Alzheimer's disease (AD), the most common form of
dementia, is a central neurodegenerative disease characterized by
progressive cognitive impairment and memory loss. The main
pathological features include intracellular neurofibrillary tangles
(1) and extracellular β-amyloid
(Aβ) peptide aggregation (2) to
form senile plaques. These pathological changes lead to clinical
manifestations characterized by memory loss, impairment of abstract
thinking, numeracy deficits, and changes in personality and
behavior (3). According to
previous studies, the pathogenesis of AD mainly includes the
following: Abnormal accumulation of Aβ (4), τ-protein hyperphosphorylation
(5), neuroinflammation (6), cardiocerebral cascade (7), cholinergic deficiency (8), excitatory amino acid toxicity
(9). At present, there is no
treatment to cure or delay the process of the disease. However,
with the increasing number of patients with AD, AD has become one
of the main medical and health societal issues. To address the lack
of effective drugs and the failure of continuous clinical trials,
the current research focus is on therapeutic strategies for
inducing the regeneration of neural stem cells (NSCs). In recent
years, research on promoting targeted neural regeneration has
received increasing attention. Therefore, the current review is
focused on the mechanisms of induced nerve regeneration in the
treatment of AD.
Neural regeneration is the process of generating new
neurons or restoring neuronal structure, which mainly refers to the
proliferation and differentiation of neural progenitor cells, and
the synaptic plasticity of neurons against memory disorders and the
ageing-related decline of learning and memory (10). In 1965, Altman and Das (11) first demonstrated that new neurons
could be generated in the adult mammalian brain. Since then,
continuous studies have found that nerve regeneration in the brains
of adult mammals mainly occurs in two regions, namely, the
subgranular zone in the dentate gyrus of the hippocampus and the
subventricular zone (12,13). Nerve regeneration may also occur in
other brain areas, such as the neocortex (14) and striatum (15). However, neurogenesis in these areas
seems to occur at lower levels. To explore whether neurogenesis in
humans lasts a lifetime, Tobin et al (16) examined the brains of 18 cadaver
donors with a mean age of 90.6 years in 2019. Among them, not only
proliferating neural precursor cells
(Nestin+/PCNA+), undifferentiated neural stem
cells (Nestin+Sox2+) and proliferating neural
stem cells (Nestin+/Sox2+/Ki67+),
but also newborn immature neurons [Doublecortin
(DCX)+/PCNA+], were found in the brains of
patients with AD, which indicated that hippocampal neurogenesis
persisted in the aged and diseased human brain (16). In addition, the study found a clear
association between the number of newborn immature neurons and the
cognitive scores of these donors. The more
DCX+/PCNA+ cells that are produced, the more
cognitive performance is improved, suggesting that the number of
neurons is positively associated with cognitive function (16). The level of cognitive function also
depends to some extent on the integrity and transmission efficiency
of synapses. Synapses are the sites where functional connections
form between neurons, and they are also the key places for
information transmission. Synapses are structurally unstable and
dynamic, which can mean that they are susceptible to nutrient
levels such as those affected by brain-derived neurotrophic factor
(BDNF) and nerve growth factor (NGF), diseases such as AD and other
factors such as inflammation and oxidative stress. An increasing
number of studies have shown that the loss of synapses in the
hippocampus and neocortex is an early symptom of AD and one of the
main causes of cognitive dysfunction (10,17).
Synaptic damage is one of the most important causes of cognitive
decline in AD. Therefore, inducing nerve regeneration might
fundamentally treat cognitive dysfunction in AD. In recent years,
research on nerve regeneration has been extensive, but generally,
it has aimed to solve two main issues: How to replenish missing or
damaged neurons, and how to re-establish neural connections. In
various transgenic (Tg) rodent models, nerve regeneration ability
could be improved by external interventions such as treatment with
different types of drugs such as escitalopram, trodusquemine,
agomelatine, silibinin, esculentoside A and butyrate (18-23),
the transplantation of human NSCs (hNSCs) (24,25)
and electrical stimulation (26),
which have been proven to increase the number of neuronal precursor
cells, and ameliorate the degree of τ-phosphorylation and
AD-related memory loss. The effects of different external
interventions on inducing nerve regeneration in various Tg rodent
models in recent years are summarized in Table I. New mature neurons induced by
external interventions from the migration of existing NSCs in
certain regions or the generation of NSCs can replenish the missing
or damaged cells, which contributes to restoring part or even all
of the neural network (18-26).
In conclusion, cognitive function is closely related
to the number of neurons and synaptic integrity. Therefore, the
recovery of damaged neurons and increase in the number of neurons
induced by nerve regeneration is expected to improve cognitive
levels, and allow more efficient treatment of AD.
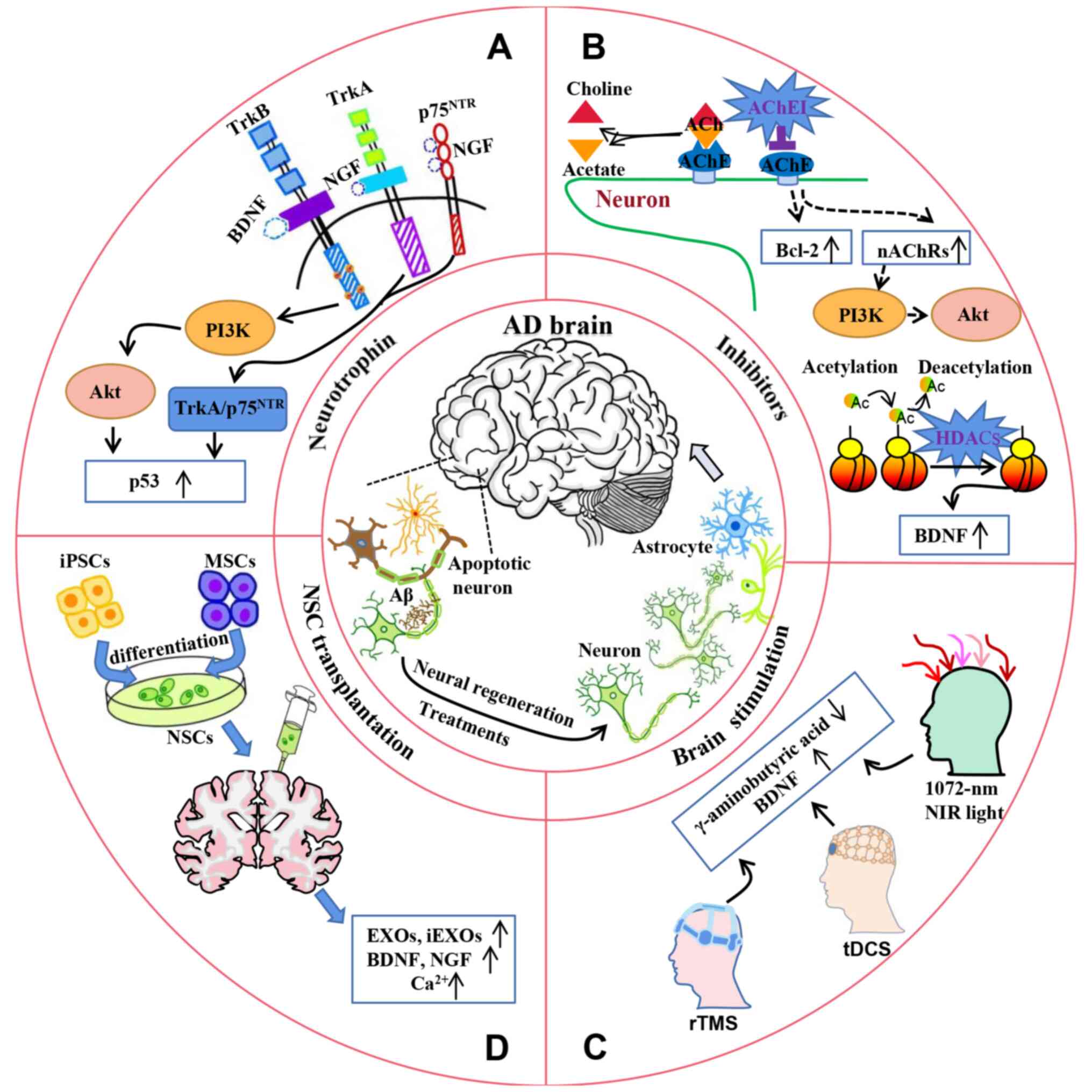
Among the neurotrophin family factors, BDNF is the
most widely distributed in the central nervous system (CNS). BDNF
mainly exists in the hippocampus, cortex and basal forebrain, and
plays a key role in synaptic plasticity, neuronal survival,
neuronal differentiation and neuronal development (27). Reduced levels of BDNF are
associated with different categories of neurological disorders,
including neurodegenerative disease, such as AD (28) and Parkinson's disease (29), neurodevelopmental disorders, such
as developmental delays (30), and
neurobehavioural and neuropsychiatric disorders (31), such as depression and anxiety.
Studies have shown that decreased BDNF levels lead to a lack of
neuronal nutritional support, resulting in the degeneration of
specific neuronal populations in the basal forebrain cholinergic
system in AD (32,33). With continuous studies on the role
of BDNF, it has been found that BDNF has a physiological effect on
the inhibition of neuronal apoptosis and the treatment of cognitive
impairment, suggesting that increasing BDNF levels might be a
promising target for the treatment of cognitive decline in patients
with AD. When BDNF nutrient solution was injected into the medial
entorhinal cortex (120 µg/side, once a day) of aged Fischer rats
with cognitive degeneration, their spatial learning and memory
abilities were markedly improved (34). In addition, invasive gene therapy
to overexpress BDNF has been successfully demonstrated to enhance
synaptic protein expression and neuronal proliferation, as well as
to attenuate amyloid load in APP/PS1 transgenic mice (35). Similar conclusions were obtained
when the studies were extended to non-human primates. Neuronal
apoptosis in the endodermis of monkeys was induced with bilateral
radiofrequency lesions in the perforant path. The monkeys were then
stereotaxically injected with a lentiviral vector expressing BDNF
into the entorhinal cortex. Stereological quantification revealed
that the lesions of the perforant path without BDNF treatment
resulted in the loss of 45.9±8.5% of neurons in layer II of the
entorhinal cortex in the lateral region compared with that in
intact monkeys. However, the monkeys in which lesions of the
perforant path were treated with BDNF were able to maintain
85.4±7.1% of the neurons on the lesion side, and their brains
continued to function normally, suggesting that BDNF can also
notably prevent neuronal death in non-human primates (34). There was little correlation between
BDNF levels and cognitive decline in patients with a clinical
diagnosis of mild cognitive impairment. However, a high level of
BDNF expression could effectively slow the rate of cognitive
decline in patients with AD (36).
