring Harb Mol Case Stud.
7(a006129)2021.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Fratangelo F, Camerlingo R, Carriero MV,
Pirozzi G, Palmieri G, Gentilcore G, Ragone C, Minopoli M, Ascierto
PA and Motti ML: Effect of ABT-888 on the apoptosis, motility and
invasiveness of BRAFi-resistant melanoma cells. Int J Oncol.
53:1149–1159. 2018.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Giunta EF, Belli V, Napolitano S, De Falco
V, Vitiello PP, Terminiello M, Caputo V, Vitale P, Zanaletti N,
Ciardiello D, et al: 13P Synergistic activity of PARP inhibitor and
ATR inhibitor in melanoma cell lines may depend on BRAF-V600
mutation status. Ann Oncol. 31:S248–S2S9. 2020.
|
|
60
|
Folkman J: Tumor angiogenesis: Therapeutic
implications. N Engl J Med. 285:1182–1186. 1971.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Ye W: The complexity of translating
Anti-angiogenesis therapy from basic science to the clinic. Dev
Cell. 37:114–125. 2016.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Claesson-Welsh L and Welsh M: VEGFA and
tumour angiogenesis. J Intern Med. 273:114–127. 2013.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Kim KB, Sosman JA, Fruehauf JP, Linette
GP, Markovic SN, McDermott DF, Weber JS, Nguyen H, Cheverton P,
Chen D, et al: BEAM: A randomized phase II study evaluating the
activity of bevacizumab in combination with carboplatin plus
paclitaxel in patients with previously untreated advanced melanoma.
J Clin Oncol. 30:34–41. 2012.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Ferrucci PF, Minchella I, Mosconi M,
Gandini S, Verrecchia F, Cocorocchio E, Passoni C, Pari C, Testori
A, Coco P and Munzone E: Dacarbazine in combination with
bevacizumab for the treatment of unresectable/metastatic melanoma:
A phase II study. Melanoma Res. 25:239–245. 2015.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Del Vecchio M, Mortarini R, Canova S, Di
Guardo L, Pimpinelli N, Sertoli MR, Bedognetti D, Queirolo P,
Morosini P, Perrone T, et al: Bevacizumab plus fotemustine as
first-line treatment in metastatic melanoma patients: Clinical
activity and modulation of angiogenesis and lymphangiogenesis
factors. Clin Cancer Res. 16:5862–5872. 2010.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Yan X, Sheng X, Chi Z, Si L, Cui C, Kong
Y, Tang B, Mao L, Wang X, Lian B, et al: Randomized phase II study
of bevacizumab in combination with carboplatin plus paclitaxel in
patients with previously untreated advanced mucosal melanoma. J
Clin Oncol. 39:881–889. 2021.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Zhou J, Mahoney KM, Giobbie-Hurder A, Zhao
F, Lee S, Liao X, Rodig S, Li J, Wu X, Butterfield LH, et al:
Soluble PD-L1 as a biomarker in malignant melanoma treated with
checkpoint blockade. Cancer Immunol Res. 5:480–492. 2017.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Algazi AP, Cha E, Ortiz-Urda SM, McCalmont
T, Bastian BC, Hwang J, Pampaloni MH, Behr S, Chong K, Cortez B, et
al: The combination of axitinib followed by paclitaxel/carboplatin
yields extended survival in advanced BRAF wild-type melanoma:
Results of a clinical/correlative prospective phase II clinical
trial. Br J Cancer. 112:1326–1331. 2015.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Fruehauf J, Lutzky J, McDermott D, Brown
CK, Meric JB, Rosbrook B, Shalinsky DR, Liau KF, Niethammer AG, Kim
S and Rixe O: Multicenter, phase II study of axitinib, a selective
second-generation inhibitor of vascular endothelial growth factor
receptors 1, 2, and 3, in patients with metastatic melanoma. Clin
Cancer Res. 17:7462–7469. 2011.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Mouriaux F, Servois V, Parienti JJ,
Lesimple T, Thyss A, Dutriaux C, Neidhart-Berard EM, Penel N,
Delcambre C, Peyro Saint Paul L, et al: Sorafenib in metastatic
uveal melanoma: Efficacy, toxicity and health-related quality of
life in a multicentre phase II study. Br J Cancer. 115:20–24.
2016.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Sheng X, Cao D, Yuan J, Zhou F, Wei Q, Xie
X, Cui C, Chi Z, Si L, Li S, et al: Sorafenib in combination with
gemcitabine plus cisplatin chemotherapy in metastatic renal
collecting duct carcinoma: A prospective, multicentre, single-arm,
phase 2 study. Eur J Cancer. 100:1–7. 2018.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Buchbinder EI, Sosman JA, Lawrence DP,
McDermott DF, Ramaiya NH, Van den Abbeele AD, Linette GP,
Giobbie-Hurder A and Hodi FS: Phase 2 study of sunitinib in
patients with metastatic mucosal or acral melanoma. Cancer.
121:4007–4015. 2015.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Mahalingam D, Malik L, Beeram M, Rodon J,
Sankhala K, Mita A, Benjamin D, Ketchum N, Michalek J, Tolcher A,
et al: Phase II study evaluating the efficacy, safety, and
pharmacodynamic correlative study of dual antiangiogenic inhibition
using bevacizumab in combination with sorafenib in patients with
advanced malignant melanoma. Cancer Chemother Pharmacol. 74:77–84.
2014.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Mittal K, Koon H, Elson P, Triozzi P,
Dowlati A, Chen H, Borden EC and Rini BI: Dual VEGF/VEGFR
inhibition in advanced solid malignancies: Clinical effects and
pharmacodynamic biomarkers. Cancer Biol Ther. 15:975–981.
