Introduction
Renal cell carcinoma (RCC) is a prevalent urologic
malignancy with incidence and fatality rates that rank fifteenth
among malignant tumors (1). RCC is
divided histologically into subtypes, with kidney renal clear cell
carcinoma (KIRC) accounting for >80% of RCCs, followed by
papillary RCC and chromophobe RCC (2,3).
Despite the fact that KIRC is a disease that may be detected early
and successfully treated with surgical or ablative techniques, up
to one-third of cases will present with or acquire metastases and
20-40% of cases will relapse after nephrectomy for localized
disease (4). KIRC is particularly
resistant to chemotherapy and the conventional treatment for
metastatic KIRC5 is targeted therapy with tyrosine kinase inhibitor
(TKI)/mammalian target of rapamycin inhibitor (mTORi) with or
without immunotherapy (5). Once
KIRC recurs, no additional treatment is successful.
The majority of patients with KIRC demonstrate
chromosomal 3p deletion and genomic mutations in the Von
Hippel-Lindau (VHL) Tumor Suppressor allele (6), followed by secondary loss of several
tumor suppressor genes, such as PBRM1, SETD2, BAP1 and/or
KDM5C (7). These genes also
contribute to genomic instability (8).
Additionally, a significant contributor to genomic
instability is homologous recombination repair deficiency (HRD)
(9). As a potent prognostic
biomarker, HRD has been discovered in recent years in a number of
types of cancer. Although identification of HRD in adrenal cortical
carcinoma predicts a worse clinical prognosis compared with non-HRD
patients, molecular characterization of HRD in ovarian malignancies
predicts more effective PARP inhibitor therapy and longer life
(10). The current clinical
detection of HRD is theoretically based on the specific,
quantifiable and permanent genomic alterations produced by HRD,
such as identifying genetic mutations, insertion/deletion patterns,
chromosomal structural abnormalities and gene copy number
variations (11). A ‘genomic scar’
made up of large-scale state transitions (LST), telomere allele
imbalance (TAI) and genomic heterozygosity loss (LOH) may result
from HRD, a functional defect in homologous recombinant DNA repair
(12). The HRD state of cells can
be partially described by LOH, TAI and LST, each of which have
unique definitions. The HRD score, which is created from the three
markers (LOH+TAI+LST), is a more reliable predictor of HRD than any
of the individual scores, even though each score has clinical
significance on its own (13,14).
The relationship between HRD and tumors has been extensively
studied in other types of cancer, such as ovarian cancer and breast
cancer, but it is still unclear how HRD affects the prognosis and
the tumor microenvironment (TME) in KIRC (15,16).
The present study discovered that HRD was a
prognostic indicator for patients with clear cell renal cell
carcinoma. HRD patients exhibited a characteristic upregulation of
immune-related and DNA damage repair pathways. Single cell RNA
sequencing (scRNA-seq) analysis revealed a higher prevalence of
exhausted T cell signatures in patients with HRD. The present study
is expected to yield novel insights into the individualized
treatment of KIRC.
Materials and methods
Sample collection
In the present study, four KIRC patients with
HRD-positive tumors were recruited between March 2021 and June 2021
in Hangzhou Hospital of Traditional Chinese Medicine (Table SI). For KIRC patients, tumor
tissues, adjacent normal tissues were collected. Written informed
consent was obtained from all participants. The present study was
performed in accordance with the ethical principles of the
Declaration of Helsinki and was approved by the Research and Ethics
Committee of Hangzhou Hospital of Traditional Chinese Medicine
(approval no. 2019KY005). The patients underwent next-generation
sequencing using whole exome sequencing (WES). Using matched normal
(blood) samples from each patient as a reference, tumor mutation
burden (TMB) was computed as the total detected missense mutations
in the pretreatment tumor samples (17). PD-L1 expression on tumor cells was
prospectively assessed using the Dako PD-L1 IHC 28-8 pharmDx test
(Agilent Technologies, Inc.) (18). Microsatellite instability is
calculated using the MSIsensor algorithm (19).
Data collection and processing
The Cancer Genome Atlas (TCGA) database, which is
open to the public and can be accessed at https://portal.gdc.cancer.gov, was used to download
the Original WES sequencing data, gene expression profile data and
associated clinical follow-up data (20). From the TCGA databases, 501 KIRC
samples with complete clinical data were obtained (https://www.cbioportal.org/study/clinicalData?id=kirc_tcga_pan_can_atlas_2018).
VarScan2 was used to identify somatic mutations, such as
insertion-deletions and single nucleotide polymorphisms (21). Synonymous single nucleotide
variants (SNV), non-synonymous SNV, stop gain SNV, non-frameshift
insertions and frameshift deletions were among the exonic
alterations that were extracted from the mutation points and
annotated using ANNOVAR (22). The
cBioPortal database (https://www.cbioportal.org/study/summary?id=kirc_tcga_pan_can_atlas_2018)
provided the somatic mutation counts, copy number variation (CNV),
fraction genome altered scores (FGA; percentage of copy number
altered chromosome regions out of measured regions) and TMB sensor
score (tumor mutation burden detection using paired tumor-normal
sequence data) (23). Lymphocytes
infiltrating HRD and homologous recombination proficient (HRP)
tumors were obtained from public database (https://doi.org/10.5522/04/16573640.v1) (24).
HRD score analysis
The number of counts of chromosomal LOH regions
longer than 15 Mb but shorter than the entire chromosome was used
to define loss of heterozygosity (LOH) (25). After smoothing and filtering out
small-scale copy number variation shorter than 3 Mb, LST were
defined as chromosome breakpoints (change in copy number or allelic
content) between adjacent regions, each of ≥10 Mb (26). The quantity of regions with allelic
imbalance that reach the sub-telomere but do not cross the
centromere is known as telomeric allelic imbalance (TAI) (27). The TAI, LST and LOH scores were
added together to create the HRD score. Table SII displays each patient's HRD
score.
Difference analysis and enrichment
analysis
Using the R-package (https://www.r-project.org/) DEseq2(28), the differences between various
molecular subtypes were examined. The cutoffs to find
differentially expressed genes (DEGs) were set as fold change >1
and adj. P<0.05. For the HRD group, the present study used gene
ontology (GO) enrichment analysis to identify specific biological
functions. All peak-related genes were first assigned to GO terms
in the GO database. Gene numbers were calculated for each term and
a hypergeometric test was used to identify GO terms that were
significantly enriched in peak-related genes compared with the
genome background. Additionally, the reference gene set for gene
set enrichment analysis (GSEA; cp.kegg.v7.0.symbols.gmt) was used
to examine the enrichment of various molecular subtypes in various
pathways (29). P<0.05 and a
false discovery rate (FDR) threshold of 0.25 were used to determine
which pathways were significantly enriched.
scRNA-seq data processing and quality
control
Mammary tissues from both benign and malignant
tumors were saved during surgery and processed in 1 h using Tissue
Storage Solution (Miltenyi Biotec GmbH). Following being rinsed in
PBS, samples were mechanically separated using a razor blade.