BDNF affects neuronal plasticity by inducing neurotransmitter
release (37), increasing vesicle
docking and enhancing the glutamate-induced postsynaptic response
(38). BDNF binds to the
high-affinity tropomyosin-related kinase B (TrkB) receptor, and
then induces Trk dimerization and autophosphorylation (39), thus activating the downstream
cascades of the phosphatidylinositol 3-kinase/protein kinase B
(PI3K/Akt) intracellular signal transduction pathways (40). Akt is phosphorylated through its
interaction with PI3K and attaches to the inner surface of the
plasma membrane (41).
Phosphorylated Akt terminates cell apoptosis and induces the
survival of nerve cells by inhibiting the activity of the tumour
suppressor gene p53 or directly blocking the activity of the
apoptosis pathway (41). Under
normal conditions, cytochrome c (cyt-c) exists in the
lacunae between the inner and outer membrane of mitochondria, and
the stimulation of apoptotic signals causes the release of cyt-c
from the mitochondrial lacunae into the cytoplasm (42). Before the release of cyt-c,
phosphorylated (p)-Akt regulates the activity of Bcl-2 family
members and controls the release of cyt-c from the mitochondria
into the cytoplasm. After the release of cyt-c, p-Akt can also
modulate the components of the apoptosome and inhibit the formation
of the apoptosome, thereby blocking the apoptosis pathway mediated
by the mitochondria (43).
Together, BDNF mediates the activity of the PI3K/Akt pathway by
binding to the membrane receptor TrkB, thereby regulating cell
survival and synaptic function, and making the BDNF/TrkB/PI3K/Akt
signalling pathway a potential therapeutic target for
neurodegeneration (44).
Furthermore, the BDNF protein is a charged, yet net hydrophobic
molecule with a low molecular weight, causing it to have a short
half-life (t1/2) in humans, which makes it a non-ideal biological
drug candidate (45). Previous
studies have shown that zinc finger E-box-binding homeobox 85, a
potent and full agonist of human TrkB, plays a role in
neuroprotection and neurorestoration (45,46).
7,8-Dihydroxyflavone (7,8-DHF), a potent TrkB agonist, has also
been shown to have therapeutic effects on AD (47). In addition, it has been
demonstrated that LMDS-1, another agonist of TrkB, can improve the
pathological phenotype of early AD mice by upregulating the
expression of BDNF. Moreover, LMDS-1 is more effective than 7,8-DHF
in alleviating the behaviour and pathological characteristics of
mice with AD (48).
In conclusion, previous studies have found that the
expression level of BDNF is positively correlated with the
cognitive level in rats, non-human primates and humans. BDNF mainly
regulates the survival of nerve cells and synaptic plasticity by
triggering the BDNF/TrkB/PI3K/Akt signalling pathway, and
ultimately improves cognitive ability. In addition, other TrkB
agonists play a functional role in neuroprotection and cognitive
function, similar to BDNF.
NGF is one of the earliest discovered and most
well-studied nerve cell growth regulators among neurotrophic
factors; it has dual biological functions of nourishing and
promoting neurite growth of neurons (49). NGF also plays an important role in
the development, differentiation, growth, regeneration and function
of central and peripheral neurons (50). Recent studies have shown that mice
lacking NGF exhibit deposits of Aβ plaques, τ-hyperphosphorylation,
synaptic dysfunction and other typical pathological features of AD
(51,52).
Treatment targeting NGF not only reduces
pathological changes in AD but also contributes to the survival and
axonal regeneration of injured neurons in AD (53). The knockout of the NGF gene
resulted in a 55% reduction in basal forebrain cholinergic neurons
and a 62% reduction in hippocampal cholinergic neurons in the CNS
of adult mice, suggesting that NGF deficiency may be an important
factor in neuron loss (54).
Increasing NGF may be an effective strategy to reduce Aβ deposition
and improve clinical cognitive impairment in AD. In the brain, NGF
is tonically secreted in its precursor form, proNGF, which is then
cleaved by the extracellular protease into mature NGF (mNGF)
(55). Interfering with NGF
transport or reducing NGF processing may lead to the overexpression
and abnormal accumulation of proNGF and a deficiency of mNGF. mNGF
is required for the growth and plasticity of cholinergic neurons
(56). NGF bioactivity is mediated
by binding to the high-affinity TrkA receptor and the low-affinity
tumour necrosis factor neurotrophin receptor (p75NTR) on the plasma
membrane (57). NGF produced by
hippocampal and cortical neurons binds to TrkA and p75NTR, and then
forms dimeric complexes that maintain the normal activity of
neurons, suggesting that NGF maintains the normal function of
neurons through the TrkA/p75NTR-mediated signalling pathway
(58). In the absence of TrkA, a
combination of NGF and p75NTR accelerates apoptosis by activating
p53, ceramide and c-Jun N-terminal kinase pathways (59). It was found that the proNGF level
in the parietal cortex of patients with AD was twice as high as
that of healthy subjects, suggesting that the degeneration of
cholinergic neurons in patients with AD may be associated with a
deficiency of mNGF (60). After
treatment with NGF, the activity of choline acetyltransferase
(ChAT) in the cerebrospinal fluid of patients with AD is markedly
enhanced, while the activity of ChAT in the cerebrospinal fluid is
associated with cognitive function, suggesting that NGF may improve
cognitive function by mediating ChAT activity (60). At present, there are animal
experiments and clinical trials of NGF with different infusion
modes, including direct intracerebral infusion of NGF (61), peripheral administration of NGF
using nasal (62) or intraocular
(63) delivery and NGF delivery
using viral vectors (53,64). Although these methods have been
proven to be effective, there are drawbacks such as pain, weight
loss and difficulty controlling the dose. It has been found that
the sustained release of NGF can be induced by redirected
implantation of NGF into the basal forebrain through encapsulated
cell biodelivery of NGF (NGF-ECB), in which the encapsulated cells
are made from fibroblasts (65).
In addition, elevated NGF can regulate the exocytosis of
presynaptic terminal vesicles and induce BFCN electrochemical
signalling, suggesting that high levels of NGF may alter AD-related
synaptic failure and neurotransmission defects (66). NGF-ECB implantation in patients
with mild to moderate AD can obviously repair the degeneration of
BFCN, and reduce the rate of brain atrophy and cognitive decline;
importantly, the safety of the treatment has been demonstrated
(67). Furthermore, the long-term
release of NGF by implanted transmitters would need to be optimized
to achieve a more predictable and stable effect in the treatment of
AD. In addition, agonists of TrkA neurotrophin receptors such as
doxycycline (68) or NGF-mimicking
drugs such as LM11A-31(69) were
reported to have NGF-like effects.
In conclusion, NGF is closely related to the
proliferation, differentiation, regeneration and
electrophysiological function of central and peripheral neurons.
Interference with NGF transport or processing may impair the normal
function of neurons. In the AD brain, targeted drug therapy or gene
therapy can induce the release of mNGF or increase the expression
level of NGF, and then NGF activates the NGF-TrkA signalling
pathway in neurons, which can ameliorate the issues of synaptic
failure and neurotransmission defects, and ultimately improve the
cognitive level of patients with AD.
In the pathological process of AD, the activity of
AChE in the brain is notably increased, which leads to the
hydrolysis of ACh and the loss of cholinergic neurons. Moreover,
the high levels of glutamate in the synaptic cleft of neurons
activate the N-methyl-D-aspartic acid receptor, which opens
Ca2+ channels and increases Ca2+ influx,
leading to neuronal necrosis (77). Most AChEIs inhibit AChE activity
and reduce the decomposition of ACh, which effectively protects
neuronal survival and alleviates AD symptoms (78,79).
Donepezil can also protect neurons from glutamate-induced
neurotoxicity by directly or indirectly activating nicotinic ACh
receptors (nAChRs). The combination of nAChR antagonists and
donepezil markedly reduced the neuroprotective effects of
donepezil, suggesting that donepezil exerts neuroprotective effects
in foetal Sprague-Dawley rats by binding to nAChRs (80). Donepezil can also prevent
Aβ-induced neural cell death by activating the PI3K pathway
(80). The activation of nAChRs
can trigger downstream PI3K and thereby catalyse the production of
p-Akt, which enhances the activation of the PI3K pathway (81). In addition, it was found that the
phosphorylation levels of Akt were notably increased in astrocytes
after treatment with donepezil (10 mM) for 6 h (82). The therapeutic effect of donepezil
was alleviated when donepezil was used in combination with a PI3K
inhibitor (30 mM) or an Akt inhibitor (1 mM), suggesting that
donepezil plays a neuroprotective role in the brain through the
PI3K/Akt signalling pathway (82).
In summary, donepezil plays a neuroprotective role by either
inhibiting AChE or activating nAChRs and their downstream PI3K/Akt
pathway. It has also been shown that the neuroprotective effect of
AChEI drugs is achieved by engaging in the upregulation of the
antiapoptotic Bcl-2 protein. When AChEIs are combined with HA14-1,
a drug that inhibits Bcl-2, the neuroprotective effect of AChEIs is
weakened, suggesting that AChEIs exert neuroprotective effects by
upregulating the target gene Bcl-2(83). The different concentrations of
AChEIs have various degrees of effectiveness in preventing
apoptosis. Galantamine, donepezil and rivastigmine show the highest
neuroprotective effects at 0.3, 1 and 3 M, respectively. With the
increase in concentration, galantamine and donepezil exhibit a
well-characterized U-shaped neuroprotective curve with the
percentage of cell death in longitudinal coordinates. High
concentrations of galantamine block nAChRs, and lead to the absence
of neuroprotective effects on cell death caused by Aβ in the human
neuroblastoma SH-SY5Y cell line (83).
