1. Introduction
Parkinson's disease (PD), which affects 6.1 million
individuals globally, is a common neurodegenerative disorder
(1). The pathological features of
this disease include aggregation of synuclein α [SNCA, also termed
α-synuclein (α-Syn)], resulting in the formation of inclusions
called Lewy neurites (LNs) and Lewy bodies (LBs), apoptosis of
dopaminergic (DA) neurons in the pars compacta of the substantia
nigra (SN) and high levels of neuroinflammation (2,3). The
main clinical manifestations of PD consist of movement disorders,
such as bradykinesia, postural instability, myotonia and static
tremor (2). Furthermore, patients
with PD may have nonmotor symptoms, including mood disorders,
hyposmia, autonomic nervous dysfunction, rapid eye movement sleep
behavior disorder and cognitive decline (3-5).
Notably, some of the aforementioned nonmotor disorders may develop
several years before the appearance of motor complications
(6).
It has previously been reported that the
pathogenesis of PD involves multiple risk factors, such as
environmental factors [for example, drugs and pesticides such as
rotenone, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and
paraquat], aging and genetic factors (such as SNCA, leucine-rich
repeat kinase 2 and PTEN-induced kinase 1) (7). Through genetic testing of patients
with early-onset PD, missense mutations of SNCA genes have been
reported (such as, A53T, A53E, H50Q, G51D, E46K and A30P) (Fig. 1C), which suggested that α-Syn may
be involved in the development of familial PD (8-10).
Two possible α-Syn pathogenic substitutions, A18T and A29S
(Fig. 1C), have also been
previously reported; however, their potential pathogenicity remains
to be further investigated (11).
Previous studies on patients with PD with triplication or
duplication of the SNCA gene have reported that overexpression of
α-Syn is causally involved in PD, which leads to rapid progression
of the disease (9,10,12).
SNCA gene mutations may cause pathological aggregation of α-Syn,
resulting in the formation of insoluble amyloidogenic fibrils
(13). Furthermore, pathological
α-Syn can be endocytosed by other neurons and then misfold normal
α-Syn proteins in these cells, which leads to the propagation of
misfolded α-Syn from cell to cell in a ‘prion-like’ fashion
(13). Previous studies have
reported that pathological α-Syn produces a variety of neurotoxic
effects, such as impairing mitochondrial function, producing
reactive oxygen species (ROS) (14), affecting neuronal membrane function
(15,16), inducing glial cells to release
inflammatory factors (17-20),
interfering with iron metabolism (21,22)
and causing neuronal presynaptic terminal function loss and
dopaminergic depletion (9,23,24),
which ultimately lead to DA neuronal death and the pathogenesis of
PD. Of note, patients with identical gene mutations generally do
not exhibit similar clinical manifestations, which indicates that
the etiology of PD is caused by complex interactions among genetic
factors, environmental influences and aging processes (25).
Neuroinflammation is an important hallmark of the
pathogenesis and development of neurodegenerative disorders,
including PD (26). There is
growing evidence that both innate and acquired immunity contribute
to the onset of PD (27,28). The innate immune reaction elicited
by microglia can lead to neuronal apoptosis and disease progression
(1). T cell infiltration has been
reported in the brain tissues of both patients with PD and a mouse
PD model (28), which suggests the
involvement of acquired immunity in PD pathogenesis (1). In PD, α-Syn interacts with glial
cells and peripheral immune cells. Microglia and astrocytes are
able to phagocytose α-Syn (29,30)
and various forms of α-Syn can activate microglia and astrocytes,
which cause a neuroinflammatory reaction (31). Nitrated α-Syn contributes to
harmful T helper cell maturation, which leads to severe neural
damage (32). Additionally,
astrocytes and oligodendrocytes are closely associated with the
inflammatory response. It has previously been reported that the
severity of neuronal loss in the SN may be related to the number of
oligodendrocytes and astrocytes with α-Syn inclusions in the brains
of patients with PD (5).
Aging is considered a major risk factor for the
onset of PD and the PD incidence rises with advancing age (26). During aging, the brain undergoes
structural and functional alterations and can suffer from chronic
low-grade neuroinflammation (33).
Neuroinflammation is also one of the features of the aging brain
(34). The aging immune system
interacts with environmental and genetic factors, resulting in an
acceleration of PD pathology (25). In the aging brain, the
intracellular clearance mechanism is impaired (35), thus, glial cells have a reduced
ability to phagocytose and degrade α-Syn (36). In SN neurons, there is an
age-associated increase in intracellular levels of α-Syn (23). Large toxic α-Syn aggregates cannot
be metabolized during aging because of the impaired
autophagy-lysosomal function, leading to chronic inflammation and
neuronal death (37). Animal
experiments have shown that aging increases the probability of
α-Syn propagation, which results in a predisposition to α-Syn
pathology (38,39). In addition, recombinant α-Syn can
induce glial cell senescence and change the expression of cellular
aging markers, such as by decreasing the expression levels of high
mobility group box 1 (HMGB1) and Lamin B1, and increasing the
expression level of p21(40).
In this review, the structure and toxicity of α-Syn,
age-related α-Syn pathogenicity in PD, the effects of α-Syn on
glial cells and peripheral immune cells and the interaction of
aging and α-Syn in glial cells are discussed. Elucidating the
relationship between α-Syn, aging and neuroinflammation is
essential for understanding the pathogenesis of PD.
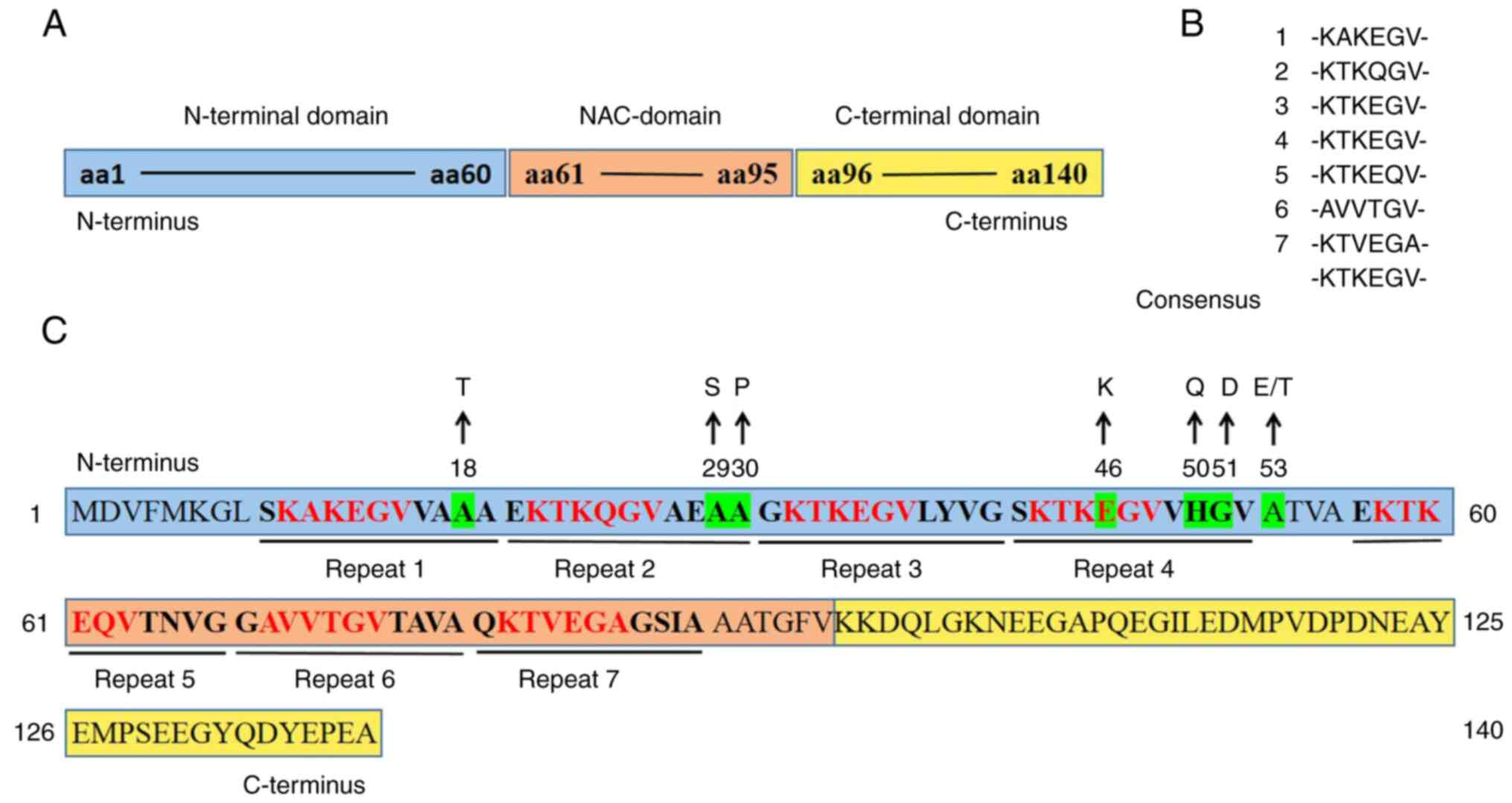
2. α-synuclein structure
α-Syn, encoded by the SNCA gene on human chromosome
4q21, is a small soluble acidic protein with an average molecular
weight of 14 kDa (16,41). The full-length α-Syn protein
contains 140 amino acids (aa) and comprises three main distinct
domains, namely, the N-terminal domain (aa, 1-60), the middle
domain (aa, 61-95) and the C-terminal domain (aa, 96-140) (Fig. 1A) (17,42,43).
A total of seven imperfect 11-aa-residue repeats can
be observed in α-Syn (44), of
which four repeats are in the N-terminal domain (23,42,43,45,46).
Each 11-aa-residue repeat (XKTKEGVXXXX) in the N-terminus contains
a highly conserved hexameric motif (KTKEGV) (9,40,42,46-48),
although certain aa sites of the hexameric motif are sometimes
replaced (Fig. 1B) (47). It has previously been reported that
the N-terminal domain, consisting almost exclusively of this
11-aa-residue repeat, is prone to form an amphipathic α-helical
structure resembling the lipid-binding domain of the exchangeable
apolipoprotein (9,10,46).
The N-terminal domain is associated with lipid membrane binding,
which may be a synergistic effect of the 11-aa-residue repeats.
Lipid binding is significantly reduced when the N-terminal domain
is truncated (10). Furthermore,
the truncation of N-terminal repeats increases the production of
β-sheet-enriched structures, while adding extra repeats results in
the opposite effect (12).
Additionally, the N-terminal domain carries an excess of seven
positive charges, which may interact with the negatively charged
C-terminal domain (49-51).
In human α-Syn, the middle domain with three
additional 11-aa-residue repeats is a highly amyloidogenic and
relatively hydrophobic region, which is also known as a
non-A-β-amyloid component (NAC) domain (23,42,43,45,46).
The NAC domain contains one extra positive charge and is slightly
electropositive (49). It has been
reported that the NAC domain serves a crucial role in α-Syn
aggregation and fibrillogenesis. The residues (aa, 71-82) in the
NAC domain, together with residues (aa, 36-42) in the N-terminal
domain, contribute to the aggregation of α-Syn (31). It has been reported that the NAC
domain can form a cross β-sheet structure (40); however, the glycosylated NAC region
may suppress the aggregation of α-Syn (10). Moreover, the NAC domain may be
involved in α-Syn aggregation, sensing membrane properties and
interactions with metal ions, proteins and vesicles (9,52).
The C-terminal domain, a highly acidic area,
includes 15 carboxylic acid groups and is rich in proline (42,53).
Due to the large numbers of acidic groups and proline residues, the
C-terminal domain has no propensity to form a specific structure;
however, it randomly forms turns and loops in solution (9,42,53).
There are certain binding sites in the C-terminal domain that are
responsible for interactions with ligands, ions and small molecules
(17,49,50).
In addition, it has previously been reported that the C-terminal
domain, enriched in negative charges, may protect the NAC domain
from aggregation by interacting with the positively charged
N-terminal domain to form an antiaggregating structure (49-51).
C-terminal truncation greatly promotes fibrillization of α-Syn due
to the charge imbalance of the N-/C-terminal domain (8,10).
The multiple posttranslational modifications in the C-terminus may
disturb long-range interactions between the N- and C-termini,
leading to exposure of the hydrophobic NAC region and subsequent
adoption of the β-sheet conformation (9). C-terminal truncation, as well as
N-terminal truncation, occur in both patients with PD and healthy
individuals (8,10).