Dissociated samples were processed in RPMI Medium 1640 (Gibco;
Thermo Fisher Scientific, Inc.) with collagenase/hyaluronidase
(20%; Stemcell Technologies, Inc.) and BSA (20 mg/ml; Beijing
Solarbio Science & Technology Co., Ltd.) at 37˚C for 1 h in
order to produce cell suspensions. In order to create single-cell
suspensions, the cells were next rinsed in a cold Hank's balanced
salt solution (Beijing Solarbio Science & Technology Co., Ltd.)
containing 0.04 percent BSA. The 10x Genomic Chromium Next GEM
Single Cell 5' Reagents kit V2 (dual index) (10x Genomics) was used
to create a 5'gene expression library. Using the High Output kit
v2.5 and the NextSeq (Illumina, Inc.), the samples were sequenced
(150 Cycles). The aggregated scRNA-seq library's low-quality cells
were eliminated (500 <nFeature <10,000; 1000 <nCount
<100,000 and 0 <pMT <0.2). Datasets were subsequently
preprocessed with DoubletFinder v2.0.2(30) to eliminate heterotypic doublets
(presuming 6% of barcodes are doublets). With SCTransform and
Harmony (31), the filtered
library was normalized and batch effects were removed. Cell
distances were then visualized in a reduced two-dimensional space
using the t-distributed stochastic neighbor embedding or uniform
manifold approximation and projection (UMAP) method (https://satijalab.org/seurat/reference/runumap).
scType was used to conduct cell type annotation and the cell
markers used in this work were taken from earlier studies (Table SIII) (32). DEseq2 was used to determine the
significance of each gene (FDR <0.01; fold change |log2FC|
>1) in order to find the DEGs between two groups of
clusters.
Single-cell copy-number variation
(CNV) evaluation
The inferCNV R package (version 1.4.0; https://github.com/broadinstitute/inferCNV/wiki) was
used to evaluate the CNV of each cell. The CNVs of cells with renal
cell carcinoma were calculated, using stromal cells as the
standard. The default hidden Markov model (HMM) settings, ‘denoise’
and a value of 0.1 for cutoff were used in the inferCNV analysis.
The default Bayesian latent mixture model was used to identify the
posterior probabilities of the CNV alterations in each cell with
the default value of 0.5 as the threshold in order to decrease the
number of false-positive CNV calls.
Data and code availability
The single cell datasets and code generated during
the present study are accessible in the Zenodo database (https://zenodo.org/record/7783618#.ZCTwZXZBy3A).
Results
Genomic scar-based homologous
recombination deficit (HRD) scores and prognostic analysis in
KIRC
The discovery cohort of the present study included
501 KIRC patients from TCGA database to examine the potential
relevance of HRD in KIRC. The top 20% of patients with the highest
HRD scores and the bottom 20% of patients with the lowest HRD
scores were determined using WES data for each KIRC sample and
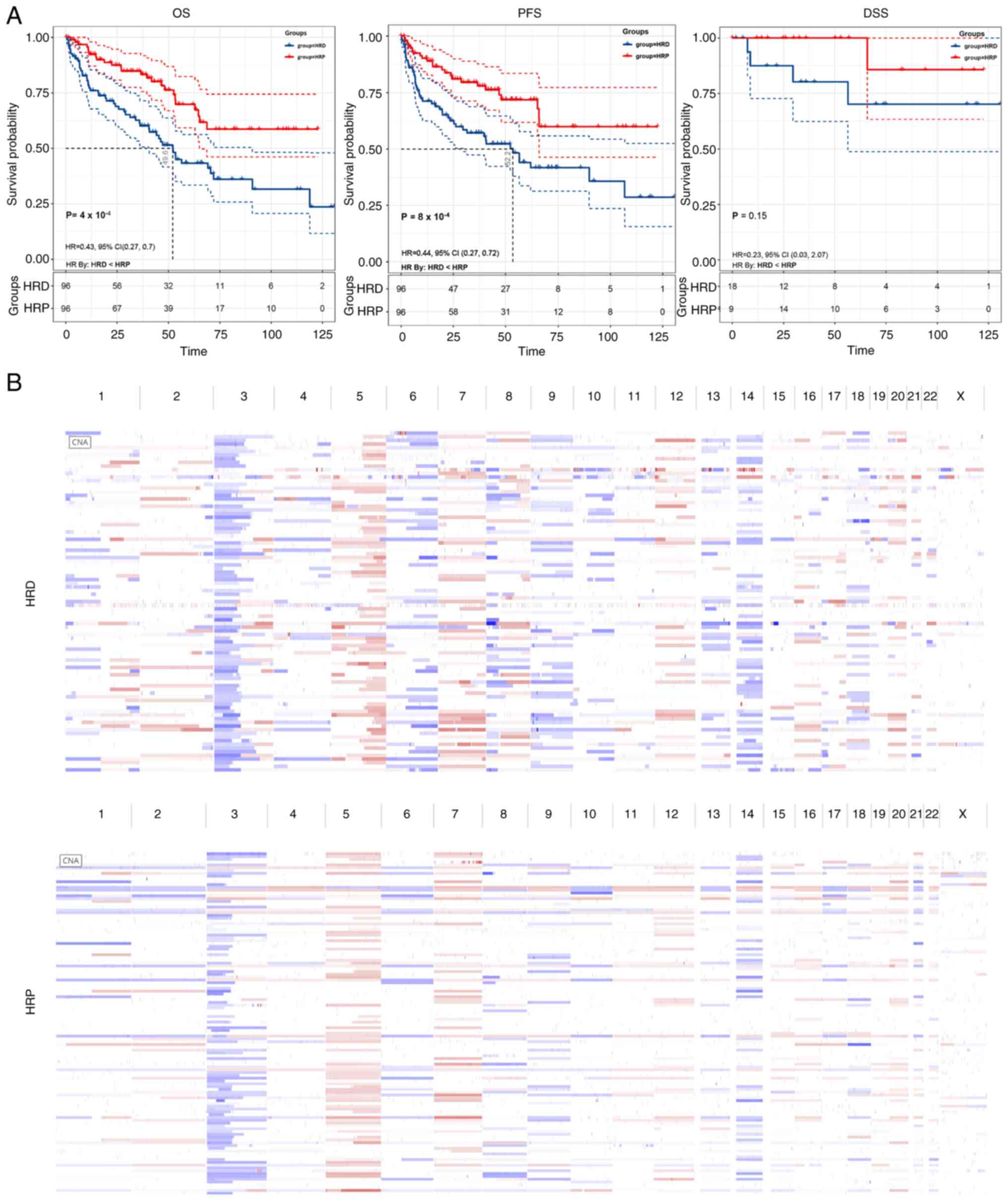
these samples were then sorted based on HRD scores. In terms of
overall survival (OS; log-rank test; P=0.0004), disease-specific
survival (DSS; log-rank test; P=0.15), or disease progression-free
survival (PFS; log-rank test; P=0.0008), patients with HRD had a
worse prognosis compared with patients with HRP, according to the
results of the survival analysis (Fig.