2014.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Carlino MS, Todd JR and Rizos H:
Resistance to c-Kit inhibitors in melanoma: Insights for future
therapies. Oncoscience. 1:423–426. 2014.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Curtin JA, Busam K, Pinkel D and Bastian
BC: Somatic activation of KIT in distinct subtypes of melanoma. J
Clin Oncol. 24:4340–4346. 2006.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Guo J, Si L, Kong Y, Flaherty KT, Xu X,
Zhu Y, Corless CL, Li L, Li H, Sheng X, et al: Phase II,
open-label, single-arm trial of imatinib mesylate in patients with
metastatic melanoma harboring c-Kit mutation or amplification. J
Clin Oncol. 29:2904–2909. 2011.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Jung S, Armstrong E, Wei AZ, Ye F, Lee A,
Carlino MS, Sullivan RJ, Carvajal RD, Shoushtari AN and Johnson DB:
Clinical and genomic correlates of imatinib response in melanomas
with KIT alterations. Br J Cancer. 127:1726–1732. 2022.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Lee SJ, Kim TM, Kim YJ, Jang KT, Lee HJ,
Lee SN, Ahn MS, Hwang IG, Lee S, Lee MH and Lee J: Phase II trial
of nilotinib in patients with metastatic malignant melanoma
Harboring KIT gene aberration: A multicenter trial of Korean cancer
study group (UN10-06). Oncologist. 20:1312–1319. 2015.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Guo J, Carvajal RD, Dummer R, Hauschild A,
Daud A, Bastian BC, Markovic SN, Queirolo P, Arance A, Berking C,
et al: Efficacy and safety of nilotinib in patients with
KIT-mutated metastatic or inoperable melanoma: Final results from
the global, single-arm, phase II TEAM trial. Ann Oncol.
28:1380–1387. 2017.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Delyon J, Chevret S, Jouary T, Dalac S,
Dalle S, Guillot B, Arnault JP, Avril MF, Bedane C, Bens G, et al:
STAT3 mediates nilotinib response in KIT-altered melanoma: A phase
II multicenter trial of the french skin cancer network. J Invest
Dermatol. 138:58–67. 2018.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Cho JH, Kim KM, Kwon M, Kim JH and Lee J:
Nilotinib in patients with metastatic melanoma harboring KIT gene
aberration. Invest New Drugs. 30:2008–2014. 2012.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Carvajal RD, Lawrence DP, Weber JS,
Gajewski TF, Gonzalez R, Lutzky J, O'Day SJ, Hamid O, Wolchok JD,
Chapman PB, et al: Phase II study of nilotinib in melanoma
Harboring KIT alterations following progression to prior KIT
inhibition. Clin Cancer Res. 21:2289–2296. 2015.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Hodi FS, Corless CL, Giobbie-Hurder A,
Fletcher JA, Zhu M, Marino-Enriquez A, Friedlander P, Gonzalez R,
Weber JS, Gajewski TF, et al: Imatinib for melanomas harboring
mutationally activated or amplified KIT arising on mucosal, acral,
and chronically sun-damaged skin. J Clin Oncol. 31:3182–3190.
2013.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 169:361–371.
2017.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Karbowniczek M, Spittle CS, Morrison T, Wu
H and Henske EP: mTOR is activated in the majority of malignant
melanomas. J Invest Dermatol. 128:980–987. 2008.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Slingluff CL Jr, Petroni GR, Molhoek KR,
Brautigan DL, Chianese-Bullock KA, Shada AL, Smolkin ME, Olson WC,
Gaucher A, Chase CM, et al: Clinical activity and safety of
combination therapy with temsirolimus and bevacizumab for advanced
melanoma: A phase II trial (CTEP 7190/Mel47). Clin Cancer Res.
19:3611–3620. 2013.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Hainsworth JD, Infante JR, Spigel DR,
Peyton JD, Thompson DS, Lane CM, Clark BL, Rubin MS, Trent DF and
Burris HA III: Bevacizumab and everolimus in the treatment of
patients with metastatic melanoma: A phase 2 trial of the Sarah
Cannon Oncology Research Consortium. Cancer. 116:4122–4129.
2010.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Krummel MF and Allison JP: CD28 and CTLA-4
have opposing effects on the response of T cells to stimulation. J
Exp Med. 182:459–465. 1995.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Chen DS, Irving BA and Hodi FS: Molecular
pathways: Next-generation immunotherapy-inhibiting programmed
death-ligand 1 and programmed death-1. Clin Cancer Res.
18:6580–6587. 2012.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Leach DR, Krummel MF and Allison JP:
Enhancement of antitumor immunity by CTLA-4 blockade. Science.
271:1734–1736. 1996.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Okazaki T, Iwai Y and Honjo T: New
regulatory co-receptors: Inducible co-stimulator and PD-1. Curr
Opin Immunol. 14:779–782. 2002.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Hodi FS, O'Day SJ, McDermott DF, Weber RW,
Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel
JC, et al: Improved survival with ipilimumab in patients with
metastatic melanoma. N Engl J Med. 363:711–723. 2010.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Eggermont AM, Chiarion-Sileni V, Grob JJ,
Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, Ascierto PA,
Richards JM, et al: Prolonged survival in stage III melanoma with
ipilimumab adjuvant therapy. N Engl J Med. 375:1845–1855.
2016.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Eggermont AM, Chiarion-Sileni V, Grob JJ,
Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, Ascierto PA,
Richards JM, et al: Adjuvant ipilimumab versus placebo after
complete resection of high-risk stage III melanoma (EORTC 18071): A
randomised, double-blind, phase 3 trial. Lancet Oncol. 16:522–530.
2015.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Bertrand A, Kostine M, Barnetche T,
Truchetet ME and Schaeverbeke T: Immune related adverse events
associated with anti-CTLA-4 antibodies: Systematic review and
meta-analysis. BMC Med. 13(211)2015.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Liao B, Shroff S, Kamiya-Matsuoka C and
Tummala S: Atypical neurological complications of ipilimumab
therapy in patients with metastatic melanoma. Neuro Oncol.