3. α-synuclein toxicity
The physiological role of α-Syn protein has not been
fully identified in normal neurons, but α-Syn may be involved in
the regulation of DA biosynthesis, neurotransmitter release,
synaptic plasticity, synaptic vesicle maintenance, axon
regeneration and mitochondrial function (1,9,10,31).
There are several possible mechanisms by which α-Syn is transferred
between neurons. As α-Syn lacks sequences needed for secretion,
α-Syn is released through a nonclassical mechanism involving
tunneling nanotubes (TNTs) and exosomes (54). Another possible mechanism for α-Syn
release is through the leakage of small amounts of the protein
between pre- and post-synapses (Fig.
2) (54).
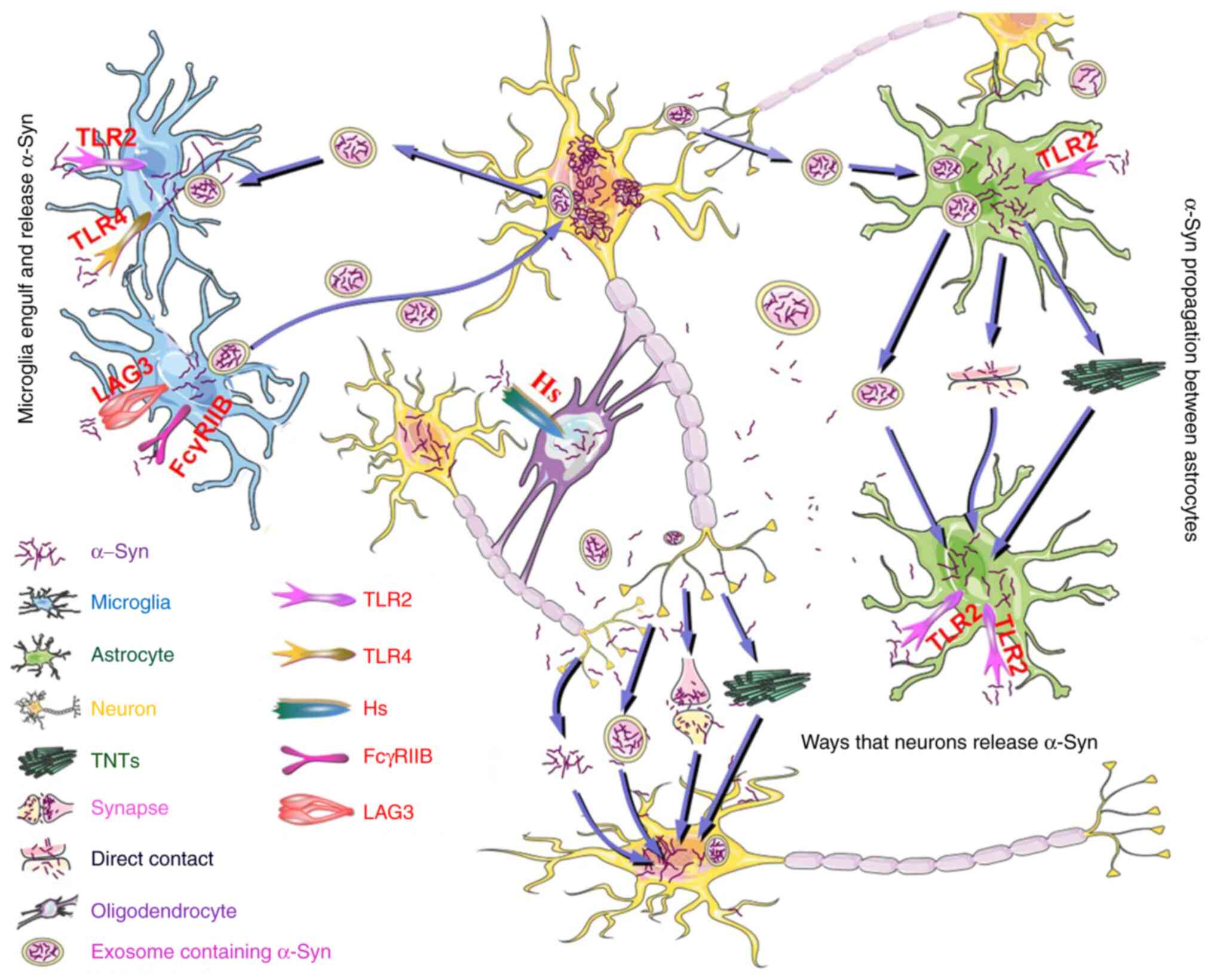 | Figure 2Possible mechanisms for intercellular
transmission of α-Syn. α-Syn is released by neurons through TNTs
and exosomes, as well as through a leaking process between pre- and
post-synapses. α-Syn also spreads to nearby neuronal cells in a
free-floating manner. Astrocytes engulf extracellular α-Syn and
transfer it to other astrocytes through direct contact between
cells, extracellular exosomes, vesicles and TNTs. The phagocytosis
of extracellular α-Syn by astrocytes depends on TLR2, while the
uptake of α-Syn by microglia is dependent on TLR2, TLR4, LAG3 and
FcγRIIB. Microglia can also endocytose exosomes containing α-Syn
and promote the transfer of α-Syn to neurons by releasing exosomal
α-Syn. Hs is implicated in oligodendroglial uptake of extracellular
α-Syn. α-Syn can be transferred from neurons to astrocytes,
microglia and oligodendrocytes and can also be transferred from
microglia to neurons, astrocytes to astrocytes and neurons to
neurons. Astrocyte to neuron transfer is rare. α-Syn, α-synuclein;
TNTs, tunneling nanotubes; TLR, Toll-like receptor; Hs, heparan
sulfate; LAG3, lymphocyte-activation gene 3; FcγRIIB, Fc gamma
receptor IIB. |
Unfolded α-Syn monomers undergo a structural change
during PD-associated pathology, sequentially forming partially
folded monomers, transient small soluble oligomers, structured
oligomers (later β-sheet structures), β-sheet-structure
protofibrillar oligomers and fibrils which accumulate in LBs
(55). A previous study reported
that soluble transient intermediate oligomers may be the critical
form that causes cytotoxicity, including mitochondrial dysfunction,
abnormal Ca2+ signaling, ROS production and neuronal
death (14). A number of
recombinant α-Syn fragments (aa, 1-95 and 61-140) have been
reported to promote the formation of aggregates from cellular
full-length α-Syn, inducing microglial toxicity and increasing the
release of inflammatory factors (17). The α-Syn protein is almost
exclusively localized to the presynaptic terminals of neurons
(3,56,57),
regulating the biosynthesis of DA (9). However, α-Syn aggregation in DA
neurons results in the depletion of soluble α-Syn, which
subsequently causes a loss of the presynaptic terminal function of
neurons and decreases DA levels in neurons in the SN-striatal
system (9,24). The aberrant accumulation of α-Syn
reduces the aggregation and fusion activity of synaptic vesicles,
which subsequently affects the release of neurotransmitters (such
as dopamine), resulting in neuronal death in the SN and movement
dysfunction (16,58). α-Syn is also reported to be found
extracellularly, which indicates that α-Syn may induce cytotoxic
effects in the extracellular space (15). Extracellular α-Syn oligomers
associate with the neuronal membrane, forming pore-like structures
and leading to enhancement of the influx of both glucose and
Ca2+ as well as membrane conductance (15,16).
This may partly explain the synaptic toxicity of α-Syn. There is a
marked increase in iron deposition in the SN of patients with PD
(21,22), which indicates that iron serves a
role in the pathology of PD. It has previously been reported that
α-Syn exerts cytotoxic effects by regulating iron metabolism.
Aggregated α-Syn inhibits ferritin release of iron and disturbs the
autophagy of iron (22). In
certain cases, such as in the presence of Cu2+, α-Syn
may perform the same function as the enzyme ferrireductase, which
is involved in the reduction of Fe3+ to Fe2+,
Fe2+/Fe3+ imbalance in PD and iron-mediated
neuronal death. The imbalance of Fe2+/Fe3+
may interfere with mitochondrial function (21). Since DA neurons consume large
amounts of ATP, mitochondrial dysfunction may lead to their death
(23). It has previously been
reported that α-Syn overexpression disrupts the fusion of
mitochondrial membranes, leading to mitochondrial fragmentation.
Overexpression of α-Syn, which reduces the contacts between the
endoplasmic reticulum and the mitochondria and interferes with
Ca2+ transfer, can lead to impaired autophagic
mechanisms, defects in mitochondrial fission and an increase in the
number of damaged mitochondria (23). The mutant forms of α-Syn or α-Syn
overexpression suppress complex I activity in mitochondria,
enhancing the generation of ROS. Furthermore, α-Syn also disrupts
the intracellular transport of mitochondria in PD (23).
α-Syn preformed fibrils (PFFs) form from recombinant
α-Syn in vitro. The α-Syn PFFs formed by aggregation in
vitro are structurally similar to the pathological α-Syn found
in vivo. α-Syn PFFs can also spread like prions between
neurons both in vivo and in vitro (59). When α-Syn PFF is injected into a
mouse brain, the serine 129 site of α-Syn PFF can be
phosphorylated, forming p-α-Syn, which is a marker of α-Syn
neurotoxicity (59).
To date, the initial trigger mechanism for the
transformation of α-Syn into pathological α-Syn is still being
explored. Different forms of α-Syn exist and the toxic mechanisms
of each α-Syn form in the pathogenesis of PD remain poorly
understood. Notably, the conformation of α-Syn may be highly
correlated with its pathological toxicity. The toxic effects of
α-Syn are multifaceted and can be severe, serving an important role
in the pathogenesis of PD. In view of this, drugs that suppress
α-Syn aggregation or promote pathological α-Syn degradation may be
a key future PD treatment.
4. Age-related α-Syn pathogenicity
The intercellular transmission of α-Syn is widely
accepted as a potential mechanism for the progression of PD
(60). Previous studies have
reported that several years before the diagnosis of PD,
pathological α-Syn is present in both the brain and many peripheral
organs, such as the skin, submandibular gland, heart, stomach and
intestines (38,61-63).
Braak et al (64,65) reported that the α-Syn protein may
originate in the peripheral nervous system, specifically in the
enteric nervous system (ENS) (66). Afterward, α-Syn spreads to the
brain tissue through prion-like mechanisms via the autonomic
nervous system (66). Other
studies have also reported that various forms of α-Syn, including
monomers, oligomers and fibrils, can be transferred from the ENS to
the brain via the vagus nerve (Fig.
3) (23,38). Upon entering the brain, α-Syn
spreads to the midbrain via interconnected neurons, leading to
PD-associated motor disorders, such as bradykinesia, postural
instability, myotonia and static tremor (Fig. 3) (39). α-Syn can also spread to nearby
neurons through extracellular vesicles (EVs) or in a free-floating
manner (Fig. 2) (67). Unlike prions, however, there is no
evidence that pathological α-Syn is infectious (68).