In conclusion, AChEI drugs have been widely used in
the clinical treatment of AD. One main capability of AChEI drugs is
to reduce the decomposition of ACh by inhibiting AChE, and the
other is to activate nAChRs and improve glutamate transport,
thereby activating the PI3K/Akt pathway and upregulating the
downstream target protein Bcl-2. Therefore, AChEI drugs induce the
survival of nerve cells, enhance the connection between synapses
and eventually attenuate AD symptoms.
Histone acetylation is an epigenetic modification
that alters gene expression without changing the DNA sequence.
HDACs catalyze not only histone deacetylation, but also a variety
of non-histone protein deacetylations, maintaining the dynamic
equilibrium between acetylation and deacetylation, and regulating
gene expression (84). The HDAC
family, composed of 18 isoforms, is divided into five groups on the
basis of phylogenetic characteristics: Class I (HDAC1, HDAC2, HDAC3
and HDAC8), class IIa (HDAC4, HDAC5, HDAC7 and HDAC9), class IIb
(HDAC6 and HDAC10), class III (silent information regulator 2) and
class IV (HDAC11). The function of HDACs is to deacetylate the
lysines on histone tails, causing the condensation and subsequent
repression of chromatin (85).
HDACs are involved in chromatin remodelling, gene
expression, and synaptic formation and plasticity, and have been
used as targets for improving synaptic function (86). The degree of histone deacetylation
is closely related to the pathological phenotype of AD. High levels
of HDAC (HDAC6) expression can be seen in the hippocampus and the
cortex of patients with AD (87).
HDAC1 plays a role in neurodegeneration, HDAC2 participates in
modulating synaptic plasticity and long-lasting changes in neural
circuits, and HDAC3-11 and class III HDACs are beneficial for their
neuroprotective effects (88). The
loss of HDAC1 in neurons causes DNA damage and cell death, while
the lack of HDAC2 in mice improves learning and memory, enhances
synaptic plasticity, and increases dendritic spine density and
synapse number (89). RGFP-966, a
selective HDAC3 inhibitor, can increase H3 and H4 acetylation and
BDNF acetylation, resulting in increased BDNF expression, reduced τ
and Aβ1-42 accumulation, and improved spatial learning and memory
in 3xTg-AD mice (90). In a
previous study, a notable increase in HDAC4 expression was
responsible for neurotoxic Aβ deposition in neuronal cells in a
dose-dependent manner (91).
Tasquinimod, a selective HDAC4 inhibitor, can partly rescue the
expression of genes related to synaptic plasticity and neuronal
memory, and shows the effect of an HDAC4-targeting treatment
(92). Owing to a notable increase
in HDAC6 expression in the hippocampus and cortex of patients with
AD and AD animal models, HDAC6 inhibitors are being investigated
for the treatment of AD (93).
Moreover, a phosphodiesterase type 5 (PDE5) inhibitor was shown to
markedly activate cAMP/cGMP responsive element binding, which
improved the long-term potentiation mechanism following hippocampal
damage in amyloid β precursor (APP)/presenilin-1 (PS1) mice. The
synergistic effect of PDE5 inhibitors and HDAC enzyme (class I
HDACs and HDAC6) inhibitors can be more effective in the treatment
of AD, which has proven to be a potential new therapeutic approach
(93). CM-414, a dual inhibitor of
PDE5 and HDACs, not only reduced the levels of Aβ and p-τ in the
brain, but also increased the density of dendritic spines in
hippocampal neurons and alleviated cognitive deficits in Tg2576
mice (93). Furthermore, 44B,
another dual compound inhibitor of PDE5 and HDACs, markedly reduced
the level of hAPP by 55% and the level of p-τ proteins by 30% in
vitro, suggesting that 44B directly inhibited hAPP and p-τ
production (94). In addition, an
in vivo study showed that 2 weeks of treatment with the dual
compound inhibitor 44B (40 mg/kg) could recover partial memory
impairment in elderly (16-month-old) Tg2576 mice (94).
In conclusion, HDACs are involved in synaptogenesis
and cognitive function by increasing deacetylation modifications of
histones or genes associated with memory such as BDNF. The
synergistic effect of PDE5 and HDAC enzyme inhibitors can not only
induce the acetylation of histones and synapse-related genes, but
also inhibit the levels of Aβ and p-τ, which has a better
therapeutic effect on AD.
In recent years, physical intervention in the
treatment of AD has gradually received widespread attention. Using
physical intervention to induce neurogenesis is expected to be an
effective therapeutic treatment for AD. Currently, there are two
major and reliable non-traumatic brain stimulations used in the
clinic, namely, repetitive transcranial magnetic stimulation (rTMS)
and transcranial direct current stimulation (tDCS).
TMS refers to the process of stimulating cortical
neurons with electromagnetic fields and then activating neuronal
circuits in the CNS (95), which
can recover the function of synaptic transmission in the cerebral
cortex, and is considered a non-invasive and painless treatment
technology (96). rTMS is the
continuous administration of repetitive electromagnetic stimulation
to reduce the imbalance between excitatory and inhibitory signals,
thereby mediating synaptic activity and neuroplasticity (97). Previous studies showed that rTMS
markedly promoted the cognitive level and language ability of
patients with AD, especially patients with mild AD, indicating that
treatment with rTMS could effectively improve the clinical
manifestations of AD (98,99). Research suggests that rTMS has
positive therapeutic effects on the nerves and psychology of
patients, as assessed by a neuropsychological quantitative scale
such as the World Health Organization-University of California
Auditory Word Learning Test (WHO-UCLAAVLT) (98), AD Assessment Scale-Cognitive
Section (ADAS-Cog) (99),
Mini-Mental State Examination (MMSE) (100) and Montreal Cognitive Assessment
Scale (MoCA) (99). These results
suggest that rTMS improved the cognitive ability and mental
behaviour of patients with mild to moderate AD. However, variations
in the intensity and location of rTMS lead to different therapeutic
effects. A randomized controlled trial showed that high-frequency
rTMS (20 Hz) markedly improved cognitive impairment, while
low-frequency rTMS (1 Hz) had no obvious effect in patients with
mild to moderate AD (101).
Moreover, studyhas proven that the cognitive function of patients
selectively deteriorates after 10 min of low-frequency rTMS (1 Hz)
(102). These results suggested
that only high-frequency rTMS has a positive effect on ameliorating
cognitive impairment. Research on different stimulus locations
found that there was a notable improvement in the cognition and
memory of patients with mild or moderate AD who received rTMS in
the right or bilateral dorsolateral prefrontal cortex (DLPFC), but
not in those who received rTMS in the left DLPFC (103).
tDCS is the direct use of a weak electrical current
(usually 1-2 mA) to stimulate specific areas of the brain, leading
to a change in resting membrane potential in neurons, and thus
inducing the excitability of brain cells. The excitability of
neurons was increased by the depolarization of anodic current
stimulation and decreased by the hyperpolarization of cathodic
current stimulation (104). The
level of γ-aminobutyric acid in the motor cortex decreased after
anodic current stimulation, whereas there was no change in the
γ-aminobutyric acid level in the motor cortex with cathodic current
or sham stimulation, suggesting that anodic current stimulation
might elicit neuronal excitation by inhibiting the level of
γ-aminobutyric acid (105). The
experimental results showed that the expression of BDNF mRNA
induced by tDCS was 9.5 times higher than that of the control
group, suggesting that tDCS might be conducive to brain health by
enhancing the level of BDNF (106). Clinical experimental results
showed that visual recognition memory scores had increased by 8.9%
when the temporal cortex of patients with AD was stimulated by tDCS
for 30 min for 5 days, but these beneficial effects lasted only 1
month after the end of stimulation regimens (107). Although increasing the strength
of an electric current might lead to excitatory changes in a larger
area of the cortex, the highest strength clinically used is 2 mA
for safety purposes. The findings of another study have suggested
that increasing the strength of tDCS did not necessarily improve
the curative effect, as tDCS enhanced electrical signals only in
the existing neural network and did not stimulate action potentials
from the resting state of neurons (108). The therapeutic effect of tDCS
depends on the current physiological state of the brain, and even
increasing the intensity of electrical current cannot form a new
neural network in the brain (109). Therefore, these results suggest
that the therapeutic effect of tDCS might be limited in patients
with advanced AD, who have a reduction in both synaptic plasticity
and long-term potentiation.
Although both rTMS and tDCS can improve the
cognitive function of patients with AD to a certain extent, they
also have limitations. First, the electric current only reaches the
cerebral cortex, and it is difficult for the current to reach the
medial prefrontal cortex, insula, cingulate gyrus and other deep
brain regions. Second, repeated use of rTMS may also induce
epilepsy; therefore, rTMS is potentially harmful to the human body
(110). rTMS stimulates a wide
range of brain areas, and therefore lacks specificity and accuracy.
Previously, light therapy has been gradually favoured by clinicians
due to its fewer side effects and deep treatment area. Studies have
shown that 1,072-nm near-infrared (NIR) light can protect immune
cells by limiting the toxic effects of ultraviolet-A on them, and
prevents cell apoptosis (111).
After 1,072-nm NIR light treatment, the expression of heat shock
proteins was markedly increased, and the levels of Aβ and p-τ
protein were notably decreased (112). Furthermore, other wavelengths of
light in the range from red to infrared light have also been shown
to improve AD symptoms (113,114). Iaccarino et al (110) found that the treatment of AD mice
with a 40-Hz light-emitting diode could enhance γ brainwaves and
reduce Aβ deposition in the brain; however, the benefit lasted for
only 1 week once therapy was discontinued. In addition,
intermittent flickering light therapy shows much promise for mild
cognitive impairment and patients with mild AD who suffer from both
sleep disturbances and cognitive deficits, including memory loss,
mental behavior abnormalities and a reduced ability to perform
daily tasks (115). The
aforementioned study showed that a 40-Hz flicker can not only
increase γ brainwaves in the visual cortex, decrease Aβ deposition,
and reduce both APP and p-tau protein expression in the
hippocampus, but that it can also have a positive impact on the
circadian rhythms of patients with AD.
