Introduction
Membranous nephropathy (MN) is a glomerular disease
characterized by glomerular basement membrane thickening, podocyte
injury and proteinuria (1,2). MN is divided into idiopathic MN (IMN)
and secondary MN (1). IMN is one
of the most common pathological types, which is an autoimmune
disease (3). The pathogenesis is
that antibodies bind to receptor-associated proteins on the surface
of glomerular podocytes, forming subepithelial in situ
immune complexes to activate complement and form the attacking
membrane complex C5b-9(3). The
effect of sublytic C5b-9 on podocytes leads to changes in podocytes
such as fusion of podocytes processes, apoptosis and abscission
from the glomerular basement membrane, resulting in structural and
functional dysfunction of glomerular filtration barrier and
ultimately proteinuria (4). The
podocyte is the epithelial cell of the renal follicle and
terminally differentiated cell. The destruction of podocyte
integrity and the reduction of podocyte numbers serve an important
role in the course of membranous nephropathy (5). The therapy of IMN mainly relies on
immune suppression, but the effects are not satisfactory.
Therefore, to find a more effective treatment for IMN, it is very
important to study the molecular mechanism of podocyte injury.
The complement system is a set of precisely
regulated protein reaction systems, including >30 soluble
proteins and membrane-bound proteins, which exist widely in serum,
tissue fluid and cell membrane surface (6). Most of the complement components
exist in the form of non-active enzyme precursors, which show
various biological activities when activated by active substances
(7). Depending on cell membrane
properties and surface inhibitors, the assembly of C5b-9 on cell
membranes has certain biological effects. When assembling on the
non-nucleated cell membrane such as that of a red blood cell, C5b-9
forms transmembrane channels to dissolve the cell. In the surface
assembly of nucleated cells, due to the presence of limiting
factors, C5b-9 shallowly inserts into the membrane and cannot cause
membrane perforation and rupture, which is called sublytic C5b-9
(8,9). Sublytic C5b-9 activates intracellular
signaling pathways, prompting cells to release inflammatory factors
and cytokines, leading to cell apoptosis, necrosis, cytolysis,
stress response and proliferation (9). Complement activation is one of the
most important mechanisms in the occurrence and development of
glomerular diseases (10,11). For instance, the end product of
complement activation sublytic C5b-9-induced immune injury of the
renal tubular by regulating the expression level of NLRP3(12). Sublytic C5b-9 could induce podocyte
injury and proteinuria in passive Heymann nephritis (PHN) rats
(13).
Autophagy is a biological process in which
cytoplasmic macromolecules and organelles degrade in membrane
vesicles. Podocytes maintain autophagy in some measure to maintain
their normal physiological functions (14). The autophagy activity of podocytes
is much higher than that of other mammalian cells, suggesting that
podocytes are essential for podocyte function. A previous study
showed that inhibition of autophagy leads to severe podocyte damage
and albuminuria production (15).
In vitro studies showed that C5b-9 could block the fusion of
autophagy lysosomes podocyte, thus causing podocyte apoptosis and
promoting IMN development (16).
Therefore, improving the autophagy activity of podocytes may be a
potential treatment method for IMN.
Transient receptor potential (TRP) family protein
mutation causes a variety of kidney diseases such as hypocalcemia
and polycystic kidney disease (17). The canonical transient receptor
potential 6 (TRPC6) belongs to the TRP superfamily of ion
channel-forming proteins. TRPC6 is a component of the slit
diaphragm and plays an important role in regulating renal function
(18,19). Changes in the expression level of
TRPC6 and channel function lead to calcium influx leading to the
development of glomerular diseases (19). Moller et al (20), found that TRPC6 expression was
increased in the glomerulus of PHN rats. The research found that
dexamethasone can resist podocyte injury by stabilizing TRPC6
expression (21). Furthermore,
TRPC6 was reported to serve an important role in cell autophagy.
For example, the knockdown of TRPC6 can enhance the autophagy of
tubular epithelial cells and thus play a protective role on the
cells in renal ischemia-reperfusion injury (18). In angiotensin II-induced podocyte
apoptosis, TRPC6 knockout can relieve the inhibition of autophagy
and play a protective role in podocytes (22). Researchers found that increased
phosphorylation levels of ERK were detected in sublytic
C5b-9-induced podocytes (23).
Moreover, activating ERK1/2 could block the autophagy of podocytes
in diabetic nephropathy (24).
TRPC6 increased intracellular Ca2+ phosphorylated ERK1/2
to activate ERK1/2 in renal ischemia/reperfusion (18). Furthermore, a previous study
reported that the expression of TRPC6 was elevated in the podocyte
induced by C5b-9(20). However,
the mechanism of TRPC6 in C5b-9-induced podocytes needs to be
further studied.
The present study established a sublytic
C5b-9-induced podocyte injury cell model, detected the effect of
TRPC6 on sublytic C5b-9-induced podocyte injury and verified
whether TRPC6 regulated sublytic C5b-9-mediated inhibition of
autophagy in the podocytes.
Materials and methods
Podocyte culture and establishment of
injury model
Mouse podocyte cell line MPC5 immortalized through
simian virus 40 treatment was purchased from the Cell Bank of the
Chinese Academic of Sciences (Shanghai, China). Cells were cultured
in RPMI 1640 medium (Gibco; Thermo Fisher Scientific, Inc.)
supplied with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.) containing 100 U/ml penicillin-streptomycin and
10 U/ml of mouse recombinant γ-interferon (R&D Systems, Inc.).
To maintain epithelial phenotype, 5x106 cells were
cultured in a humidified atmosphere with 95% air and 5%
CO2 at 33˚C. When reaching 70% confluence, cells were
maintained for 10-14 days on a plate coated with type I collagen
(Gibco; Thermo Fisher Scientific, Inc.) without γ-interferon at
37˚C for differentiation (23).
The fully differentiated cells stopped growing and arborized cells
were kept, which were used in the following experiments.
To establish sublytic C5b-9-induced podocyte injury,
normal human serum (NHS, complement source), obtained from healthy
volunteers in Weifang Yidu Central Hospital, was stimulated with
zymosan to form the C5b-9 membrane attack complex. The method as
described by Ishikawa et al (25) was used in the present study. A
total of 1x107 differentiated podocytes were inoculated
with different concentrations (2, 4, 6, 8, 10 and 12%) of
zymosan-activated serum (ZAS) for 6 h. Heat-inactivated human serum
treated was used as a control. Written informed consent was signed
by all participants in the study.