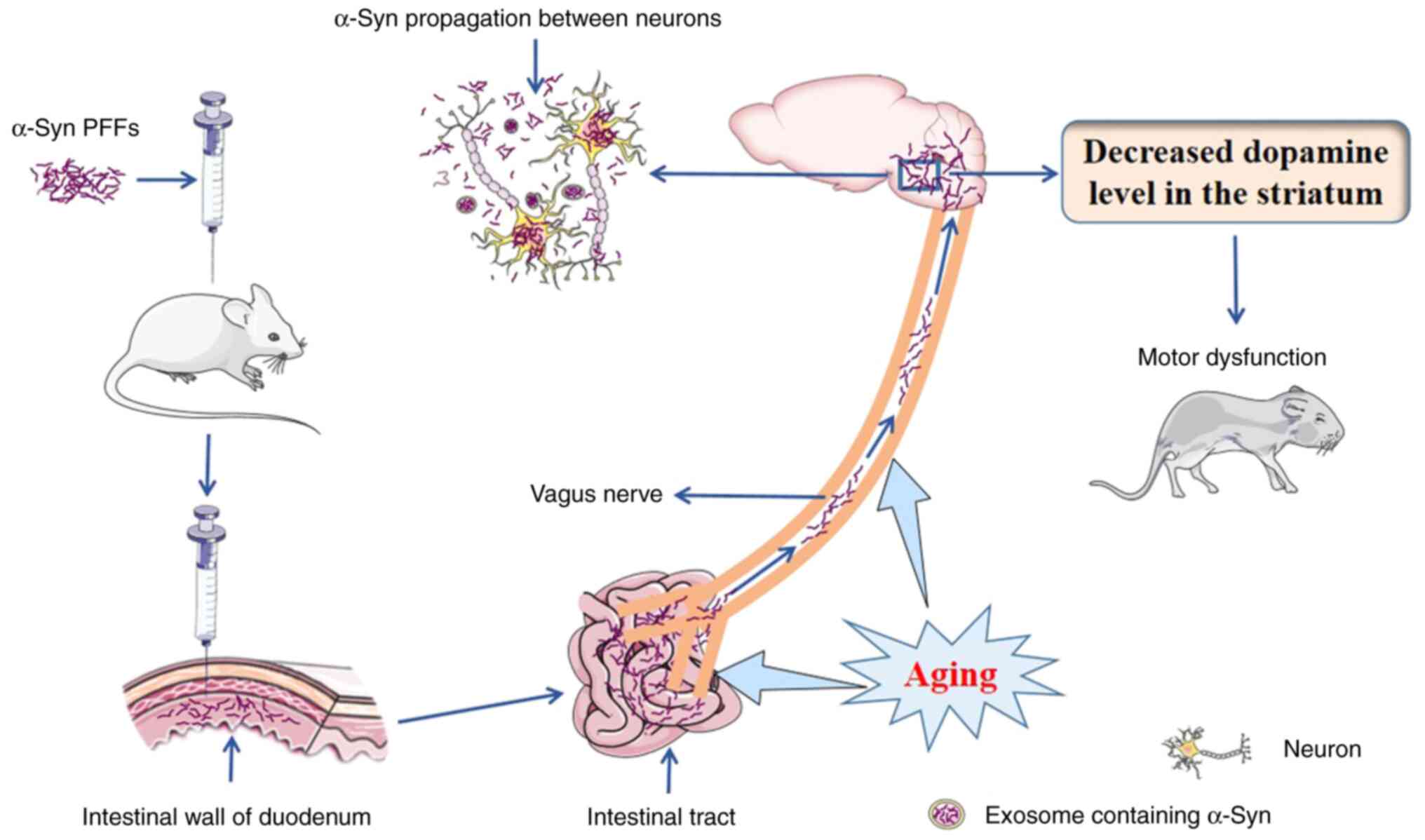
Van Den Berge et al (38) injected recombinant human α-Syn PFFs
(hPFFs) or mouse α-Syn PFFs (mPFFs) into the intestinal wall of the
duodenum and pylorus in wild-type rats and demonstrated that age is
a key factor in efficient α-Syn propagation along the
intestine-brain axis (Fig. 3). In
older mice, when compared with younger mice, after inoculation of
α-Syn PFFs, α-Syn pathology is transmitted from the ENS to the
brain stem and this transmission is accompanied by a decreased
dopamine level in the striatum (Fig.
3) (39). Van Den Berge et
al (38) also reported that:
i) Sympathetic denervation in the heart and vagal denervation in
the stomach are age-related and associated with the amount of α-Syn
derived from α-Syn PFF after injection into wild-type rats; ii)
deposits of phosphorylated α-Syn in aged rats are much denser and
more resistant to proteinase K, a nonspecific serine protease used
for protein digestion, compared with those in younger rats; iii)
mPFFs are more efficient than hPFFs at inducing α-Syn transmission
from the intestine to the brain in young rats, which suggests that
a species barrier does exist; and iv) the pathological outcomes of
old rats treated with hPFFs closely resemble, or are worse than,
those of young rats treated with mPFFs, which indicates that aging
lowers the species barrier (Fig.
3). Glucocerebrosidase (GCase) is a lysosomal enzyme and
impairment of GCase function leads to aberrant accumulation of
α-Syn. Injection of α-Syn PFFs into the mouse duodenum can activate
inflammation, decrease GCase production and impair GCase function
and ENS physiology. However, upregulation of the production of
GCase alleviates the ENS functional deficit and α-Syn-associated
pathology. The expression of GCase is markedly decreased in the
duodenum in older mice compared with younger mice and this decrease
is correlated with an increase in p-α-Syn, which indicates that
aged mice are more vulnerable to α-Syn pathology in the intestinal
tract. During aging, intestinal protein homeostasis (including
maintenance of the function of GCase) and the ability to eliminate
α-Syn aggregates decline (39).
This negative variation leads to an increased vulnerability to
α-Syn pathology and dysfunction of the sensorimotor system
(Fig. 3) (39). Conversely, overexpression of α-Syn
can lead to an imbalance in intestinal homeostasis and accelerate
the occurrence of intestinal aging (13).
Iba et al (69) explored the role of aging and the
inflammatory response in synucleinopathies by injecting α-Syn PFFs
into the striata of young or aged mice. It was observed that aged
mice showed enhanced α-Syn accumulation in specific brain regions
and had more pronounced behavioral deficits compared with young
mice. The loss of DA neurons and DA nerve endings in the striata of
mice inoculated with α-Syn PFFs increased with aging. Compared with
young mice inoculated with α-Syn PFFs, aged mice injected with
α-Syn PFFs showed enhanced T cell infiltration and sustained active
microglia in the brain tissue. Transcriptome analysis suggested
that inflammation in the brain was greater in aged mice when
compared with young mice inoculated with α-Syn PFFs. Rauschenberger
et al (70) employed
hm2α-Syn-39 mice, a PD mouse model carrying a
double-mutant human α-Syn gene (A30P/A53T), to explore the effects
of the inflammatory response and aging on DA neurons. The
aforementioned study reported that the striatal DA terminals,
striatal dopamine levels and SN DA neurons were reduced in
hm2α-Syn-39 PD mice at the age of 16-17 months compared
with age-matched control mice. In the striata of
hm2α-Syn-39 PD mice, there was an age-related
association between striatal DA terminal loss and the number of
infiltrating clusters of differentiation 4+
(CD4+) and CD8+ T cells. Such a correlation,
however, was not observed in the wild-type mice. No meaningful
age-dependent changes in the numbers of CD8+ T cells,
CD4+ T cells or B220+ B cells were reported
in the nigrostriatal tracts of the wild-type group of mice. By
comparison, in the SN of hm2α-Syn-39 PD mice, marked
age-dependent increases in the number of CD8+ T cells,
glial fibrillary acidic protein (GFAP)+ astrocytes and
CD11b+ microglia were observed. Furthermore,
age-dependent impairment of motor function was also observed in
hm2α-Syn-39 PD mice.
In addition, a previous study reported that the
expression levels of the α-Syn-related complex were correlated with
age. Nerve globins are O2-binding proteins that
participate in the etiopathogenesis of neurodegenerative disorders.
The levels of neuronal hemoglobin (nHb), a member of the nerve
globin protein family, are decreased in α-Syn deposits, DA neurons
and LBs of patients with PD. In the brain tissue of elderly
individuals, a complex consisting of Hb and α-Syn
(nHbα-Syn) has been reported. In the human
striatum, the nHbα-Syn complex level in
mitochondria decreases with age, while in the cytoplasm, the
nHbα-Syn level increases with age (71). In the SN, a similar expression
pattern of nHbα-Syn is observed (71). Of note, the
nHbα-Syn complex also exists in red blood
cells (RBCs). The levels of nHbα-Syn in RBCs
significantly increase with age and significantly increase in
patients with PD (71). However,
the specific mechanism by which nHbα-Syn or
pHbα-Syn expression changes with age remains
to be further investigated.
The aforementioned studies indicate that the
pathogenicity of α-Syn may be age-related. Aging promotes the
development of α-Syn pathology in the intestinal tract and α-Syn
PFF transmission along the intestine-brain axis. The pathological
changes caused by α-Syn, including the loss of DA nerve endings,
behavioral defects and inflammation in the brain, are aggravated
with age. This may explain the susceptibility of elderly
individuals to PD. Furthermore, maintaining normal intestinal
function may be beneficial for PD patients. Therefore, the effects
of diet and the gastrointestinal microbiome on PD should be
evaluated.
5. α-synuclein and astrocytes
In the central nervous system (CNS), astrocytes are
the most numerous non-nerve cell population, serving important
roles in neurotrophic support, neurotransmitter transmission,
synaptic development and neuroinflammation (16,18,57).
There are two different phenotypes of astrocytes, namely, the A1
phenotype (proinflammatory phenotype) and the A2 phenotype
(anti-inflammatory phenotype) (72). A1 astrocytes can release neurotoxic
cytokines, such as complement C3, which are harmful to
oligodendrocytes and neurons (19,73-75);
hence, A1 astrocytes are considered a characteristic of normal
aging and neurodegenerative diseases (16). In contrast to A1 astrocytes, A2
astrocytes are reported to be neuroprotective and capable of
producing neurotrophic cytokines, including nerve growth factor and
brain-derived neurotrophic factor (BDNF), to promote neuronal
growth and survival (16,73). Depending on the changes taking
place in the brain environment, astrocytes may shift between A1 and
A2 types in a stimulus-specific manner (18,72,75).
For example, A1 astrocytes can be induced by TNF, complement
component 1q and IL-1α, which are secreted by activated microglia
(76). Notably, in another
previous study, the numbers of astrocytes of both phenotypes were
increased in animal models of PD (77); however, no or only slight increases
in the numbers of astroglial cells were observed in the brain
tissues of patients with PD (16).
In astrocytes, α-Syn expression levels are normally
low (29,78), whereas α-Syn accumulates within
astrocytes of patients with PD (20). Thus, it is reported that α-Syn in
astrocytes may originate from neurons (57). Neurons release α-Syn in a variety
of conformations, such as monomers, oligomers and fibril species
(30). Previous studies have
reported that astrocytes engulf extracellular α-Syn released by
neurons and are involved in α-Syn transmission (16,29,74).
Astrocytes take up α-Syn and transfer it to other astrocytes
through direct contact between cells, extracellular exosomes and
vesicles (Fig. 2) (16,57,79).
In addition, TNTs are considered an important pathway for α-Syn
transmission. TNTs are transient tubular structures used for
long-distance cell-to-cell communication (79). Through TNTs, astrocytes transfer
excessive α-Syn oligomers to other astrocytes (Fig. 2) (57). α-Syn can be transferred from
neurons to neurons, neurons to astrocytes and astrocytes to
astrocytes (79) but is rarely
transferred from astrocytes to neurons (Fig. 2) (18).
Astrocytes express Toll-like receptors (TLRs),
which are involved in astrocyte activation, neuroinflammation and
α-Syn uptake (18,29,75).
In astrocytes, the uptake of extracellular α-Syn relies on TLR2
rather than TLR4 (Fig. 2)
(18), while intracellular α-Syn
relies on TLR2, TLR3 and TLR4 to induce inflammatory responses in
astrocytes (18,80). Notably, α-Syn entry into microglia
is dependent on TLR2 and TLR4(78). Astrocytes can degrade α-Syn fibrils
more efficiently compared with neurons (79). Neurons and microglia can also
degrade α-Syn fibrils. TLR2 stimulation accelerates the
phagocytosis of α-Syn fibrils by neurons, astrocytes and microglia
(30,78) and TLR2 stimulation, rather than
TLR4 stimulation, significantly inhibits intracellular α-Syn fibril
degradation in astrocytes and neurons (30). However, with or without stimulation
of TLR2, α-Syn fibrils are degraded efficiently in microglia
(30). It has also been reported
that astrocytes phagocytose and eliminate extracellular recombinant
human α-Syn through the autophagy and ubiquitin-proteasome pathways
in vitro (81).
Overexpression of wild-type α-Syn and expression of mutated forms
of α-Syn can impair autophagy and the ubiquitin-proteasome system
(23).
α-Syn can potentiate the activation of astrocytes
and trigger their inflammatory response (18). Previous studies have reported that
monomeric α-Syn, oligomeric α-Syn and fibrillar α-Syn can activate
astrocytes, which leads to neuronal death (78,82).
However, these α-Syn forms differ in their capacity for the
activation and induction of cytotoxicity in astrocytes. In
astrocytes, α-Syn oligomers can cause significant mitochondrial
dysfunction, whereas the effects of monomers and fibrils on
mitochondria are mild. Fibrillary, oligomeric and monomeric forms
of α-Syn can all markedly increase IL-1β and TNF-α mRNA expression
levels in astrocytes, while only oligomers induce an increase in
extracellular hydrogen peroxide (82). Intracellular α-Syn induces
neurotoxic proinflammatory responses, including expression of IL-6,
C-X3-C motif chemokine ligand 1 (CX3CL1), chemokine (C-C motif)
ligand 5 (CCL5) and TNF-α in astrocytes via the p38 MAPK and NF-κB
signaling pathways (Fig. 4)
(18,20).