In summary, as physical therapies for AD, rTMS and
tDCS can improve the cognitive function of patients with AD by
activating the synaptic activity of neuronal circuits in the CNS.
However, the treatment effects of those electrical stimulations are
transient, and the stimulations have numerous side effects. Light
therapy has gradually attracted the interest of researchers.
Research suggests that infrared light and wavelengths in the range
from red to infrared light are effective in the treatment of AD
with the advantage of having few side effects.
Nerve damage and insufficient endogenous nerve
regeneration in the brains of patients with AD lead to neuronal
depletion and the disruption of neural circuits, ultimately causing
cognitive decline (116).
Although the induction of nerve regeneration has been proven to
fundamentally solve the issue of neuronal loss in AD, the number of
endogenous NSCs in the adult brain is limited; therefore, it is
anticipated that NSC transplantation will induce nerve regeneration
(117). Stem cell therapy emerged
in the early 2000s and has been explored as a potential treatment
for various diseases, especially neurological diseases, including
AD, Parkinson's disease, Huntington's disease, multiple sclerosis
and stroke (118). In recent
years, successes in the culture and transplantation of NSCs have
provided a new vision for brain injury repair and AD treatment
(119). NSCs have the
characteristics of self-renewal and multidirectional
differentiation potential (120).
Therefore, after NSCs are implanted into the brain, surviving NSCs
migrate and differentiate into neurons and glial cells. Newborn
neurons integrate into functional neural circuits, which can
alleviate learning and memory disorders (121). Exogenous NSCs are mainly derived
from three sources: Direct extraction from embryonic or foetal
nerve tissue (122),
transdifferentiation of induced multipotent stem cells (iPSCs)
(123) and the differentiation of
mesenchymal stem cells (MSCs) (124). Some issues are encountered after
the transplantation of NSCs extracted from embryonic or foetal
nerve tissue, such as immune rejection and teratomas, and these
problems, along with various ethical issues, limit the application
of these NSCs in vivo. iPSCs are induced by the transduction
of four reprogramming factors, Qct3/4, Sox2, Klf4 and c-Myc, in the
dermal fibroblasts of the patient (125). NSCs originating from iPSCs lack
ethical and histocompatibility problems, but there are still issues
such as a low induction rate and the risk of triggering tumours
(126). MSCs are derived from
bone marrow, umbilical cord, foetal blood and adipose tissue. MSCs
have the ability to differentiate into different tissue types and
cells, such as nerve cells, osteocytes and cardiomyocytes. The
advantages of MSCs are their wide range of sources, easy isolation
and culture, and lack of ethical issues and immune rejection, but
they also have issues such as difficulty differentiating into
functional neurons in vivo (127).
A large number of studies have confirmed that the
transplantation of exogenous NSCs can induce nerve regeneration
(128-130),
but the mechanism is not fully understood. Studies have found that
exogenous NSCs transplanted into the brain not only have the role
of replacing lost and injured neurons, but also have the auxiliary
role of secreting neurotrophic factors to improve nerve
regeneration and immunity (131,132). Secreted neurotrophins play
important roles in the proliferation, survival, migration and
differentiation of NSCs, thereby participating in neuroprotective
and repair functions (132).
However, there are still existing issues with the low survival rate
and neuronal differentiation rate of transplanted NSCs due to the
harsh environment in the brain (133). Both BDNF and NGF are conducive to
neuroprotection and the differentiation of endogenous or exogenous
NSCs into neurons. Therefore, a combined method of BDNF and NSCs or
NGF and NSCs for the treatment of AD has been explored.
Intracerebral injection is not ideal for this combined method due
to the short half-life of BDNF and NGF. Therefore, a nano-delivery
system of BDNF or NGF encapsulated by liposomes, polymer micelles
or nanoparticles could be used to establish a long-term,
continuous, slow-release and controlled drug delivery mode.
Although some results have been obtained with this system, it is
still in the animal phase of research testing, and a clinical
application is thus lacking (134,135). There is a double therapeutic
effect for gene therapy using NSCs as a carrier. Exogenous genes
for BDNF or NGF are transfected into NSCs, and BDNF and NGF are
thus continuously overexpressed with the proliferation of NSCs. On
the one hand, BDNF and NGF are widely expressed during cell
migration. On the other hand, the expression of BDNF and NGF
contributes to promoting the differentiation of NSCs into neurons
(136,137). Exosomes (EXOs) are one of the
smallest extracellular vesicles released by cells. Transplanted
exogenous MSCs can secrete EXOs to regulate the pathological
microenvironment and neural plasticity, indicating that EXOs are
involved in nerve repair and regeneration (138). The aforementioned study found
that NSCs derived from human iPSCs secreted EXOs, which could
reduce the inflammatory response, ameliorate oxidative stress and
facilitate NSC differentiation, suggesting that NSC-derived EXOs
could be used as a supportive adjuvant for NSC transplantation
(139). Another study found that
induced neural progenitor cells (iNPCs) from mouse fibroblasts and
astrocytes abundantly released EXOs, which could promote the
proliferation of neural progenitors and release of growth factors
via activating the downstream extracellular signal-regulated kinase
pathways, indicating that iNPC-derived EXOs played a critical role
in functional rehabilitation of brain lesions (140). Moreover, the combination of
exogenous NSCs with induced EXOs could alleviate the injury of
brain tissue, and promote the recovery of motor function,
suggesting that NSCs together with EXOs have potent therapeutic
effects in neurological disorders (139). Furthermore, the involvement of
microRNAs (miRNAs/miR) in biological activities, including cell
growth and metabolism, has been well documented. EXOs have been
used as novel biological vehicles to transfer different miRNAs, and
the delivery of EXO-mediated miRNAs had an effect on treating
neurological diseases (141).
miR-21a is enriched in EXOs at high levels, and has key roles in
the generation of neurons and mediating the neurogenic potential of
EXOs (138). A One study
demonstrated that EXOs with miR-21a overexpression had a greater
capacity for the promotion of neuronal differentiation and the
inhibition of gliogenesis, indicating that EXOs may achieve a
therapeutic effect in neurogenesis promotion via the transference
of miR-21a (138). miR-455-3p was
found to have protective effects on the regulation of APP, levels
of amyloid-β, mitochondrial biogenesis and dynamics, synaptic
activity and the viability or apoptosis of the cell (142). EXOs released from MSCs alleviated
hippocampal neuronal injury through transferring miR-455-3p
(143). Injecting miR-133b EXOs
preserved neurons and promoted the regeneration of axons (144), and EXOs harvested from
miR-133b-overexpressing MSCs were shown to improve neural
plasticity and functional recovery (145), suggesting that the transfer of
EXO-mediated miR-133b represents a novel therapeutic approach for
the treatment of neurological diseases. An animal study indicated
that transplantation of NSCs could protect basal forebrain
cholinergic neurons and restore synaptic impairment, eventually
leading to improvements in learning and memory functions in APP/PS1
Tg mice (146). Stem cell therapy
for human subjects with AD has been conducted since 2011. In a
phase I clinical trial in Korea, 9 patients with mild to moderate
AD were studied to assess the safety and dose-limiting toxicity of
a stereotactic brain injection of human umbilical cord
blood-derived MSCs (147). There
were no serious adverse events, such as fever, during the follow-up
period of 24 months (147).
Despite this lack of adverse events, the feasibility and safety of
stem cell therapy need to be evaluated further in larger
trials.
In conclusion, transplanted NSCs can not only
directly replenish lost neurons, but also indirectly ameliorate the
pathological environment by secreting neurotrophic factors or EXOs,
which affect the survival, proliferation, differentiation and
synaptic density of neurons. Treatment with BDNF combined with NSCs
or NGF combined with NSCs has been proven to efficiently induce
nerve regeneration, the level of which is better than that with NSC
transplantation alone. However, in clinical applications, issues
with brain mechanical injury, graft location, efficacy and safety
are still encountered in NSC transplantation.
Although nerve regeneration was first proven to
exist in the mammalian CNS in the 1960s (11), the enhancement of nerve
regeneration as a treatment strategy for AD has been gaining
traction only in recent years. A growing number of studies have
shown that inducing nerve regeneration can fundamentally improve
the self-care ability and cognitive impairment of patients with AD
(103,107). However, inducing nerve generation
is still a difficult, yet key point of intervention methods in the
clinical treatment of diseases. Previous research in AD has shown
that endogenous nerve regeneration can be improved by increasing
the expression levels of BDNF and NGF, inhibiting AChE, activating
nAChRs, increasing acetylation modifications of histone and
memory-related genes such as BDNF, increasing the synaptic activity
of neuronal circuits by electrical stimulation, and transplanting
exogenous NSCs (Fig. 1).
Currently, there is no clear conclusion on the therapeutic effect
of different interventions due to the lack of comparison and rating
scale evaluations. Moreover, the conclusions on interventions are
mostly based on the results of animal experiments, and the clinical
therapeutic effects need to be further confirmed. Furthermore,
physical therapies such as rTMS, tDCS and light therapy show a
curative effect, but also have side effects on the human body, and
the optimal stimulation intensity and duration need to be further
investigated. Therefore, further studies are needed to determine
the effectiveness of different interventions and elucidate the
mechanisms for interventions inducing nerve regeneration. In
addition, the combination of interventions with different modes of
action, such as chemical stimulation and physical stimulation, may
be a more robust and effective treatment plan.
Not applicable.
Funding: The present review was supported by the Zhejiang
Provincial Natural Science Foundation of China (grant no.
