Introduction
Pneumonia, a common inflammatory disease of the
lung, is a huge burden on both society and individuals, and is
associated with high incidence and mortality rates, with ~120
million new cases and 1.3 million deaths every year (1), especially in children and seniors
>65 years old. It has been reported that ~921 000 children under
the age of 5 years died from pneumonia in 2015 globally (2). Pneumonia, the typical clinical
symptoms of which include cough, dyspnea, fever and chest pain, may
develop into respiratory distress syndrome, accompanied by multiple
complications, such as heart failure and parapneumonic pleurisy
(3-5).
Pneumonia is caused by various types of infection, with viruses
such as Staphylococcus aureus and Streptococcus
pneumoniae, bacteria such as respiratory syncytial virus
parainfluenza virus, fungi and other pathogens (6,7). In
the last few years, superinfection with difficult-to-treat fungi
such as Fusarium and the resistance of germs to common
antibiotics has made treating and curing pneumonia difficult,
despite the clinical advantages of the latest generation
antibiotics such as cefiderocol (8-11).
Furthermore, the novel coronavirus (severe acute respiratory
syndrome coronavirus 2) causing the COVID-19 disease pandemic,
which can induce respiratory damage and progress to pneumonia or
damage to the whole body, has infected millions of individuals,
leading to >8,000,000 cases and >400,000 deaths (12-15).
Thus, pneumonia is a severe clinical problem and an improved
understanding of the pathological mechanism of pneumonia is
urgently required for the exploration of effective therapeutic
modalities.
Forkhead box protein A2 (FOXA2; also referred to as
hepatocyte nuclear factor 3β), belongs to the forkhead
transcription factor family, which contains a winged-helix
DNA-binding domain (16). FOXA2
was initially identified in liver tissues by Jackson et al
(17), who found that FOXA2 could
control the expression of liver-specific genes. At present,
accumulating evidence has revealed that FOXA2 is widely distributed
in multiple tissues, and abnormal FOXA2 expression is associated
with various diseases. For instance, high expression levels of
FOXA2 are observed in various types of cancer, including esophageal
squamous cell carcinoma, prostate cancer and lung cancer, and are
associated with tumorigenesis or tumor metastasis (18-20).
In addition, FOXA2 is depleted by microbial infection and
proinflammatory responses in diseased airways, causing the
dysregulation of mucin biosynthesis and goblet cell development,
and ultimately leading to acute deterioration of pulmonary
functions (21). FOXA2 is also
downregulated in chronic obstructive pulmonary disease, and FOXA2
overexpression serves a protective role against cigarette
smoke-induced cellular senescence and lung inflammation (22). Although the aforementioned evidence
suggests the critical regulatory role of FOXA2 in multiple
pulmonary diseases, the functional effects and molecular mechanism
of FOXA2 in pneumonia remain to be determined.
Therefore, the present study aimed to explore the
expression and regulatory role of FOXA2 in pneumonia, and attempted
to elucidate the regulatory mechanism of FOXA2, in order to provide
novel strategies for the treatment of pneumonia.
Materials and methods
Cell culture and treatment
WI-38 human embryonic fibroblast cells were obtained
from Procell Life Science & Technology Co., Ltd., and cultured
in minimum essential medium (Procell Life Science & Technology
Co., Ltd.) supplemented with 10% FBS (Procell Life Science &
Technology Co., Ltd.) and 1% penicillin/streptomycin mixture in a
humified incubator with 5% CO2 at 37˚C. WI-38 cells were
treated with increasing doses (5, 10 and 15 µg/ml) of
lipopolysaccharide (LPS; cat. no. L2880; MilliporeSigma) for 24 h
at 37˚C to establish an in vitro infantile pneumonia cell
model as described previously (23,24).
In addition, to investigate the molecular mechanism, WI-38 cells
were pretreated with U46619 (2 mM; cat. no. sc-201242; Santa Cruz
Biotechnology, Inc.), an activator of p38 signaling (25), for 4 h at 37˚C prior to LPS
exposure. Normally cultured WI-38 cells were used as the control
group.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells using the
Eastep® Super Total RNA Extraction Kit (Promega
Corporation) according to the manufacturer's instructions.
Following detection of the concentration and purity of the
extracted RNA using a NanoDrop 3000 Spectrophotometer (Thermo
Fisher Scientific, Inc.), total RNA was reverse transcribed into
cDNA using EasyScript First-Strand cDNA Synthesis SuperMix
(TransGen Biotech Co., Ltd.). The conditions for RT were as
follows: 37˚C for 10 min, followed by 85˚C for 5 sec and holding at
4˚C. Subsequently, qPCR was performed using ChamQ SYBR Master Mix
(Vazyme Biotech Co., Ltd.) and a LightCycler 96 (Roche Applied
Science). The following thermocycling conditions were used for the
qPCR: Initial denaturation at 95˚C for 10 min; followed by 40
cycles of 95˚C for 10 sec, 60˚C for 60 sec and 95˚C for 15 sec. The
sequences of the primers used in the present study were as follows:
FOXA2 forward, 5'-CGACTGGAGCAGCTACTATGC-3' and reverse,
5'-ATGTACGTGTTCATGCCGTTC-3'; and GAPDH forward,
5'-CAGGAGGCATTGCTGATGAT-3' and reverse, 5'-GAAGGCTGGGGCTCATTT-3'.
Data were analyzed using the 2-ΔΔCq method (26) and normalized to GAPDH.
Western blotting
Total protein was extracted from cells using RIPA
lysis buffer (Thermo Fisher Scientific, Inc.). Following detection
of the protein concentration using a BCA kit (Thermo Fisher
Scientific, Inc.), equal amounts of protein (30 µg per lane) were
separated by SDS-PAGE on 12% gels and then transferred onto PVDF
membranes (MilliporeSigma). The membranes were blocked with 5%
skimmed milk for 2 h at room temperature, followed by probing with
primary antibodies against FOXA2 (cat. no. ab23630; 1:1,000;
Abcam), cyclooxygenase-2 (Cox2; cat. no. ab179800; 1:1,000; Abcam),
inducible nitric oxide synthase (iNOS; cat. no. ab178945; 1:1,000;
Abcam), NADPH oxidase (Nox)2 (cat. no. ab129068; 1:5,000; Abcam),
Nox4 (cat. no. ab154244; 1:1,000; Abcam), Bcl-2 (cat. no. ab32124;
1:1,000; Abcam), Bax (cat. no. ab32503; 1:1,000; Abcam),
Cleaved-poly(ADP-ribose) polymerase 1 (PARP1; cat. no. ab32064;
1:1,000; Abcam), PARP1 (cat. no. ab227244; 1:1,000; Abcam),
phosphorylated (p)-p38 (cat. no. 9215; 1:1,000; Cell Signaling
Technology, Inc.), p38 (cat. no. 9212; 1:1,000; Cell Signaling
Technology, Inc.), p-STAT3 (cat. no. 94994; 1:1,000; Cell Signaling
Technology, Inc.), STAT3 (cat. no. ab109085; 1:1,000; Abcam),
glucose-regulated protein 78 (GRP78; cat. no. ab21685; 1:1,000;
Abcam), CHOP (cat. no. 5554; 1:1,000; Cell Signaling Technology,
Inc.), X-box binding protein 1 (XBP1; cat. no. ab37152; 1:1,000;
Abcam), activating transcription factor 6 (ATF6; cat. no.
orb381900; 1:1,000; Biorbyt, Ltd.), p-eukaryotic translation
initiation factor 2 subunit α (eIF2α; cat. no. orb15000; 1:1,000;
Biorbyt, Ltd.), eIF2α (cat. no. orb480065; 1:1,000; Biorbyt, Ltd.)
and GAPDH (cat. no. ab9485; 1:2,500; Abcam) at 4˚C overnight.
Subsequently, the membranes were incubated with horseradish
peroxidase-conjugated goat-anti-rabbit IgG secondary antibody (cat.
no. ab6721; 1:2,000; Abcam) for 1 h at room temperature. The blot
signals were developed using Immobilon ECL Ultra Western HRP
Substrate (MilliporeSigma), and were analyzed using ImageJ 1.52a
software (National Institutes of Health).