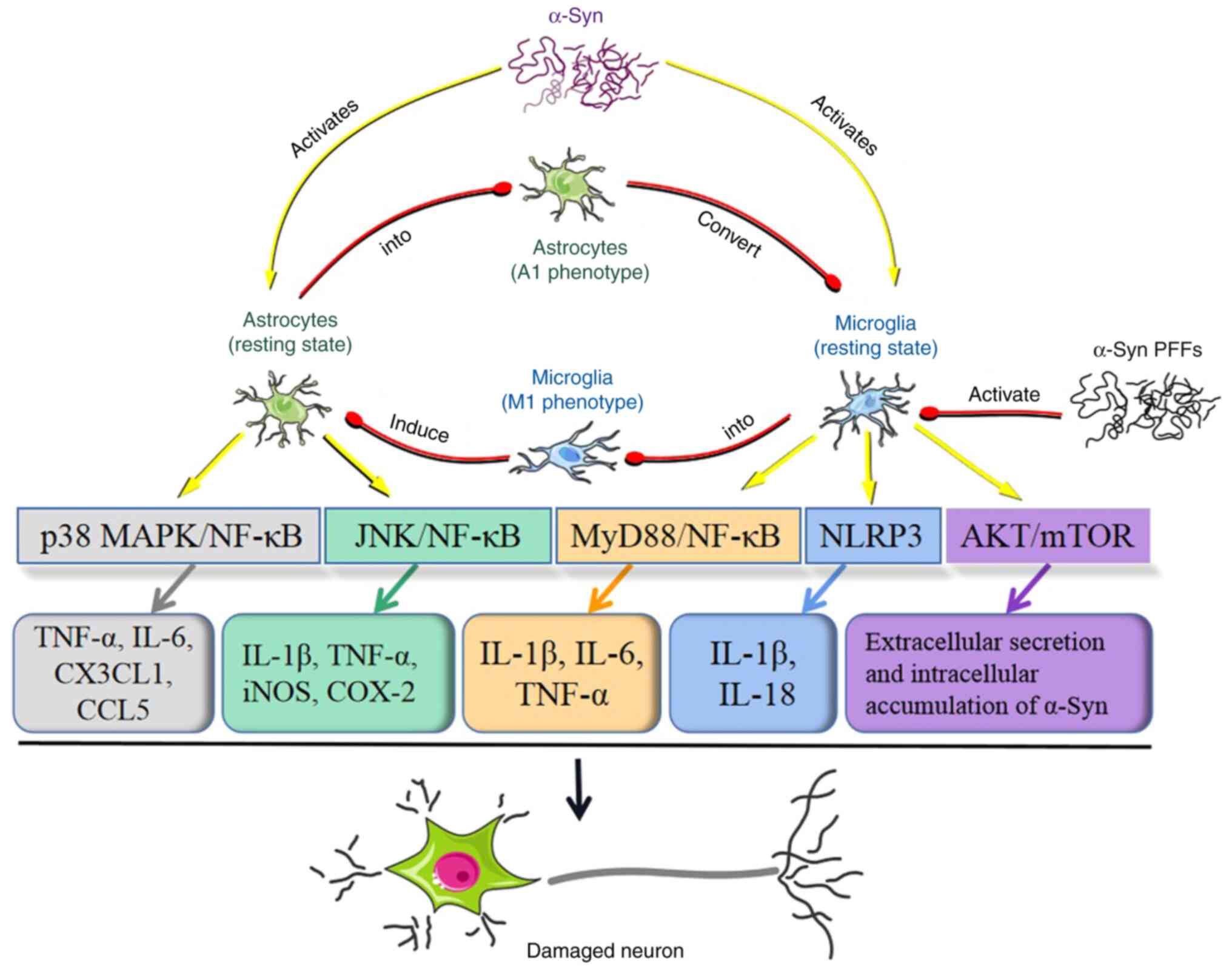 | Figure 4Inflammatory activation of astrocytes
and microglia by α-Syn and phenotypic switching between astrocytes
and microglia. α-Syn induces the expression of CX3CL1, CCL5, IL-6
and TNF-α in astrocytes through the p38 MAPK and NF-κB signaling
pathways. α-Syn aggregates also activate astrocytes and trigger
neuroinflammation through the JNK and NF-κB signaling pathways.
This leads astrocytes to express COX-2, iNOS, TNF-α and IL-1β. In
microglia, α-Syn activates the MyD88-NF-κB signaling pathway,
leading microglia to generate proinflammatory factors, including
IL-6, IL-1β and TNF-α. α-Syn can prime and activate the microglial
NLRP3 inflammasome, leading to the generation of IL-18 and IL-1β by
microglia. Exosomal α-Syn suppresses autophagy in microglia through
activation of the AKT/mTOR signaling pathway, which leads to
accelerated extracellular α-Syn secretion and increased
intracellular α-Syn accumulation. These inflammatory responses
induced by α-Syn are conducive to DA neuronal death. α-Syn PFFs
activate microglia into the M1 phenotype, which in turn induces
astrocytes to develop into a neurotoxic A1 phenotype. The
inflammatory responses in astrocytes are also capable of converting
microglia into an M1-like phenotype. α-Syn, α-synuclein; PFFs,
preformed fibrils; CX3CL1, C-X3-C motif chemokine ligand 1; CCL5,
chemokine (C-C motif) ligand 5; COX-2, cyclooxygenase-2; iNOS,
inducible nitric oxide synthase; NLRP3, nucleotide-binding
oligomerization domain leucine-rich repeat and pyrin
domain-containing 3; MyD88, myeloid differentiation factor 88; DA,
dopaminergic. |
Wild-type or A53T mutated α-Syn markedly enhances
the expression of TLR2 and TLR3 in astrocytes and TLR2 or TLR3 may
mediate extracellular inflammatory pathways and intracellular
inflammatory pathways, respectively (80). A53T mutant α-Syn aggregates
activate astrocytes and triggers neuroinflammation through the JNK
and NF-κB signaling pathways. Subsequently, astrocytes express
cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS),
TNF-α and IL-1β (Fig. 4) (19). Heat shock protein 70, a molecular
chaperone, has been reported to be an important modulator that
suppresses the JNK and NF-κB signaling pathways in astrocytes and
inhibits inflammation induced by α-Syn, exhibiting an
anti-inflammatory effect (19). A
previous study reported that co-expression of α-Syn and IFN-γ may
be one of the mechanisms leading to the pathogenesis of PD
(83). In astrocytes, IFN-γ alone
increases TNF-α and TLR3 mRNA expression levels. Wild-type or
A53T-mutant α-Syn alone enhances the expression levels of TNF-α,
NF-κB, IL-1β, TLR2 and TLR3. Further mechanistic study confirmed
that IFN-γ amplifies the wild-type or A53T mutant α-Syn-inducing
effect, except in the case of IL-1β in astrocytes (84). IFN-γ enhances the stimulation of
α-Syn and the inflammation of astrocytes through TNF-α, TLR2 and
TLR3, which indicates that the participation of IFN-γ in the innate
immunity, induced by α-Syn, is necessary to initiate and maintain
astrocyte activation (84).
Astrocytes are not a type of immune cells, but they
are involved in brain inflammation. α-Syn can trigger an
inflammatory response in astrocytes and the inflammatory factors
released by astrocytes can have a toxic effect on neurons. In
addition, α-Syn oligomers cause mitochondrial dysfunction in
astrocytes. Astrocytes that undergo inflammatory responses and
astrocytes with mitochondrial dysfunction may reduce neurotrophic
and metabolic support for neurons, exacerbating neurodegeneration.
Inhibiting the inflammatory response of astrocytes and enhancing
their neurotrophic and metabolic support for neurons may contribute
to neuronal survival and alleviate PD symptoms.
6. α-synuclein and microglia
Microglia are a type of tissue-specific macrophages
that reside in the CNS (57).
Microglia are implicated in brain development, protein aggregate
elimination, neuronal survival, neuronal apoptosis,
immunosurveillance, synaptic plasticity maintenance, synaptic
pruning, neural circuit shaping and injury repair (16,57,85,86).
In numerous neurodegenerative diseases (including PD, Alzheimer's
disease and amyotrophic lateral sclerosis), microglia can be
polarized into two major functional subtypes, including the M1
(proinflammatory) phenotype and the M2 (anti-inflammatory)
phenotype (37,73,87).
M1 microglia mediate brain immunological reactions by secreting
proinflammatory substances, including TNF-α, IL-12, IFN-γ, IL-1β
and IL-6, which contribute to neuronal death (73,85).
In addition, M1 microglia also express major histocompatibility
complex class (MHC)-I and MHC-II molecules, which are associated
with antigen presentation (16,88-90).
Unlike the M1 phenotype, M2 microglia exert neuroprotective effects
(37). Neurotrophic factors, such
as insulin-like growth factor-1 (IGF-1) and BDNF and
anti-inflammatory cytokines, such as IL-13, IL-4, TGF-β and IL-10
are released by M2 microglia, promoting neuronal survival (37,73,85).
It has been previously reported that some microglial regulatory
mediators, such as triggering receptors expressed on myeloid cells
2 (TREM2), TGF-β1, IFN-β and IL-10, have the potential to convert
microglia from the M1 state to the M2 state (85), which may delay PD progression
(37,91). During chronic inflammation,
microglia lose their M2 anti-inflammatory phenotypic
characteristics and preferentially differentiate into the
proinflammatory M1 state (92).
Interactions between α-Syn and microglia may serve
a pivotal role in PD pathogenesis (85). Similar to astrocytes, microglia are
able to take up α-Syn released by neurons (30,57,78).
It has been previously reported that the phagocytosis of α-Syn by
microglia is dependent on TLR2 and TLR4 (Fig. 2) (36,78).
TLR2 accounts for the microglial uptake of soluble α-Syn oligomers;
however, the specific α-Syn forms internalized by microglia via
TLR4 remain to be further investigated (78). TLR4 promotes extracellular α-Syn
clearance and DA neuronal survival in the SN, while elimination of
TLR4 impairs the ability of microglia to phagocytose α-Syn,
enhancing dyskinesia and death of nigral DA neurons (93). It been reported that TLR2-deficient
microglia can also endocytose α-Syn monomers and low concentrations
of α-Syn fibrils and oligomers. A previous study indicated that the
phagocytosis of cellular α-Syn by microglia depends not only on
TLR2 receptors but also on numerous other types of receptors
(30). For instance, the uptake of
extracellular α-Syn fibrils by microglia is also reliant on
receptors including lymphocyte-activation gene 3 and Fc gamma
receptor IIB (Fig. 2) (78). It is possible that each type of
receptor only recognizes one specific α-Syn conformation (30). EVs, including exosomes and
microvesicles, have been reported as possible routes of α-Syn
transmission in the CNS. The α-Syn oligomers in EVs are more easily
endocytosed and cause more damage to recipient cells than
non-EV-encapsulated α-Syn oligomers (87). Another study has confirmed that
microglia may achieve efficient uptake of exosomes containing
monomeric and oligomeric α-Syn and that microglia can be activated
by these exosomes. Exosomal α-Syn suppresses autophagy in microglia
through activation of the AKT/mTOR signaling pathway, which leads
to accelerated extracellular α-Syn secretion and increased
intracellular α-Syn accumulation (Fig.
4) (94). Additionally,
microglia may promote the transfer of α-Syn to neurons by releasing
exosomal α-Syn (Fig. 2) (94).
TLRs, as receptors for extracellular α-Syn, account
for not only the binding and internalization of α-Syn but also
signal conduction and the inflammatory response (36,57,78,85).
TLR1, TLR2, TLR4 and TLR5 are responsible for α-Syn-induced
inflammatory activation of microglia (57,85,95,96).
Previous studies have reported that TLR7 and TLR8 may also account
for microglial activation (96,97).
A previous study also reported that α-Syn oligomers convert
microglia into the M1 phenotype via the TLR1 and TLR7 signaling
pathways (98). In addition, α-Syn
can activate microglia through other receptors, such as purinergic
receptor P2X ligand-gated ion channel 7 (P2X7 receptor), CD36 and
Fcγ receptors expressed in microglia (57,99).