1A).
The present study examined genome-wide copy number
variation to show differences in genomic instability between HRD
and HRP patients because HRD is an assessment of genome-wide
traces. As expected, HRD patients had more genomic instability
(Fig. 1B).
The mutation landscape in KIRC
An integrated analysis of the WES data was conducted
in the study population (The top 20% of patients with the highest
HRD scores and the bottom 20% of patients with the lowest HRD
scores). The five most frequently mutated genes, VHL, PBRM1,
TTN, SETD2 and BAP1, all displayed mutation rates of
>15% in both groups, as shown in Fig. 2A, which was consistent with the
findings of other cohort studies. Next, the additional genetic
variations between the two categories was assessed, including
mutation count and fraction genome altered (FGA). In the HRD group
compared with the HRP group, the signs of genomic instability were
significantly more prevalent (Wilcoxon signed-rank test,
P<0.0001, Fig. 2B).
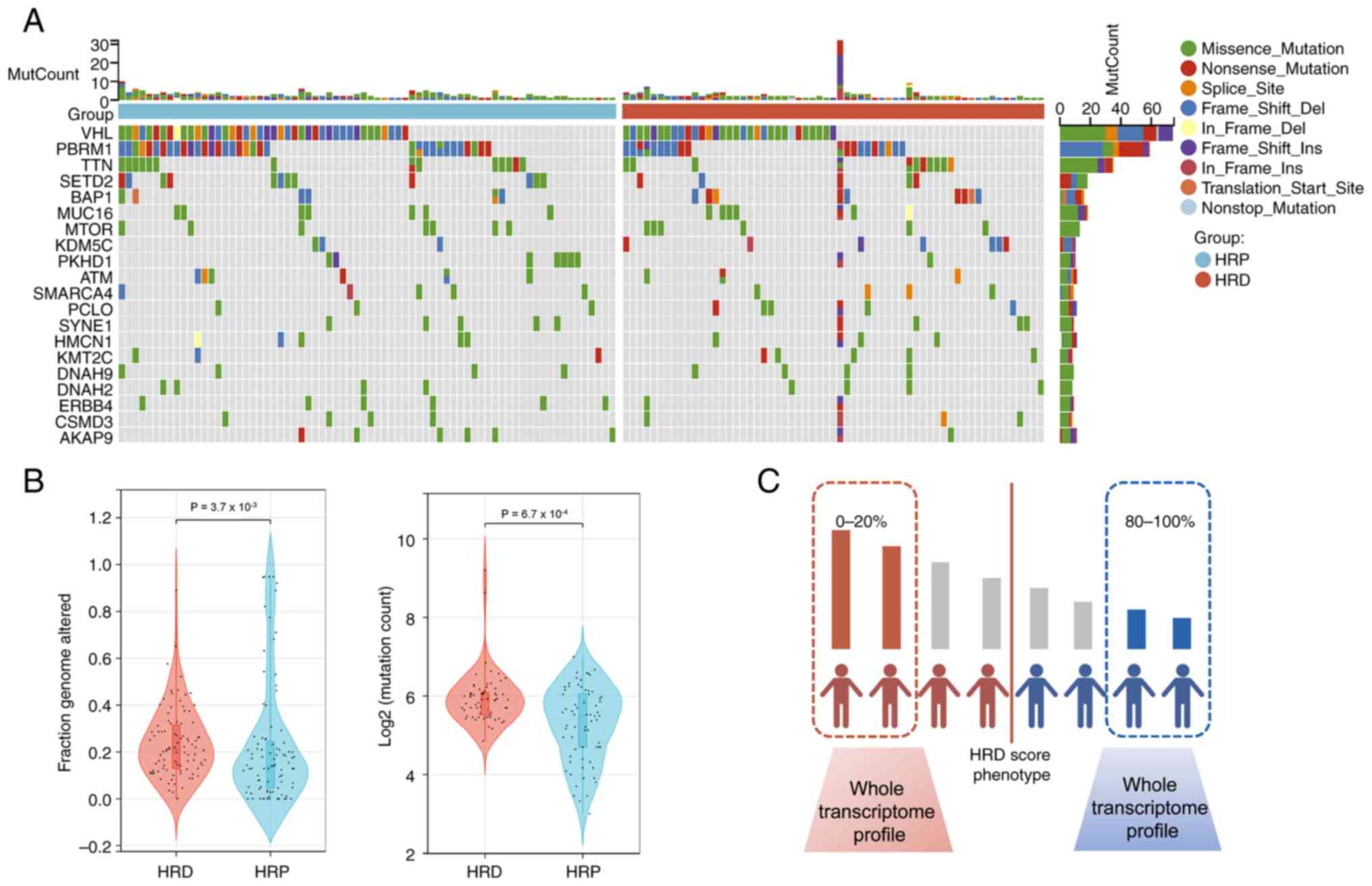 | Figure 2The mutation landscape in the
TCGA-KIRC cohort. (A) Summary of the most prevalent genomic
alterations in different HRD group. The mutational matrix shows
Missense_Mutations (green), Nonsense_Mutations (red),
Frame_shift_Deletions (blue), Splice_site mutations (orange),
Frame_shift_Insertions (purple), In_Frame_Dels (yellow),
In_Frame_Ins (brownness), Translation_Start_Site mutations (light
brown), Nonstop_Mutations (light blue). (B) Violin plot of fraction
of genome altered in HRD and HRP groups (Wilcoxon signed-rank
test;); Violin plot of mutation count in the HRD and the HRP groups
(Wilcoxon signed-rank test;). (C) WES calculated HRD scores for
each KIRC patient, with the 20% of patients with the highest HRD
scores constituting the HRD group and the 20% of patients with the
lowest HRD scores constituting the HRP group. TCGA, The Cancer