Cell viability assay
Cell viability was examined using a Cell Counting
Kit-8 (CCK-8) assay. In brief, WI-38 cells were seeded into 96-well
plates at the density of 4x104 cells/well and incubated
with different concentrations of LPS for 24 h at 37˚C.
Subsequently, 10 µl CCK-8 reagent (Beijing Solarbio Science &
Technology Co., Ltd.) was added to each well and the cells were
incubated at 37˚C for 2 h. Finally, the absorbance of each well at
450 nm was measured using a full wavelength microplate reader
(Thermo Fisher Scientific, Inc.). The cell viability was calculated
as follows: Cell viability (%)=experimental group OD/control group
OD x100.
Cell transfection
FOXA2 overexpression vector (oe-FOXA2) was
established by inserting the FOXA2 gene into the pcDNA3.1 vector
(Shanghai GenePharma Co., Ltd.), referring to the empty vector as
the negative control (overexpression-negative control; oe-NC) were
obtained from Shanghai GenePharma Co., Ltd. After indicated
treatment, WI-38 cells seeded in 6-well culture plates
(2x105 cells/well) were transfected with 5 µg/ml oe-NC
or oe-FOXA2 using Lipofectamine® 3000 (Invitrogen;
Thermo Fisher Scientific, Inc.) at 37˚C for 48 h. Cells were
harvested for the subsequent assays 48 h after transfection.
Measurement of cytokines
In brief, the cell medium was collected and the
supernatant was collected after being centrifuged at 2,000 x g for
10 min at 4˚C. The concentrations of inflammatory cytokines,
including TNF-α, IL-6 and IL-1β, in the supernatants from WI-38
cells were assessed using ELISA kits (cat. nos. STA00D, S6050 and
SLB50, respectively; R&D Systems, Inc.) according to the
manufacturer's protocols.
Measurement of reactive oxygen species
(ROS) and oxidative stress markers
The cellular ROS levels were evaluated using a
2',7'-dichlorofluorescein diacetate (DCFH-DA) probe. In brief,
WI-38 cells at a density of 5x105 cells/ml were treated
with 10 µM DCFH-DA (Cayman Chemical Company) for 45 min in the dark
at 37˚C. Afterwards, the images were captured under a fluorescence
microscope (Olympus Corporation).
In brief, the cell medium was collected and the
supernatant was collected after being centrifuged at 2,000 x g for
10 min at 4˚C. The levels of malondialdehyde (MDA), and the
activities of superoxide dismutase (SOD) and catalase (CAT) in the
supernatants from WI-38 cells were assessed using their
corresponding kits from Nanjing Jiancheng Bioengineering Institute
(cat. nos. A003-1, A001-3 and A007-1, respectively) according to
the manufacturer's protocols.
Flow cytometry
WI-38 cells were digested with trypsin, washed with
ice-cold PBS and resuspended with 1X binding buffer (Beyotime
Institute of Biotechnology). Subsequently, Annexin V-FITC (Beyotime
Institute of Biotechnology) and PI (Beyotime Institute of
Biotechnology) were added to stain cells for 30 min in the dark at
4˚C. The apoptotic cells were detected by flow cytometry
(FACSCalibur; BD Biosciences) and FlowJo™ VX10 software (FlowJo
LLC). The apoptosis rate was calculated as follows: Apoptosis rate
%=(number of early apoptotic cells + number of late apoptotic
cells)/total number of cells x100.
Caspase3 activity assay
Total protein was extracted from WI-38 cells using
RIPA lysis buffer (Thermo Fisher Scientific, Inc.) and its
concentration was determined using a BCA kit (Thermo Fisher
Scientific, Inc.). Subsequently, the caspase3 activity was assessed
using a Caspase-3 Activity Assay Kit (cat. no. BC3830; Beijing
Solarbio Science & Technology Co., Ltd.) according to the
manufacturer's protocol. The absorbance at 405 nm was measured
using a full wavelength microplate reader (Thermo Fisher
Scientific, Inc.). According to absorbance at 405 nm of standard
sample in each concentration, the standard curve was drawn. The
caspase-3 activity in each sample was calculated according to the
standard curve, and the average was taken.
Statistical analysis
All data were analyzed utilizing GraphPad Prism
version 8.0 (Dotmatics) and presented as the mean ± standard
deviation of three independent experiments. Comparisons among
groups were performed using one-way ANOVA with Tukey's post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
FOXA2 expression is downregulated in
LPS-induced WI-38 cells
To study the role of FOXA2 in pneumonia, WI-38 cells
were induced by LPS to simulate the inflammatory injury in
pneumonia in vitro, and the expression levels of FOXA2 were
examined. As shown in Fig. 1A and
B, both the protein and mRNA
expression levels of FOXA2 were gradually decreased with increasing
concentrations of LPS, suggesting the downregulation of FOXA2 in an
LPS-induced in vitro model of pneumonia in WI-38 cells.
Additionally, the cell viability was decreased by LPS in a
concentration-dependent manner (Fig.
1C). In particular, 15 µg/ml LPS led to a significant decrease
in WI-38 cell viability and 5 µg/ml LPS led to a slight decrease in
WI-38 cell viability, while 10 µg/ml LPS induced ~50% cell
viability, thus 10 µg/ml LPS was used in subsequent experiments.
Subsequently, oe-FOXA2 was transfected into WI-38 cells. The
results in Fig. 1D and E demonstrated the transfection efficacy
of the FOXA2 overexpression plasmids. Significant elevation of
FOXA2 mRNA and protein expression was observed in the oe-FOXA2
group compared with the oe-NC group. Subsequently, LPS-induced
WI-38 cells were also transfected with oe-FOXA2, and the data
indicated that the decreased FOXA2 expression following LPS
exposure was partly restored by transfection with oe-FOXA2
(Fig. 1F and G).
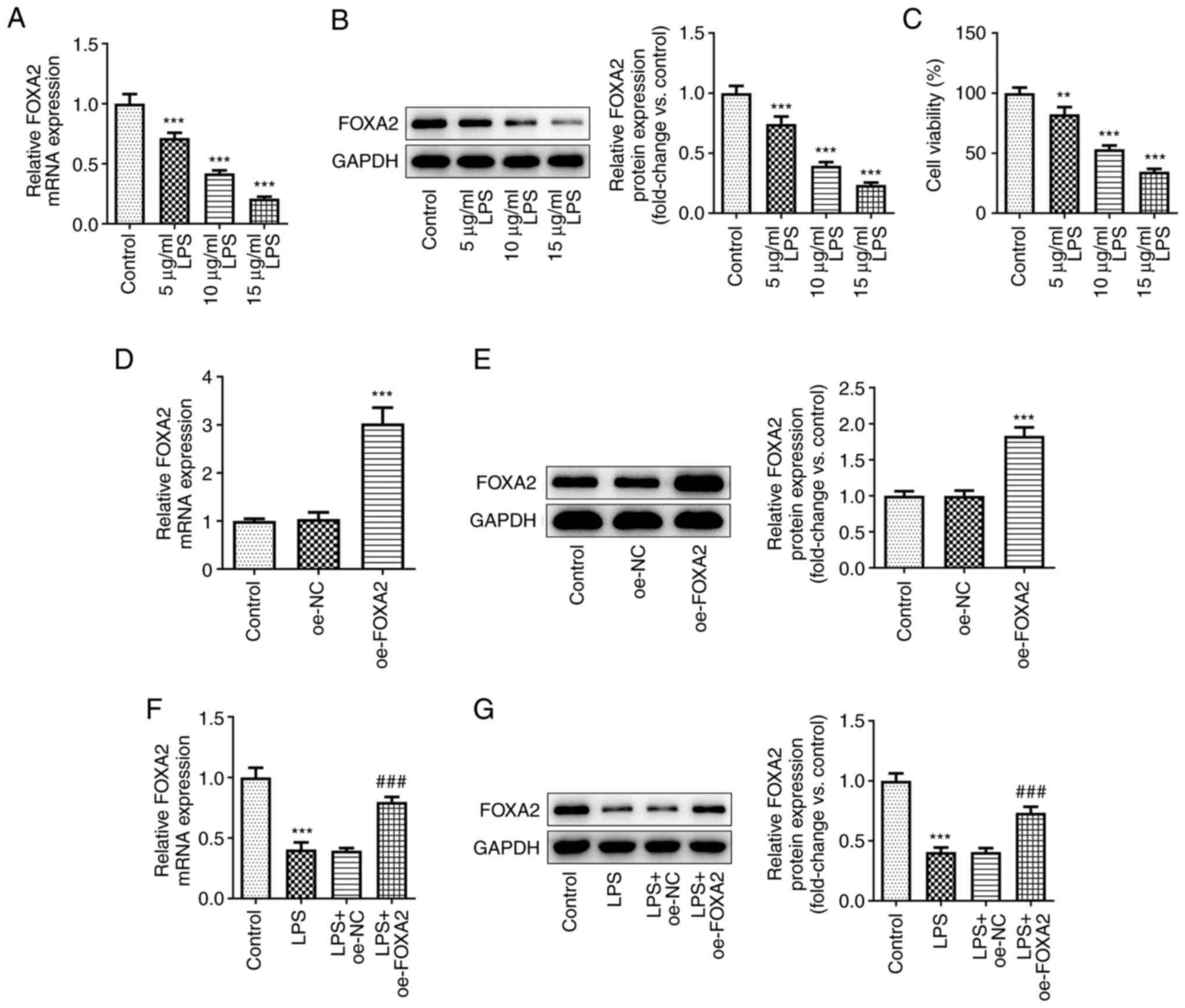 | Figure 1FOXA2 is downregulated in LPS-induced
WI-38 cells. WI-38 cells were induced by different concentrations
(5, 10 and 15 µg/ml) of LPS to simulate the inflammatory injury in
pneumonia in vitro. (A) mRNA and (B) protein expression
levels of FOXA2 were examined using RT-qPCR and western blotting,
respectively. ***P<0.001 vs. Control. (C) Cell
viability was detected using a Cell Counting Kit-8 assay.