LY23H090004), the Fundamental Research Funds for the Provincial
Universities of Zhejiang (grant no. SJLY2023008), the National
Natural Science Foundation of China (grant no. 82001155), the
Natural Science Foundation of Ningbo (grant no. 2023J068), the
College Students' Scientific and Technological Innovation Project
(Xin Miao Talent Plan) of Zhejiang Province (grant no.
2022R405A045) and the K. C. Wong Magna Fund in Ningbo
University.
Not applicable.
LL conceived and planned the article. JG drafted the
manuscript, drew the figure and created the table. JG and LL
carried out the literature review, and contributed equally in
revising the manuscript. All authors read and approved the final
version of the manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Soria Lopez JA, González HM and Léger GC:
Alzheimer's disease. Handb Clin Neurol. 167:231–255.
2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tiwari S, Atluri V, Kaushik A, Yndart A
and Nair M: Alzheimer's disease: Pathogenesis, diagnostics, and
therapeutics. Int J Nanomedicine. 14:5541–5554. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ayers D and Scerri C: Non-coding RNA
influences in dementia. Noncoding RNA Res. 3:188–194.
2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Panza F, Lozupone M, Logroscino G and
Imbimbo BP: A critical appraisal of amyloid-β-targeting therapies
for Alzheimer disease. Nat Rev Neurol. 15:73–88. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Gao Y and Tan L, Yu JT and Tan L: Tau in
Alzheimer's disease: Mechanisms and therapeutic strategies. Curr
Alzheimer Res. 15:283–300. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Fernández-Calle R, Konings SC,
Frontiñán-Rubio J, García-Revilla J, Camprubí-Ferrer L, Svensson M,
Martinson I, Boza-Serrano A, Venero JL, Nielsen HM, et al: APOE in
the bullseye of neurodegenerative diseases: Impact of the APOE
genotype in Alzheimer's disease pathology and brain diseases. Mol
Neurodegener. 17(62)2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Honjo K, Black SE and Verhoeff NPLG:
Alzheimer's disease, cerebrovascular disease, and the β-amyloid
cascade. Can J Neurol Sci. 39:712–728. 2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Sharma K: Cholinesterase inhibitors as
Alzheimer's therapeutics (review). Mol Med Rep. 20:1479–1487.
2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hynd MR, Scott HL and Dodd PR:
Glutamate-mediated excitotoxicity and neurodegeneration in
Alzheimer's disease. Neurochem Int. 45:583–595. 2004.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sun MK: Roles of neural regeneration in
memory pharmacology. Neural Regen Res. 13:406–407. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Altman J and Das GD: Autoradiographic and
histological evidence of postnatal hippocampal neurogenesis in
rats. J Comp Neurol. 124:319–335. 1965.PubMed/NCBI View Article : Google Scholar
|
|
12
|
von Bohlen Und Halbach O:
Immunohistological markers for staging neurogenesis in adult
hippocampus. Cell Tissue Res. 329:409–420. 2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Low VF, Faull RL, Bennet L, Gunn AJ and
Curtis MA: Neurogenesis and progenitor cell distribution in the
subgranular zone and subventricular zone of the adult sheep brain.
Neuroscience. 244:173–187. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Gould E, Reeves AJ, Graziano MS and Gross
CG: Neurogenesis in the neocortex of adult primates. Science.
286:548–552. 1999.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Farzanehfar P: Comparative review of adult
midbrain and striatum neurogenesis with classical neurogenesis.
Neurosci Res. 134:1–9. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tobin MK, Musaraca K, Disouky A, Shetti A,
Bheri A, Honer WG, Kim N, Dawe RJ, Bennett DA, Arfanakis K and
Lazarov O: Human hippocampal neurogenesis persists in aged adults
and Alzheimer's disease patients. Cell Stem Cell. 24:974–982.e3.
2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Scheff SW, Price DA, Schmitt FA and Mufson
EJ: Hippocampal synaptic loss in early Alzheimer's disease and mild
cognitive impairment. Neurobiol Aging. 27:1372–1384.
2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wang YJ, Gong WG, Ren QG and Zhang ZJ:
Escitalopram alleviates Alzheimer's disease-type tau pathologies in
the aged P301L tau transgenic mice. J Alzheimers Dis. 77:807–819.
2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang L, Qin Z, Sharmin F, Lin W, Ricke
KM, Zasloff MA, Stewart AFR and Chen HH: Tyrosine phosphatase PTP1B
impairs presynaptic NMDA receptor-mediated plasticity in a mouse
model of Alzheimer's disease. Neurobiol Dis.
156(105402)2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Yang XB, Zu HB, Zhao YF and Yao K:
Agomelatine prevents amyloid plaque deposition, tau
phosphorylation, and neuroinflammation in APP/PS1 mice. Front Aging
Neurosci. 13(766410)2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Duan S, Guan X, Lin R, Liu X, Yan Y, Lin
R, Zhang T, Chen X, Huang J, Sun X, et al: Silibinin inhibits
acetylcholinesterase activity and amyloid β peptide aggregation: A
dual-target drug for the treatment of Alzheimer's disease.
Neurobiol Aging. 36:1792–1807. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
He Z, Li X, Wang Z, Tu S, Feng J, Du X, Ni
J, Li N and Liu Q: Esculentoside A alleviates cognitive deficits
and amyloid pathology through peroxisome proliferator-activated
receptor γ-dependent mechanism in an Alzheimer's disease model.
Phytomedicine. 98(153956)2022.PubMed/NCBI View Article : Google Scholar : (Epub ahead of
print).
|
|
23
|
Wang C, Zheng D, Weng F, Jin Y and He L:
Sodium butyrate ameliorates the cognitive impairment of Alzheimer's
disease by regulating the metabolism of astrocytes.
Psychopharmacology (Berl). 239:215–227. 2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhang HA, Yuan CX, Liu KF, Yang QF, Zhao
J, Li H, Yang QH, Song D, Quan ZZ and Qing H: Neural stem cell
transplantation alleviates functional cognitive deficits in a mouse
model of tauopathy. Neural Regen Res. 17:152–162. 2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lilja AM, Malmsten L, Röjdner J, Voytenko
L, Verkhratsky A, Ögren SO, Nordberg A and Marutle A: Neural stem
cell transplant-induced effect on neurogenesis and cognition in
Alzheimer Tg2576 mice is inhibited by concomitant treatment with
amyloid-lowering or cholinergic α7 nicotinic receptor drugs. Neural
Plast. 2015(370432)2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Li W, Kong LH, Wang H, Shen F, Wang YW,
Zhou H and Sun GJ: High-frequency electroacupuncture evidently
reinforces hippocampal synaptic transmission in Alzheimer's disease
rats. Neural Regen Res. 11:801–806. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Riolo G, Ricci C, De Angelis N, Marzocchi
C, Guerrera G, Borsellino G, Giannini F and Battistini S: BDNF and
pro-BDNF in amyotrophic lateral sclerosis: A new perspective for
biomarkers of neurodegeneration. Brain Sci. 12(617)2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Levey AI, Qiu D, Zhao L, Hu WT, Duong DM,
Higginbotham L, Dammer EB, Seyfried NT, Wingo TS, Hales CM, et al:
A phase II study repurposing atomoxetine for neuroprotection in
mild cognitive impairment. Brain. 145:1924–1938. 2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zoladz JA, Majerczak J, Zeligowska E,
Mencel J, Jaskolski A, Jaskolska A and Marusiak J:
Moderate-intensity interval training increases serum brain-derived
neurotrophic factor level and decreases inflammation in Parkinson's
disease patients. J Physiol Pharmacol. 65:441–448. 2014.PubMed/NCBI
|
|
30
|
Eyileten C, Sharif L, Wicik Z, Jakubik D,
Jarosz-Popek J, Soplinska A, Postula M, Czlonkowska A,
Kaplon-Cieslicka A and Mirowska-Guzel D: The relation of the
brain-derived neurotrophic factor with MicroRNAs in
neurodegenerative diseases and ischemic stroke. Mol Neurobiol.
58:329–347. 2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ball S, Marangell LB, Lipsius S and
Russell JM: Brain-derived neurotrophic factor in generalized
anxiety disorder: results from a duloxetine clinical trial. Prog
Neuropsychopharmacol Biol Psychiatry. 43:217–221. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Shekari A and Fahnestock M: Retrograde
axonal transport of BDNF and proNGF diminishes with age in basal
forebrain cholinergic neurons. Neurobiol Aging. 84:131–140.
2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Thoenen H, Zafra F, Hengerer B and
Lindholm D: The synthesis of nerve growth factor and brain-derived
neurotrophic factor in hippocampal and cortical neurons is
regulated by specific transmitter systems. Ann N Y Acad Sci.
640:86–90. 1991.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Nagahara AH, Merrill DA, Coppola G,
Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner
JM, et al: Neuroprotective effects of brain-derived neurotrophic
factor in rodent and primate models of Alzheimer's disease. Nat
Med. 15:331–337. 2009.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Arora S, Kanekiyo T and Singh J:
Functionalized nanoparticles for brain targeted BDNF gene therapy
to rescue Alzheimer's disease pathology in transgenic mouse model.
Int J Biol Macromol. 208:901–911. 2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Beeri MS and Sonnen J: Brain BDNF
expression as a biomarker for cognitive reserve against Alzheimer
disease progression. Neurology. 86:702–703. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Dong BE, Chen H and Sakata K: BDNF
deficiency and enriched environment treatment affect
neurotransmitter gene expression differently across ages. J
Neurochem. 154:41–55. 2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Paraskevopoulou F, Herman MA and Rosenmund
C: Glutamatergic innervation onto striatal neurons potentiates
GABAergic synaptic output. J Neurosci. 39:4448–4460.
2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Miyazaki S, Oikawa H, Takekoshi H,
Hoshizaki M, Ogata M and Fujikawa T: Anxiolytic effects of
acanthopanax senticosus HARMS occur via regulation of autonomic
function and activate hippocampal BDNF-TrkB signaling. Molecules.
24(132)2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Li Y, Xiang L, Wang C, Song Y, Miao J and
Miao M: Protection against acute cerebral ischemia/reperfusion
injury by leonuri herba total alkali via modulation of
BDNF-TrKB-PI3K/Akt signaling pathway in rats. Biomed Pharmacother.