Lactate dehydrogenase (LDH)
detection
The concentrations of LDH were used to detect
podocyte damage induced by the C5b-9 membrane attack complex
(16). After 1x106
podocytes were treated with ZAS, the medium supernatant was
collected and the released LDH was tested using Quantitative
Lactate Dehydrogenase Assay Kits (Sigma-Aldrich; Merck KGaA)
according to the manufacturer's instructions.
Cytotoxicity=(experimental LDH release values-background
values)/(maximum LDH release values-background values) x100%
Immunofluorescence assay
A total of 1x104 podocytes were fixed
into 4% paraformaldehyde for 30 min at room temperature and
permeabilized with 0.2% Triton X-100 for 15 min at room
temperature. After blocking with 5% BSA (Thermo Fisher Scientific,
Inc.) for 1 h at room temperature, the cells were cultured with
primary antibody anti-C5b-9 (1:100; cat. no. #ab55811; Abcam),
anti-LC3 (1:100; cat. no. #ab192890; Abcam), anti-p62 (1:100; cat.
no. #ab109012; Abcam) at 4˚C overnight. On the second day, the
cells were incubated with Fluorescein-AffiniPure goat anti-rabbit
IgG (1:200; cat. no. #ab150077; Abcam) for 1 h at room temperature.
The nuclei were counterstained with 49,69-diamidino-2-phenylindole
hydrochloride (DAPI) for 15 min at 37˚C in the dark. The
fluorescence intensity image was acquired through a confocal
microscope (Zeiss AG).
Western blotting
Podocytes' total protein was extracted using a lysis
buffer [0.1% Triton X-100, 50 mM Tris (pH 7.0), 100 Mm NaCl, 1 mM
EDTA, 1 mM PMSF] and protease inhibitor cocktail (Sigma-Aldrich;
Merck KGaA). The total protein in the supernatant was quantified
using a BCA Protein Assay kit (cat. no. 23235; Pierce; Thermo
Fisher Scientific, Inc.). Total protein (25 µg loaded per lane) was
separated by 10% SDS-PAGE and transferred onto polyvinylidene
difluoride membranes (Sigma-Aldrich; Merck KGaA). After blocking
with 5% free-fat milk for 1 h at room temperature, the membranes
were incubated with the following primary antibodies: Rabbit
anti-TRPC6 (1:1,000; cat. no. #ab105845; Abcam); rabbit anti-LC3B
(1:1,000; cat. no. #ab192890; Abcam); rabbit anti-p62 (1:10,000;
cat. no. #ab109012; Abcam); rabbit anti-p-ERK (1:1,000; cat. no.
#ab201015; Abcam); rabbit anti-t-ERK (1:5,000; cat. no. #ab184699;
Abcam); and rabbit anti-GAPDH (1:2,500; cat. no. #ab9485; Abcam)
antibody at 4˚C overnight. Following primary incubation, the
membranes were incubated with HRP-conjugated goat anti-rabbit IgG
(1:2,000, Abcam # ab6721) for 2 h at room temperature. Protein
bands were visualized using an enhanced chemiluminescence detection
system (MilliporeSigma) and densitometry quantitative analysis was
carried out using Image J software version 1.46r (National
Institutes of Health).
Cell transfection and treatment
The TRPC6 small interfering (si)RNA (si-TRPC6;
sense, 5'-ATTGATCCTGGATCTAGAGTG-3) and negative control siNC
(sense, 5'-UUCUCCGAACGUGUCACGUTT-3') were synthesized by Guangzhou
RiboBio Co., Ltd. When reaching 7-80% confluence, cells were
transfected with 50 nM si-TRPC6/si-NC using
Lipofectamine® 3000 Transfection Reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) at 37˚C, and 48 h later,
transfected cells were used for further experimentation.
After transfection with si-TRPC6, the cells were
treated with ERK1/2 activator [12-O-tetradecanoylphorbol-13-acetate
(TPA)] for 12 h at 37˚C to activate ERK1/2 in podocytes.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Podocytes' total RNA was harvested using Trizol™
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Total RNA was reverse-transcribed into
cDNA using TaqMan Reverse Transcription Kit (Takara Bio, Inc.),
according to the manufacturer's protocol. qPCR was performed using
the SYBR Green PCR kit (Thermo Fisher Scientific, Inc.), according
to the manufacturer's protocol and analyzed using the ABI Prism
7500 Software version 2.0.6 (Thermo Fisher Scientific, Inc.). PCR
amplification conditions were: 95˚C for 10 min, followed by 40
cycles of denaturation at 95˚C for 15 sec, 60˚C for 20 sec and 72˚C
for 30 sec. The following primer pairs were used for qPCR: TRPC6
forward, 5'-GTTAACTGCGATGATCAATAGTT-3' and reverse,
5'-GACTTGGTACAAGATTGAAGG-3'; GAPDH forward,
5'-CTGCCCAGAACATCATCC-3' and reverse, 5'-CTCAGATGCCTGCTTCAC-3'. The
RNA expression level of TRPC6 was quantified using the
2-∆∆Cq method (26) and
normalized to the internal reference gene GAPDH.
Phalloidin staining
Sublytic C5b-9 treated podocytes were fixed into 4%
paraformaldehyde for 40 min at room temperature. Whereafter, the
cells were permeabilized in 0.5% Triton X-100 for 10 min at room
temperature. Subsequently, podocytes were incubated in 200 µl
rhodamine phalloidin reagent (Sigma-Aldrich; Merck KGaA) for 30 min
in the dark at room temperature. And DAPI was applied to stain the
nucleus. The images were observed under a confocal microscope
(Zeiss AG).
Cell Counting Kit-8 (CCK-8) assay
The viability of podocytes transfected with siTRPC6
was detected using a CCK-8 (Dojindo Laboratories, Inc.). In brief,
5x103 cells/well were seeded into a 96-well plate. After
incubating, 10 µl CCK-8 was added to the medium for 2 h. The
absorbance values were tested in a microplate reader (Guangzhou
RiboBio Co., Ltd.) at 450 nm.
Flow cytometry analysis
The cell apoptosis rate of damaged podocytes was
examined through double staining with Annexin V-FITC Apoptosis
Detection kit (cat. no. #C1062M; Beyotime Institute of
Biotechnology). Cells were collected and cultured with 5 µl annexin
V-FITC for 15 min at room temperature, and continually cultured
with 10 µl PI for 10 min in the dark at room temperature.
Subsequently, cell apoptosis was analyzed through the CytoFLEX
(Beckman Coulter, Inc.).