16:589–593. 2014.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Ribas A, Camacho LH, Lopez-Berestein G,
Pavlov D, Bulanhagui CA, Millham R, Comin-Anduix B, Reuben JM, Seja
E, Parker CA, et al: Antitumor activity in melanoma and anti-self
responses in a phase I trial with the anti-cytotoxic T
lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J
Clin Oncol. 23:8968–8977. 2005.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Kirkwood JM, Lorigan P, Hersey P,
Hauschild A, Robert C, McDermott D, Marshall MA, Gomez-Navarro J,
Liang JQ and Bulanhagui CA: Phase II trial of tremelimumab
(CP-675,206) in patients with advanced refractory or relapsed
melanoma. Clin Cancer Res. 16:1042–1048. 2010.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Ribas A, Kefford R, Marshall MA, Punt CJ,
Haanen JB, Marmol M, Garbe C, Gogas H, Schachter J, Linette G, et
al: Phase III randomized clinical trial comparing tremelimumab with
standard-of-care chemotherapy in patients with advanced melanoma. J
Clin Oncol. 31:616–622. 2013.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Larkin J, Minor D, D'Angelo S, Neyns B,
Smylie M, Miller WH Jr, Gutzmer R, Linette G, Chmielowski B, Lao
CD, et al: Overall survival in patients with advanced melanoma who
received nivolumab versus Investigator's choice chemotherapy in
CheckMate 037: A randomized, controlled, open-label phase III
trial. J Clin Oncol. 36:383–390. 2018.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Ribas A, Puzanov I, Dummer R, Schadendorf
D, Hamid O, Robert C, Hodi FS, Schachter J, Pavlick AC, Lewis KD,
et al: Pembrolizumab versus investigator-choice chemotherapy for
ipilimumab-refractory melanoma (KEYNOTE-002): A randomised,
controlled, phase 2 trial. Lancet Oncol. 16:908–918.
2015.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Ott PA, Hodi FS and Robert C: CTLA-4 and
PD-1/PD-L1 blockade: New immunotherapeutic modalities with durable
clinical benefit in melanoma patients. Clin Cancer Res.
19:5300–5309. 2013.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Hodi FS, Chesney J, Pavlick AC, Robert C,
Grossmann KF, McDermott DF, Linette GP, Meyer N, Giguere JK,
Agarwala SS, et al: Combined nivolumab and ipilimumab versus
ipilimumab alone in patients with advanced melanoma: 2-year overall
survival outcomes in a multicentre, randomised, controlled, phase 2
trial. Lancet Oncol. 17:1558–1568. 2016.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Wolchok JD, Chiarion-Sileni V, Gonzalez R,
Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J,
Dummer R, et al: Long-term outcomes with nivolumab plus ipilimumab
or nivolumab alone versus ipilimumab in patients with advanced
melanoma. J Clin Oncol. 40:127–137. 2022.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Larkin J, Chiarion-Sileni V, Gonzalez R,
Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J,
Dummer R, et al: Five-year survival with combined nivolumab and
ipilimumab in advanced melanoma. N Engl J Med. 381:1535–1546.
2019.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Long GV, Atkinson V, Lo S, Sandhu S,
Guminski AD, Brown MP, Wilmott JS, Edwards J, Gonzalez M, Scolyer
RA, et al: Combination nivolumab and ipilimumab or nivolumab alone
in melanoma brain metastases: A multicentre randomised phase 2
study. Lancet Oncol. 19:672–681. 2018.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Weber J, Mandala M, Del Vecchio M, Gogas
HJ, Arance AM, Cowey CL, Dalle S, Schenker M, Chiarion-Sileni V,
Marquez-Rodas I, et al: Adjuvant Nivolumab versus ipilimumab in
resected stage III or IV melanoma. N Engl J Med. 377:1824–1835.
2017.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Larkin J, Del Vecchio M, Mandala M, Gogas
H, Arance AM, Dalle S, Cowey CL, Schenker M, Grob JJ,
Chiarion-Sileni V, et al: Adjuvant nivolumab versus ipilimumab in
resected stage III/IV melanoma: 5-year efficacy and biomarker
results from CheckMate 238. Clin Cancer Res: Apr 14, 2023 doi:
10.1158/1078-0432.CCR-22-3145 (Epub ahead of print).
|
|
110
|
Eggermont AMM, Blank CU, Mandala M, Long
GV, Atkinson VG, Dalle S, Haydon AM, Meshcheryakov A, Khattak A,
Carlino MS, et al: Longer Follow-up confirms recurrence-free
survival benefit of adjuvant pembrolizumab in high-risk stage III
melanoma: Updated results from the EORTC 1325-MG/KEYNOTE-054 trial.
J Clin Oncol. 38:3925–3936. 2020.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Eggermont AMM, Blank CU, Mandalà M, Long
GV, Atkinson VG, Dalle S, Haydon AM, Meshcheryakov A, Khattak A,
Carlino MS, et al: Adjuvant pembrolizumab versus placebo in
resected stage III melanoma (EORTC 1325-MG/KEYNOTE-054): Distant
metastasis-free survival results from a double-blind, randomised,
controlled, phase 3 trial. Lancet Oncol. 22:643–654.
2021.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Grossmann KF, Othus M, Patel SP, Tarhini
AA, Sondak VK, Knopp MV, Petrella TM, Truong TG, Khushalani NI,
Cohen JV, et al: Adjuvant pembrolizumab versus IFNα2b or ipilimumab
in resected high-risk melanoma. Cancer Discov. 12:644–653.
2022.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Luke JJ, Rutkowski P, Queirolo P, Del
Vecchio M, Mackiewicz J, Chiarion-Sileni V, de la Cruz Merino L,
Khattak MA, Schadendorf D, Long GV, et al: Pembrolizumab versus
placebo as adjuvant therapy in completely resected stage IIB or IIC
melanoma (KEYNOTE-716): A randomised, double-blind, phase 3 trial.