**P<0.01 and ***P<0.001 vs. Control.
WI-38 cells were transfected with oe-FOXA2 or oe-NC, and the (D)
mRNA and (E) protein expression levels of FOXA2 were examined using
RT-qPCR and western blotting, respectively.
***P<0.001 vs. oe-NC. The untransfected or
transfected WI-38 cells were induced by LPS (10 µg/ml), and the (F)
mRNA and (G) protein expression levels of FOXA2 were examined using
RT-qPCR and western blotting, respectively.
***P<0.001 vs. Control; ###P<0.001 vs.
LPS + oe-NC. FOXA2, forkhead box protein A2; LPS,
lipopolysaccharide; oe-FOXA2, FOXA2 overexpression vector; oe-NC,
overexpression negative control; RT-qPCR, reverse
transcription-quantitative PCR. |
FOXA2 overexpression alleviates
LPS-induced inflammation and oxidative stress in WI-38 cells
To explore the regulatory role of FOXA2 in
pneumonia, inflammation and oxidative stress levels were examined.
According to the ELISA, the levels of pro-inflammatory cytokines,
including TNF-α, IL-6 and IL-1β, were increased in LPS-exposed
WI-38 cells, whereas FOXA2 overexpression suppressed this increase
(Fig. 2A). In addition, high ROS
activity in the LPS group was observed based on the fluorescence
images in Fig. 2B, and this was
partly abolished by FOXA2 overexpression. Furthermore, LPS also
upregulated MDA levels, and downregulated SOD and CAT activities in
WI-38 cells, which was reversed by FOXA2 overexpression (Fig. 2C), suggesting that FOXA2
overexpression could inhibit LPS-induced oxidative stress in WI-38
cells. Furthermore, western blotting revealed that the expression
levels of inflammation- and oxidative stress-related proteins
(Cox2, iNOS, Nox2 and Nox4) were significantly increased following
LPS treatment, which were then depleted again by FOXA2
overexpression (Fig. 2D).
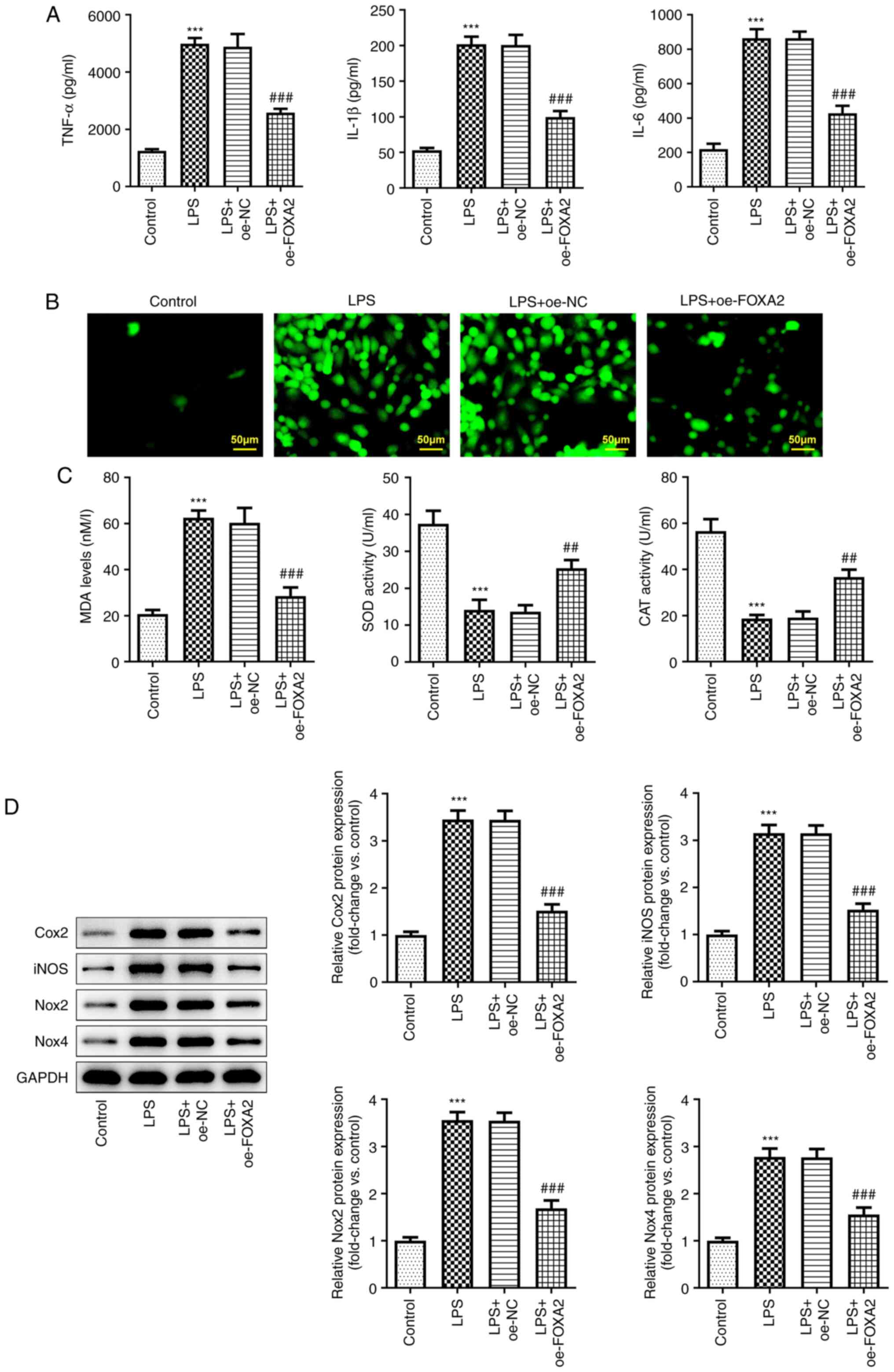 | Figure 2FOXA2 overexpression alleviates
LPS-induced inflammation and oxidative stress in WI-38 cells. (A)
Production of pro-inflammatory cytokines, including TNF-α, IL-6 and
IL-1β, was examined by ELISA. (B) Cellular reactive oxygen species
levels were detected using the 2',7'-dichlorofluorescein diacetate
method. Magnification, x200. Scale bar, 50 µM. (C) MDA levels, SOD
activity and CAT activity were assessed using their corresponding
kits. (D) Protein expression levels of Cox2, iNOS, Nox2 and Nox4
were examined using western blotting. ***P<0.001 vs.