Transmission electron microscopy
(TEM)
TEM was performed according to a previous method
(18). Podocytes were fixed first
in 2.5% glutaraldehyde for 1 h at 4˚C and then in 1%
OsO4 for 1 h at room temperature, and dehydrated using
30, 50, 70, 80, 90, 95 and 100% ethanol in series. Cells were
stained with 2% uranyl acetate for 20 min at room temperature and
lead citrate for 5 min at room temperature. After drying, the
autophagosomes were visualized using an Hitachi 7500 transmission
electron microscope aperture grid with 100 µm at 80 kV, x10K in
HC-1 mode (Hitachi, Ltd.).
Lysosomal enzyme activity
detection
The effect of TRPC6 on the lysosomal fusion stage of
autophagy was evaluated through the lysosomal enzyme activity. The
lysosomal enzyme activity was detected using Cathepsin B Activity
Assay kit and Cathepsin L Activity Assay kit (cat. nos. #MAK387 and
#CBA023; Sigma-Aldrich; Merck KGaA) according to the manufacturer's
instructions. A total of 1x105 podocytes treated with 10
µM Chloroquine (CQ) for 48 h was used as positive control.
Statistical analysis
Data are presented as means ± standard deviation.
Statistical analysis was carried out using GraphPad Prism 8.3
(Dotmatics). The Student's t-test was used to compare the
difference between the two groups, whilst one-way ANOVA followed by
Bonferroni post hoc test was used to analyze the difference between
>2 groups. All experiments were repeated three times. P<0.05
was considered to indicate a statistically significant
difference.
Results
The expression of TRPC6 is increased
in sublytic C5b-9-induced podocytes
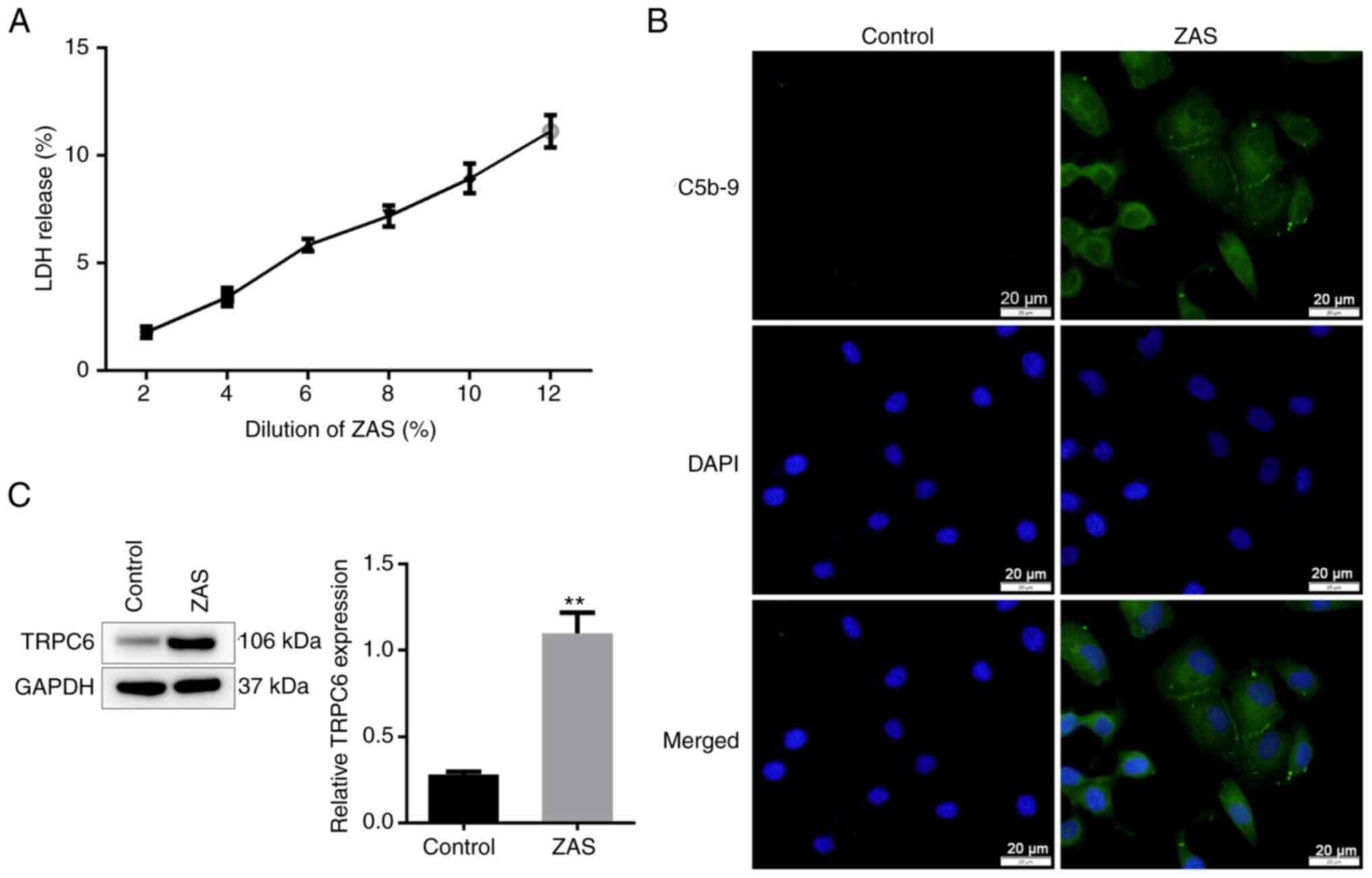
To investigate the specific damage mechanism of
C5b-9 to podocytes, ZAS was used to establish the podocyte injury
model. The LDH release increased following treatment with ZAS in a
dose-dependent manner, indicating damage to the podocytes'
membranes. The LDH level was <10% when ZAS was at 10% dilution
and this condition was chosen to establish a C5b-9-induced
podocytes injury model in subsequent experiments (Fig. 1A). To investigate whether sublytic
C5b-9 accumulated in podocytes, an immunofluorescent assay was
performed to evaluate the level of C5b-9. Compared with the cells
treated with heat-inactivated human serum, C5b-9 was successfully
deposited in podocytes treated with ZAS (Fig. 1B). Subsequently, the results of
western blotting showed that the expression of TRPC6 was enhanced
in sublytic C5b-9-induced podocytes (Fig. 1C).