Notably, each α-Syn receptor may only interact with a specific
conformation of α-Syn for the uptake of α-Syn by microglia, as
previously described (30). α-Syn
receptors involved in the activation of microglia may also be
conformationally sensitive. A previous study reported that the TLR2
ligand activity of α-Syn possesses conformational sensitivity and
that TLR2 is activated only by particular forms of oligomers
(100). Adenoviral vector-treated
differentiated SY5Y neuroblastoma cells (AVT-dSY5Y) overexpress
human α-Syn and the α-Syn in the cytoplasm of these cells is
predominantly monomeric. The extracellular protein outside the
cytosol contains many stable α-Syn oligomers. The α-Syn released by
AVT-dSY5Y, not the intracytoplasmic α-Syn fraction, activates
microglia through a TLR2-dependent mechanism (100). Moreover, misfolded α-Syn
increases the expression levels of TLR2 and TLR3 in microglial
culture (80). It has also been
reported that treatment of microglia with α-Syn leads to elevated
expression of TLR1 and TLR7(101). TLR1, TLR2 and TLR4 on the surface
of microglia are able to respond to monomeric, oligomeric and
fibrillar forms of α-Syn and activate the myeloid differentiation
factor 88 (MyD88)-NF-κB signaling pathway, which leads to
α-Syn-induced neuroinflammation (57). Activation of NF-κB in the
MyD88-dependent pathway produces various proinflammatory factors,
including IL-6, IL-1β and TNF-α (Fig.
4) (85). Monomeric and
oligomeric α-Syn are able to prime and activate the microglial
nucleotide-binding oligomerization domain leucine-rich repeat and
pyrin domain-containing 3 (NLRP3) inflammasome, a multiunit protein
complex, through TLR2 and TLR5 ligation (95). The NLRP3 inflammasome recruits the
small adaptor molecule apoptosis-associated speck-like protein,
which subsequently triggers cysteine-aspartate protease-1
(caspase-1) activation (74,86).
Caspase-1 is capable of producing the mature proinflammatory
factors IL-18 and IL-1β by cleaving their precursor forms (Fig. 4) (16,57,86).
In addition, activated caspase-1 results in C-terminal truncation
of α-Syn, which leads to the formation of more aggregates and an
increased inflammatory response (16,17).
Microglia in patients with PD may be overactivated and inflammatory
factors generated by microglia are one of the causes of DA neuronal
death (27).
Interactions between microglia and astrocytes may
also take place. α-Syn PFFs can activate microglia into the M1
phenotype, which in turn induces astrocytes to acquire a neurotoxic
A1 phenotype. This microglia-mediated induction of astrocytes to an
A1 subtype is directly blocked by glucagon-like peptide-1 receptor
agonists (102). Notably, it has
previously been reported that the inflammatory response in
astrocytes is also capable of converting microglia into an M1-like
phenotype through paracrine action (Fig. 4) (18).
Membrane receptors expressed by glial cells,
including astrocytes and microglia, are structurally sensitive and
recognize only specific forms of α-Syn. Notably, certain membrane
receptors on the surfaces of glial cells are involved in α-Syn
endocytosis, while others are implicated in α-Syn-mediated
inflammatory responses. Although several α-Syn-related receptors
and their corresponding α-Syn forms have been identified, the
specific α-Syn form binding to TLR4 remains unclear. Moreover,
potential receptors for α-Syn, which have not yet been discovered,
may exist. Further research is needed to develop therapeutic
interventions to selectively inhibit glial receptors from binding
to α-Syn, thereby preventing glial inflammatory responses.
7. α-synuclein and oligodendrocytes
Oligodendrocytes are glial cells of the CNS that
possess numerous functions, including myelin generation, metabolic
support, immunomodulation and production of neurotrophins, such as
glial cell line-derived neurotrophic factor, BDNF and IGF-1
(73,103).
The expression of α-Syn in oligodendroglial cells
is low under normal physiological conditions (73). The amount of endogenous α-Syn
protein expressed by rat oligodendrocyte lineage cells is ~20% of
that expressed by neurons (104).
In multiple system atrophy (MSA), a rare neurodegenerative
disorder, pathological α-Syn accumulates in oligodendrocytes,
forming glial cytoplasmic inclusions (GCIs), which is a unique
hallmark of this disease (31,103). In PD, by contrast, misfolded
α-Syn mainly appears in neurons (105), forming LNs and LBs. A previous
study reported that α-Syn inclusions are present in
oligodendrocytes in the midbrains of patients with PD but that they
differ in topography and antigenicity from GCIs in MSA
oligodendrocytes (106).
Oligodendrocytes in certain parts of the brain in patients with PD
appear to be unaffected by α-Syn. Several years before the onset of
PD motor dysfunction, α-Syn is already present in the anterior
olfactory nucleus (AON) of the olfactory bulb (5). In AON regions of postmortem patients
with PD, α-Syn is present in some neurons, astrocytes, microglia
and pericytes, whereas no α-Syn is present in oligodendrocytes
(5). In MSA, oligodendrocytes are
affected by α-Syn and α-Syn aggregates to form GCIs, while in PD,
α-Syn mainly affects neurons and aggregates to form LNs and LBs
(2,3,31,103,105). This is a future research
direction which requires further study, as related studies may be
of significance for revealing the toxicity of α-Syn and the
pathogenesis of synucleinopathies, including MSA and PD.
Oligodendrocytes are able to take up monomeric and
oligomeric forms of α-Syn and small amounts of α-Syn fibrils in a
dynamin-dependent manner both in vivo and in vitro
(107). Exogenous α-Syn PFFs can
also be phagocytosed by oligodendrocyte precursor cells (OPCs),
which increases the expression of endogenous α-Syn and triggers
impairment of autophagy in OPCs, which can result in endogenous
α-Syn accumulation (104). In
addition, it has been reported that α-Syn can be transferred from
neurons to oligodendrocytes (Fig.
2) (107). α-Syn uptake by
oligodendrocytes is dependent on clathrin and is inhibited when
clathrin expression is silenced (108). In addition, the phagocytosis of
α-Syn by oligodendrocytes is both time- and dose-dependent
(108). Finally, it has been
previously identified that heparan sulfate and exosomes are
involved in oligodendroglial phagocytosis of extracellular α-Syn
fibrils (Fig. 2) (31,109).
A previous study reported that TLR4 expression is
upregulated in α-Syn-transgenic MSA mice, an effect that is related
to oligodendroglial overexpression of α-Syn (110). However, it remains unclear
whether there is an interaction between TLR4 and α-Syn in
oligodendrocytes in PD. In OPCs treated with α-Syn PFFs, the mRNA
expression levels of myelinization-inhibiting cytokines, such as
IL-1β and Sirtuin2, are increased, while the mRNA expression levels
of myelinization-promoting cytokines, such as Contactin 1 and
chemokine C-X-C-motif receptor 7, are decreased (104). Overexpression of α-Syn by
oligodendrocytes may lead to neuroinflammation associated with
nitrogen stress in a transgenic MSA mouse model (110). Certain studies have indicated
that oligodendrocytes are able to produce a range of chemokines and
cytokines, including CCL2/5, chemokine (CXC motif) ligand 9/10 and
IL1-β (104,111). Oligodendrocytes also express
receptors capable of transducing immune-related signals (111). However, whether α-Syn triggers an
inflammatory response in oligodendrocytes in PD remains largely
unknown.
8. α-synuclein and acquired immunity
The brain was previously thought to be an
immune-privileged organ that was not easily infiltrated by
peripheral immune cells because of the blood-cerebrospinal fluid
barrier and blood-brain barrier (BBB) (16,112). However, it has been reported that
there exists bidirectional communication between the CNS and the
immune system (113,114).
Further evidence indicates that acquired immunity,
also known as adaptive immunity, serves a role in the pathogenesis
of PD (25,27). In animal models of PD, T cells have
been reported to infiltrate the SN (115). In α-Syn PFF-inoculated mouse
brains, the relative percentages of natural killer cells,
CD8+ T cells, CD19+ B cells, CD4+
T cells and activated myeloid cells
(CD11b+CD45high) are increased (116). In patients with PD, peripheral T
cells can access the brain and invade the SN (37). For instance, CD3+,
CD4+ and CD8+ T cells have been reported in
the brains of patients with PD (25). Furthermore, α-Syn proteins from
dead cells are reported to be capable of activating microglia
(37). The activated microglia
secrete a series of inflammatory factors, including IL-1β and TNF-α
(73,85), which act on microvascular
endothelial cells, leading to increased BBB permeability that
facilitates T cell entry into the brain (Fig. 5) (37). Likewise, astrocytes around blood
vessels produce inflammatory factors under pathological conditions,
such as IL-6, IL-1β and TNF-α, also resulting in an increase in BBB
permeability (Fig. 5) (33). Additionally, microglia take up
α-Syn proteins and process them in endosomes (117). Then, the microglia express MHC-II
proteins that present α-Syn antigenic peptides to CD4+ T
cells, therefore initiating acquired immunity (Fig. 5) (117-119).
CD4+ T cells can be activated into proinflammatory T
helper (Th)-type cells, such as Th17 and Th1 cells and
anti-inflammatory T cells, such as regulatory T (Treg) cells, which
have different functions (Fig. 5)
(28). Th17 and Th1 cells are
involved in enhancing the inflammatory response, mediating PD
pathology and inducing DA neuronal death (Fig. 5) (28). On the other hand, Treg cells are
involved in the maintenance of immune homeostasis, reduction of
inflammation and secretion of protective factors, such as IL-10 and
TGF-β (37). Notably, nitrated
α-Syn is able to induce harmful Th cell maturation, which can cause
severe neural damage (32).
Degradation of α-Syn may produce potential antigenic peptides,
which can be loaded onto MHC-I proteins and ultimately presented to
CD8+ T cells (Fig. 5)
(90,117). CD8+ T cells are
involved in neuronal death and α-Syn aggregation in PD (120). Astrocytes with accumulated α-Syn
also generate large amounts of MHC-II molecules in the brains of
patients with PD (Fig. 5). These
astrocytes are present near CD4+ T cells in the brain,
implying that there may exist a cross-interaction between
CD4+ T cells and astrocytes (18). A previous study reported that
astrocytes containing accumulated α-Syn are more likely to activate
CD4+ T cells via the MHC-II signaling pathway in
vitro compared with microglia containing accumulated α-Syn
(18,121). Furthermore, it has been reported
that DA neurons also express MHC in PD, which suggests that α-Syn
may be an antigenic substance capable of facilitating T cell
activation (27). A number of
antigenic regions have been identified in α-Syn. Of these regions,
one consists of aa 31-45 and 32-46 is located near the N-terminus,
while the other antigenic region, comprising aa 116-140, is close
to the C-terminus (122). The
researchers examined the response of T cells to the α-Syn antigenic
peptides in 9 patients with PD. For the majority of patients, it is
mainly CD4+ T cells secreting IL-4 or IFN-γ that respond
to α-Syn antigenic peptides, while for only 1 patient, the response
to α-Syn antigenic peptides is mediated by IFNγ-producing
CD8+ T cells (122). A
previous study also suggested that the reactivity of
CD4+ T cells against α-Syn could be detected ~10 years
before the onset of motor disorders associated with PD and is
positively correlated with age (123).