Control; ##P<0.01 and ###P<0.001 vs.
LPS + oe-NC. CAT, catalase; Cox2, cyclooxygenase-2; FOXA2, forkhead
box protein A2; iNOS, inducible nitric oxide synthase; LPS,
lipopolysaccharide; MDA, malondialdehyde; Nox, NADPH oxidase;
oe-FOXA2, FOXA2 overexpression vector; oe-NC, overexpression
negative control; SOD, superoxide dismutase. |
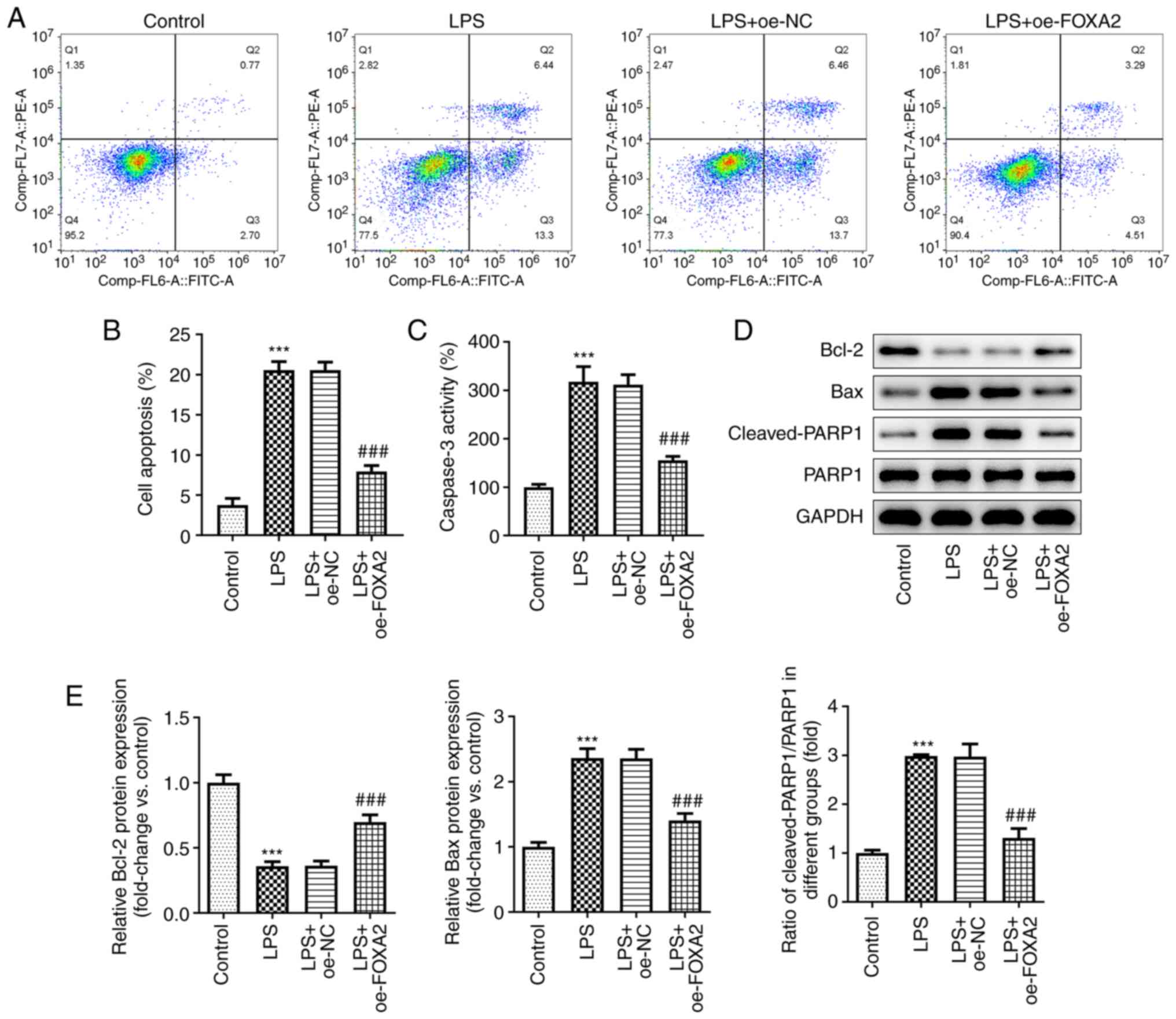
FOXA2 overexpression alleviates
LPS-induced apoptosis in WI-38 cells
The effect of FOXA2 on apoptosis was assessed in
LPS-induced WI-38 cells. As shown in Fig. 3A and B, LPS treatment led to increased
apoptosis in WI-38 cells, which was partially reversed by FOXA2
overexpression, suggesting an anti-apoptotic activity of FOXA2,
which was then verified by the decreased Caspase3 activity
following FOXA2 overexpression in LPS-induced WI-38 cells since
Caspase3 is considered a primary executioner of apoptosis (Fig. 3C). Furthermore, the protein
expression levels of the anti-apoptotic protein Bcl-2 were
downregulated and the protein expression levels of the
pro-apoptotic proteins Bax and Cleaved-PARP1/PARP1 were
significantly upregulated following LPS exposure in WI-38 cells,
and these were then partly reversed by FOXA2 overexpression
(Fig. 3D and E).
FOXA2 restricts endoplasmic reticulum
stress (ERS) by blocking p38/STAT3 signaling
Through western blotting, it was revealed that the
increased p-/total-(t)-p38 and p-/t-STAT3 levels in LPS-treated
WI-38 cells were both decreased after FOXA2 was overexpressed
(Fig. 4A), implying that p38/STAT3
signaling was activated by LPS stimulation in WI-38 cells, but was
inactivated by FOXA2 overexpression. p38 MAPK and STAT3 signaling
are important signal transduction pathways mediating ERS (27,28),
thus whether FOXA2 influenced ERS via p38/STAT3 signaling was then
assessed. As shown in Fig. 4B, LPS
treatment triggered ERS in WI-38 cells, as evidenced by the
upregulated protein expression levels of GRP78, CHOP, XBP1, ATF6
and p/t-eIF2α, which were reduced by FOXA2 overexpression. However,
the inhibitory effect of FOXA2 on GRP78, CHOP, XBP1 and p/t-eIF2α
protein expression levels in LPS-induced WI-38 cells was partly
reversed by additional treatment with U46619, an activator of p38
signaling, indicating that FOXA2 may inhibit LPS-triggered ERS in
WI-38 cells partly through inactivating p38 signaling.