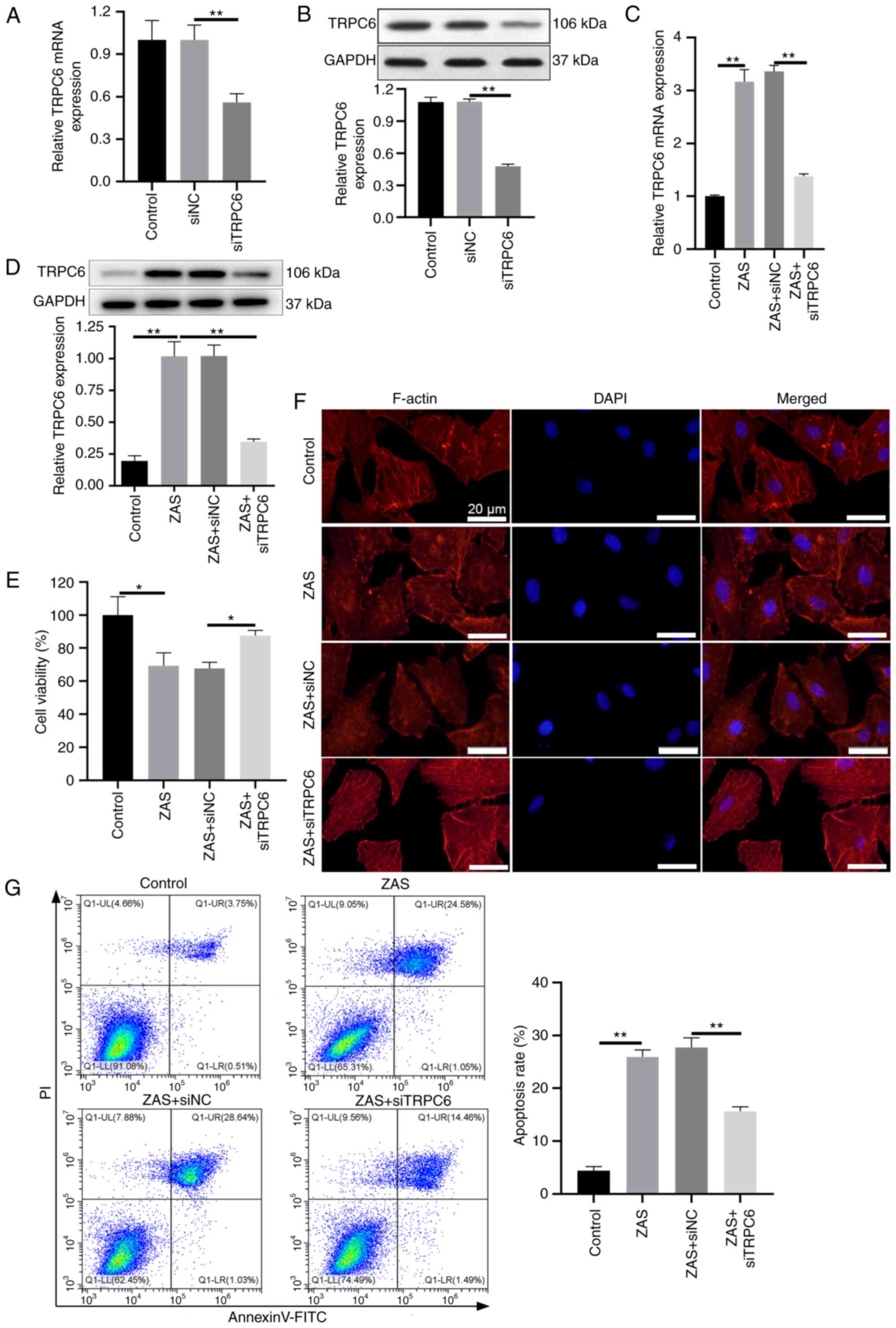
Knockdown of TRPC6 attenuates the
damage of podocytes induced by C5b-9
To determine whether TRPC6 serves a role in sublytic
C5b-9-induced podocyte damage, TRPC6 was knocked down through
siTRPC6 transfection in podocytes before sublytic C5b-9 induction.
As shown in Fig. 2A and B, the mRNA and protein levels of TRPC6
were significantly decreased in podocytes transfected with siTRPC6.
Moreover, transfection of siTRPC6 decreased the expression of TRPC6
in sublytic C5b-9-induced podocytes, compared with that in
C5b-9-induced podocytes transfected with siNC (Fig. 2C and D). Subsequently, the viability of
podocytes was detected using CCK-8 assay. C5b-9 decreased the
viability of podocytes and the knockdown of TRPC6 reversed the
reduced viability in sublytic C5b-9-induced podocytes (Fig. 2E). Moreover, the results of
phalloidin staining showed that sublytic C5b-9 visibly destroyed
actin stress fiber and the loss of actin stress fiber was relieved
by TRPC6 inhibition in sublytic C5b-9-induced podocytes (Fig. 2F). In addition, flow cytometry
analysis was applied to examine the effect of TRPC6 on the
apoptosis of sublytic C5b-9-induced podocytes and the result
revealed that the apoptosis rate was 25.63% in the C5b-9 group,
whilst it was reduced to 9.68% in C5b-9 + siTRPC6 group (Fig. 2G). Together, the results suggested
that TRPC6 knockdown inhibited sublytic C5b-9-induced podocyte
injury and cell apoptosis.