133(111021)2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Yamaguchi A, Tamatani M, Matsuzaki H,
Namikawa K, Kiyama H, Vitek MP, Mitsuda N and Tohyama M: Akt
activation protects hippocampal neurons from apoptosis by
inhibiting transcriptional activity of p53. J Biol Chem.
276:5256–5264. 2001.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Chimenti MS, Sunzini F, Fiorucci L, Botti
E, Fonti GL, Conigliaro P, Triggianese P, Costa L, Caso F, Giunta
A, et al: Potential role of cytochrome c and tryptase in psoriasis
and psoriatic arthritis pathogenesis: Focus on resistance to
apoptosis and oxidative stress. Front Immunol.
9(2363)2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Zhou LJ, Mo YB, Bu X, Wang JJ, Bai J,
Zhang JW, Cheng AB, Ma JH, Wang YW and Xie YX: Erinacine
facilitates the opening of the mitochondrial permeability
transition pore through the inhibition of the PI3K/Akt/GSK-3β
signaling pathway in human hepatocellular carcinoma. Cell Physiol
Biochem. 50:851–867. 2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Jiang T, Wang XQ, Ding C and Du XL:
Genistein attenuates isoflurane-induced neurotoxicity and improves
impaired spatial learning and memory by regulating cAMP/CREB and
BDNF-TrkB-PI3K/Akt signaling. Korean J Physiol Pharmacol.
21:579–589. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Merkouris S, Barde YA, Binley KE, Allen
ND, Stepanov AV, Wu NC, Grande G, Lin CW, Li M, Nan X, et al: Fully
human agonist antibodies to TrkB using autocrine cell-based
selection from a combinatorial antibody library. Proc Natl Acad Sci
USA. 115:E7023–E7032. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Tacke C, DiStefano PS, Lindsay RM,
Metzdorf K, Zagrebelsky M and Korte M: Actions of the TrkB agonist
antibody ZEB85 in regulating the architecture and synaptic
plasticity in hippocampal neurons. Front Mol Neurosci.
15(945348)2022.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Chen C, Wang Z, Zhang Z, Liu X, Kang SS,
Zhang Y and Ye K: The prodrug of 7,8-dihydroxyflavone development
and therapeutic efficacy for treating Alzheimer's disease. Proc
Natl Acad Sci USA. 115:578–583. 2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Fan CH, Lin CW, Huang HJ, Lee-Chen GJ, Sun
YC, Lin W, Chen CM, Chang KH, Su MT and Hsieh-Li HM: LMDS-1, a
potential TrkB receptor agonist provides a safe and neurotrophic
effect for early-phase Alzheimer's disease. Psychopharmacology
(Berl). 237:3173–3190. 2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Tuszynski MH, Yang JH, Barba D, U HS,
Bakay RA, Pay MM, Masliah E, Conner JM, Kobalka P, Roy S and
Nagahara AH: Nerve growth factor gene therapy: Activation of
neuronal responses in Alzheimer disease. JAMA Neurol. 72:1139–1147.
2015.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Rocco ML, Soligo M, Manni L and Aloe L:
Nerve growth factor: Early studies and recent clinical trials. Curr
Neuropharmacol. 16:1455–1465. 2018.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Ding XW, Li R, Geetha T, Tao YX and Babu
JR: Nerve growth factor in metabolic complications and Alzheimer's
disease: Physiology and therapeutic potential. Biochim Biophys Acta
Mol Basis Dis. 1866(165858)2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Tiveron C, Fasulo L, Capsoni S, Malerba F,
Marinelli S, Paoletti F, Piccinin S, Scardigli R, Amato G, Brandi
R, et al: ProNGF\NGF imbalance triggers learning and memory
deficits, neurodegeneration and spontaneous epileptic-like
discharges in transgenic mice. Cell Death Differ. 20:1017–1030.
2013.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Mitra S, Behbahani H and Eriksdotter M:
Innovative therapy for Alzheimer's disease-with focus on
biodelivery of NGF. Front Neurosci. 13(38)2019.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Ruberti F, Capsoni S, Comparini A, Di
Daniel E, Franzot J, Gonfloni S, Rossi G, Berardi N and Cattaneo A:
Phenotypic knockout of nerve growth factor in adult transgenic mice
reveals severe deficits in basal forebrain cholinergic neurons,
cell death in the spleen, and skeletal muscle dystrophy. J
Neurosci. 20:2589–2601. 2000.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Soligo M, Albini M, Bertoli FL, Marzano V,
Protto V, Bracci-Laudiero L, Minnone G, De Benedetti F, Chiaretti
A, Mantuano E and Manni L: Different responses of PC12 cells to
different pro-nerve growth factor protein variants. Neurochem Int.
129(104498)2019.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Isaev NK, Stelmashook EV and Genrikhs EE:
Role of nerve growth factor in plasticity of forebrain cholinergic
neurons. Biochemistry (Mosc). 82:291–300. 2017.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Delivanoglou N, Boziki M, Theotokis P,
Kesidou E, Touloumi O, Dafi N, Nousiopoulou E, Lagoudaki R,
Grigoriadis N, Charalampopoulos I and Simeonidou C: Spatio-temporal
expression profile of NGF and the two-receptor system, TrkA and
p75NTR, in experimental autoimmune encephalomyelitis. J
Neuroinflammation. 17(41)2020.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Yan T, Zhang Z and Li D: NGF receptors and
PI3K/AKT pathway involved in glucose fluctuation-induced damage to
neurons and α-lipoic acid treatment. BMC Neurosci.
21(38)2020.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Ioannou MS and Fahnestock M: ProNGF, but
not NGF, switches from neurotrophic to apoptotic activity in
response to reductions in TrkA receptor levels. Int J Mol Sci.
18(599)2017.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Karami A, Eyjolfsdottir H, Vijayaraghavan
S, Lind G, Almqvist P, Kadir A, Linderoth B, Andreasen N, Blennow
K, Wall A, et al: Changes in CSF cholinergic biomarkers in response
to cell therapy with NGF in patients with Alzheimer's disease.
Alzheimers Dement. 11:1316–1328. 2015.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Pakzaban P and Chiocca EA: Nerve growth
factor protects against herpes simplex virus type 1 neurotoxicity
in the rat striatum. Neuroreport. 5:993–996. 1994.PubMed/NCBI View Article : Google Scholar
|
|
62
|
De Rosa R, Garcia AA, Braschi C, Capsoni
S, Maffei L, Berardi N and Cattaneo A: Intranasal administration of
nerve growth factor (NGF) rescues recognition memory deficits in
AD11 anti-NGF transgenic mice. Proc Natl Acad Sci USA.
102:3811–3816. 2005.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Lambiase A, Pagani L, Di Fausto V, Sposato
V, Coassin M, Bonini S and Aloe L: Nerve growth factor eye drop
administrated on the ocular surface of rodents affects the nucleus
basalis and septum: Biochemical and structural evidence. Brain Res.
1127:45–51. 2007.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Hohsfield LA, Geley S, Reindl M and Humpel
C: The generation of NGF-secreting primary rat monocytes: A
comparison of different transfer methods. J Immunol Methods.
391:112–124. 2013.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Eriksdotter M, Navarro-Oviedo M, Mitra S,
Wahlberg L, Linderoth B, Tjernberg LO and Behbahani H:
Cerebrospinal fluid from Alzheimer patients affects cell-mediated
nerve growth factor production and cell survival in vitro. Exp Cell
Res. 371:175–184. 2018.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Moyano P, Flores A, Garcia J, García JM,
Anadon MJ, Frejo MT, Sola E, Pelayo A and Del Pino J: Bisphenol A
single and repeated treatment increases HDAC2, leading to
cholinergic neurotransmission dysfunction and SN56 cholinergic
apoptotic cell death through AChE variants overexpression and
NGF/TrkA/P75NTR signaling disruption. Food Chem Toxicol.
157(112614)2021.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Eyjolfsdottir H, Eriksdotter M, Linderoth
B, Lind G, Juliusson B, Kusk P, Almkvist O, Andreasen N, Blennow K,
Ferreira D, et al: Targeted delivery of nerve growth factor to the
cholinergic basal forebrain of Alzheimer's disease patients:
Application of a second-generation encapsulated cell biodelivery
device. Alzheimers Res Ther. 8(30)2016.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Amaral LD, Santos NAGD, Sisti FM, Del Bel
E and Santos ACD: The antibiotic doxycycline mimics the NGF
signaling in PC12 cells: A relevant mechanism for neuroprotection.
Chem Biol Interact. 341(109454)2021.PubMed/NCBI View Article : Google Scholar
|
|
69
|
James ML, Belichenko NP, Shuhendler AJ,
Hoehne A, Andrews LE, Condon C, Nguyen TV, Reiser V, Jones P, Trigg
W, et al: [18F]GE-180 PET detects reduced microglia activation
after LM11A-31 therapy in a mouse model of Alzheimer's disease.
Theranostics. 7:1422–1436. 2017.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Bartus RT, Dean RL III, Beer B and Lippa
AS: The cholinergic hypothesis of geriatric memory dysfunction.
Science. 217:408–414. 1982.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Moss DE: Improving anti-neurodegenerative
benefits of acetylcholinesterase inhibitors in Alzheimer's disease:
Are irreversible inhibitors the future? Int J Mol Sci.
21(3438)2020.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Korabecny J, Spilovska K, Mezeiova E,
Benek O, Juza R, Kaping D and Soukup O: A systematic review on
donepezil-based derivatives as potential cholinesterase inhibitors
for Alzheimer's disease. Curr Med Chem. 26:5625–5648.
2019.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Parsons CG, Danysz W, Dekundy A and Pulte
I: Memantine and cholinesterase inhibitors: Complementary
mechanisms in the treatment of Alzheimer's disease. Neurotox Res.