Genome Atlas; KIRC, kidney renal clear cell carcinoma; HRD,
homologous recombination deficit; HRP, homologous recombination
proficient; WES, whole exome sequencing. |
Discovering the HRD transcriptome
signature in KIRC
To explore the transcriptomic signatures associated
with HRD, the KIRC gene expression profile data was examined to
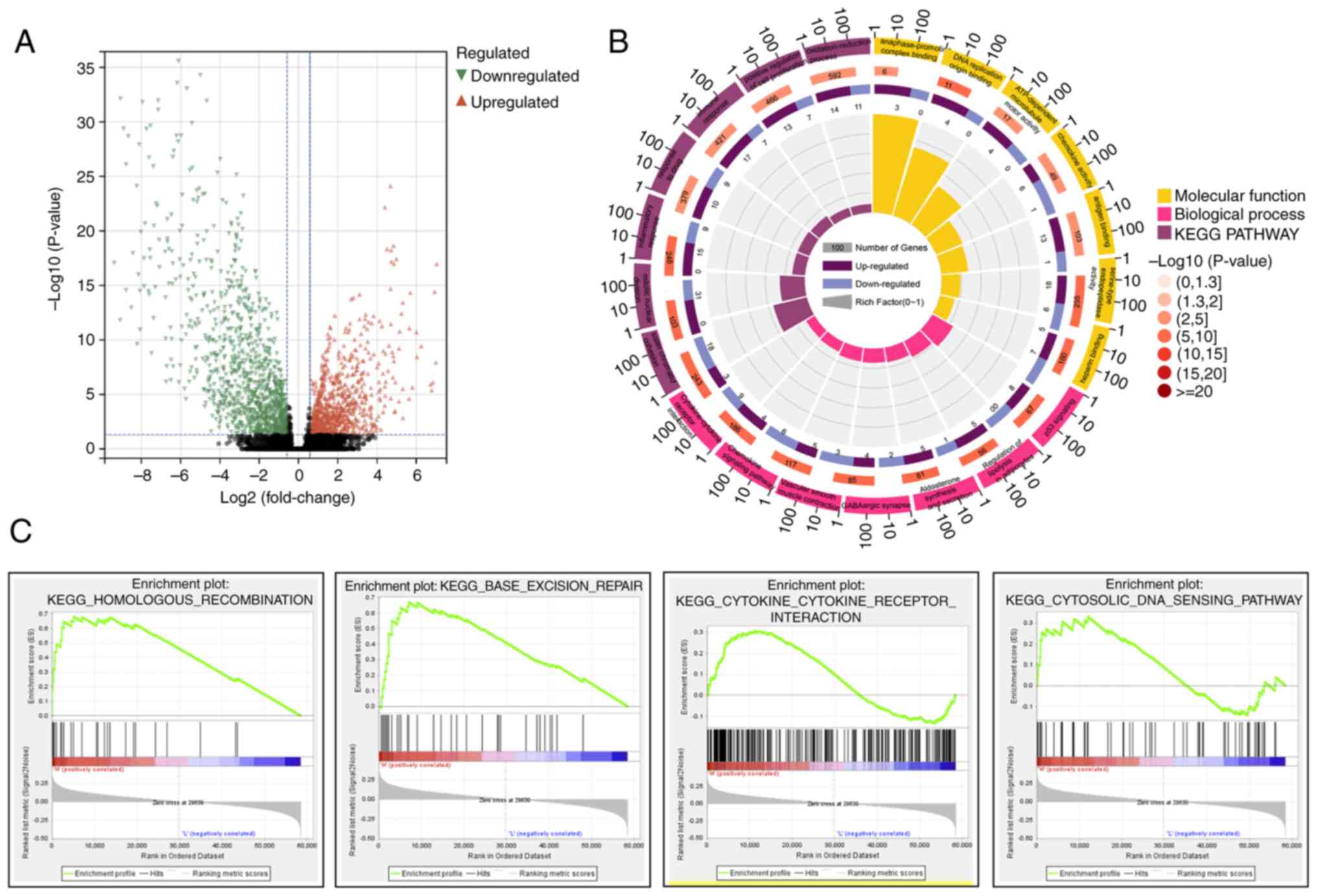
understand HRD-specific transcriptome markers (Fig. 2C). A total of 1,540 genes were
found to be differentially expressed between HRD and HRP patients
according to the DEseq2 analysis of gene expression profiling data
(|logFC| >1.5; FDR 0.05). Specifically, out of the 1,540 DEGs,
630 DEGs were upregulated and 910 were downregulated (Fig. 3A). Using the online analytical tool
DAVID, Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling
pathway enrichment analysis and Gene Ontology (GO) functional
annotation were performed to obtain a thorough and in-depth
understanding of the biological characteristics of these DEGs.
According to GO analysis, sister chromatid cohesion, mitotic
nuclear division, inflammatory response and other biological
processes are among the enriched biological processes (BP) of
upregulated DEGs. The enriched molecular function (MF) of
upregulated DEGs included chemokine activity, DNA replication
origin binding and anaphase-promoting complex binding (Fig. 3B). The regulation of lipolysis in
adipocytes, aldosterone synthesis and secretion, GABAergic
synapses, vascular smooth muscle contraction, chemokine signaling
pathway and Chemokine-Chemokine receptor interaction are among the
enriched pathways of upregulated DEGs, according to KEGG analysis
(Fig. 3B). The homologous
recombination pathway, the base-excision repair pathway, Chemokine
receptor interaction and the cytosolic DNA sensing pathway were all
significantly enriched in the HRD group, according to the GSEA
analysis (Fig. 3C).