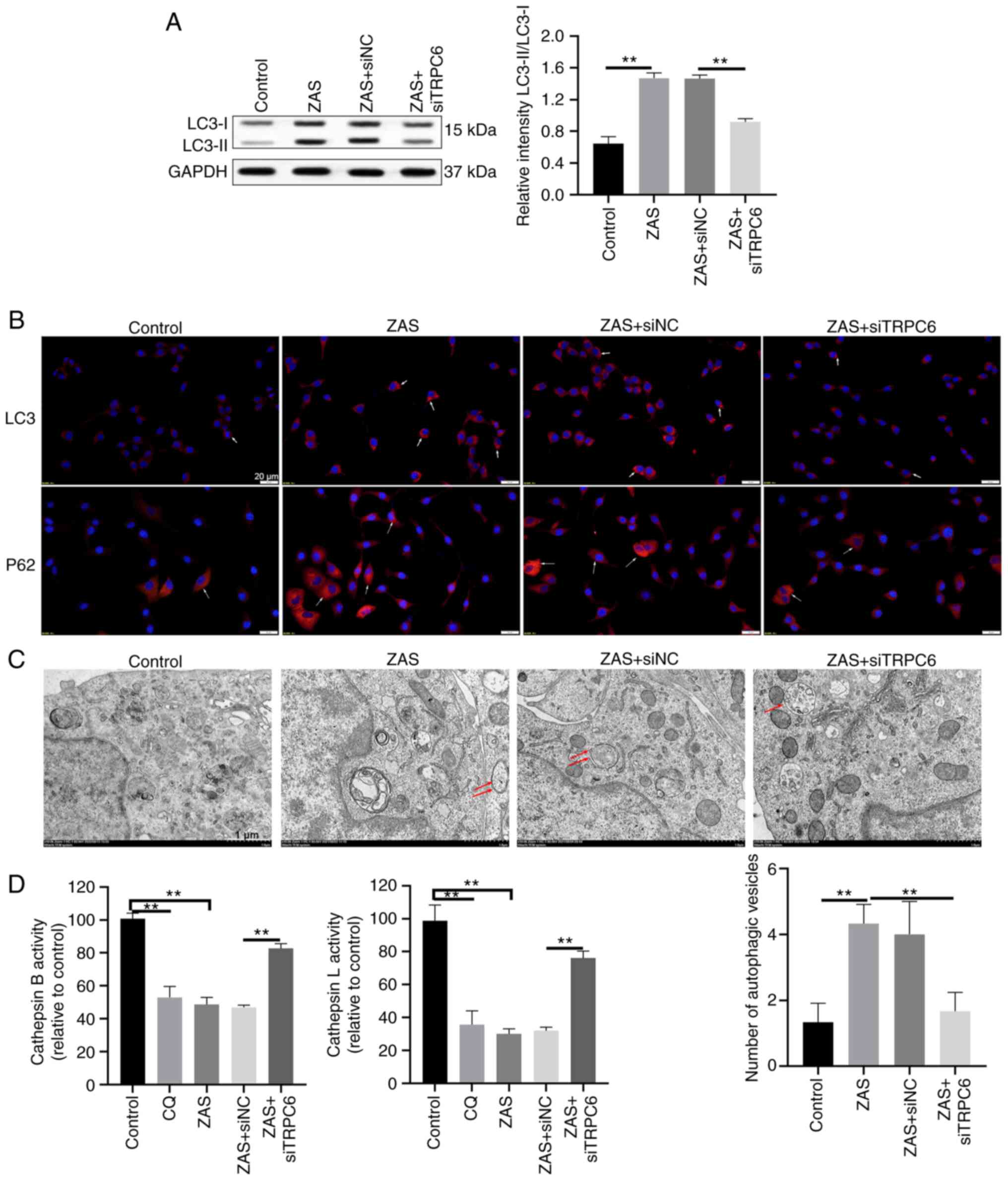
TRPC6 inhibition promotes the
autophagy of C5b-9-induced podocytes
Subsequently, it was evaluated whether TRPC6
regulates autophagy in sublytic C5b-9-induced podocytes. As shown
in Fig. 3A, sublytic C5b-9
improved the level of LC3-II and the knockdown of TRPC6 suppressed
LC3-II turnover mediated by sublytic C5b-9 in podocytes. Similarly,
the positive puncta of LC3 raised in sublytic C5b-9-treated
podocytes, whereas it weakened in sublytic C5b-9-treated podocytes
transfected with siTRPC6 (Fig.
3B). The effect of TRPC6 on the autophagy of sublytic
C5b-9-treated podocytes was also evaluated through fluorescence
staining of p62. After podocytes were exposed to ZAS, the positive
staining of p62 was increased (Fig.
3B), which indicated that sublytic C5b-9 suppressed the
autophagy of podocytes. Knockdown of TRPC6 partially bated the
level of p62 in sublytic C5b-9-treated podocytes (Fig. 3B). In addition, autophagic vesicles
were observed under TEM in sublytic C5b-9-treated podocytes. The
number of autophagosomes and damaged lysosomes increased in the
C5b-9 group (Fig. 3C). The number
of autophagy-lysosomes raised in the C5b-9 + siTRPC6 group,
compared with that in the C5b-9 and C5b-9 + siNC groups (Fig. 3C). CQ was shown to inhibit the
fusion between autophagosome and lysosome and the degradation of
lysosome protein by increasing the pH value of lysosome (27). Subsequently, lysosome enzyme
activity in podocytes was evaluated. The activity of both cathepsin
B and cathepsin L was decreased in sublytic C5b-9-treated podocytes
and chloroquine-treated podocytes, while the knockdown of TRPC6
reversed the effect on the activity of cathepsin B and cathepsin L
in sublytic C5b-9-treated podocytes (Fig. 3D). The above results indicated that
knockdown of TRPC6 facilitated autophagy in sublytic C5b-9-induced
podocytes.