Lancet. 399:1718–1729. 2022.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Lao CD, Khushalani NI, Angeles C and
Petrella TM: Current state of adjuvant therapy for melanoma: Less
is more, or more is better? Am Soc Clin Oncol Educ Book. 42:1–7.
2022.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Blank CU, Rozeman EA, Fanchi LF, Sikorska
K, van de Wiel B, Kvistborg P, Krijgsman O, van den Braber M,
Philips D, Broeks A, et al: Neoadjuvant versus adjuvant ipilimumab
plus nivolumab in macroscopic stage III melanoma. Nat Med.
24:1655–1661. 2018.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Rozeman EA, Menzies AM, van Akkooi ACJ,
Adhikari C, Bierman C, van de Wiel BA, Scolyer RA, Krijgsman O,
Sikorska K, Eriksson H, et al: Identification of the optimal
combination dosing schedule of neoadjuvant ipilimumab plus
nivolumab in macroscopic stage III melanoma (OpACIN-neo): A
multicentre, phase 2, randomised, controlled trial. Lancet Oncol.
20:948–960. 2019.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Rozeman EA, Hoefsmit EP, Reijers ILM, Saw
RPM, Versluis JM, Krijgsman O, Dimitriadis P, Sikorska K, van de
Wiel BA, Eriksson H, et al: Survival and biomarker analyses from
the OpACIN-neo and OpACIN neoadjuvant immunotherapy trials in stage
III melanoma. Nat Med. 27:256–263. 2021.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Versluis JM, Menzies AM, Sikorska K,
Rozeman EA, Saw RPM, van Houdt WJ, Eriksson H, Klop WMC, Ch'ng S,
van Thienen JV, et al: Survival update of neoadjuvant ipilimumab
plus nivolumab in macroscopic stage III melanoma in the OpACIN and
OpACIN-neo trials. Ann Oncol. 34:420–430. 2023.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Reijers ILM, Menzies AM, van Akkooi ACJ,
Versluis JM, van den Heuvel NMJ, Saw RPM, Pennington TE, Kapiteijn
E, van der Veldt AAM, Suijkerbuijk KPM, et al: Personalized
response-directed surgery and adjuvant therapy after neoadjuvant
ipilimumab and nivolumab in high-risk stage III melanoma: The PRADO
trial. Nat Med. 28:1178–1188. 2022.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Long GV, Menzies AM and Scolyer RA:
Neoadjuvant checkpoint immunotherapy and melanoma: The time is now.
J Clin Oncol. 41:3236–3248. 2023.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Lucas M, Lijnsvelt J, Pulleman S, Scolyer
RA, Menzies AM, Van Akkooi ACJ, van Houdt WJ, Shannon KF,
Pennington T, Suijkerbuijk K, et al: The NADINA trial: A
multicenter, randomised, phase 3 trial comparing the efficacy of
neoadjuvant ipilimumab plus nivolumab with standard adjuvant
nivolumab in macroscopic resectable stage III melanoma. J Clin
Oncol. 40(TPS9605)2022.
|
|
122
|
Qin S, Xu L, Yi M, Yu S, Wu K and Luo S:
Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol
Cancer. 18(155)2019.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Woo SR, Turnis ME, Goldberg MV, Bankoti J,
Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, et
al: Immune inhibitory molecules LAG-3 and PD-1 synergistically
regulate T-cell function to promote tumoral immune escape. Cancer
Res. 72:917–927. 2012.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Ascierto PA, Bono P, Bhatia S, Melero I,
Nyakas MS, Svane IM, Gomez-Roca C, Larkin J, Schadendorf D, Dummer
R, et al: LBA18 Efficacy of BMS-986016, a monoclonal antibody that
targets lymphocyte activation gene-3 (LAG-3), in combination with
nivolumab in pts with melanoma who progressed during prior
anti–PD-1/PD-L1 therapy (mel prior IO) in all-comer and
biomarker-enriched populations. Ann Oncol. 28:611–612. 2017.
|
|
125
|
Tawbi HA, Schadendorf D, Lipson EJ,
Ascierto PA, Matamala L, Castillo Gutiérrez E, Rutkowski P, Gogas
HJ, Lao CD, De Menezes JJ, et al: Relatlimab and nivolumab versus
nivolumab in untreated advanced melanoma. N Engl J Med. 386:24–34.
2022.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Amaria RN, Postow M, Burton EM, Tetzlaff
MT, Ross MI, Torres-Cabala C, Glitza IC, Duan F, Milton DR, Busam
K, et al: Neoadjuvant relatlimab and nivolumab in resectable
melanoma. Nature. 611:155–160. 2022.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Rosenberg SA, Lotze MT, Muul LM, Leitman
S, Chang AE, Ettinghausen SE, Matory YL, Skibber JM, Shiloni E,
Vetto JT, et al: Observations on the systemic administration of
autologous lymphokine-activated killer cells and recombinant
interleukin-2 to patients with metastatic cancer. N Engl J Med.
313:1485–1492. 1985.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Rosenberg SA, Spiess P and Lafreniere R: A
new approach to the adoptive immunotherapy of cancer with
tumor-infiltrating lymphocytes. Science. 233:1318–1321.
1986.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Rosenberg SA, Yang JC, Sherry RM, Kammula
US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF,
Wunderlich JR, et al: Durable complete responses in heavily
pretreated patients with metastatic melanoma using T-cell transfer
immunotherapy. Clin Cancer Res. 17:4550–4557. 2011.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Sarnaik AA, Hamid O, Khushalani NI, Lewis
KD, Medina T, Kluger HM, Thomas SS, Domingo-Musibay E, Pavlick AC,
Whitman ED, et al: Lifileucel, a Tumor-infiltrating lymphocyte
therapy, in metastatic melanoma. J Clin Oncol. 39:2656–2666.