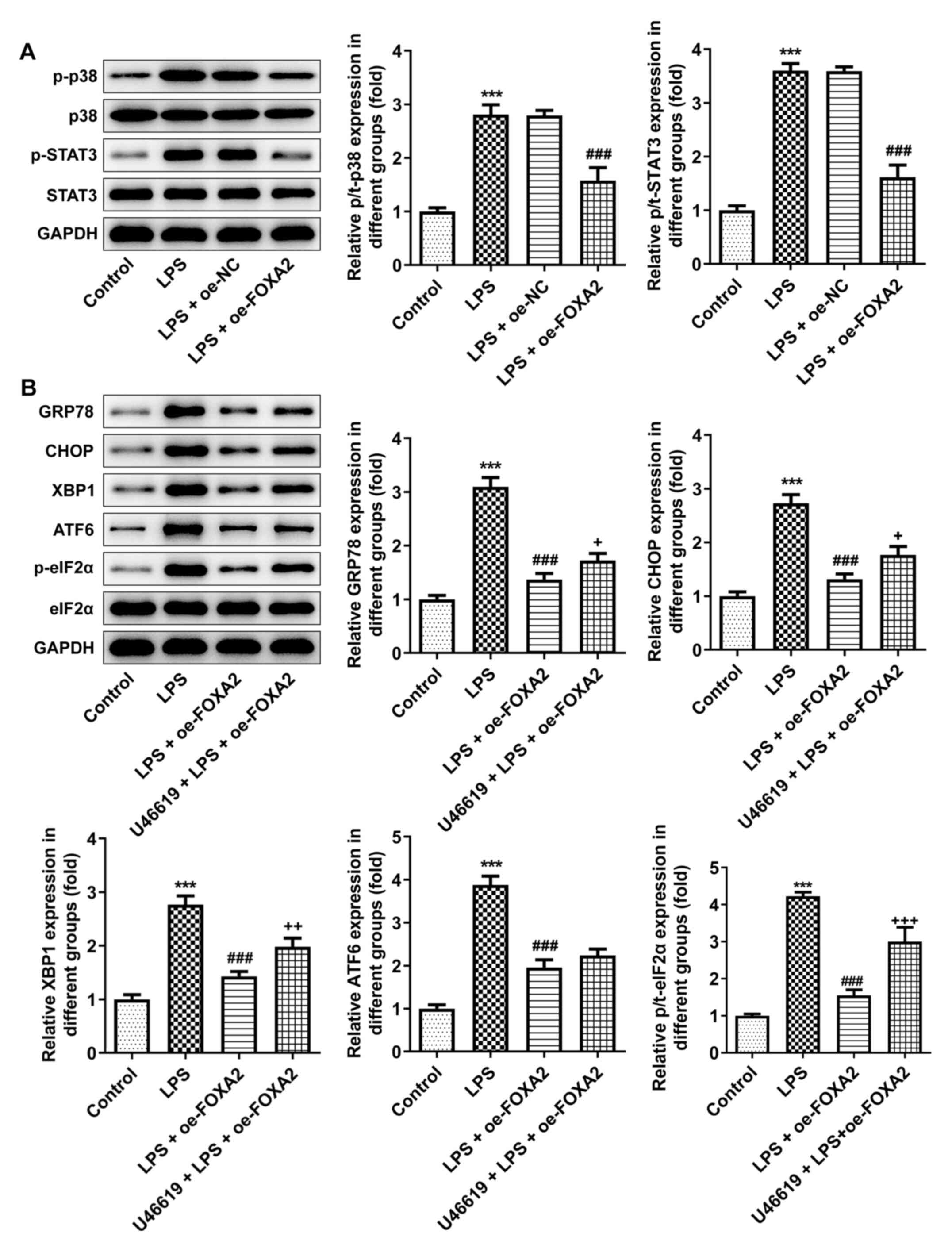 | Figure 4FOXA2 decreases ERS by blocking
p38/STAT3 signaling. (A) Expression levels of p38/STAT3
signaling-related proteins were examined using western blotting.
***P<0.001 vs. Control; ###P<0.001 vs.
LPS + oe-NC. (B) FOXA2-overexpressing WI-38 cells were pretreated
with U46619, an activator of p38 signaling, and then induced by
LPS. The expression levels of ERS-related proteins were detected
using western blotting. ***P<0.001 vs. Control;
###P<0.001 vs. LPS; +P<0.05,
++P<0.01 and +++P<0.001 vs. LPS +
oe-FOXA2. ATF6, activating transcription factor 6; eIF2α,
eukaryotic translation initiation factor 2 subunit α; ERS,
endoplasmic reticulum stress; FOXA2, forkhead box protein A2;
GRP78, glucose-regulated protein 78; LPS, lipopolysaccharide;
oe-FOXA2, FOXA2 overexpression vector; oe-NC, overexpression
negative control; p-, phosphorylated; t-, total; XBP1, X-box
binding protein 1. |
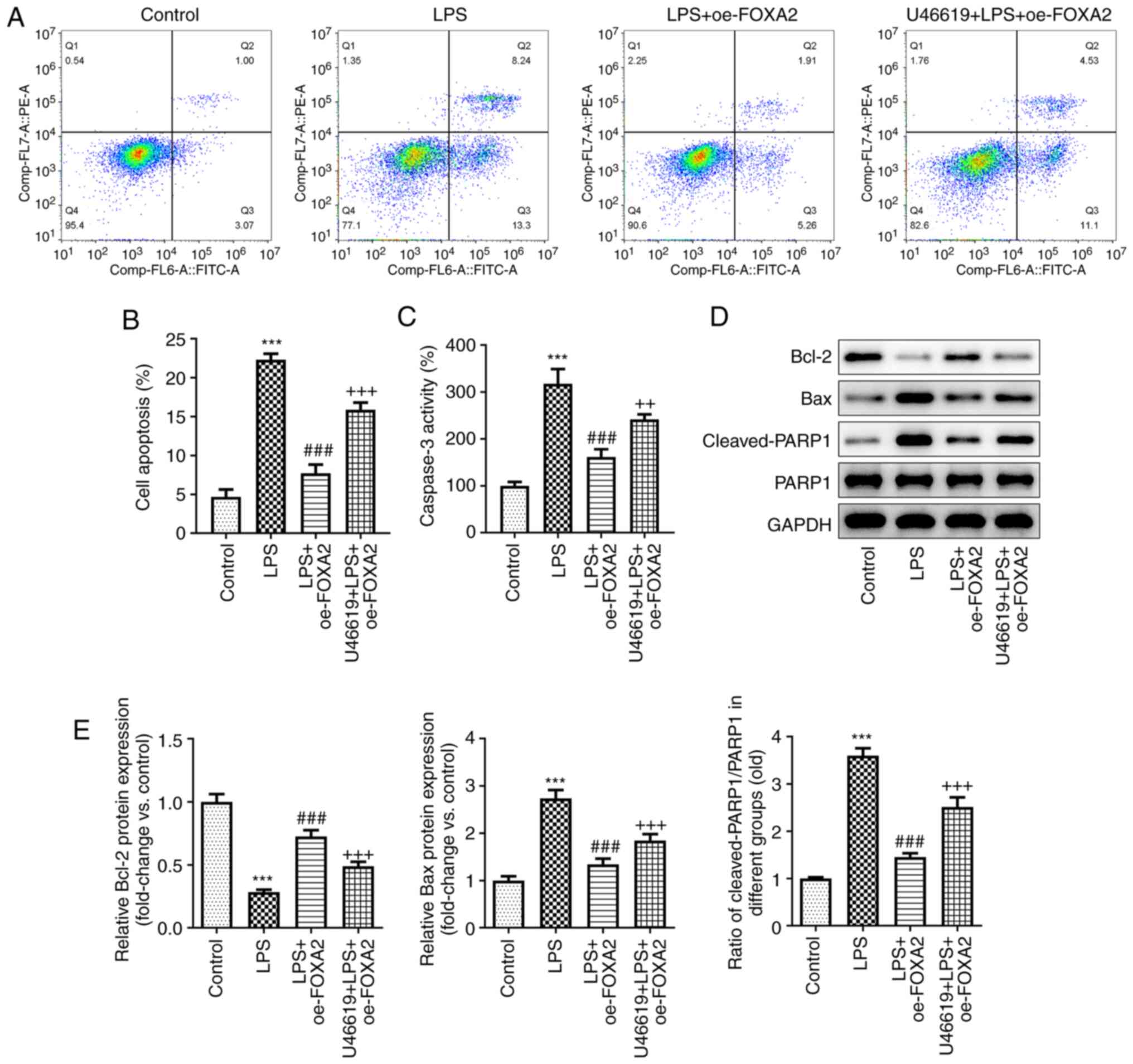
FOXA2 protects WI-38 cells against
LPS-induced oxidative stress, inflammation and apoptosis by
inactivating p38/STAT3 signaling
Finally, to clarify whether FOXA2 attenuated
LPS-induced cell injury by inactivating p38/STAT3 signaling,
oxidative stress, inflammation and apoptosis were examined in
FOXA2-overexpressing WI-38 cells with or without U46619 treatment.