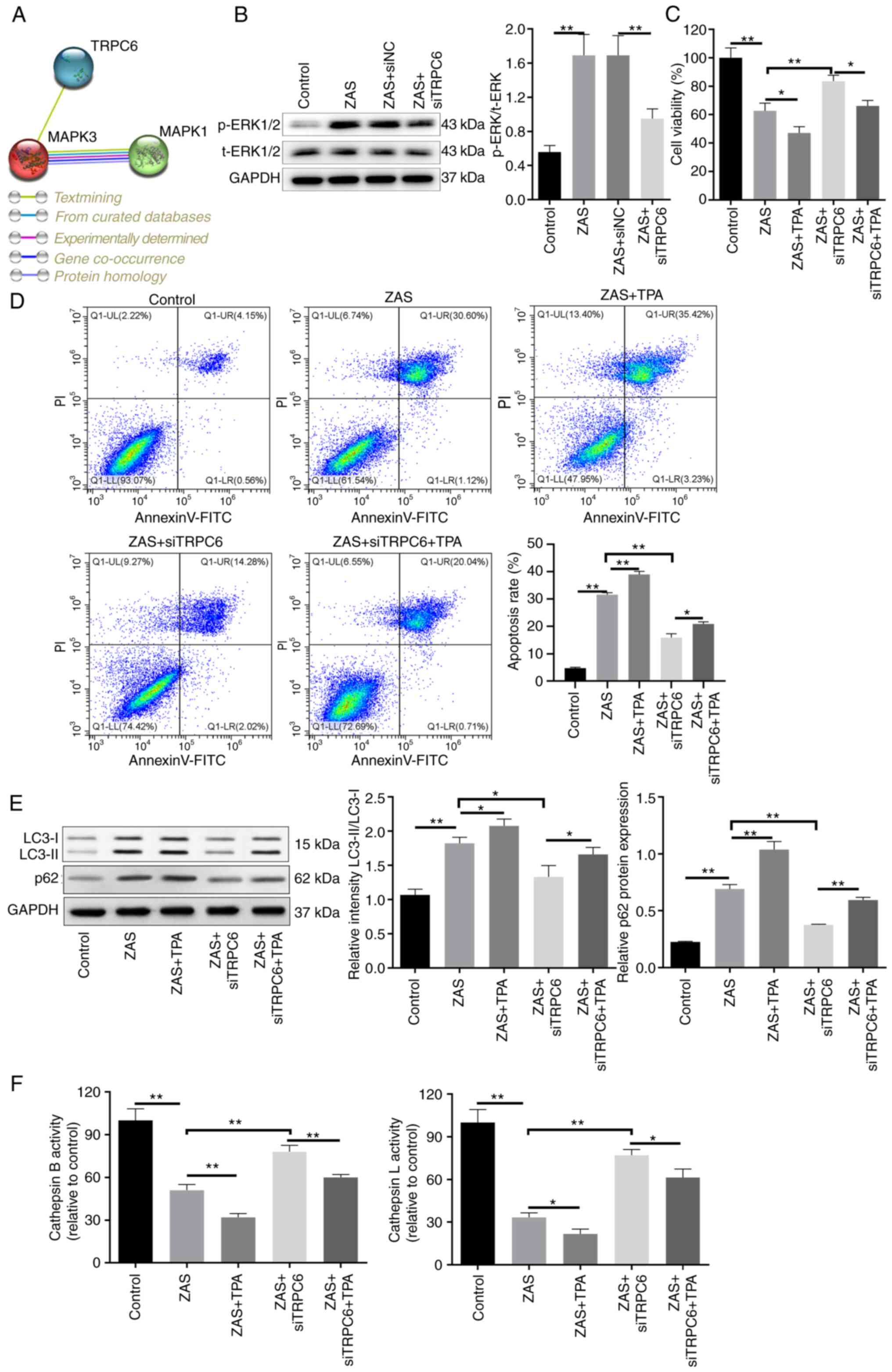
Knockdown of TRPC6 protects sublytic
C5b-9-induced podocytes via inhibiting phosphorylation of
ERK1/2
Activated ERK was found in sublytic C5b-9-induced
podocytes (23) and the regulatory
relationship between TRPC6 and ERK1/2 (MAPK3/1) was predicted using
the STRING database (Fig. 4A).
Subsequently, whether TRPC6 regulated the activation of ERK1/2 in
sublytic C5b-9-induced podocytes was elucidated. It was revealed
through western blot analysis that sublytic C5b-9 enhanced the
phosphorylation level of ERK1/2 and TRPC6 knockdown decreased the
level of p-ERK1/2 (Fig. 4B).
Subsequently, TPA was used to treat siTRPC6 transfected podocytes
and CCK-8 assay and flow cytometry analysis showed that TPA
inhibited viability and increased the apoptosis of sublytic
C5b-9-induced podocytes (Fig. 4C
and D). The addition of TPA partly
reversed the protective effect of TRPC6 knockdown on sublytic
C5b-9-induced podocytes (Fig. 4C
and D). To deeply investigate the
effect of ERK1/2 on the autophagy of TRPC6 inhibitor transfected
and sublytic C5b-9-induced podocytes, the protein levels of LC3 and
p62 were assessed. It was presented that TPA improved LC3-II and
p62 levels in sublytic C5b-9-induced podocytes (Fig. 4E). Likewise, compared with those in
the ZAS + siTRPC6 group, the levels of LC3-II and p62 were elevated
in ZAS + siTRPC6+TPA group (Fig.
4E). TPA also partially reversed the effect of TRPC6 on the
activity of cathepsin B and cathepsin L (Fig. 4F). All of these findings
illustrated that TRPC6 regulated autophagy by activating
phosphorylation ERK1/2 in sublytic C5b-9-induced podocytes.
Discussion
MN accounts for 25-40% of nephrotic syndromes and
IMN accounts for about two-thirds of MN (2). The ineffective treatment of patients
with IMN results in chronic urinary protein, hypoproteinemia and
metabolic disorders, which eventually lead to end-stage renal
failure (2). Therefore, it is very
important to further study the pathogenesis of IMN and to find
effective therapeutic targets and methods. A previous study
reported that the level of the C5b-9 membrane attack complex, a
product of complement activation, was higher than that of the
control in the urine of MN animal model (28). The sublytic C5b-9 can directly
cause podocyte injury (29). The
podocytes, basement membrane and endodermal cells form the
glomerular filtration barrier. The damage of podocytes can lead to
proteinuria (5). So podocyte
damage is the direct cause of renal glomerular disease. In the
current study, normal human serum was stimulated with zymosan to
obtain a sublytic C5b-9 complex and established a sublytic
C5b-9-induced podocyte injury model. The deposition of C5b-9 was
observed in the podocyte injury model. Meanwhile, cell viability
was reduced and the apoptosis rate was raised in sublytic
C5b-9-induced podocytes. These results were consistent with the
finding of Zheng et al (30) which reported that sublytic C5b-9
induced podocyte injury. In addition, F-actin is the main support
of the foot process structure of podocytes. Sublytic C5b-9 could
dissolve F-actin and adhesion plaque complexes (29) and the present research verified
that F-actin was detected in sublytic C5b-9-induced podocytes.
TRPC6 participates in several physiological and
pathological processes. For example, appropriate mechanical force
activated TRPC6 to regulate periodontal tissue reconstruction
(31). TRPC6 promoted LPS-induced
inflammatory response in bronchial epithelial cells (32). TRPC6 interacts with podocin,
nephrin, actin-4 and CD2-associated proteins to maintain the
structure and function of glomerular podocytes (19). Enhanced expression of TRPC6 was
found in human urinary protein nephropathy such as IMN (19), while sublytic C5b-9 mediated
elevated expression of TRPC6 in cultured podocytes in vitro
(20). The combination of PLA2R on
podocytes and anti-PLA2R antibodies activated the TRPC6 channel and
increased the expression of TRPC6, resulting in the structural and
functional impairment of podocytes (33). In the present study, increased
expression of TRPC6 was detected in sublytic C5b-9-induced
podocytes. After siTRPC6 transfection in sublytic C5b-9-induced
podocytes, the cell damage was weakened.