2021.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Seitter SJ, Sherry RM, Yang JC, Robbins
PF, Shindorf ML, Copeland AR, McGowan CT, Epstein M, Shelton TE,
Langhan MM, et al: Impact of prior treatment on the efficacy of
adoptive transfer of tumor-infiltrating lymphocytes in patients
with metastatic melanoma. Clin Cancer Res. 27:5289–5298.
2021.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Sarnaik AA, Hamid O, Khushalani NI, Lewis
KD, Medina T, Kluger HM, Thomas SS, Domingo-Musibay E, Pavlick AC,
Whitman ED, et al: Lifileucel, a Tumor-infiltrating lymphocyte
therapy, in metastatic melanoma. J Clin Oncol. 39:2656–2666.
2021.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Larkin J, Sarnaik A, Chesney JA,
Khushalani NI, Kirkwood JM, Weber JS, Lewis KD, Medina TM, Kluger
HM, Thomas SS, et al: Lifileucel (LN-144), a cryopreserved
autologous tumor infiltrating lymphocyte (TIL) therapy in patients
with advanced melanoma: Evaluation of impact of prior anti-PD-1
therapy. J Clin Oncol. 39 (15_suppl)(9505)2021.
|
|
134
|
David OM, Sylvia L, Amanda P, Sukari A,
Thomas S, Wenham R, Gogas H, Jazaeri A, Monk B, Rose P, et al: 492
Phase 2 efficacy and safety of autologous tumor-infiltrating
lymphocyte (TIL) cell therapy in combination with pembrolizumab in
immune checkpoint inhibitor-naïve patients with advanced cancers. J
Immuno Therapy Cancer. 9 (Suppl 2)(A523)2021.
|
|
135
|
Puzanov I, Milhem MM, Minor D, Hamid O, Li
A, Chen L, Chastain M, Gorski KS, Anderson A, Chou J, et al:
Talimogene laherparepvec in combination with ipilimumab in
previously untreated, unresectable stage IIIB-IV melanoma. J Clin
Oncol. 34:2619–2626. 2016.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Long G, Dummer R, Johnson D, Michielin O,
Martin-Algarra S, Treichel S, Chan E, Diede S and Ribas A: 429
Long-term analysis of MASTERKEY-265 phase 1b trial of talimogene
laherparepvec (T-VEC) plus pembrolizumab in patients with
unresectable stage IIIB-IVM1c melanoma. J ImmunoTherapy Cancer. 8
(Suppl 3)(A261)2020.
|
|
137
|
Chesney J, Puzanov I, Collichio F, Singh
P, Milhem MM, Glaspy J, Hamid O, Ross M, Friedlander P, Garbe C, et
al: Randomized, Open-label phase II study evaluating the efficacy
and safety of talimogene laherparepvec in combination with
ipilimumab versus ipilimumab alone in patients with advanced,
unresectable melanoma. J Clin Oncol. 36:1658–1667. 2018.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Chesney JA, Ribas A, Long GV, Kirkwood JM,
Dummer R, Puzanov I, Hoeller C, Gajewski TF, Gutzmer R, Rutkowski
P, et al: Randomized, Double-blind, placebo-controlled, global
phase III trial of talimogene laherparepvec combined with
pembrolizumab for advanced melanoma. J Clin Oncol. 41:528–540.
2022.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Lowe KL, Cole D, Kenefeck R, I OK, Lepore
M and Jakobsen BK: Novel TCR-based biologics: Mobilising T cells to
warm ‘cold’ tumours. Cancer Treat Rev. 77:35–43. 2019.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Oates J, Hassan NJ and Jakobsen BK:
ImmTACs for targeted cancer therapy: Why, what, how, and which. Mol
Immunol. 67:67–74. 2015.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Carvajal RD, Nathan P, Sacco JJ, Orloff M,
Hernandez-Aya LF, Yang J, Luke JJ, Butler MO, Stanhope S, Collins
L, et al: Phase I study of safety, tolerability, and efficacy of
tebentafusp using a Step-up dosing regimen and expansion in
patients with metastatic uveal melanoma. J Clin Oncol.
40:1939–1948. 2022.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Nathan P, Hassel JC, Rutkowski P, Baurain
JF, Butler MO, Schlaak M, Sullivan RJ, Ochsenreither S, Dummer R,
Kirkwood JM, et al: Overall survival benefit with tebentafusp in
metastatic uveal melanoma. N Engl J Med. 385:1196–1206.
2021.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Piulats JM, Espinosa E, de la Cruz Merino
L, Varela M, Alonso Carrión L, Martín-Algarra S, López Castro R,
Curiel T, Rodríguez-Abreu D, Redrado M, et al: Nivolumab plus
ipilimumab for treatment-naïve metastatic uveal melanoma: An
open-label, multicenter, phase II trial by the spanish
multidisciplinary melanoma group (GEM-1402). J Clin Oncol.
39:586–598. 2021.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Pelster MS, Gruschkus SK, Bassett R,
Gombos DS, Shephard M, Posada L, Glover MS, Simien R, Diab A, Hwu
P, et al: Nivolumab and ipilimumab in metastatic uveal melanoma:
Results from a single-arm phase II study. J Clin Oncol. 39:599–607.
2021.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Carlino MS, Larkin J and Long GV: Immune
checkpoint inhibitors in melanoma. Lancet. 398:1002–1014.
2021.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Begley J and Ribas A: Targeted therapies
to improve tumor immunotherapy. Clin Cancer Res. 14:4385–4391.
2008.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Koya RC, Mok S, Otte N, Blacketor KJ,
Comin-Anduix B, Tumeh PC, Minasyan A, Graham NA, Graeber TG, Chodon
T and Ribas A: BRAF inhibitor vemurafenib improves the antitumor
activity of adoptive cell immunotherapy. Cancer Res. 72:3928–3937.