An ELISA revealed that the downregulated production of TNF-α, IL-6
and IL-1β following FOXA2 overexpression in LPS-induced WI-38 cells
was partly elevated by U46619 treatment (Fig. 5A). Furthermore, FOXA2
overexpression was observed to reduce ROS and MDA levels, but
increased SOD and CAT activities in LPS-induced WI-38 cells,
whereas the impact of FOXA2 overexpression on ROS, MDA and CAT
levels were partly reversed by U46619 treatment (Fig. 5B and C). The upregulated protein expression
levels of Cox2, iNOS, Nox2 and Nox4 in the U46619 + LPS + oe-FOXA2
group compared with the LPS + oe-FOXA2 group further demonstrated
that FOXA2 may protect WI-38 cells against LPS-triggered oxidative
stress and inflammation partly through inactivating p38/STAT3
signaling (Fig. 5D). In addition,
the anti-apoptotic activity of FOXA2 in LPS-induced WI-38 cells was
weakened by U46619 treatment, as demonstrated by the elevated
apoptosis rate and Caspase3 activity, accompanied by elevated
protein expression levels of Bax and Cleaved-PARP1/PARP1, and
reduced Bcl-2 protein expression levels in the U46619 + LPS +
oe-FOXA2 group compared with the LPS + oe-FOXA2 group (Fig. 6).
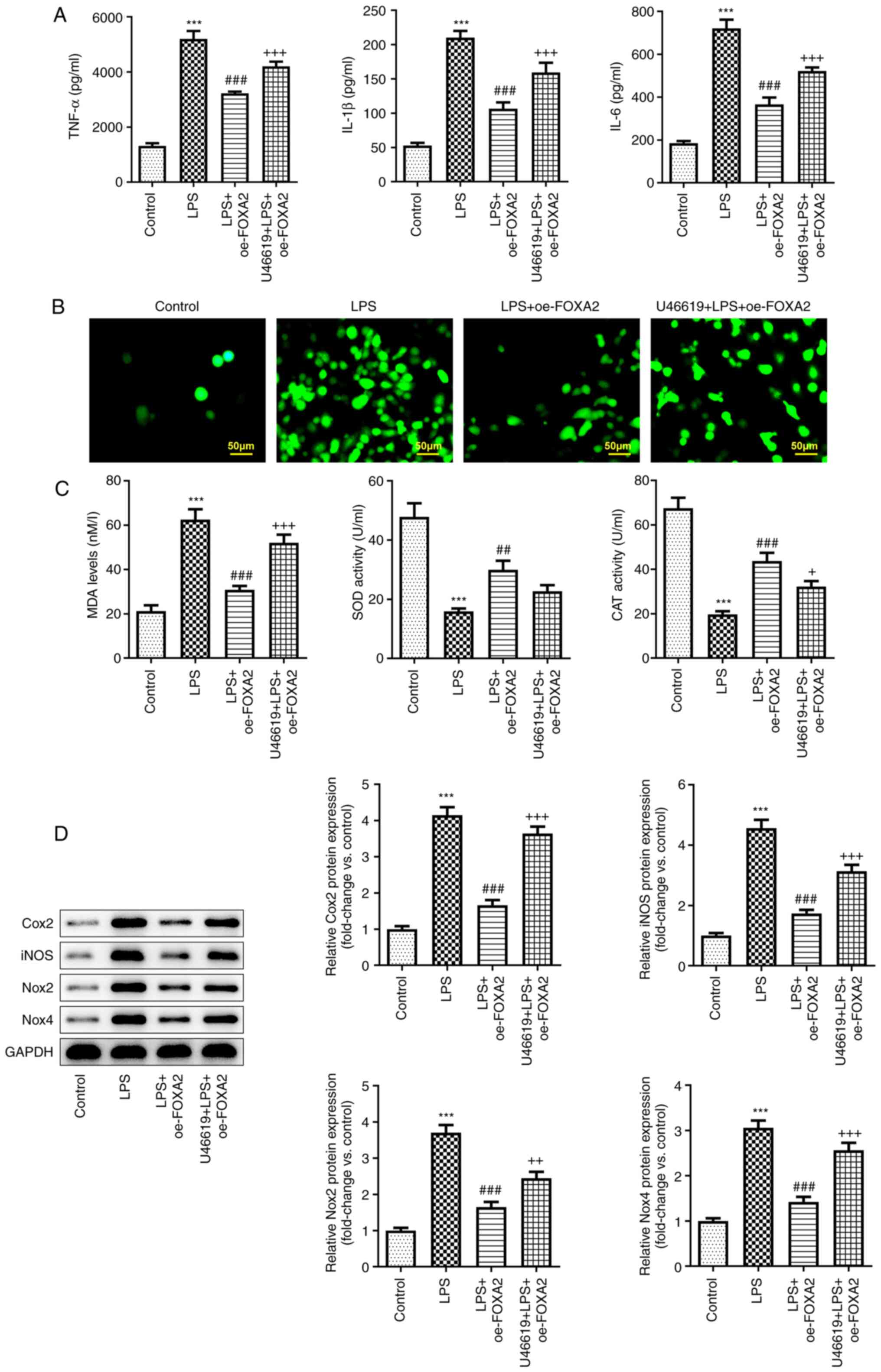 | Figure 5FOXA2 protects WI-38 cells against
LPS-induced oxidative stress and inflammation via inactivation of
p38/STAT3 signaling. (A) Production of pro-inflammatory cytokines,
including TNF-α, IL-6 and IL-1β, was measured using ELISA. (B)
Cellular reactive oxygen species levels were detected using the
2',7'-dichlorofluorescein diacetate method. Magnification, x200.
Scale bar, 50 µM. (C) MDA levels, SOD activity and CAT activity
were assessed using their corresponding kits. (D) Protein
expression levels of Cox2, iNOS, Nox2 and Nox4 were examined using
western blotting. ***P<0.001 vs. Control;
##P<0.01 and ###P<0.001 vs. LPS;
+P<0.05, ++P<0.01 and
+++P<0.001 vs. LPS + oe-FOXA2. CAT, catalase; Cox2,
cyclooxygenase-2; FOXA2, forkhead box protein A2; iNOS, inducible
nitric oxide synthase; LPS, lipopolysaccharide; MDA,
malondialdehyde; Nox, NADPH oxidase; oe-FOXA2, FOXA2 overexpression
vector; SOD, superoxide dismutase. |
Discussion
Pneumonia is a common, complicated and serious
inflammatory disease of the lung, affecting numerous individuals
worldwide, and is associated with a threat to life quality and an
economic burden (29). LPS, a
gram-negative bacterial endotoxin, serves a critical role in the
initiation of pneumonia, and LPS-induced acute lung injury
contributes to the development of pneumonia (30). Therefore, alleviating LPS-induced
lung injury has been widely recognized as an effective strategy for
the treatment of pneumonia (23,31,32).
An in vitro pneumonia model was induced by LPS in WI-38
cells in the present study. Following LPS exposure, the expression
levels of FOXA2 were reduced. Thereafter, gain-of-function
experiments were conducted, which confirmed that FOXA2
overexpression exerted inhibitory effects on the LPS-triggered
inflammatory response, oxidative stress, apoptosis and ERS in WI-38
cells, suggesting FOXA2 as a positive regulator for alleviating
pneumonia. In terms of the mechanism, the protective role of FOXA2
against pneumonia was partially abolished by a p38 activator,
indicating that FOXA2 blocked the progression of pneumonia
partially through blocking of the p38/STAT3 signaling pathway.
Therefore, we hypothesized that FOXA2 could serve as a promising
target for the treatment of pneumonia.
Pneumonia can result in the release of
pro-inflammatory cytokines, neutrophil infiltration and lung tissue
destruction, and the activated neutrophils may further aggravate
the inflammatory response by generating ROS, which in turn triggers
oxidative stress, ultimately leading to apoptosis (33-36).