Autophagy is a biological process of degradation of
cytoplasmic macromolecules and organelles in capsular vesicles
(27). Cell removes cellular
wastes and carries out structural reconstruction to maintain
protein metabolism balance and cellular environment stability
through autophagy (27).
Podocytes, glomerular endothelial cells and renal tubular
epithelial cells all maintain basal autophagy to keep the normal
physiological functions of the cells (34,35).
In lupus nephritis, autophagy exhibited a cell-protective effect on
antibody-induced podocyte damage (36). The synergistic action of autophagy
in epithelial and podocyte cells inhibits diabetic
glomerulosclerosis (37). A
previous study found that in sublytic C5b-9-treated podocytes, the
number of autophagosomes was increased but the number of autophagic
lysosomes was decreased and the lysosomes were destroyed, which
indicated that sublytic C5b-9 inhibited the autophagy process
(16). In addition, previous
research reported that TRPC6 promoted oxidative stress-mediated
renal tubular epithelial cell apoptosis by inhibiting autophagy and
suppressing autophagy in Ang-II-induced podocyte apoptosis
(22,38). Knockdown of TRPC6 improved renal
ischemia/reperfusion by enhancing autophagy (18). The results of the present study
showed that the knockdown of TRPC6 could weaken podocyte apoptosis
by facilitating the progress of autophagy.
Subsequently, the present authors deeply
investigated the mechanism of TRPC6 in the regulation autophagy of
sublytic C5b-9 treated podocytes. By using the STRING database, it
was predicted that ERK1/2 and TRPC6 had a regulatory relationship.
Moreover, the result of the western blot assay revealed that the
level of phosphorylation of ERK1/2 was elevated in sublytic C5b-9
treated podocytes and reduced due to the transfection of siTRPC6.
The present findings were consistent with the discovery of Zheng
et al (30) that sublytic
C5b-9 promoted podocyte injury by activating the phosphorylation of
ERK1/2. TRPC6-increased intracellular Ca2+ could
activate ERK1/2 phosphorylation to promote inflammation in
bronchial epithelial cells (32)
and TRPC6 interacted with ERK1/2(39). In addition, it was also shown that
apelin blocked the autophagy of podocytes in diabetic nephropathy
by activating ERK1/2(24). In the
present study, an ERK1/2 activator was applied to activate the
phosphorylation of ERK1/2 in siTRPC6 transfected podocytes induced
by sublytic C5b-9. The results demonstrated that the ERK1/2
activation partially reversed the effect of TRPC6 on sublytic
C5b-9-treated podocytes. However, whether the specific regulatory
mechanism between TRPC6 and ERK1/2 was directly or indirectly
acting through the involvement of other molecules in podocytes
needs further investigation.
The present data revealed that sublytic C5b-9
induced the injury of podocytes as well as knockdown of TRPC6
weakened the injury of sublytic C5b-9-induced podocytes.
Furthermore, the knockdown of TRPC6 promoted the progression of
autophagy by inhibiting the phosphorylation of ERK1/2 in sublytic
C5b-9-induced podocytes. The present study provided important
evidence for the regulatory roles of TRPC6 on autophagy in sublytic
C5b-9-induced podocytes which might provide a new basal mechanism
for the treatment of IMN.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YYL and YFF conceived and designed the project, and
wrote the paper. JL acquired and analyzed the data. YFF modified
the manuscript. YYL and YFF confirm the authenticity of all the raw
data. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
This research was approved by the Ethical Committee
of Weifang Yidu Central Hospital (approval no. 2017-032 and obeyed
the principles of the Declaration of Helsinki. Written informed
consent was signed by all participants in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Keri KC, Blumenthal S, Kulkarni V, Beck L
and Chongkrairatanakul T: Primary membranous nephropathy:
Comprehensive review and historical perspective. Postgrad Med J.
95:23–31. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Safar-Boueri L, Piya A, Beck LH Jr and
Ayalon R: Membranous nephropathy: Diagnosis, treatment, and
monitoring in the post-PLA2R era. Pediatr Nephrol. 36:19–30.
2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Liu W, Gao C, Liu Z, Dai H, Feng Z, Dong
Z, Zheng Y, Gao Y, Tian X and Liu B: Idiopathic membranous
nephropathy: Glomerular pathological pattern caused by extrarenal
immunity activity. Front Immunol. 11(1846)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ma H, Sandor DG and Beck LH Jr: The role
of complement in membranous nephropathy. Semin Nephrol. 33:531–542.
2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Nagata M: Podocyte injury and its
consequences. Kidney Int. 89:1221–1230. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Galindo-Izquierdo M and Pablos Alvarez JL:
Complement as a therapeutic target in systemic autoimmune diseases.
Cells. 10(148)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yang P, Skiba NP, Tewkesbury GM, Treboschi
VM, Baciu P and Jaffe GJ: Complement-mediated regulation of
apolipoprotein E in cultured human RPE cells. Invest Ophthalmol Vis
Sci. 58:3073–3085. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Fishelson Z and Kirschfink M: Complement
C5b-9 and Cancer: Mechanisms of cell damage, cancer counteractions,
and approaches for intervention. Front Immunol.
10(752)2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Takano T, Elimam H and Cybulsky AV:
Complement-mediated cellular injury. Semin Nephrol. 33:586–601.
2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Bateman RM, Sharpe MD, Jagger JE, Ellis
CG, Solé-Violán J, López-Rodríguez M, Herrera-Ramos E,
Ruíz-Hernández J, Borderías L, Horcajada J, et al: 36th
International symposium on intensive care and emergency medicine:
Brussels, Belgium. 15-18 March 2016. Crit Care. 20 (Suppl
2)(S94)2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Tan Y and Zhao MH: Complement in
glomerular diseases. Nephrology (Carlton). 23 (Suppl 4):S11–S15.
2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Xie H, Yang L, Yang Y, Jiang W, Wang X,
Huang M, Zhang J and Zhu Q: C5b-9 membrane attack complex activated
NLRP3 inflammasome mediates renal tubular immune injury in
trichloroethylene sensitized mice. Ecotoxicol Environ Saf.
208(111439)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wang X, Liu J, Tian R, Zheng B, Li C,
Huang L, Lu Z, Zhang J, Mao W, Liu B, et al: Sanqi oral solution
mitigates proteinuria in rat passive heymann nephritis and blocks
podocyte apoptosis via Nrf2/HO-1 Pathway. Front Pharmacol.