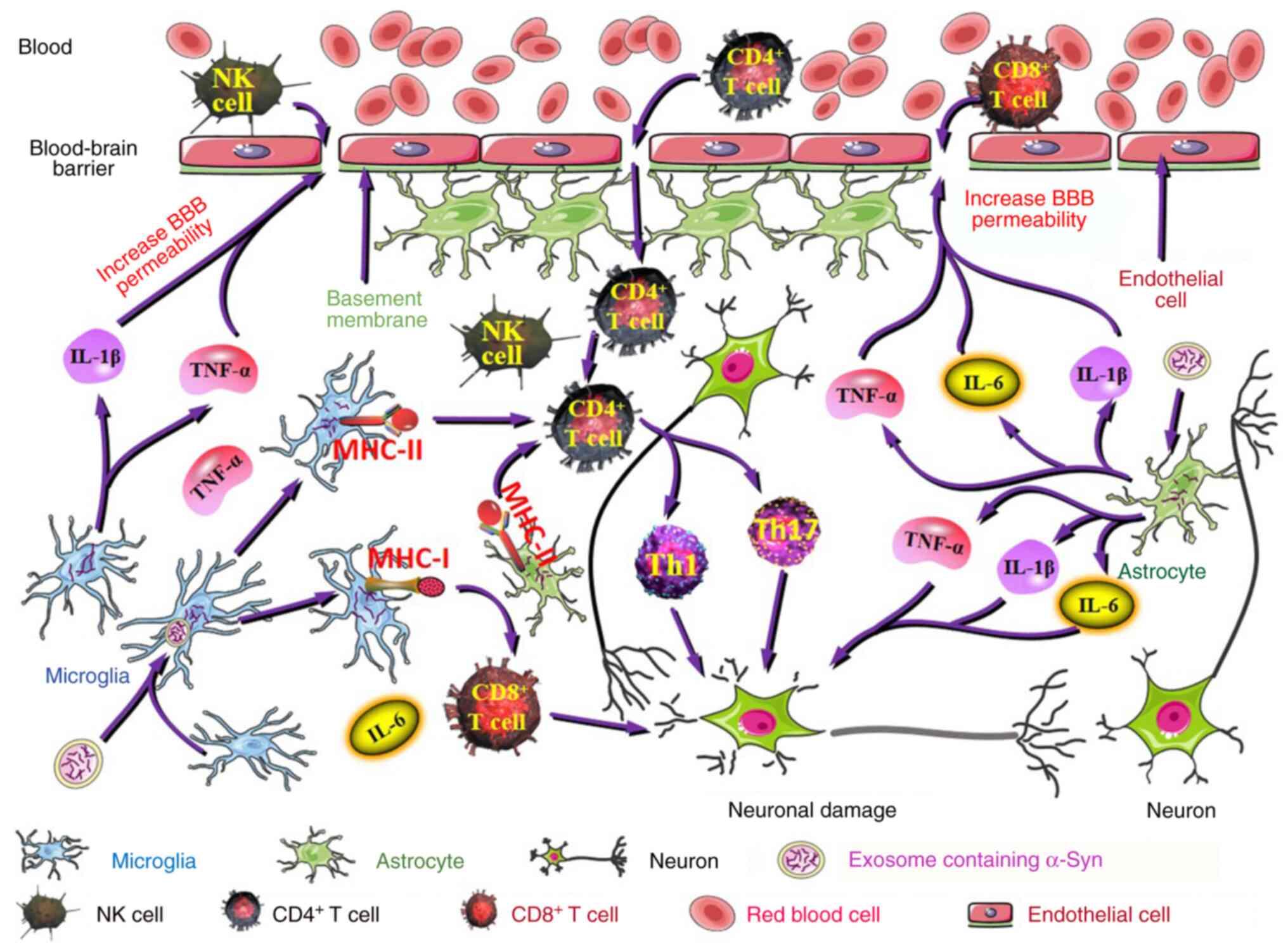 | Figure 5Adaptive immunity is implicated in PD
pathogenesis. The inflammatory factors (IL-1β and TNF-α) produced
by activated microglia act on microvascular endothelial cells,
resulting in increased BBB permeability that facilitates NK cell,
CD8+ T cell and CD4+ T cell entry into the
brain. Likewise, astrocytes around the blood vessels produce
inflammatory factors under pathological conditions, such as IL-6,
IL-1β and TNF-α, which results in an increase in BBB permeability.
Degradation of α-Syn may produce potentially antigenic peptides.
Microglia and astrocytes express MHC-II proteins that present α-Syn
antigenic peptides to CD4+ T cells, which are
subsequently activated into proinflammatory Th17 and Th1 cells.
α-Syn antigenic peptides can also be loaded onto MHC-I proteins and
ultimately presented to CD8+ T cells by microglia. Th17
and Th1 cells are involved in enhancing inflammatory reactions,
mediating PD pathology and inducing DA neuronal death.
CD8+ T cells are involved in neuronal death and α-Syn
aggregation in PD. PD, Parkinson's disease; BBB, blood-brain
barrier; NK cells, natural killer cells; CD4, cluster of
differentiation 4; CD8, cluster of differentiation 8; MHC-I, major
histocompatibility complex class I; MHC-II, major
histocompatibility complex class II; Th1 cells, type 1 T helper
cells; Th17 cells, type 17 T helper cells; DA, dopaminergic. |
Overall, through studies on animal PD models and PD
patients, it has been established that peripheral immune cell
infiltration in the brain exists in PD, suggesting an imbalance of
immune homeostasis in the pathological state. This imbalanced
immune homeostasis may be, at least in part, caused by a complex
interaction among α-Syn, DA neurons, microglia, astrocytes and
peripheral immune cells.
9. α-synuclein and glial cell
senescence
Senescence may occur in certain types of brain
cells, such as endothelial cells, astrocytes, microglia, neurons
and oligodendroglial progenitor cells (27). Senescent cells exhibit a
senescence-associated secretory phenotype (SASP), the main feature
of which is the generation of multiple factors, including IL-1β and
IL-6 (124,125). During aging, cells such as
astrocytes, express the SASP phenotype, exhibiting changes in
nuclear ultrastructure and increased expression of vimentin
filaments, GFAP, HMGB1 (a nuclear protein), IL-1β, IL-6 and TNF-α
(33). IL-1β, IL-6 and TNF-α may
also be biological markers of PD (126), which suggests a possible
relationship between aging and PD. The SASP contributes to immune
monitoring and clearance of senescent cells, which may be
responsible for leukocyte infiltration and death of DA neurons in
the SN (27). Previous research
indicated that aging and the SASP may be important promoters of PD
pathology (40). However, not all
SASPs of senescent cells cause apoptosis, inflammation and
fibrosis. The SASP may entail regenerative and growth cytokines in
certain senescent cells (127).
When senescent cells cannot be cleared in time, after a certain
threshold is exceeded, the number of proinflammatory or
proapoptotic senescent cells continues to increase, which leads to
tissue damage and the progression of age-related diseases (127).
Both cellular aging and α-Syn are involved in PD
pathology. Microglial senescence is an important factor in the
development of PD (28). Microglia
can engulf and degrade α-Syn in normal physiological states;
however, the phagocytic ability of microglia decreases under aging
conditions, leading to α-Syn accumulation and neurotoxicity
(36). The α-Syn ingested by
microglia is then degraded in autophagosomes (128). There is a reduction in the
expression of autophagic protein during aging, which directly
influences α-Syn clearance (57).
Previous research has reported that decreased autophagy is
associated with aggregation of α-Syn (31). Compared with microglia isolated
from young mice, microglia isolated from adult mice exhibit a lower
capacity to phagocytose α-Syn and secrete more inflammatory factors
(57). Senescent microglia
generate TNF-α, which promotes α-Syn deposition (28). A previous study reported that
selective clearance of senescent microglia from the brain can
relieve dyskinesia in animal PD models and significantly reduce the
level of α-Syn in cerebrospinal fluid (125). In addition to senescent
microglia, activated microglia can also produce TNF-α, which
enhances neuronal α-Syn secretion and thus promotes intercellular
α-Syn propagation. Moreover, microglia-derived TNF-α promotes the
SASP of neurons and causes neuronal aging (60).
α-Syn PFFs induce senescence in microglia and
astrocytes, which results in altered expression of cellular
senescence markers (40).
Specifically, in microglia and astrocytes of α-Syn PFF-injected
mice, α-Syn PFF-induced toxicity downregulates the expression of
cellular senescence markers, such as HMGB1 and Lamin B1, whilst
upregulating the expression of the p21 aging marker (40). These results are consistent with
the observations in substantia nigra pars compacta tissues of
postmortem patients with PD (40).
In general, the expression levels of HMGB1 and Lamin B1 are
decreased in senescent cells, while the expression level of p21 is
increased (129-131).
However, other animal experiments have reported that HMGB1
expression increases in astrocytes and decreases in neurons with
advancing age (33,132). Therefore, the mechanism of α-Syn
regulation of senescence markers and the phenotypes of distinct
senescent cells should be further elucidated. HMGB1, which is
located mainly in the nucleus, is involved in various physiological
processes associated with DNA (including DNA repair, replication
and transcription) (133). In
senescent cells, HMGB1 is secreted into the extracellular space
from the nucleus (129),
resulting in DNA double-strand breaks (132). In certain cases, senescent
astrocytes can release HMGB1 and this extracellular HMGB1 induces
healthy astrocytes to develop a senescent-like phenotype (134). In addition, HMGB1 actively
released into the extracellular space by aging cells, may stimulate
target cells to secrete TNF-α, IL-6 and IL-1β by activating
relevant receptors of target cells, such as TLR4(134). Therefore, it can be suggested
that α-Syn-induced glial cell senescence may affect the surrounding
cells and tissue microenvironment by secreting related proteins,
which may explain the toxic effects of α-Syn.
α-Syn may induce senescence in microglia and
astrocytes; however, whether it can induce senescence in other
types of cells requires further research. Senescent cells lose
their normal physiological functions and produce harmful
substances, which can affect the function of tissues. It is
generally difficult to reverse the process of cellular aging,
although progress has been made (135-137).
Transplantation of exogenous stem cells may be a future treatment
for PD. Stem cells have previously been reported to have
anti-inflammatory effects and regenerative abilities, which may
benefit patients with PD (92).
10. Conclusion
Various risk factors are implicated in the onset of
PD, including the expression of pathological α-Syn. Pathological
α-Syn can induce a variety of toxic effects that result in DA
neuronal death and pathogenesis associated with PD. Drugs that
suppress α-Syn aggregation or target α-Syn for degradation may have
potential in the future for the treatment of patients with PD. PD
is also an age-related neurodegenerative disorder and several
characteristics of PD are similar to certain manifestations of
aging. Previous research has shown that α-Syn spreads more easily,
for example from the intestine to the brain, in aged animals. Under
aging conditions, the ability of cells to take up and degrade α-Syn
decreases, which leads to α-Syn accumulation and neuroinflammation.
Additionally, α-Syn has the potential to induce senescence in glial
cells, which is associated with changes in the expression of
cellular senescence markers. Given that the toxic effects of α-Syn
are associated with aging, the possibility of treating PD with
antiaging drugs should be explored. In addition, as α-Syn
expression may originate in the intestines, further research should
focus on the intestinal function, intestinal symptoms and the
intestinal microbiota of patients with PD. Research on such topics
may be helpful to understand the pathogenesis of PD and provide a
basis for the development of new therapeutic drugs. Drugs that
prolong life and reduce age-related decline in intestinal function,
such as rapamycin, also warrant further research. Both cellular
aging and inflammation are implicated in PD pathology.
Transplantation of exogenous stem cells with anti-inflammatory and
regenerative properties may be a worthwhile therapeutic approach.
Moreover, PD is caused by the selective death of DA neurons; hence,
transplantation of DA neurons may replace these dead DA neurons.
However, cell transplantation is associated with a number of
issues, such as problems associated with the source of transplanted
cells, ethical issues, survival of transplanted cells and the
ability of transplanted cells to establish functional synaptic
connections with other neurons. The excessive inflammatory response
of glial cells triggered by α-Syn mediates the death of DA neurons.
In addition, activated glial cells secrete inflammatory factors,
which lead to an increase in BBB permeability, which causes T cell
infiltration into the brain. Certain T cell subtypes enhance the
inflammatory response and induce DA neuronal death. Therefore,
blocking the binding of α-Syn to the relevant receptors of glial
cells or regulating the inflammatory response of glial cells could
be a promising strategy for PD treatment.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the Medical and Health
Science and Technology Development Planning Project of Shandong
Province (grant no. 2019WS582) and the Major Basic Research Project
of Natural Science Foundation of Shandong Province (grant no.
ZR2019ZD23).
Availability of data and materials
Not applicable.
Authors' contributions
NZ wrote the original draft of the manuscript. ZY
and HX reviewed and edited the manuscript. RC reviewed and improved
the language of this manuscript. SS and SX created the figures. SW
reviewed and edited the manuscript, supervised the project and
obtained funding. Data authentication is not applicable. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhu B, Yin D, Zhao H and Zhang L: The
immunology of Parkinson's disease. Semin Immunopathol. 44:659–672.
2022.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Jang JH, Yeom MJ, Ahn S, Oh JY, Ji S, Kim
TH and Park HJ: Acupuncture inhibits neuroinflammation and gut
microbial dysbiosis in a mouse model of Parkinson's disease. Brain
Behav Immun. 89:641–655. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Fields CR, Bengoa-Vergniory N and
Wade-Martins R: Targeting alpha-synuclein as a therapy for
Parkinson's disease. Front Mol Neurosci. 12(299)2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zhao Y, Zhang Z, Qin S, Fan W, Li W, Liu
J, Wang S, Xu Z and Zhao M: Acupuncture for Parkinson's disease:
Efficacy evaluation and mechanisms in the dopaminergic neural
circuit. Neural Plast. 2021(9926445)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Stevenson TJ, Murray HC, Turner C, Faull
RLM, Dieriks BV and Curtis MA: α-synuclein inclusions are abundant
in non-neuronal cells in the anterior olfactory nucleus of the
Parkinson's disease olfactory bulb. Sci Rep.