2012.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Wilmott JS, Long GV, Howle JR, Haydu LE,
Sharma RN, Thompson JF, Kefford RF, Hersey P and Scolyer RA:
Selective BRAF inhibitors induce marked T-cell infiltration into
human metastatic melanoma. Clin Cancer Res. 18:1386–1394.
2012.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Frederick DT, Piris A, Cogdill AP, Cooper
ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, et
al: BRAF inhibition is associated with enhanced melanoma antigen
expression and a more favorable tumor microenvironment in patients
with metastatic melanoma. Clin Cancer Res. 19:1225–1231.
2013.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Boni A, Cogdill AP, Dang P, Udayakumar D,
Njauw CN, Sloss CM, Ferrone CR, Flaherty KT, Lawrence DP, Fisher
DE, et al: Selective BRAFV600E inhibition enhances T-cell
recognition of melanoma without affecting lymphocyte function.
Cancer Res. 70:5213–5219. 2010.PubMed/NCBI View Article : Google Scholar
|
|
151
|
Ribas A, Lawrence D, Atkinson V, Agarwal
S, Miller WH Jr, Carlino MS, Fisher R, Long GV, Hodi FS, Tsoi J, et
al: Combined BRAF and MEK inhibition with PD-1 blockade
immunotherapy in BRAF-mutant melanoma. Nat Med. 25:936–940.
2019.PubMed/NCBI View Article : Google Scholar
|
|
152
|
Ascierto PA, Ferrucci PF, Fisher R, Del
Vecchio M, Atkinson V, Schmidt H, Schachter J, Queirolo P, Long GV,
Di Giacomo AM, et al: Dabrafenib, trametinib and pembrolizumab or
placebo in BRAF-mutant melanoma. Nat Med. 25:941–946.
2019.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Ferrucci PF, Di Giacomo AM, Del Vecchio M,
Atkinson V, Schmidt H, Schachter J, Queirolo P, Long GV, Stephens
R, Svane IM, et al: KEYNOTE-022 part 3: A randomized, double-blind,
phase 2 study of pembrolizumab, dabrafenib, and trametinib in
BRAF-mutant melanoma. J Immunother Cancer.
8(e001806)2020.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Maio M, Carlino MS, Joshua AM, McWhirter
E, Ribas A, Ascierto PA, Miller WH Jr, Butler MO, Ferrucci PF,
Zielinski RR, et al: KEYNOTE-022: Pembrolizumab with trametinib in
patients with BRAF wild-type melanoma or advanced solid tumours
irrespective of BRAF mutation. Eur J Cancer. 160:1–11.
2022.PubMed/NCBI View Article : Google Scholar
|
|
155
|
Dummer R, Lebbé C, Atkinson V, Mandalà M,
Nathan PD, Arance A, Richtig E, Yamazaki N, Robert C, Schadendorf
D, et al: Combined PD-1, BRAF and MEK inhibition in advanced
BRAF-mutant melanoma: Safety run-in and biomarker cohorts of
COMBI-i. Nat Med. 26:1557–1563. 2020.PubMed/NCBI View Article : Google Scholar
|
|
156
|
Tawbi HA, Robert C, Brase JC, Gusenleitner
D, Gasal E, Garrett J, Savchenko A, Görgün G, Flaherty KT, Ribas A,
et al: Spartalizumab or placebo in combination with dabrafenib and
trametinib in patients with BRAF V600-mutant melanoma: Exploratory
biomarker analyses from a randomized phase 3 trial (COMBI-i). J
Immunother Cancer. 10(e004226)2022.PubMed/NCBI View Article : Google Scholar
|
|
157
|
Johnson DB, Pectasides E, Feld E, Ye F,
Zhao S, Johnpulle R, Merritt R, McDermott DF, Puzanov I, Lawrence
D, et al: Sequencing treatment in BRAFV600 mutant melanoma:
Anti-PD-1 before and after BRAF inhibition. J Immunother. 40:31–35.
2017.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Saab KR, Mooradian MJ, Wang DY, Chon J,
Xia CY, Bialczak A, Abbate KT, Menzies AM, Johnson DB, Sullivan RJ
and Shoushtari AN: Tolerance and efficacy of BRAF plus MEK
inhibition in patients with melanoma who previously have received
programmed cell death protein 1-based therapy. Cancer. 125:884–891.
2019.PubMed/NCBI View Article : Google Scholar
|
|
159
|
Atkins MB, Lee SJ, Chmielowski B, Tarhini
AA, Cohen GI, Truong TG, Moon HH, Davar D, O'Rourke M, Stephenson
JJ, et al: Combination dabrafenib and trametinib versus combination
nivolumab and ipilimumab for patients with advanced BRAF-mutant
melanoma: The DREAMseq trial-ECOG-ACRIN EA6134. J Clin Oncol.
41:186–197. 2023.PubMed/NCBI View Article : Google Scholar
|
|
160
|
Ascierto PA, Mandalà M, Ferrucci PF,
Guidoboni M, Rutkowski P, Ferraresi V, Arance A, Guida M, Maiello
E, Gogas H, et al: Sequencing of ipilimumab plus nivolumab and
encorafenib plus binimetinib for untreated BRAF-mutated metastatic
melanoma (SECOMBIT): A randomized, Three-arm, Open-label phase II
Trial. J Clin Oncol. 41:212–221. 2023.PubMed/NCBI View Article : Google Scholar
|
|
161
|
Ziogas DC, Theocharopoulos C, Koutouratsas
T, Haanen J and Gogas H: Mechanisms of resistance to immune
checkpoint inhibitors in melanoma: What we have to overcome? Cancer
Treat Rev. 113(102499)2023.PubMed/NCBI View Article : Google Scholar
|
|
162
|
Schmittnaegel M, Rigamonti N, Kadioglu E,
Cassará A, Wyser Rmili C, Kiialainen A, Kienast Y, Mueller HJ, Ooi
CH, Laoui D and De Palma M: Dual angiopoietin-2 and VEGFA
inhibition elicits antitumor immunity that is enhanced by PD-1
checkpoint blockade. Sci Transl Med. 9(eaak9670)2017.PubMed/NCBI View Article : Google Scholar
|
|
163
|
Schoenfeld J, Jinushi M, Nakazaki Y,
Wiener D, Park J, Soiffer R, Neuberg D, Mihm M, Hodi FS and Dranoff
G: Active immunotherapy induces antibody responses that target
tumor angiogenesis. Cancer Res. 70:10150–10160. 2010.PubMed/NCBI View Article : Google Scholar
|
|
164
|
Yi M, Jiao D, Qin S, Chu Q, Wu K and Li A:
Synergistic effect of immune checkpoint blockade and
anti-angiogenesis in cancer treatment. Mol Cancer.