24:358–369. 2013.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Liang J, Li J, Jia R, Wang Y, Wu R, Zhang
H, Hang L and Xu Y: Identification of the optimal cognitive drugs
among Alzheimer's disease: A Bayesian meta-analytic review. Clin
Interv Aging. 13:2061–2073. 2018.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Aguglia E, Onor ML, Saina M and Maso E: An
open-label, comparative study of rivastigmine, donepezil and
galantamine in a real-world setting. Curr Med Res Opin.
20:1747–1752. 2004.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Nordberg A, Darreh-Shori T, Peskind E,
Soininen H, Mousavi M, Eagle G and Lane R: Different cholinesterase
inhibitor effects on CSF cholinesterases in Alzheimer patients.
Curr Alzheimer Res. 6:4–14. 2009.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Joshi S and Kapur J: N-methyl-D-aspartic
acid receptor activation downregulates expression of δ
subunit-containing GABAA receptors in cultured hippocampal neurons.
Mol Pharmacol. 84:1–11. 2013.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Shih CC, Chen PY, Chen MF and Lee TJF:
Differential blockade by huperzine A and donepezil of sympathetic
nicotinic acetylcholine receptor-mediated nitrergic neurogenic
dilations in porcine basilar arteries. Eur J Pharmacol.
868(172851)2020.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Ito T, Inden M, Ueda T, Asaka Y, Kurita H
and Hozumi I: The neuroprotective effects of activated alpha7
nicotinic acetylcholine receptor against mutant copper-zinc
superoxide dismutase 1-mediated toxicity. Sci Rep.
10(22157)2020.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Noh MY, Koh SH, Kim Y, Kim HY, Cho GW and
Kim SH: Neuroprotective effects of donepezil through inhibition of
GSK-3 activity in amyloid-beta-induced neuronal cell death. J
Neurochem. 108:1116–1125. 2009.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Kihara T, Shimohama S, Sawada H, Honda K,
Nakamizo T, Shibasaki H, Kume T and Akaike A: alpha 7 nicotinic
receptor transduces signals to phosphatidylinositol 3-kinase to
block A beta-amyloid-induced neurotoxicity. J Biol Chem.
276:13541–13546. 2001.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Makitani K, Nakagawa S, Izumi Y, Akaike A
and Kume T: Inhibitory effect of donepezil on bradykinin-induced
increase in the intracellular calcium concentration in cultured
cortical astrocytes. J Pharmacol Sci. 134:37–44. 2017.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Arias E, Gallego-Sandin S, Villarroya M,
García AG and López MG: Unequal neuroprotection afforded by the
acetylcholinesterase inhibitors galantamine, donepezil, and
rivastigmine in SH-SY5Y neuroblastoma cells: Role of nicotinic
receptors. J Pharmacol Exp Ther. 315:1346–1353. 2005.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Zhao S, Zhang X and Li H: Beyond histone
acetylation-writing and erasing histone acylations. Curr Opin
Struct Biol. 53:169–177. 2018.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Ganai SA, Ramadoss M and Mahadevan V:
Histone deacetylase (HDAC) inhibitors-emerging roles in neuronal
memory, learning, synaptic plasticity and neural regeneration. Curr
Neuropharmacol. 14:55–71. 2016.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Fuller NO, Pirone A, Lynch BA, Hewitt MC,
Quinton MS, McKee TD and Ivarsson M: CoREST complex-selective
histone deacetylase inhibitors show prosynaptic effects and an
improved safety profile to enable treatment of synaptopathies. ACS
Chem Neurosci. 10:1729–1743. 2019.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Xu K, Dai XL, Huang HC and Jiang ZF:
Targeting HDACs: A promising therapy for Alzheimer's disease. Oxid
Med Cell Longev. 2011(143269)2011.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Kumar V, Kundu S, Singh A and Singh S:
Understanding the role of histone deacetylase and their inhibitors
in neurodegenerative disorders: Current targets and future
perspective. Curr Neuropharmacol. 20:158–178. 2022.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Guan JS, Haggarty SJ, Giacometti E,
Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X,
Mazitschek R, et al: HDAC2 negatively regulates memory formation
and synaptic plasticity. Nature. 459:55–60. 2009.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Janczura KJ, Volmar CH, Sartor GC, Rao SJ,
Ricciardi NR, Lambert G, Brothers SP and Wahlestedt C: Inhibition
of HDAC3 reverses Alzheimer's disease-related pathologies in vitro
and in the 3xTg-AD mouse model. Proc Natl Acad Sci USA.
115:E11148–E11157. 2018.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Chen YA, Lu CH, Ke CC, Chiu SJ, Chang CW,
Yang BH, Gelovani JG and Liu RS: Evaluation of class IIa histone
deacetylases expression and in vivo epigenetic imaging in a
transgenic mouse model of Alzheimer's disease. Int J Mol Sci.
22(8633)2021.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Li Y, Sang S, Ren W, Pei Y, Bian Y, Chen Y
and Sun H: Inhibition of histone deacetylase 6 (HDAC6) as a
therapeutic strategy for Alzheimer's disease: A review (2010-2020).
Eur J Med Chem. 226(113874)2021.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Cuadrado-Tejedor M, Garcia-Barroso C,
Sánchez-Arias JA, Rabal O, Pérez-González M, Mederos S, Ugarte A,
Franco R, Segura V, Perea G, et al: A first-in-class small-molecule
that acts as a dual inhibitor of HDAC and PDE5 and that rescues
hippocampal synaptic impairment in Alzheimer's disease mice.
Neuropsychopharmacology. 42:524–539. 2017.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Rabal O, Sánchez-Arias JA,
Cuadrado-Tejedor M, de Miguel I, Pérez-González M, García-Barroso
C, Ugarte A, Estella-Hermoso de Mendoza A, Sáez E, Espelosin M, et
al: Design, synthesis, biological evaluation and in vivo testing of
dual phosphodiesterase 5 (PDE5) and histone deacetylase 6
(HDAC6)-selective inhibitors for the treatment of Alzheimer's
disease. Eur J Med Chem. 150:506–524. 2018.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Koch G, Martorana A and Caltagirone C:
Transcranial magnetic stimulation: Emerging biomarkers and novel
therapeutics in Alzheimer's disease. Neurosci Lett.
719(134355)2020.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Bursali C, Özkan FÜ, Kaysin MY, Dortcan N,
Aktas I and Külcü DG: Effectiveness of repetitive transcranial
magnetic stimulation in patients with failed back surgery syndrome:
A double-blind randomized placebo-controlled study. Pain Physician.
24:E23–E30. 2021.PubMed/NCBI
|
|
97
|
Minzenberg MJ and Leuchter AF: The effect
of psychotropic drugs on cortical excitability and plasticity
measured with transcranial magnetic stimulation: Implications for
psychiatric treatment. J Affect Disord. 253:126–140.
2019.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Zhao J, Li Z, Cong Y, Zhang J, Tan M,
Zhang H, Geng N, Li M, Yu W and Shan P: Repetitive transcranial
magnetic stimulation improves cognitive function of Alzheimer's
disease patients. Oncotarget. 8:33864–33871. 2017.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Sabbagh M, Sadowsky C, Tousi B, Agronin
ME, Alva G, Armon C, Bernick C, Keegan AP, Karantzoulis S, Baror E,
et al: Effects of a combined transcranial magnetic stimulation
(TMS) and cognitive training intervention in patients with
Alzheimer's disease. Alzheimers Dement. 16:641–650. 2020.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Saitoh Y, Hosomi K, Mano T, Takeya Y,
Tagami S, Mori N, Matsugi A, Jono Y, Harada H, Yamada T and Miyake
A: Randomized, sham-controlled, clinical trial of repetitive
transcranial magnetic stimulation for patients with Alzheimer's
dementia in Japan. Front Aging Neurosci. 14(993306)2022.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Ahmed MA, Darwish ES, Khedr EM, El Serogy
YM and Ali AM: Effects of low versus high frequencies of repetitive
transcranial magnetic stimulation on cognitive function and
cortical excitability in Alzheimer's dementia. J Neurol. 259:83–92.
2012.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Trojano L, Conson M, Maffei R and Grossi
D: Categorical and coordinate spatial processing in the imagery
domain investigated by rTMS. Neuropsychologia. 44:1569–1574.
2006.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Turriziani P, Smirni D, Zappalà G, Mangano
GR, Oliveri M and Cipolotti L: Enhancing memory performance with
rTMS in healthy subjects and individuals with mild cognitive
Impairment: The role of the right dorsolateral prefrontal cortex.
Front Hum Neurosci. 6(62)2012.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Sanches C, Levy R, Benisty S, Volpe-Gillot
L, Habert MO, Kas A, Ströer S, Pyatigorskaya N, Kaglik A, Bourbon
A, et al: Testing the therapeutic effects of transcranial direct
current stimulation (tDCS) in semantic dementia: A double blind,
sham controlled, randomized clinical trial. Trials.
20(632)2019.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Bunai T, Hirosawa T, Kikuchi M, Fukai M,
Yokokura M, Ito S, Takata Y, Terada T and Ouchi Y: tDCS-induced
modulation of GABA concentration and dopamine release in the human
brain: A combination study of magnetic resonance spectroscopy and
positron emission tomography. Brain Stimul. 14:154–160.
2021.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Lefebvre S and Liew SL: Anatomical
parameters of tDCS to modulate the motor system after stroke: A
review. Front Neurol. 8(29)2017.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Boggio PS, Ferrucci R, Mameli F, Martins
D, Martins O, Vergari M, Tadini L, Scarpini E, Fregni F and Priori
A: Prolonged visual memory enhancement after direct current
stimulation in Alzheimer's disease. Brain Stimul. 5:223–230.
2012.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Batsikadze G, Moliadze V, Paulus W, Kuo MF
and Nitsche MA: Partially non-linear stimulation
intensity-dependent effects of direct current stimulation on motor
cortex excitability in humans. J Physiol. 591:1987–2000.
2013.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Woods AJ, Antal A, Bikson M, Boggio PS,
Brunoni AR, Celnik P, Cohen LG, Fregni F, Herrmann CS, Kappenman
ES, et al: A technical guide to tDCS, and related non-invasive
brain stimulation tools. Clin Neurophysiol. 127:1031–1048.