Analysis of tissue samples from
HRD-positive KIRC patients using single-cell sequencing
As standard bulk RNA-sequencing assumes that each
gene is expressed identically in every cell (33), it is obviously unable to explore
intratumoral heterogeneity at the cell-type level. Single-cell
RNA-sequencing (scRNA-seq) has made feasible the profiling of the
transcriptome of a single cell. Consequently, the scRNA-seq data of
four KIRC patients were analyzed. A total of four KIRC patients
undergoing radical nephrectomy provided a total of eight tissue
samples for this investigation. The Materials and methods section
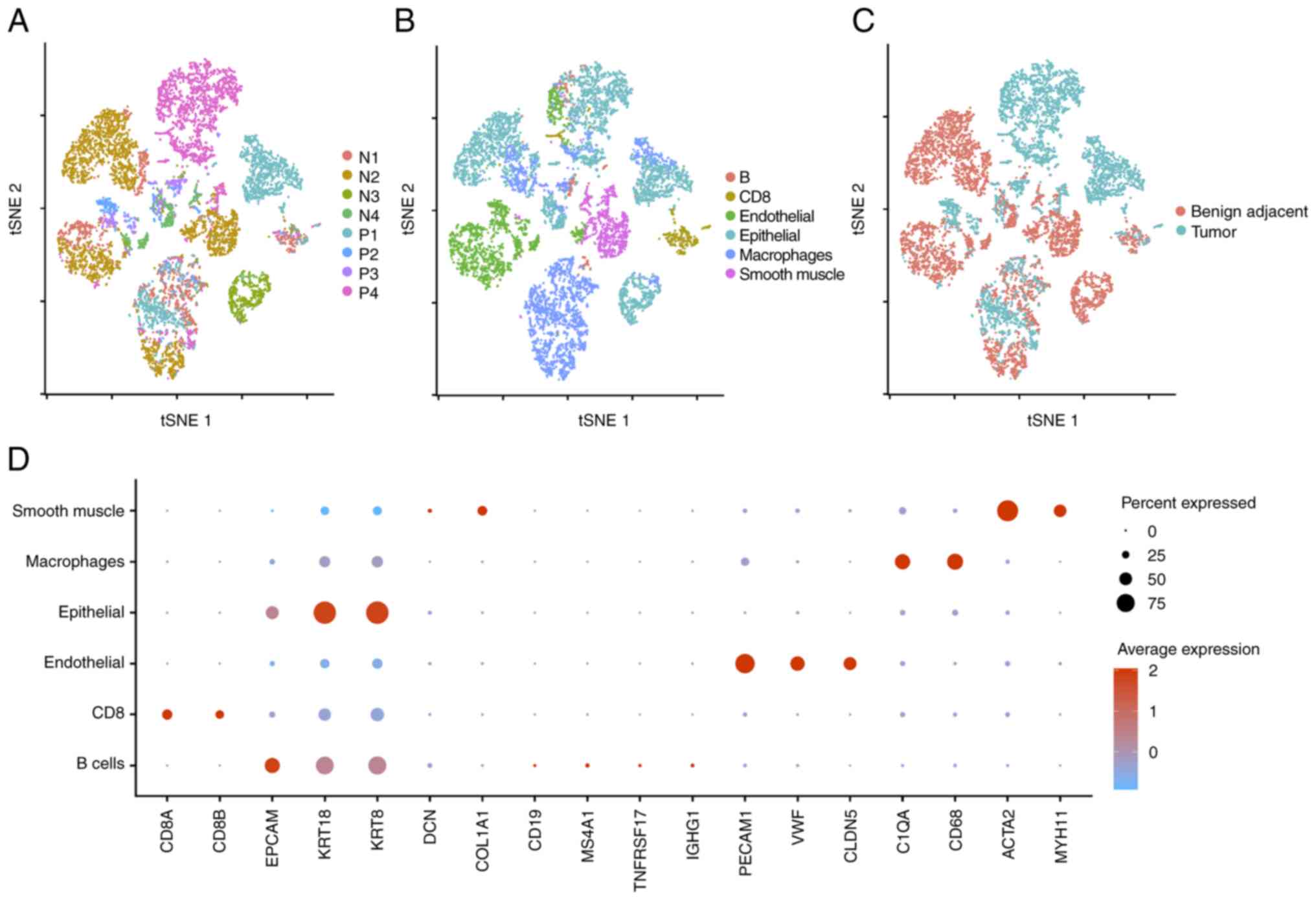
describes the quality control criteria. A total of five separate
clusters with diverse gene profiles were identified using
unsupervised clustering of these cells and their distributions were
comparable across patients (Fig.
4A). After removing batch effects and regressing out the
influence of the number of unique molecular identifiers (UMIs) and
percentage of mitochondrion-derived UMI counts, 9,333 cells with
≥200 UMIs passed the quality filtering. These cells were separated
into six major cell lineages: B cells, CD8 T cells, endothelium
cells, epithelial cells, macrophage cells and smooth muscle cells
(Fig. 4B). The annotation of
various clusters in Fig. 4C was
performed in accordance with the type of tissue sample. In
different cases, the boundaries between neighboring benight tumor
tissue cells are unclear, although tumor tissue cells are
relatively autonomous (Fig. 4C).
Fig. 4D is a dot diagram
illustrating marker genes for each cell subgroup.
The present study estimated and identified
large-scale chromosomal CNV by inferCNV for each sample based on
transcriptomes to identify malignant cells. Epithelial cells from
tumor tissues generated an inferCNV clustered heatmap that matches
to the normalized expression levels of epithelial cells in benight
neighboring tumor tissue. The resultant CNV heatmap depicted
regions of increase in red and regions of loss in blue. As there
were not enough epithelial cells in some samples, epithelial cells
from the normal tissues of patients 2 and 3 were chosen as the
control group and the tumor tissues of patients 1 and 4 were chosen
as the case group. It was discovered that the epithelial cells of
tumor tissues underwent significant CNV occurrences compared with
control cells (Fig. 5A). These
findings indicated that the epithelial cells were cancerous cells.
Compared with conventional bulk RNA-seq, scRNA-seq may assess TME
with greater precision. Therefore, KEGG enrichment analysis was
conducted on the upregulated and downregulated DEGs of malignant
and normal epithelial cells. As indicated in Fig. 5B, epithelial cells of tumor tissue
were enriched in the DNA repair, cell cycle, Chemokine-Chemokine
receptor interaction and innate immune response pathways.