10(6682)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Campo F, Carletti R, Fusconi M, Pellicano
C, Pontieri FE, Di Gioia CR and de Vincentiis M: Alpha-synuclein in
salivary gland as biomarker for Parkinson's disease. Rev Neurosci.
30:455–462. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Jansen van Rensburg Z, Abrahams S, Bardien
S and Kenyon C: Toxic feedback loop involving iron, reactive oxygen
species, α-synuclein and neuromelanin in Parkinson's disease and
intervention with turmeric. Mol Neurobiol. 58:5920–5936.
2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Guerrero-Ferreira R, Taylor NM, Mona D,
Ringler P, Lauer ME, Riek R, Britschgi M and Stahlberg H: Cryo-EM
structure of alpha-synuclein fibrils. Elife.
7(e36402)2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Vasquez V, Mitra J, Wang H, Hegde PM, Rao
KS and Hegde ML: A multi-faceted genotoxic network of
alpha-synuclein in the nucleus and mitochondria of dopaminergic
neurons in Parkinson's disease: Emerging concepts and challenges.
Prog Neurobiol. 185(101729)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Burré J, Sharma M and Südhof TC: Cell
biology and pathophysiology of α-synuclein. Cold Spring Harb
Perspect Med. 8(a024091)2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Fujioka S, Ogaki K, Tacik PM, Uitti RJ,
Ross OA and Wszolek ZK: Update on novel familial forms of
Parkinson's disease and multiple system atrophy. Parkinsonism Relat
Disord. 20 (Suppl 1):S29–S34. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Koo HJ, Lee HJ and Im H: Sequence
determinants regulating fibrillation of human alpha-synuclein.
Biochem Biophys Res Commun. 368:772–778. 2008.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Liu W, Lim KL and Tan EK:
Intestine-derived α-synuclein initiates and aggravates pathogenesis
of Parkinson's disease in Drosophila. Transl Neurodegener.
11(44)2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Choi ML, Chappard A, Singh BP, Maclachlan
C, Rodrigues M, Fedotova EI, Berezhnov AV, De S, Peddie CJ, Athauda
D, et al: Pathological structural conversion of α-synuclein at the
mitochondria induces neuronal toxicity. Nat Neurosci. 25:1134–1148.
2022.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Pacheco CR, Morales CN, Ramírez AE, Muñoz
FJ, Gallegos SS, Caviedes PA, Aguayo LG and Opazo CM: Extracellular
α-synuclein alters synaptic transmission in brain neurons by
perforating the neuronal plasma membrane. J Neurochem. 132:731–741.
2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Cardinale A, Calabrese V, de Iure A and
Picconi B: Alpha-synuclein as a prominent actor in the inflammatory
synaptopathy of Parkinson's disease. Int J Mol Sci.
22(6517)2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chakroun T, Evsyukov V, Nykänen NP,
Höllerhage M, Schmidt A, Kamp F, Ruf VC, Wurst W, Rösler TW and
Höglinger GU: Alpha-synuclein fragments trigger distinct
aggregation pathways. Cell Death Dis. 11(84)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wang C, Yang T, Liang M, Xie J and Song N:
Astrocyte dysfunction in Parkinson's disease: From the perspectives
of transmitted α-synuclein and genetic modulation. Transl
Neurodegener. 10(39)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yu WW, Cao SN, Zang CX, Wang L, Yang HY,
Bao XQ and Zhang D: Heat shock protein 70 suppresses
neuroinflammation induced by α-synuclein in astrocytes. Mol Cell
Neurosci. 86:58–64. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Kim C, Spencer B, Rockenstein E, Yamakado
H, Mante M, Adame A, Fields JA, Masliah D, Iba M, Lee HJ, et al:
Immunotherapy targeting toll-like receptor 2 alleviates
neurodegeneration in models of synucleinopathy by modulating
α-synuclein transmission and neuroinflammation. Mol Neurodegener.
13(43)2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Sian-Hulsmann J and Riederer P: The role
of alpha-synuclein as ferrireductase in neurodegeneration
associated with Parkinson's disease. J Neural Transm (Vienna).
127:749–754. 2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Riederer P, Monoranu C, Strobel S,
Iordache T and Sian-Hülsmann J: Iron as the concert master in the
pathogenic orchestra playing in sporadic Parkinson's disease. J
Neural Transm (Vienna). 128:1577–1598. 2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Melo TQ, Copray SJCVM and Ferrari MFR:
Alpha-synuclein toxicity on protein quality control, mitochondria
and endoplasmic reticulum. Neurochem Res. 43:2212–2223.
2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ninkina N, Tarasova TV, Chaprov KD, Roman
AY, Kukharsky MS, Kolik LG, Ovchinnikov R, Ustyugov AA, Durnev AD
and Buchman VL: Alterations in the nigrostriatal system following
conditional inactivation of α-synuclein in neurons of adult and
aging mice. Neurobiol Aging. 91:76–87. 2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Tansey MG, Wallings RL, Houser MC, Herrick
MK, Keating CE and Joers V: Inflammation and immune dysfunction in
Parkinson disease. Nat Rev Immunol. 22:657–673. 2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhao YF, Qiong-Zhang Zhang JF, Lou ZY, Zu
HB, Wang ZG, Zeng WC and Kai-Yao and Xiao BG: The synergy of aging
and LPS exposure in a mouse model of Parkinson's disease. Aging
Dis. 9:785–797. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Russo T and Riessland M: Age-related
midbrain inflammation and senescence in Parkinson's disease. Front
Aging Neurosci. 14(917797)2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Su R and Zhou T: Alpha-synuclein induced
immune cells activation and associated therapy in Parkinson's
disease. Front Aging Neurosci. 13(769506)2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Tremblay ME, Cookson MR and Civiero L:
Glial phagocytic clearance in Parkinson's disease. Mol
Neurodegener. 14(16)2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kim C, Kwon S, Iba M, Spencer B,
Rockenstein E, Mante M, Adame A, Shin SJ, Fields JA, Rissman RA, et
al: Effects of innate immune receptor stimulation on extracellular
α-synuclein uptake and degradation by brain resident cells. Exp Mol
Med. 53:281–290. 2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Fellner L, Gabassi E, Haybaeck J and
Edenhofer F: Autophagy in α-synucleinopathies-an overstrained
system. Cells. 10(3143)2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Caggiu E, Arru G, Hosseini S, Niegowska M,
Sechi G, Zarbo IR and Sechi LA: Inflammation, infectious triggers,
and Parkinson's disease. Front Neurol. 10(122)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Salminen A, Ojala J, Kaarniranta K,
Haapasalo A, Hiltunen M and Soininen H: Astrocytes in the aging
brain express characteristics of senescence-associated secretory
phenotype. Eur J Neurosci. 34:3–11. 2011.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Coleman C and Martin I: Unraveling
Parkinson's disease neurodegeneration: Does aging hold the clues? J
Parkinsons Dis. 12:2321–2338. 2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Pang SY, Ho PW, Liu HF, Leung CT, Li L,
Chang EES, Ramsden DB and Ho SL: The interplay of aging, genetics
and environmental factors in the pathogenesis of Parkinson's
disease. Transl Neurodegener. 8(23)2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wendimu MY and Hooks SB: Microglia
phenotypes in aging and neurodegenerative diseases. Cells.
11(2091)2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Nasrolahi A, Safari F, Farhoudi M,
Khosravi A, Farajdokht F, Bastaminejad S, Sandoghchian Shotorbani S
and Mahmoudi J: Immune system and new avenues in Parkinson's
disease research and treatment. Rev Neurosci. 30:709–727.
2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Van Den Berge N, Ferreira N, Mikkelsen TW,
Alstrup AKO, Tamgüney G, Karlsson P, Terkelsen AJ, Nyengaard JR,
Jensen PH and Borghammer P: Ageing promotes pathological
alpha-synuclein propagation and autonomic dysfunction in wild-type
rats. Brain. 144:1853–1868. 2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Challis C, Hori A, Sampson TR, Yoo BB,
Challis RC, Hamilton AM, Mazmanian SK, Volpicelli-Daley LA and
Gradinaru V: Gut-seeded α-synuclein fibrils promote gut dysfunction
and brain pathology specifically in aged mice. Nat Neurosci.
23:327–336. 2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Verma DK, Seo BA, Ghosh A, Ma SX,
Hernandez-Quijada K, Andersen JK, Ko HS and Kim YH: Alpha-synuclein
preformed fibrils induce cellular senescence in Parkinson's disease
models. Cells. 10(1694)2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Rotter A, Lenz B, Pitsch R,
Richter-Schmidinger T, Kornhuber J and Rhein C: Alpha-synuclein RNA
expression is increased in major depression. Int J Mol Sci.
20(2029)2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Phan HTM, Bartz JC, Ayers J, Giasson BI,
Schubert M, Rodenhausen KB, Kananizadeh N, Li Y and Bartelt-Hunt
SL: Adsorption and decontamination of α-synuclein from medically
and environmentally-relevant surfaces. Colloids Surf B
Biointerfaces. 166:98–107. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
D'Onofrio M, Munari F and Assfalg M:
Alpha-synuclein-nanoparticle interactions: Understanding,
controlling and exploiting conformational plasticity. Molecules.
25(5625)2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Makasewicz K, Wennmalm S, Stenqvist B,
Fornasier M, Andersson A, Jönsson P, Linse S and Sparr E:
Cooperativity of α-synuclein binding to lipid membranes. ACS Chem
Neurosci. 12:2099–2109. 2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Davidson WS, Jonas A, Clayton DF and
George JM: Stabilization of alpha-synuclein secondary structure
upon binding to synthetic membranes. J Biol Chem. 273:9443–9449.
1998.PubMed/NCBI View Article : Google Scholar
|
|
46
|
George JM, Jin H, Woods WS and Clayton DF:
Characterization of a novel protein regulated during the critical
period for song learning in the zebra finch. Neuron. 15:361–372.
1995.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Uéda K, Fukushima H, Masliah E, Xia Y,
Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y and Saitoh T:
Molecular cloning of cDNA encoding an unrecognized component of
amyloid in Alzheimer disease. Proc Natl Acad Sci USA.
90:11282–11286. 1993.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Weinreb PH, Zhen W, Poon AW, Conway KA and
Lansbury PT Jr: NACP, a protein implicated in Alzheimer's disease
and learning, is natively unfolded. Biochemistry. 35:13709–13715.
1996.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Acquasaliente L, Pontarollo G, Radu CM,
Peterle D, Artusi I, Pagotto A, Uliana F, Negro A, Simioni P and De
Filippis V: Exogenous human α-synuclein acts in vitro as a mild
platelet antiaggregant inhibiting α-thrombin-induced platelet
activation. Sci Rep. 12(9880)2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Dedmon MM, Lindorff-Larsen K,
Christodoulou J, Vendruscolo M and Dobson CM: Mapping long-range
interactions in alpha-synuclein using spin-label NMR and ensemble
molecular dynamics simulations. J Am Chem Soc. 127:476–477.
2005.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Bogale TA, Faustini G, Longhena F, Mitola
S, Pizzi M and Bellucci A: Alpha-synuclein in the regulation of
brain endothelial and perivascular cells: Gaps and future
perspectives. Front Immunol. 12(611761)2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Bozelli JC Jr, Kamski-Hennekam E, Melacini
G and Epand RM: α-Synuclein and neuronal membranes: Conformational
flexibilities in health and disease. Chem Phys Lipids.
235(105034)2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Chakraborty R and Chattopadhyay K:
Cryo-electron microscopy uncovers key residues within the core of
alpha-synuclein fibrils. ACS Chem Neurosci. 10:1135–1136.
2019.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Walsh DM and Selkoe DJ: A critical
appraisal of the pathogenic protein spread hypothesis of
neurodegeneration. Nat Rev Neurosci. 17:251–260. 2016.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Choi ML and Gandhi S: Crucial role of
protein oligomerization in the pathogenesis of Alzheimer's and
Parkinson's diseases. FEBS J. 285:3631–3644. 2018.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Liu G, Aliaga L and Cai H: α-Synuclein,
LRRK2 and their interplay in Parkinson's disease. Future Neurol.