2016.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Iaccarino HF, Singer AC, Martorell AJ,
Rudenko A, Gao F, Gillingham TZ, Mathys H, Seo J, Kritskiy O,
Abdurrob F, et al: Gamma frequency entrainment attenuates amyloid
load and modifies microglia. Nature. 540:230–235. 2016.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Bradford A, Barlow A and Chazot PL:
Probing the differential effects of infrared light sources IR1072
and IR880 on human lymphocytes: Evidence of selective
cytoprotection by IR1072. J Photochem Photobiol B. 81:9–14.
2005.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Grillo SL, Duggett NA, Ennaceur A and
Chazot PL: Non-invasive infra-red therapy (1072 nm) reduces
β-amyloid protein levels in the brain of an Alzheimer's disease
mouse model, TASTPM. J Photochem Photobiol B. 123:13–22.
2013.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Wang M, Cao J, Amakye WK, Gong C, Li Q and
Ren J: Mid infrared light treatment attenuates cognitive decline
and alters the gut microbiota community in APP/PS1 mouse model.
Biochem Biophys Res Commun. 523:60–65. 2020.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Huang N, Yao D, Jiang W, Wei C, Li M, Li
W, Mu H, Gao M, Ma Z, Lyu J and Tong Z: Safety and efficacy of
630-nm red light on cognitive function in older adults with mild to
moderate Alzheimer's disease: Protocol for a randomized controlled
study. Front Aging Neurosci. 12(143)2020.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Figueiro MG and Leggett S: Intermittent
light exposures in humans: A case for dual entrainment in the
treatment of Alzheimer's disease. Front Neurol.
12(625698)2021.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Ying Y and Wang JZ: Illuminating neural
circuits in Alzheimer's disease. Neurosci Bull. 37:1203–1217.
2021.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Ourednik J, Ourednik V, Lynch WP,
Schachner M and Snyder EY: Neural stem cells display an inherent
mechanism for rescuing dysfunctional neurons. Nat Biotechnol.
20:1103–1110. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
118
|
Alessandrini M, Preynat-Seauve O, De Bruin
K and Pepper MS: Stem cell therapy for neurological disorders. S
Afr Med J. 109:70–77. 2019.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Kang JM, Yeon BK, Cho SJ and Suh YH: Stem
cell therapy for Alzheimer's disease: A review of recent clinical
trials. J Alzheimers Dis. 54:879–889. 2016.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Zhang B, Yan W, Zhu Y, Yang W, Le W, Chen
B, Zhu R and Cheng L: Nanomaterials in neural-stem-cell-mediated
regenerative medicine: Imaging and treatment of neurological
diseases. Adv Mater. 30(e1705694)2018.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Shu H, Guo Z, Chen X, Qi S, Xiong X, Xia
S, Huang Q, Lan L, Gong J, Huang S, Yang B, et al: Intracerebral
transplantation of neural stem cells restores manganese-induced
cognitive deficits in mice. Aging Dis. 12:371–385. 2021.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Csobonyeiova M, Polak S, Zamborsky R and
Danisovic L: Recent progress in the regeneration of spinal cord
injuries by induced pluripotent stem cells. Int J Mol Sci.
20(3838)2019.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Banda E and Grabel L: Directed
differentiation of human embryonic stem cells into neural
progenitors. Methods Mol Biol. 1307:289–298. 2016.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Shahbazi E, Mirakhori F, Ezzatizadeh V and
Baharvand H: Reprogramming of somatic cells to induced neural stem
cells. Methods. 133:21–28. 2018.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Daadi MM: Generation of neural stem cells
from induced pluripotent stem cells. Methods Mol Biol. 1919:1–7.
2019.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Matsui T, Akamatsu W, Nakamura M and Okano
H: Regeneration of the damaged central nervous system through
reprogramming technology: Basic concepts and potential application
for cell replacement therapy. Exp Neurol. 260:12–18.
2014.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Zhang L, Dong ZF and Zhang JY:
Immunomodulatory role of mesenchymal stem cells in Alzheimer's
disease. Life Sci. 246(117405)2020.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Yip S, Aboody KS, Burns M, Imitola J,
Boockvar JA, Allport J, Park KI, Teng YD, Lachyankar M, McIntosh T,
et al: Neural stem cell biology may be well suited for improving
brain tumor therapies. Cancer J. 9:189–204. 2003.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Park KI: Transplantation of neural stem
cells: Cellular & gene therapy for hypoxic-ischemic brain
injury. Yonsei Med J. 41:825–835. 2000.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Zhao L, Liu JW, Kan BH, Shi HY, Yang LP
and Liu XY: Acupuncture accelerates neural regeneration and
synaptophysin production after neural stem cells transplantation in
mice. World J Stem Cells. 12:1576–1590. 2020.PubMed/NCBI View Article : Google Scholar
|
|
131
|
De Feo D, Merlini A, Laterza C and Martino
G: Neural stem cell transplantation in central nervous system
disorders: From cell replacement to neuroprotection. Curr Opin
Neurol. 25:322–333. 2012.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Yu JH, Seo JH, Lee JY, Lee MY and Cho SR:
Induction of Neurorestoration From Endogenous Stem Cells. Cell
Transplant. 25:863–882. 2016.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Wu K, Zhang R, Lu Y, Wen L, Li Y, Duan R,
Yao Y and Jia Y: Lin28B regulates the fate of grafted mesenchymal
stem cells and enhances their protective effects against
Alzheimer's disease by upregulating IGF-2. J Cell Physiol.
234:21860–21876. 2019.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Sun B, Taing A, Liu H, Nie G, Wang J, Fang
Y, Liu L, Xue Y, Shi J, Liao YP, et al: Nerve growth
factor-conjugated mesoporous silica nanoparticles promote
neuron-like PC12 cell proliferation and neurite growth. J Nanosci
Nanotechnol. 16:2390–2393. 2016.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Mili B, Das K, Kumar A, Saxena AC, Singh
P, Ghosh S and Bag S: Preparation of NGF encapsulated chitosan
nanoparticles and its evaluation on neuronal differentiation
potentiality of canine mesenchymal stem cells. J Mater Sci Mater
Med. 29(4)2017.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Mortazavi Y, Sheikhsaran F, Khamisipour
GK, Soleimani M, Teimuri A and Shokri S: The evaluation of nerve
growth factor over expression on neural lineage specific genes in
human mesenchymal stem cells. Cell J. 18:189–196. 2016.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Lee HJ, Lim IJ, Park SW, Kim YB, Ko Y and
Kim SU: Human neural stem cells genetically modified to express
human nerve growth factor (NGF) gene restore cognition in the mouse
with ibotenic acid-induced cognitive dysfunction. Cell Transplant.
21:2487–2496. 2012.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Ma Y, Li C, Huang Y, Wang Y, Xia X and
Zheng JC: Exosomes released from neural progenitor cells and
induced neural progenitor cells regulate neurogenesis through
miR-21a. Cell Commun Signal. 17(96)2019.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Zhang R, Mao W, Niu L, Bao W, Wang Y, Wang
Y, Zhu Y, Yang Z, Chen J, Dong J, et al: NSC-derived exosomes
enhance therapeutic effects of NSC transplantation on cerebral
ischemia in mice. Elife. 12(e84493)2023.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Ma Y, Wang K, Pan J, Fan Z, Tian C, Deng
X, Ma K, Xia X, Huang Y and Zheng JC: Induced neural progenitor
cells abundantly secrete extracellular vesicles and promote the
proliferation of neural progenitors via extracellular
signal-regulated kinase pathways. Neurobiol Dis. 124:322–334.
2019.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Zhang J, Li S, Li L, Li M, Guo C, Yao J
and Mi S: Exosome and exosomal microRNA: Trafficking, sorting, and
function. Genomics Proteomics Bioinformatics. 13:17–24.
2015.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Xin H, Wang F, Li Y, Lu QE, Cheung WL,
Zhang Y, Zhang ZG and Chopp M: Secondary release of exosomes from
astrocytes contributes to the increase in neural plasticity and
improvement of functional recovery after stroke in rats treated
with exosomes harvested from MicroRNA 133b-overexpressing
multipotent mesenchymal stromal cells. Cell Transplant. 26:243–257.
2017.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Gan C and Ouyang F: Exosomes released from
bone-marrow stem cells ameliorate hippocampal neuronal injury
through transferring miR-455-3p. J Stroke Cerebrovasc Dis.
31(106142)2022.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Li D, Zhang P, Yao X, Li H, Shen H, Li X,
Wu J and Lu X: Exosomes derived from miR-133b-modified mesenchymal
stem cells promote recovery after spinal cord injury. Front
Neurosci. 12(845)2018.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Takata T, Nonaka W, Iwama H, Kobara H,
Deguchi K, Masugata H, Touge T, Miyamoto O, Nakamura T, Itano T and
Masaki T: Light exercise without lactate elevation induces ischemic
tolerance through the modulation of microRNA in the gerbil
hippocampus. Brain Res. 1732(146710)2020.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Zhu Q, Zhang N, Hu N, Jiang R, Lu H, Xuan
A, Long D and Chen Y: Neural stem cell transplantation improves
learning and memory by protecting cholinergic neurons and restoring
synaptic impairment in an amyloid precursor protein/presenilin 1
transgenic mouse model of Alzheimer's disease. Mol Med Rep.
21:1172–1180. 2020.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Kim HJ, Seo SW, Chang JW, Lee JI, Kim CH,
Chin J, Choi SJ, Kwon H, Yun HJ, Lee JM, et al: Stereotactic brain
injection of human umbilical cord blood mesenchymal stem cells in
patients with Alzheimer's disease dementia: A phase 1 clinical
trial. Alzheimers Dement (N Y). 1:95–102. 2015.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Gauthier S, Rosa-Neto P, Morais JA and
Webster C: World Alzheimer Report, 2021: Journey through the
diagnosis of dementia. Alzheimer's Disease International. 2021.
|