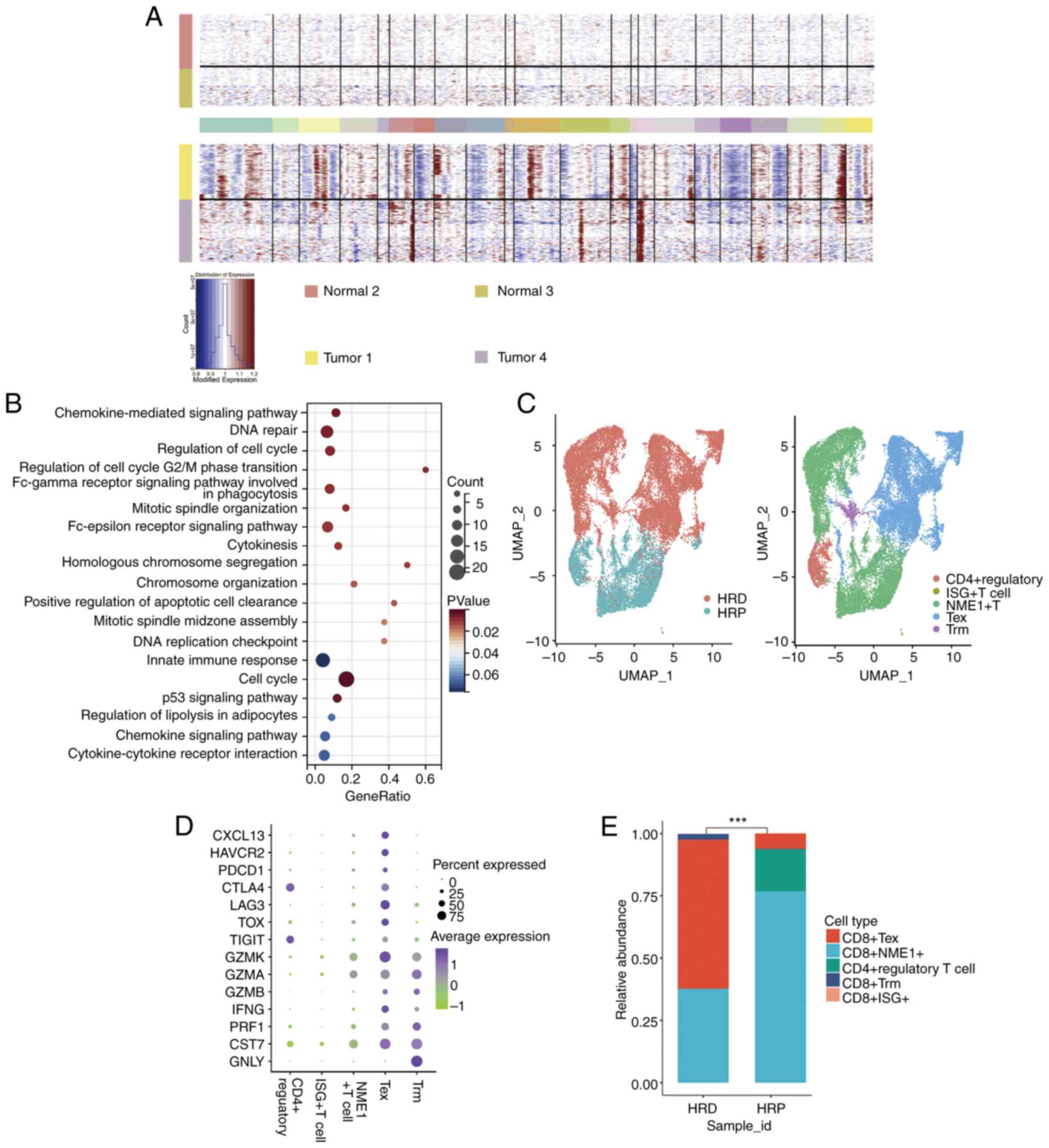 | Figure 5Single-cell sequencing analysis of
the T-cell characteristics of HRD-positive KIRC patients. (A) Top,
the hierarchical heatmap displaying large-scale CNVs in normal
tissues from the epithelial cells of two KIRC patients; bottom, the
hierarchical heatmap displaying large-scale CNVs in tumor tissues
from the epithelial cells of two KIRC patients. (B) Analysis of
differential genes in malignant epithelial cells based on KEGG and
GO functional enrichment. (C) Left, UMAP projection of cells
colored by HRD and HRP patients. Right, the tSNE plot of the five
major T cell types isolated from tumor tissues. (D) The bubble
chart displays the 14 signature gene expressions across the five
cellular clusters. The size of the dots represents the percentage
of cells that express a particular marker and the color spectrum
indicates the average expression levels of the markers (log1p
transformed). (E) Relative proportion of each cell cluster from HRD
and HRP patients as indicated. ***P<0.01. HRD,
homologous recombination deficit; KIRC, kidney renal clear cell
carcinoma; CNVs, single-cell copy-number variation; KEGG, Kyoto
Encyclopedia of Genes and Genomes; UMAP, uniform manifold
approximation and projection; HRP, homologous recombination
proficient; tSNE, t-distributed stochastic neighbor embedding. |
Analysis of the T cell characteristics
of HRD-positive KIRC patients using single-cell sequencing
Chemokine-related signaling pathways play a crucial
role in the migration of T cells (34,35).
The present study speculated that HRD-positive patients may be
characterized by the presence of loss-of-function mutations in
homologous recombination repair (HRR)-related genes, whereas
patients with HRP may lack mutations in genes involved in the HRR
signaling pathway. The present study found that in HRD-positive
cancers, the chemokine-related signaling pathways were activated
(Fig. 5B). To further characterize
T cells in HRD tumors, the present study evaluated scRNA-seq data
recovered from HRD and HRP tumor-infiltrating T lymphocyte
suspensions . scType algorithm classified T cells as
CD8+ (ISG+, NME1+, Tex and Trm) and CD4+ regulatory
(Fig. 5C). Expression of
cytotoxicity marker genes, such as GZMA/B/K and IFNG and
immunological checkpoint marker genes, such as LAG3 and PDCD1,
distinguished CD8+ Tex cells (Fig. 5D). The percentage of lymphocytes
infiltrating HRD and HRP tumors were then compared. As depicted in
Fig. 5E, when HRD tumors were
compared with HRP cancers, CD8+ Tex cells were
dramatically increased (60% vs. 6.2%, P<0.001) and CD4 +
regulatory cells were significantly decreased (0.1 percent vs.
17.2%, P<0.001).
Discussion
Malignancies frequently exhibit HRD, a common
genetic abnormality (36). The
improvement of our understanding of malignancies is facilitated by
HRD research. Despite the fact that few research have addressed the
impact and importance of HRD in KIRC, genomic instability is
associated to the prevalence of the disease (37,38).
While KIRC has additional genomic instability characteristics, it
has a lower prevalence of BRCA1/2 mutations than breast or ovarian
cancer. Additionally, KIRC had not offered a gene panel gold
standard for HRD detection. A mutational signature-based technique
for predicting HRD is the HRD score. It can more correctly predict
HRD in KIRC since it focuses on the consequences of HRD rather than
its root cause. The present study was the first to look into the
prognostic value of HRD in KIRC, to the best of the authors'
knowledge.
Patients with KIRC frequently experience recurring
tumors as distant metastases and only 10% of them respond to
chemotherapy (5). This may be due
to the absence of biomarkers that stratify chemotherapy treatment
and HRD may be an excellent biomarker for predicting the efficacy
of platinum treatment in KIRC. Obviously, additional research is
required to determine the relationship between HRD-positive KIRC
patients and the efficacy of platinum treatment.