12(727874)2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhou XJ, Klionsky DJ and Zhang H:
Podocytes and autophagy: A potential therapeutic target in lupus
nephritis. Autophagy. 15:908–912. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Yoshibayashi M, Kume S, Yasuda-Yamahara M,
Yamahara K, Takeda N, Osawa N, Chin-Kanasaki M, Nakae Y, Yokoi H,
Mukoyama M, et al: Protective role of podocyte autophagy against
glomerular endothelial dysfunction in diabetes. Biochem Biophys Res
Commun. 525:319–325. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Liu WJ, Li ZH, Chen XC, Zhao XL, Zhong Z,
Yang C, Wu HL, An N, Li WY and Liu HF: Blockage of the
lysosome-dependent autophagic pathway contributes to complement
membrane attack complex-induced podocyte injury in idiopathic
membranous nephropathy. Sci Rep. 7(8643)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Liu X, Yao X and Tsang SY:
Post-Translational modification and natural mutation of TRPC
Channels. Cells. 9(135)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hou X, Huang M, Zeng X, Zhang Y, Sun A, Wu
Q, Zhu L, Zhao H and Liao Y: The Role of TRPC6 in renal
ischemia/reperfusion and cellular hypoxia/reoxygenation injuries.
Front Mol Biosci. 8(698975)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Dryer SE, Roshanravan H and Kim EY: TRPC
channels: Regulation, dysregulation and contributions to chronic
kidney disease. Biochim Biophys Acta Mol Basis Dis. 1865:1041–1066.
2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Moller CC, Wei C, Altintas MM, Li J, Greka
A, Ohse T, Pippin JW, Rastaldi MP, Wawersik S, Schiavi S, et al:
Induction of TRPC6 channel in acquired forms of proteinuric kidney
disease. J Am Soc Nephrol. 18:29–36. 2007.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yu S and Yu L: Dexamethasone resisted
podocyte injury via stabilizing TRPC6 expression and distribution.
Evid Based Complement Alternat Med. 2012(652059)2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Shengyou Y and Li Y: The effects of
siRNA-silenced TRPC6 on podocyte autophagy and apoptosis induced by
AngII. J Renin Angiotensin Aldosterone Syst. 16:1266–1273.
2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhang MH, Fan JM, Xie XS, Deng YY, Chen
YP, Zhen R, Li J, Cheng Y and Wen J: Ginsenoside-Rg1 protects
podocytes from complement mediated injury. J Ethnopharmacol.
137:99–107. 2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Liu Y, Zhang J, Wang Y and Zeng X: Apelin
involved in progression of diabetic nephropathy by inhibiting
autophagy in podocytes. Cell Death Dis. 8(e3006)2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ishikawa S, Tsukada H and Bhattacharya J:
Soluble complex of complement increases hydraulic conductivity in
single microvessels of rat lung. J Clin Invest. 91:103–109.
1993.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Schmittgen TD and Livak KJ: Analyzing
Real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Pasquier B: Autophagy inhibitors. Cell Mol
Life Sci. 73:985–1001. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Jefferson JA, Pippin JW and Shankland SJ:
Experimental models of membranous nephropathy. Drug Discov Today
Dis Models. 7:27–33. 2010.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Papagianni AA, Alexopoulos E, Leontsini M
and Papadimitriou M: C5b-9 and adhesion molecules in human
idiopathic membranous nephropathy. Nephrol Dial Transplant.
17:57–63. 2002.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zheng R, Deng Y, Chen Y, Fan J, Zhang M,
Zhong Y, Zhu R and Wang L: Astragaloside IV attenuates complement
membranous attack complex induced podocyte injury through the MAPK
pathway. Phytother Res. 26:892–898. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
31
|
Wang L, Liang H, Sun B, Mi J, Tong X, Wang
Y, Chen M, Yu L, Pan J, Liu S, et al: Role of TRPC6 in periodontal
tissue reconstruction mediated by appropriate stress. Stem Cell Res
Ther. 13(401)2022.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhou LF, Chen QZ, Yang CT, Fu ZD, Zhao ST,
Chen Y, Li SN, Liao L, Zhou YB, Huang JR and Li JH: TRPC6
contributes to LPS-induced inflammation through ERK1/2 and p38
pathways in bronchial epithelial cells. Am J Physiol Cell Physiol.
314:C278–C288. 2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Putta P, Smith AH, Chaudhuri P,
Guardia-Wolff R, Rosenbaum MA and Graham LM: Activation of the
cytosolic calcium-independent phospholipase A2 β isoform
contributes to TRPC6 externalization via release of arachidonic
acid. J Biol Chem. 297(101180)2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Hartleben B, Godel M, Meyer-Schwesinger C,
Liu S, Ulrich T, Kobler S, Wiech T, Grahammer F, Arnold SJ,
Lindenmeyer MT, et al: Autophagy influences glomerular disease
susceptibility and maintains podocyte homeostasis in aging mice. J
Clin Invest. 120:1084–1096. 2010.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Fang L, Zhou Y, Cao H, Wen P, Jiang L, He
W, Dai C and Yang J: Autophagy attenuates diabetic glomerular
damage through protection of hyperglycemia-induced podocyte injury.
PLoS One. 8(e60546)2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Qi YY, Zhou XJ, Cheng FJ, Hou P, Ren YL,
Wang SX, Zhao MH, Yang L, Martinez J and Zhang H: Increased
autophagy is cytoprotective against podocyte injury induced by
antibody and interferon-α in lupus nephritis. Ann Rheum Dis.
77:1799–1809. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lenoir O, Jasiek M, Henique C, Guyonnet L,
Hartleben B, Bork T, Chipont A, Flosseau K, Bensaada I, Schmitt A,
et al: Endothelial cell and podocyte autophagy synergistically
protect from diabetes-induced glomerulosclerosis. Autophagy.
11:1130–1145. 2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hou X, Xiao H, Zhang Y, Zeng X, Huang M,
Chen X, Birnbaumer L and Liao Y: Transient receptor potential
channel 6 knockdown prevents apoptosis of renal tubular epithelial
cells upon oxidative stress via autophagy activation. Cell Death
Dis. 9(1015)2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Farmer LK, Rollason R, Whitcomb DJ, Ni L,
Goodliff A, Lay AC, Birnbaumer L, Heesom KJ, Xu SZ, Saleem MA and
Welsh GI: TRPC6 binds to and activates calpain, independent of its
channel activity, and regulates podocyte cytoskeleton, cell
adhesion, and motility. J Am Soc Nephrol. 30:1910–1924.
2019.PubMed/NCBI View Article : Google Scholar
|