Therefore, potential drugs or targets that possess
anti-inflammatory, antioxidant and anti-apoptotic properties may be
effective for the treatment of pneumonia. Increasing evidence has
verified FOXA2 as a promising molecular target in pulmonary
diseases through reducing inflammation, oxidative stress and
apoptosis. For example, Yánez et al (37) revealed that FOXA2 deletion in T
cells aggravated the T helper 2 inflammatory response in allergic
airway inflammation. Increase in FOXA2 can also weaken cigarette
smoke extract-induced cell inflammation to delay the progression of
chronic obstructive pulmonary disease (22). Long non-coding RNA-NEF has been
shown to inhibit hyperoxia-induced oxidative stress, inflammation
and apoptosis in lung epithelial tissues by upregulating
FOXA2(38). Consistentlyin the
present study, FOXA2 was found to be aberrantly decreased following
stimulation with increasing concentrations of LPS, suggesting that
FOXA2 mRNA and protein expression was decreased in pneumonia.
Subsequently, FOXA2 overexpression was observed to attenuate the
LPS-induced inflammatory response, oxidative stress and apoptosis
in WI-38 cells, demonstrating that FOXA2 may exert
anti-inflammatory, anti-oxidative stress and anti-apoptotic
activities to protect against pneumonia. In addition, previous
evidence has confirmed the crosstalk between ERS and LPS-induced
lung inflammation (39).
Furthermore, FOXA2 has been demonstrated to serve a vital role in
oxidative stress and ERS, and to exert anti-apoptotic activity by
regulating the cellular inhibitor of apoptosis protein 1 signaling
pathway (40). In the present
study, ERS was observed to be triggered by LPS in WI-38 cells,
whereas FOXA2 overexpression inhibited the activation of ERS, thus
indicating that FOXA2-induced suppression of inflammation,
oxidative stress and apoptosis in pneumonia may be associated with
inactivation of ERS.
The p38 MAPK signaling pathway is a crucial
signaling pathway for the induction of the inflammatory response
and cell apoptosis (41,42). STAT3 is a member of the STAT family
of transcription factors involved in cellular responses to various
cytokines (43). It has been
reported that STAT3 is a substrate of p38 MAPK and inhibition of
p38 suppresses the activation of STAT3(44). p38/STAT3 signaling has been
recognized as a critical pathway participating in
inflammation-related diseases. For instance, nilotinib has been
shown to modulate LPS-induced neuroinflammatory responses by
regulating p38/STAT3 signaling (45). Thymic stromal lymphopoietin may
promote asthmatic airway remodeling by activating p38/STAT3
signaling (44). In agreement with
the aforementioned studies, p38/STAT3 signaling was demonstrated to
be activated by LPS stimulation in WI-38 cells in the present
study, suggesting that p38/STAT3 signaling was activated in
pneumonia. However, this effect was partly weakened by FOXA2
overexpression, suggesting that FOXA2 may exert an inhibitory
effect on p38/STAT3 signaling, which was consistent with the
previous evidence that FOXA2 overexpression could suppress p38 MAPK
signaling to attenuate cigarette smoke-induced lung inflammation
(46). In addition, to verify
whether FOXA2 also functioned in LPS-induced in vitro model
of pneumonia via inactivation of the p38/STAT3 pathway, a p38
activator U46619 was introduced in the present study. Treatment
with U46619 partly weakened the inhibitory effects of FOXA2 on the
LPS-triggered inflammatory response, oxidative stress and
apoptosis, as well as ERS in WI-38 cells, indicating that FOXA2 may
serve a protective role against pneumonia partly via regulation of
the p38/STAT3 signaling pathway.
The present study had some limitations. The
expression profile of FOXA2 was not validated in clinical patients
with pneumonia. The regulatory role of FOXA2 in an animal model of
pneumonia also needs to be verified. In addition, the present study
only used a single embryonic fibroblast cell line and only
addressed a small part of the potential regulatory mechanism of
FOXA2, thus more cell lines should be used for in-depth research of
the molecular mechanism of FOXA2 in pneumonia. All these
limitations need to be addressed in future studies.
In conclusion, to the best of our knowledge, the
present study was the first to reveal that FOXA2 exerted a
protective effect against pneumonia by inhibiting the inflammatory
response, oxidative stress and apoptosis, which may be partially
achieved via modulation of the p38/STAT3 signaling pathway and ERS.
These findings may provide a novel idea for the development of
targeted therapeutic strategies for pneumonia.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Natural Science
Foundation Project of Fujian Province (grant no. 2022J011439).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JX designed the study. ZX, YL, SX and HZ conducted
the experiments to collect data. ZX, YL and SX analyzed and
interpreted the data. ZX drafted the manuscript and JX revised the
manuscript. ZX and JX confirm the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kulkarni D, Wang X, Sharland E, Stansfield
D, Campbell H and Nair H: The global burden of hospitalisation due
to pneumonia caused by Staphylococcus aureus in the under-5
years children: A systematic review and meta-analysis.
EClinicalMedicine. 44(101267)2022.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Liu L, Oza S, Hogan D, Chu Y, Perin J, Zhu
J, Lawn JE, Cousens S, Mathers C and Black RE: Global, regional,
and national causes of under-5 mortality in 2000-15: An updated
systematic analysis with implications for the sustainable
development goals. Lancet. 388:3027–3035. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ma C, Zhang D, Ma Q, Liu Y and Yang Y:
Arbutin inhibits inflammation and apoptosis by enhancing autophagy
via SIRT1. Adv Clin Exp Med. 30:535–544. 2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lee JK, Lee J, Park YS, Lee CH, Yim JJ,
Yoo CG, Kim YW, Han SK and Lee SM: Clinical manifestations of
pneumonia according to the causative organism in patients in the
intensive care unit. Korean J Intern Med. 30:829–836.
2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Calina D RL, Rosu AF, Ianoşi G, Ianoşi S,
Zlatian O, Mitruț R, Docea A, Rogoveanu O and Mitruț P: Etiological
diagnosis and pharmacotherapeutic management of parapneumonic
pleuresy. Farmacia. 64:946–952. 2016.
|
|
6
|
Carvalhaes CG, Sader HS, Rhomberg PR and
Mendes RE: Tedizolid activity against a multicentre worldwide
collection of Staphylococcus aureus and Streptococcus pneumoniae
recovered from patients with pneumonia (2017-2019). Int J Infect
Dis. 107:92–100. 2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Fieldhouse JK, Toh TH, Lim WH, Ting J, Ha
SJ, Hii KC, Kong CI, Wong TM, Wong SC, Warkentien TE and Gray GC:
Surveillance for respiratory syncytial virus and parainfluenza
virus among patients hospitalized with pneumonia in Sarawak,
Malaysia. PLoS One. 13(e0202147)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tanase A, Colita A, Ianosi G, Neagoe D,
Branisteanu DE, Calina D, Docea AO, Tsatsakis A and Ianosi SL: Rare
case of disseminated fusariosis in a young patient with graft vs.
host disease following an allogeneic transplant. Exp Ther Med.
12:2078–2082. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Taheri Y, Jokovic N, Vitorovic J,
Grundmann O, Maroyi A and Calina D: The burden of the serious and
difficult-to-treat infections and a new antibiotic available:
Cefiderocol. Front Pharmacol. 11(578823)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ungureanu A, Zlatian O, Mitroi G, Drocas
A, Tirca T, Calina D, Dehelean C, Docea AO, Izotov BN, Rakitskii
VN, et al: Staphylococcus aureus colonisation in patients from a
primary regional hospital. Mol Med Rep. 16:8771–8780.
2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zlatian O, Balasoiu AT, Balasoiu M,
Cristea O, Docea AO, Mitrut R, Spandidos DA, Tsatsakis AM, Bancescu
G and Calina D: Antimicrobial resistance in bacterial pathogens
among hospitalised patients with severe invasive infections. Exp
Ther Med. 16:4499–4510. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Islam MT, Nasiruddin M, Khan IN, Mishra
SK, Kudrat EZM, Riaz TA, Ali ES, Rahman MS, Mubarak MS, Martorell
M, et al: A perspective on emerging therapeutic interventions for
COVID-19. Front Public Health. 8(281)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Calina D, Sarkar C, Arsene AL, Salehi B,
Docea AO, Mondal M, Islam MT, Zali A and Sharifi-Rad J: Recent
advances, approaches and challenges in targeting pathways for
potential COVID-19 vaccines development. Immunol Res. 68:315–324.