18(60)2019.PubMed/NCBI View Article : Google Scholar
|
|
165
|
Chen PL, Roh W, Reuben A, Cooper ZA,
Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V,
Wani K, et al: Analysis of immune signatures in longitudinal tumor
samples yields insight into biomarkers of response and mechanisms
of resistance to immune checkpoint blockade. Cancer Discov.
6:827–837. 2016.PubMed/NCBI View Article : Google Scholar
|
|
166
|
Adachi Y, Kamiyama H, Ichikawa K,
Fukushima S, Ozawa Y, Yamaguchi S, Goda S, Kimura T, Kodama K,
Matsuki M, et al: Inhibition of FGFR reactivates IFNγ signaling in
tumor cells to enhance the combined antitumor activity of
lenvatinib with Anti-PD-1 antibodies. Cancer Res. 82:292–306.
2022.PubMed/NCBI View Article : Google Scholar
|
|
167
|
Kato Y, Tabata K, Kimura T,
Yachie-Kinoshita A, Ozawa Y, Yamada K, Ito J, Tachino S, Hori Y,
Matsuki M, et al: Lenvatinib plus anti-PD-1 antibody combination
treatment activates CD8+ T cells through reduction of
tumor-associated macrophage and activation of the interferon
pathway. PLoS One. 14(e0212513)2019.PubMed/NCBI View Article : Google Scholar
|
|
168
|
O'Day S, Gonzalez R, Kim K, Chmielowski B,
Kefford R, Loquai GLC, Cowey CL, Hauschild A, Hainsworth JD, Hersey
P, et al: A phase II study of the multitargeted kinase inhibitor
lenvatinib in patients with advanced BRAF wild-type melanoma. J
Clin Oncol. 31(9026)2013.
|
|
169
|
Taylor MH, Lee CH, Makker V, Rasco D,
Dutcus CE, Wu J, Stepan DE, Shumaker RC and Motzer RJ: Phase IB/II
trial of lenvatinib plus pembrolizumab in patients with advanced
renal cell carcinoma, endometrial cancer, and other selected
advanced solid tumors. J Clin Oncol. 38:1154–1163. 2020.PubMed/NCBI View Article : Google Scholar
|
|
170
|
Arance A, de la Cruz-Merino L, Petrella
TM, Jamal R, Ny L, Carneiro A, Berrocal A, Márquez-Rodas I,
Spreafico A, Atkinson V, et al: Phase II LEAP-004 study of
lenvatinib plus pembrolizumab for melanoma with confirmed
progression on a programmed cell death Protein-1 or programmed
death ligand 1 inhibitor given as monotherapy or in combination. J
Clin Oncol. 41:75–85. 2023.PubMed/NCBI View Article : Google Scholar
|
|
171
|
Tarhini AA, Frankel P, Margolin KA,
Christensen S, Ruel C, Shipe-Spotloe J, Gandara DR, Chen A and
Kirkwood JM: Aflibercept (VEGF Trap) in inoperable stage III or
stage IV melanoma of cutaneous or uveal origin. Clin Cancer Res.
17:6574–6581. 2011.PubMed/NCBI View Article : Google Scholar
|
|
172
|
Strickland KC, Howitt BE, Shukla SA, Rodig
S, Ritterhouse LL, Liu JF, Garber JE, Chowdhury D, Wu CJ, D'Andrea
AD, et al: Association and prognostic significance of
BRCA1/2-mutation status with neoantigen load, number of
tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high
grade serous ovarian cancer. Oncotarget. 7:13587–13598.
2016.PubMed/NCBI View Article : Google Scholar
|
|
173
|
Shen J, Zhao W, Ju Z, Wang L, Peng Y,
Labrie M, Yap TA, Mills GB and Peng G: PARPi Triggers the
STING-dependent immune response and enhances the therapeutic
efficacy of immune checkpoint blockade independent of BRCAness.
Cancer Res. 79:311–319. 2019.PubMed/NCBI View Article : Google Scholar
|
|
174
|
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen
MK, Hsu JM, Hsu JL, Yu WH, Du Y, Lee HH, et al: PARP inhibitor
upregulates PD-L1 expression and enhances cancer-associated
immunosuppression. Clin Cancer Res. 23:3711–3720. 2017.PubMed/NCBI View Article : Google Scholar
|
|
175
|
Russo V and Protti MP: Tumor-derived
factors affecting immune cells. Cytokine Growth Factor Rev.
36:79–87. 2017.PubMed/NCBI View Article : Google Scholar
|
|
176
|
Showalter A, Limaye A, Oyer JL, Igarashi
R, Kittipatarin C, Copik AJ and Khaled AR: Cytokines in immunogenic
cell death: Applications for cancer immunotherapy. Cytokine.
97:123–132. 2017.PubMed/NCBI View Article : Google Scholar
|
|
177
|
Ribas A, Medina T, Kummar S, Amin A,
Kalbasi A, Drabick JJ, Barve M, Daniels GA, Wong DJ, Schmidt EV, et
al: SD-101 in combination with pembrolizumab in advanced melanoma:
Results of a phase Ib, multicenter study. Cancer Discov.