By analyzing the expression profile of patients with
different HRD scores, the present study identified DNA damage
repair (DDR) signaling pathways and the innate immune related
pathways were enriched in patients with high HRD scores. The
widespread activation of DDR signaling pathways supports the notion
that HRD serves as a biomarker for assessing genomic instability
(39). Additionally, HRD patients
exhibit activation of innate immune-related signaling pathways. To
confirm that these activated immune-related signaling pathways
originate from HRD tumor cells, the present study conducted
scRNA-seq analyses. The results of scRNA-seq directly proved that
HRD tumor cells upregulated the Chemokine-related signaling
pathways. Chemokines are a class of proteins that attract
leukocytes to the site of infection and serve an important role in
the inflammatory response (40).
Researchers believe that chemokines also play a key role in
tumorigenesis and development. For example, some studies have found
that chemokines mediate a variety of immune cells into the tumor
microenvironment, help T cells enter tumors and affect tumor
immunity and therapeutic effects (40-42).
The activation of Chemokine-related signals causes changes in TME
(37), as evidenced by scRNA-seq
results of T lymphocytes: intratumoral T cells in the HRD patients
were distinct from T cells in the HRP patients. The present study
found that KIRC patients with HRD had the highest proportion of
CXCL13+ Tex. Previous studies have shown that
CXCL13 is a signature gene of tumor-specific T cells
(43,44) and these results suggest that KIRC
patients with HRD may have a higher proportion of tumor-specific T
lymphocytes in their tumors and that immunotherapy may benefit
these patients. Furthermore, the present study discovered that
intratumoral T cells in HRP had a larger percentage of Treg cells.
Further investigation is necessary to determine the cause of this
event.
It was expected that the results of the present
study will provide important insights and lead to a more effective
therapeutic treatment and prognosis for KIRC. As the present study
was conducted on a cohort from a public database, it did not
collect comprehensive clinical data regarding the clinical care of
patients and treatment options. More research is needed on the
prognostic mechanism of genomic instability in KIRC. The analysis
and discussion of the HRD-related genome, transcriptome and TME in
this review would provide a new method for identifying therapy- and
prognosis-related biomarkers.
Supplementary Material
Molecular pathological information of
the tumors.
HRD score of patients.
Single-cell annotation markers.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by Key Medical Discipline
of Hangzhou City (grant no. 2021-21), Key Medical Discipline of
Zhejiang Province (grant no. 2018-2-3) and Key Laboratory of
Clinical Cancer Pharmacology and Toxicology Research of Zhejiang
Province (grant no. 2020E10021).
Availability of data and materials
The single cell datasets and code generated during
the present study this investigation are accessible in the Zenodo
database (https://zenodo.org/record/7783618#.ZCTwZXZBy3A).
Authors' contributions
LH, FG, JZ and QX designed the present study. YH and
LH collected data. FG, MY and QY analyzed data. YH, QX, MY and JZ
prepared the manuscript and MY and QY checked the grammar. YH and
LH supervised the present study. YH was responsible for funding
acquisition. YH and LH confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate.
Experiments with human participants were approved by
the Research Ethics Committee of Hangzhou Hospital of Traditional
Chinese Medicine (approval number: 2019KY005) and performed
according to the Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hsieh JJ, Purdue MP, Signoretti S, Swanton
C, Albiges L, Schmidinger M, Heng DY, Larkin J and Ficarra V: Renal
cell carcinoma. Nat Rev Dis Primers. 3(17009)2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Sanchez DJ and Simon MC: Genetic and
metabolic hallmarks of clear cell renal cell carcinoma. Biochim
Biophys Acta Rev Cancer. 1870:23–31. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lobo J, Ohashi R, Amin MB, Berney DM,
Compérat EM, Cree IA, Gill AJ, Hartmann A, Menon S, Netto GJ, et
al: The WHO 2022 landscape of papillary and chromophobe renal cell
carcinoma. Histopathology. 81:426–438. 2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Choueiri TK and Motzer RJ: Systemic
therapy for metastatic renal-cell carcinoma. N Eng J Med.
376:354–366. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Makhov P, Joshi S, Ghatalia P, Kutikov A,
Uzzo RG and Kolenko VM: Resistance to systemic therapies in clear
cell renal cell carcinoma: Mechanisms and management strategies.
Mol Cancer Ther. 17:1355–1364. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Hsieh JJ, Le VH, Oyama T, Ricketts CJ, Ho
TH and Cheng EH: Chromosome 3p loss-orchestrated VHL, HIF and
epigenetic deregulation in clear cell renal cell carcinoma. J Clin
Oncol. 36(JCO2018792549)2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Liao L, Liu ZZ, Langbein L, Cai W, Cho EA,
Na J, Niu X, Jiang W, Zhong Z, Cai WL, et al: Multiple tumor
suppressors regulate a HIF-dependent negative feedback loop via
ISGF3 in human clear cell renal cancer. Elife.
7(e37925)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
De Cubas AA and Rathmell WK: Epigenetic
modifiers: Activities in renal cell carcinoma. Nat Rev Urol.
15:599–614. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ledermann JA, Drew Y and Kristeleit RS:
Homologous recombination deficiency and ovarian cancer. Eur J
Cancer. 60:49–58. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Shi Z, Zhao Q, Lv B, Qu X, Han X, Wang H,
Qiu J and Hua K: Identification of biomarkers complementary to
homologous recombination deficiency for improving the clinical
outcome of ovarian serous cystadenocarcinoma. Clin Transl Med.
11(e399)2021.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Marquard AM, Eklund AC, Joshi T,
Krzystanek M, Favero F, Wang ZC, Richardson AL, Silver DP, Szallasi
Z and Birkbak NJ: Pan-cancer analysis of genomic scar signatures
associated with homologous recombination deficiency suggests novel
indications for existing cancer drugs. Biomark Res. 3:1–10.
2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Telli ML, Timms KM, Reid J, Hennessy B,
Mills GB, Jensen KC, Szallasi Z, Barry WT, Winer EP, Tung NM, et
al: Homologous recombination deficiency (HRD) score predicts
response to platinum-containing neoadjuvant chemotherapy in
patients with triple-negative breast cancerHRD predicts response to
platinum therapy in TNBC. Clin Cancer Res. 22:3764–3773.
2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sztupinszki Z, Diossy M, Krzystanek M,
Reiniger L, Csabai I, Favero F, Birkbak NJ, Eklund AC, Syed A and
Szallasi Z: Migrating the SNP array-based homologous recombination
deficiency measures to next generation sequencing data of breast
cancer. NPJ Breast Cancer. 4(16)2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Takaya H, Nakai H, Takamatsu S, Mandai M
and Matsumura N: Homologous recombination deficiency status-based
classification of high-grade serous ovarian carcinoma. Sci Rep.