2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Islam MT, Quispe C, Martorell M, Docea AO,
Salehi B, Calina D, Reiner Ž and Sharifi-Rad J: Dietary
supplements, vitamins and minerals as potential interventions
against viruses: Perspectives for COVID-19. Int J Vitam Nutr Res.
92:49–66. 2022.PubMed/NCBI View Article : Google Scholar
|
|
15
|
COVID-19 Excess Mortality Collaborators.
Estimating excess mortality due to the COVID-19 pandemic: A
systematic analysis of COVID-19-related mortality, 2020-21. Lancet.
399:1513–1536. 2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li J, Machado AC, Guo M, Sagendorf JM,
Zhou Z, Jiang L, Chen X, Wu D, Qu L, Chen Z, et al: Structure of
the forkhead domain of FOXA2 bound to a complete DNA consensus
site. Biochemistry. 56:3745–3753. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Jackson DA, Rowader KE, Stevens K, Jiang
C, Milos P and Zaret KS: Modulation of liver-specific transcription
by interactions between hepatocyte nuclear factor 3 and nuclear
factor 1 binding DNA in close apposition. Mol Cell Biol.
13:2401–2410. 1993.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Orstad G, Fort G, Parnell TJ, Jones A,
Stubben C, Lohman B, Gillis KL, Orellana W, Tariq R, Klingbeil O,
et al: FoxA1 and FoxA2 control growth and cellular identity in
NKX2-1-positive lung adenocarcinoma. Dev Cell. 57:1866–1882 e10.
2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Gao H, Yan Z, Sun H and Chen Y: FoXA2
promotes esophageal squamous cell carcinoma progression by ZEB2
activation. World J Surg Oncol. 19(286)2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Connelly ZM, Jin R, Zhang J, Yang S, Cheng
S, Shi M, Cates JM, Shi R, DeGraff DJ, Nelson PS, et al: FOXA2
promotes prostate cancer growth in the bone. Am J Transl Res.
12:5619–5629. 2020.PubMed/NCBI
|
|
21
|
Choi W, Choe S and Lau GW: Inactivation of
FOXA2 by respiratory bacterial pathogens and dysregulation of
pulmonary mucus homeostasis. Front Immunol. 11(515)2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhou Y, Liu L, Tao S, Yao Y, Wang Y, Wei
Q, Shao A and Deng Y: Parthanatos and its associated components:
Promising therapeutic targets for cancer. Pharmacol Res.
163(105299)2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Bai D, Han A and Cong S: The effect of
down-regulation of CCL5 on lipopolysaccharide-induced WI-38
fibroblast injury: A potential role for infantile pneumonia. Iran J
Basic Med Sci. 21:449–454. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yu Y, Yang T, Ding Z and Cao Y:
Circ_0026579 alleviates LPS-induced WI-38 cells inflammation injury
in infantile pneumonia. Innate Immun. 28:37–48. 2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ren H, Meng Q, Yepuri N, Du X, Sarpong JO
and Cooney RN: Protective effects of glutathione on oxidative
injury induced by hydrogen peroxide in intestinal epithelial cells.
J Surg Res. 222:39–47. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hotamisligil GS and Davis RJ: Cell
signaling and stress responses. Cold Spring Harb Perspect Biol.
8(a006072)2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Tang AC, Saferali A, He G, Sandford AJ,
Strug LJ and Turvey SE: Endoplasmic reticulum stress and chemokine
production in cystic fibrosis airway cells: Regulation by STAT3
modulation. J Infect Dis. 215:293–302. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Bjarnason A, Westin J, Lindh M, Andersson
LM, Kristinsson KG, Love A, Baldursson O and Gottfredsson M:
Incidence, etiology, and outcomes of community-acquired pneumonia:
A population-based study. Open Forum Infect Dis.
5(ofy010)2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Liu Y, Bao C, Deng G and Ouyang Y:
Arid2-IR downregulates miR-132-3p through methylation to promote
LPS-induced ALI in pneumonia. Inhal Toxicol. 34:297–303.
2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
An X, Sun X, Hou Y, Yang X, Chen H, Zhang
P and Wu J: Protective effect of oxytocin on LPS-induced acute lung
injury in mice. Sci Rep. 9(2836)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Shi J, Wang H, Liu J, Zhang Y, Luo J, Li
Y, Yang C and Jiang J: Ganoderic acid B attenuates LPS-induced lung
injury. Int Immunopharmacol. 88(106990)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Wang S, Lu B, Liu J and Gu Y: TRIM27
suppresses inflammation injuries in pediatric pneumonia by
targeting TLR4/NF-kappaB signaling pathway. Allergol Immunopathol
(Madr). 50:33–39. 2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Cao X and Wan H and Wan H: Urolithin A
induces protective autophagy to alleviate inflammation, oxidative
stress, and endoplasmic reticulum stress in pediatric pneumonia.
Allergol Immunopathol (Madr). 50:147–153. 2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Cui H, Zhang S, Wu Z, Xu C, Xu D and Jin
Z: Insulin-like growth factor-1 reduces hyperoxia-induced lung
inflammation and oxidative stress and inhibits cell apoptosis
through PERK/eIF2alpha/ATF4/CHOP signaling. Exp Lung Res.
48:187–197. 2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Xu C, Song L, Zhang W, Zou R and Zhu M:
6'-O-galloylpaeoniflorin alleviates inflammation and oxidative
stress in pediatric pneumonia through activating Nrf2 activation.
Allergol Immunopathol (Madr). 50:71–76. 2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yánez DC, Lau CI, Papaioannou E, Chawda
MM, Rowell J, Ross S, Furmanski A and Crompton T: The pioneer
transcription factor Foxa2 modulates T helper differentiation to
reduce mouse allergic airway disease. Front Immunol.
13(890781)2022.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Mei M, Nie J, Sun H, Wang H and Rong L:
LncRNA-NEF regulated the hyperoxia-induced injury of lung
epithelial cells by FOXA2. Am J Transl Res. 12:5563–5574.
2020.PubMed/NCBI
|
|
39
|
Kim HJ, Jeong JS, Kim SR, Park SY, Chae HJ
and Lee YC: Inhibition of endoplasmic reticulum stress alleviates
lipopolysaccharide-induced lung inflammation through modulation of
NF-κB/HIF-1α signaling pathway. Sci Rep. 3(1142)2013.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Wang K, Brems JJ, Gamelli RL and Holterman
AX: Foxa2 may modulate hepatic apoptosis through the cIAP1 pathway.
Cell Signal. 25:867–874. 2013.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zou X and Blank M: Targeting p38 MAP
kinase signaling in cancer through post-translational
modifications. Cancer Lett. 384:19–26. 2017.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Santana FPR, da Silva RC, Ponci V,
Pinheiro A, Olivo CR, Caperuto LC, Arantes-Costa FM, Claudio SR,
Ribeiro DA, Tibério IFLC, et al: Dehydrodieugenol improved lung
inflammation in an asthma model by inhibiting the STAT3/SOCS3 and
MAPK pathways. Biochem Pharmacol. 180(114175)2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Tolomeo M and Cascio A: The multifaced
role of STAT3 in cancer and its implication for anticancer therapy.
Int J Mol Sci. 22(603)2021.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Cao L, Liu F, Liu Y, Liu T, Wu J, Zhao J,
Wang J, Li S, Xu J and Dong L: TSLP promotes asthmatic airway
remodeling via p38-STAT3 signaling pathway in human lung
fibroblast. Exp Lung Res. 44:288–301. 2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Kim J, Lee HJ, Park JH, Cha BY and Hoe HS:
Nilotinib modulates LPS-induced cognitive impairment and
neuroinflammatory responses by regulating P38/STAT3 signaling. J
Neuroinflammation. 19(187)2022.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Tao Y, Sun Y, Wu B, Xu D, Yang J, Gu L and
Du C: Overexpression of FOXA2 attenuates cigarette smoke-induced
cellular senescence and lung inflammation through inhibition of the
p38 and Erk1/2 MAPK pathways. Int Immunopharmacol.
94(107427)2021.PubMed/NCBI View Article : Google Scholar
|