8:1250–1257. 2018.PubMed/NCBI View Article : Google Scholar
|
|
178
|
Ribas A, Medina T, Kirkwood JM, Zakharia
Y, Gonzalez R, Davar D, Chmielowski B, Campbell KM, Bao R, Kelley
H, et al: Overcoming PD-1 Blockade resistance with CpG-A toll-like
receptor 9 agonist vidutolimod in patients with metastatic
melanoma. Cancer Discov. 11:2998–3007. 2021.PubMed/NCBI View Article : Google Scholar
|
|
179
|
Matson V, Chervin CS and Gajewski TF:
Cancer and the Microbiome-influence of the commensal microbiota on
cancer, immune responses, and immunotherapy. Gastroenterology.
160:600–613. 2021.PubMed/NCBI View Article : Google Scholar
|
|
180
|
Routy B, Le Chatelier E, Derosa L, Duong
CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C,
Roberti MP, et al: Gut microbiome influences efficacy of PD-1-based
immunotherapy against epithelial tumors. Science. 359:91–97.
2018.PubMed/NCBI View Article : Google Scholar
|
|
181
|
Matson V, Fessler J, Bao R, Chongsuwat T,
Zha Y, Alegre ML, Luke JJ and Gajewski TF: The commensal microbiome
is associated with anti-PD-1 efficacy in metastatic melanoma
patients. Science. 359:104–108. 2018.PubMed/NCBI View Article : Google Scholar
|
|
182
|
Davar D, Dzutsev AK, McCulloch JA,
Rodrigues RR, Chauvin JM, Morrison RM, Deblasio RN, Menna C, Ding
Q, Pagliano O, et al: Fecal microbiota transplant overcomes
resistance to anti-PD-1 therapy in melanoma patients. Science.
371:595–602. 2021.PubMed/NCBI View Article : Google Scholar
|
|
183
|
Gok Yavuz B, Gunaydin G, Gedik ME,
Kosemehmetoglu K, Karakoc D, Ozgur F and Guc D: Cancer associated
fibroblasts sculpt tumour microenvironment by recruiting monocytes
and inducing immunosuppressive PD-1+ TAMs. Sci Rep.
9(3172)2019.PubMed/NCBI View Article : Google Scholar
|
|
184
|
Baruch EN, Youngster I, Ben-Betzalel G,
Ortenberg R, Lahat A, Katz L, Adler K, Dick-Necula D, Raskin S,
Bloch N, et al: Fecal microbiota transplant promotes response in
immunotherapy-refractory melanoma patients. Science. 371:602–609.
2021.PubMed/NCBI View Article : Google Scholar
|
|
185
|
Carreira S, Goodall J, Denat L, Rodriguez
M, Nuciforo P, Hoek KS, Testori A, Larue L and Goding CR: Mitf
regulation of Dia1 controls melanoma proliferation and
invasiveness. Genes Dev. 20:3426–3439. 2006.PubMed/NCBI View Article : Google Scholar
|
|
186
|
Arozarena I and Wellbrock C: Phenotype
plasticity as enabler of melanoma progression and therapy
resistance. Nat Rev Cancer. 19:377–391. 2019.PubMed/NCBI View Article : Google Scholar
|
|
187
|
Smith MP, Brunton H, Rowling EJ, Ferguson
J, Arozarena I, Miskolczi Z, Lee JL, Girotti MR, Marais R, Levesque
MP, et al: Inhibiting drivers of non-mutational drug tolerance is a
salvage strategy for targeted melanoma therapy. Cancer Cell.
29:270–284. 2016.PubMed/NCBI View Article : Google Scholar
|
|
188
|
Haq R, Yokoyama S, Hawryluk EB, Jönsson
GB, Frederick DT, McHenry K, Porter D, Tran TN, Love KT, Langer R,
et al: BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene
that confers resistance to BRAF inhibition. Proc Natl Acad Sci USA.
110:4321–4326. 2013.PubMed/NCBI View Article : Google Scholar
|
|
189
|
Smith MP, Sanchez-Laorden B, O'Brien K,
Brunton H, Ferguson J, Young H, Dhomen N, Flaherty KT, Frederick
DT, Cooper ZA, et al: The immune microenvironment confers
resistance to MAPK pathway inhibitors through macrophage-derived
TNFα. Cancer Discov. 4:1214–1229. 2014.PubMed/NCBI View Article : Google Scholar
|
|
190
|
Müller J, Krijgsman O, Tsoi J, Robert L,
Hugo W, Song C, Kong X, Possik PA, Cornelissen-Steijger PD, Geukes
Foppen MH, et al: Low MITF/AXL ratio predicts early resistance to
multiple targeted drugs in melanoma. Nat Commun.
5(5712)2014.PubMed/NCBI View Article : Google Scholar
|
|
191
|
Nyakas M, Fleten KG, Haugen MH, Engedal N,
Sveen C, Farstad IN, Flørenes VA, Prasmickaite L, Mælandsmo GM and
Seip K: AXL inhibition improves BRAF-targeted treatment in
melanoma. Sci Rep. 12(5076)2022.PubMed/NCBI View Article : Google Scholar
|
|
192
|
Lee JH, Shklovskaya E, Lim SY, Carlino MS,
Menzies AM, Stewart A, Pedersen B, Irvine M, Alavi S, Yang JYH, et
al: Transcriptional downregulation of MHC class I and melanoma
de-differentiation in resistance to PD-1 inhibition. Nat Commun.
11(1897)2020.PubMed/NCBI View Article : Google Scholar
|
|
193
|
Falcone I, Conciatori F, Bazzichetto C,
Ferretti G, Cognetti F, Ciuffreda L and Milella M: Tumor
microenvironment: Implications in melanoma resistance to targeted
therapy and immunotherapy. Cancers (Basel). 12(2870)2020.PubMed/NCBI View Article : Google Scholar
|