10(2757)2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Timms KM, Abkevich V, Hughes E, Neff C,
Reid J, Morris B, Kalva S, Potter J, Tran TV, Chen J, et al:
Association of BRCA1/2 defects with genomic scores predictive of
DNA damage repair deficiency among breast cancer subtypes. Breast
Cancer Res. 16(475)2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Shi Z, Shen J, Qiu J, Zhao Q, Hua K and
Wang H: CXCL10 potentiates immune checkpoint blockade therapy in
homologous recombination-deficient tumors. Theranostics.
11(7175)2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Merino DM, McShane LM, Fabrizio D, Funari
V, Chen SJ, White JR, Wenz P, Baden J, Barrett JC, Chaudhary R, et
al: Establishing guidelines to harmonize tumor mutational burden
(TMB): In silico assessment of variation in TMB quantification
across diagnostic platforms: Phase I of the friends of cancer
research TMB harmonization project. J Immunother Cancer.
8(e000147)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Gainor JF, Rizvi H, Jimenez Aguilar E,
Skoulidis F, Yeap BY, Naidoo J, Khosrowjerdi S, Mooradian M, Lydon
C, Illei P, et al: Clinical activity of programmed cell death 1
(PD-1) blockade in never, light and heavy smokers with
non-small-cell lung cancer and PD-L1 expression ≥ 50%. Ann Oncol.
31:404–411. 2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Niu B, Ye K, Zhang Q, Lu C, Xie M,
McLellan MD, Wendl MC and Ding L: MSIsensor: microsatellite
instability detection using paired tumor-normal sequence data.
Bioinformatics. 30:1015–1016. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tomczak K, Czerwińska P and Wiznerowicz M:
Review the cancer genome atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 2015:A68–A77. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Koboldt DC, Zhang Q, Larson DE, Shen D,
McLellan MD, Lin L, Miller CA, Mardis ER, Ding L and Wilson RK:
VarScan 2: Somatic mutation and copy number alteration discovery in
cancer by exome sequencing. Genome Res. 22:568–576. 2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38(e164)2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6(pl1)2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Au L, Hatipoglu E, Robert de Massy M,
Litchfield K, Beattie G, Rowan A, Schnidrig D, Thompson R, Byrne F,
Horswell S, et al: Determinants of anti-PD-1 response and
resistance in clear cell renal cell carcinoma. Cancer Cell.
39:1497–1518.e11. 2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Abkevich V, Timms KM, Hennessy BT, Potter
J, Carey MS, Meyer LA, Smith-McCune K, Broaddus R, Lu KH, Chen J,
et al: Patterns of genomic loss of heterozygosity predict
homologous recombination repair defects in epithelial ovarian
cancer. Br J Cancer. 107:1776–1782. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Popova T, Manié E, Rieunier G,
Caux-Moncoutier V, Tirapo C, Dubois T, Delattre O, Sigal-Zafrani B,
Bollet M, Longy M, et al: Ploidy and large-scale genomic
instability consistently identify basal-like breast carcinomas with
BRCA1/2 inactivation. Cancer Res. 72:5454–5462. 2012.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Birkbak NJ, Wang ZC, Kim JY, Eklund AC, Li
Q, Tian R, Bowman-Colin C, Li Y, Greene-Colozzi A, Iglehart JD, et
al: Telomeric allelic imbalance indicates defective DNA repair and
sensitivity to DNA-damaging agents. Cancer Discov. 2:366–375.
2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15(550)2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005.PubMed/NCBI View Article : Google Scholar
|
|
30
|
McGinnis CS, Murrow LM and Gartner ZJ:
DoubletFinder: Doublet detection in single-cell RNA sequencing data
using artificial nearest neighbors. Cell Syst. 8:329–337.e4.
2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Korsunsky I, Millard N, Fan J, Slowikowski
K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR and Raychaudhuri
S: Fast, sensitive and accurate integration of single-cell data
with Harmony. Nat Methods. 16:1289–1296. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ianevski A, Giri AK and Aittokallio T:
Fully-automated and ultra-fast cell-type identification using
specific marker combinations from single-cell transcriptomic data.
Nature Commun. 13(1246)2022.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Li X and Wang CY: From bulk, single-cell
to spatial RNA sequencing. Int J Oral Sci. 13(36)2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Poznansky MC, Olszak IT, Foxall R, Evans
RH, Luster AD and Scadden DT: Active movement of T cells away from
a chemokine. Nat Med. 6:543–548. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
35
|
Greener JG, Kandathil SM, Moffat L and
Jones DT: A guide to machine learning for biologists. Nat Rev Mol
Cell Biol. 23:40–55. 2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Alhmoud JF, Woolley JF, Al Moustafa AE and
Malki MI: DNA damage/repair management in cancers. Cancers (Basel).
12(1050)2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wang Y, Yan K, Wang L and Bi J: Genome
instability-related long non-coding RNA in clear renal cell
carcinoma determined using computational biology. BMC Cancer.
21:1–13. 2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Niu S, Liu K, Xu Y, et al: Genomic
landscape of Chinese clear cell renal cell carcinoma patients with
venous tumor thrombus identifies chromosome 9 and 14 deletions and
related immunosuppressive microenvironment. Front Oncol.
11(646338)2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Fakouri NB, Hou Y, Demarest TG,
Christiansen LS, Okur MN, Mohanty JG, Croteau DL and Bohr VA:
Toward understanding genomic instability, mitochondrial dysfunction
and aging. FEBS J. 286:1058–1073. 2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kohli K, Pillarisetty VG and Kim TS: Key
chemokines direct migration of immune cells in solid tumors. Cancer
Gene Ther. 29:10–21. 2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Dangaj D, Bruand M, Grimm AJ, Ronet C,
Barras D, Duttagupta PA, Lanitis E, Duraiswamy J, Tanyi JL,
Benencia F, et al: Cooperation between constitutive and inducible
chemokines enables T cell engraftment and immune attack in solid
tumors. Cancer Cell. 35:885–900.e10. 2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Mollica Poeta V, Massara M, Capucetti A
and Bonecchi R: Chemokines and chemokine receptors: New targets for
cancer immunotherapy. Front Immunol. 10(379)2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Lowery FJ, Krishna S, Yossef R, Parikh NB,
Chatani PD, Zacharakis N, Parkhurst MR, Levin N, Sindiri S, Sachs
A, et al: Molecular signatures of antitumor neoantigen-reactive T
cells from metastatic human cancers. Science. 375:877–884.
2022.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zheng C, Fass JN, Shih YP, Gunderson AJ,
Sanjuan Silva N, Huang H, Bernard BM, Rajamanickam V, Slagel J,
Bifulco CB, et al: Transcriptomic profiles of neoantigen-reactive T
cells in human gastrointestinal cancers. Cancer Cell.
40:410–423.e17. 2022.PubMed/NCBI View Article : Google Scholar
|