7:145–153. 2012.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Wang R, Ren H, Kaznacheyeva E, Lu X and
Wang G: Association of glial activation and α-synuclein pathology
in Parkinson's disease. Neurosci Bull. 39:479–490. 2022.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Imbriani P, Schirinzi T, Meringolo M,
Mercuri NB and Pisani A: Centrality of early synaptopathy in
Parkinson's disease. Front Neurol. 9(103)2018.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Kam TI, Mao X, Park H, Chou SC,
Karuppagounder SS, Umanah GE, Yun SP, Brahmachari S, Panicker N,
Chen R, et al: Poly(ADP-ribose) drives pathologic α-synuclein
neurodegeneration in Parkinson's disease. Science.
362(eaat8407)2018.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Bae EJ, Choi M, Kim JT, Kim DK, Jung MK,
Kim C, Kim TK, Lee JS, Jung BC, Shin SJ, et al: TNF-α promotes
α-synuclein propagation through stimulation of
senescence-associated lysosomal exocytosis. Exp Mol Med.
54:788–800. 2022.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Gelpi E, Navarro-Otano J, Tolosa E, Gaig
C, Compta Y, Rey MJ, Martí MJ, Hernández I, Valldeoriola F, Reñé R
and Ribalta T: Multiple organ involvement by alpha-synuclein
pathology in Lewy body disorders. Mov Disord. 29:1010–1018.
2014.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Donadio V, Incensi A, Piccinini C,
Cortelli P, Giannoccaro MP, Baruzzi A and Liguori R: Skin nerve
misfolded α-synuclein in pure autonomic failure and Parkinson
disease. Ann Neurol. 79:306–316. 2016.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Beach TG, Adler CH, Sue LI, Vedders L, Lue
L, White Iii CL, Akiyama H, Caviness JN, Shill HA, Sabbagh MN, et
al: Multi-organ distribution of phosphorylated alpha-synuclein
histopathology in subjects with Lewy body disorders. Acta
Neuropathol. 119:689–702. 2010.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Braak H, Rüb U, Gai WP and Del Tredici K:
Idiopathic Parkinson's disease: Possible routes by which vulnerable
neuronal types may be subject to neuroinvasion by an unknown
pathogen. J Neural Transm (Vienna). 110:517–536. 2003.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Braak H, Ghebremedhin E, Rüb U, Bratzke H
and Del Tredici K: Stages in the development of Parkinson's
disease-related pathology. Cell Tissue Res. 318:121–134.
2004.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Liu B, Fang F, Pedersen NL, Tillander A,
Ludvigsson JF, Ekbom A, Svenningsson P, Chen H and Wirdefeldt K:
Vagotomy and Parkinson disease: A Swedish register-based
matched-cohort study. Neurology. 88:1996–2002. 2017.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Ho PW, Leung CT, Liu H, Pang SY, Lam CS,
Xian J, Li L, Kung MH, Ramsden DB and Ho SL: Age-dependent
accumulation of oligomeric SNCA/α-synuclein from impaired
degradation in mutant LRRK2 knockin mouse model of Parkinson
disease: Role for therapeutic activation of chaperone-mediated
autophagy (CMA). Autophagy. 16:347–370. 2020.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Henderson MX, Trojanowski JQ and Lee VMY:
α-Synuclein pathology in Parkinson's disease and related
α-synucleinopathies. Neurosci Lett. 709(134316)2019.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Iba M, McDevitt RA, Kim C, Roy R,
Sarantopoulou D, Tommer E, Siegars B, Sallin M, Kwon S, Sen JM, et
al: Aging exacerbates the brain inflammatory micro-environment
contributing to α-synuclein pathology and functional deficits in a
mouse model of DLB/PD. Mol Neurodegener. 17(60)2022.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Rauschenberger L, Behnke J, Grotemeyer A,
Knorr S, Volkmann J and Ip CW: Age-dependent neurodegeneration and
neuroinflammation in a genetic A30P/A53T double-mutated α-synuclein
mouse model of Parkinson's disease. Neurobiol Dis.
171(105798)2022.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Yang W, Li X, Li X and Yu S:
Hemoglobin-α-synuclein complex exhibited age-dependent alterations
in the human striatum and peripheral RBCs. Neurosci Lett.
736(135274)2020.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Ma M, Li H, Wu J, Zhang Y, Shen H, Li X,
Wang Z and Chen G: Roles of prokineticin 2 in subarachnoid
hemorrhage-induced early brain injury via regulation of phenotype
polarization in astrocytes. Mol Neurobiol. 57:3744–3758.
2020.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Jeon YM, Kwon Y, Jo M, Lee S, Kim S and
Kim HJ: The role of glial mitochondria in alpha-synuclein toxicity.
Front Cell Dev Biol. 8(548283)2020.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Li Q and Haney MS: The role of glia in
protein aggregation. Neurobiol Dis. 143(105015)2020.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Wang P and Ye Y: Astrocytes in
neurodegenerative diseases: A perspective from tauopathy and
α-synucleinopathy. Life (Basel). 11(938)2021.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Liddelow SA, Guttenplan KA, Clarke LE,
Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS,
Peterson TC, et al: Neurotoxic reactive astrocytes are induced by
activated microglia. Nature. 541:481–487. 2017.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Diniz LP, Araujo APB, Matias I, Garcia MN,
Barros-Aragão FGQ, de Melo Reis RA, Foguel D, Braga C, Figueiredo
CP, Romão L and Gomes FCA: Astrocyte glutamate transporters are
increased in an early sporadic model of synucleinopathy. Neurochem
Int. 138(104758)2020.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Chavarria C, Ivagnes R and Souza JM:
Extracellular alpha-synuclein: Mechanisms for glial cell
internalization and activation. Biomolecules.
12(655)2022.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Vargas JY, Grudina C and Zurzolo C: The
prion-like spreading of α-synuclein: From in vitro to in vivo
models of Parkinson's disease. Ageing Res Rev. 50:89–101.
2019.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Wang J, Chen Z, Walston JD, Gao P, Gao M
and Leng SX: α-Synuclein activates innate immunity but suppresses
interferon-γ expression in murine astrocytes. Eur J Neurosci:
10.1111/ejn.13956, 2018 (Epub ahead of print).
|
|
81
|
Hua J, Yin N, Xu S, Chen Q, Tao T, Zhang
J, Ding J, Fan Y and Hu G: Enhancing the astrocytic clearance of
extracellular α-synuclein aggregates by ginkgolides attenuates
neural cell injury. Cell Mol Neurobiol. 39:1017–1028.
2019.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Chavarría C, Rodríguez-Bottero S, Quijano
C, Cassina P and Souza JM: Impact of monomeric, oligomeric and
fibrillar alpha-synuclein on astrocyte reactivity and toxicity to
neurons. Biochem J. 475:3153–3169. 2018.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Liscovitch N and French L: Differential
co-expression between α-synuclein and IFN-γ signaling genes across
development and in Parkinson's disease. PLoS One.
9(e115029)2014.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Wang J, Chen Z, Walston JD, Gao P, Gao M
and Leng SX: Interferon-γ potentiates α-synuclein-induced
neurotoxicity linked to toll-like receptors 2 and 3 and tumor
necrosis factor-α in murine astrocytes. Mol Neurobiol.
56:7664–7679. 2019.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Liu CY, Wang X, Liu C and Zhang HL:
Pharmacological targeting of microglial activation: New therapeutic
approach. Front Cell Neurosci. 13(514)2019.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Li Y, Xia Y, Yin S, Wan F, Hu J, Kou L,
Sun Y, Wu J, Zhou Q, Huang J, et al: Targeting microglial
α-synuclein/TLRs/NF-kappaB/NLRP3 inflammasome axis in Parkinson's
disease. Front Immunol. 12(719807)2021.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Zhao Y and Yang G: Potential of
extracellular vesicles in the Parkinson's disease-pathological
mediators and biomarkers. Neurochem Int. 144(104974)2021.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Sarlus H and Heneka MT: Microglia in
Alzheimer's disease. J Clin Invest. 127:3240–3249. 2017.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Neefjes J, Jongsma ML, Paul P and Bakke O:
Towards a systems understanding of MHC class I and MHC class II
antigen presentation. Nat Rev Immunol. 11:823–836. 2011.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Chhatbar C and Prinz M: The roles of
microglia in viral encephalitis: From sensome to therapeutic
targeting. Cell Mol Immunol. 18:250–258. 2021.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Kong W, Wang X, Yang X, Huang W, Han S,
Yin J, Liu W, He X and Peng B: Activation of TRPV1 contributes to
recurrent febrile seizures via inhibiting the microglial M2
phenotype in the immature brain. Front Cell Neurosci.
13(442)2019.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Gordon J, Lockard G, Monsour M, Alayli A,
Choudhary H and Borlongan CV: Sequestration of inflammation in
Parkinson's Disease via stem cell therapy. Int J Mol Sci.
23(10138)2022.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Stefanova N, Fellner L, Reindl M, Masliah
E, Poewe W and Wenning GK: Toll-like receptor 4 promotes
α-synuclein clearance and survival of nigral dopaminergic neurons.
Am J Pathol. 179:954–963. 2011.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Xia Y, Zhang G, Han C, Ma K, Guo X, Wan F,
Kou L, Yin S, Liu L, Huang J, et al: Microglia as modulators of
exosomal alpha-synuclein transmission. Cell Death Dis.
10(174)2019.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Scheiblich H, Bousset L, Schwartz S, Griep
A, Latz E, Melki R and Heneka MT: Microglial NLRP3 inflammasome
activation upon TLR2 and TLR5 ligation by distinct α-synuclein
assemblies. J Immunol. 207:2143–2154. 2021.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Watson MB, Richter F, Lee SK, Gabby L, Wu
J, Masliah E, Effros RB and Chesselet MF: Regionally-specific
microglial activation in young mice over-expressing human wildtype
alpha-synuclein. Exp Neurol. 237:318–334. 2012.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Wallach T, Raden M, Hinkelmann L, Brehm M,
Rabsch D, Weidling H, Krüger C, Kettenmann H, Backofen R and
Lehnardt S: Distinct SARS-CoV-2 RNA fragments activate Toll-like
receptors 7 and 8 and induce cytokine release from human
macrophages and microglia. Front Immunol.
13(1066456)2023.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Du XY, Xie XX and Liu RT: The role of
α-synuclein oligomers in Parkinson's disease. Int J Mol Sci.
21(8645)2020.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Zhang YN, Fan JK, Gu L, Yang HM, Zhan SQ
and Zhang H: Metabotropic glutamate receptor 5 inhibits
α-synuclein-induced microglia inflammation to protect from
neurotoxicity in Parkinson's disease. J Neuroinflammation.
18(23)2021.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Kim C, Ho DH, Suk JE, You S, Michael S,
Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ and Lee SJ:
Neuron-released oligomeric α-synuclein is an endogenous agonist of
TLR2 for paracrine activation of microglia. Nat Commun.
4(1562)2013.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Heidari A, Yazdanpanah N and Rezaei N: The
role of Toll-like receptors and neuroinflammation in Parkinson's
disease. J Neuroinflammation. 19(135)2022.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Yun SP, Kam TI, Panicker N, Kim S, Oh Y,
Park JS, Kwon SH, Park YJ, Karuppagounder SS, Park H, et al: Block
of A1 astrocyte conversion by microglia is neuroprotective in
models of Parkinson's disease. Nat Med. 24:931–938. 2018.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Kermorgant M, Fernagut PO, Meissner WG,
Arvanitis DN, N'Guyen D, Senard JM and Pavy-Le Traon A: Age and
Gender differences in cardiovascular autonomic failure in the
transgenic PLP-syn mouse, a model of multiple system atrophy. Front
Neurol. 13(874155)2022.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Kaji S, Maki T, Kinoshita H, Uemura N,
Ayaki T, Kawamoto Y, Furuta T, Urushitani M, Hasegawa M, Kinoshita
Y, et al: Pathological endogenous α-synuclein accumulation in
oligodendrocyte precursor cells potentially induces inclusions in
multiple system atrophy. Stem Cell Reports. 10:356–365.
2018.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Dutta S, Hornung S, Kruayatidee A, Maina
KN, Del Rosario I, Paul KC, Wong DY, Duarte Folle A, Markovic D,
Palma JA, et al: α-Synuclein in blood exosomes immunoprecipitated
using neuronal and oligodendroglial markers distinguishes
Parkinson's disease from multiple system atrophy. Acta Neuropathol.
142:495–511. 2021.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Wakabayashi K, Hayashi S, Yoshimoto M,
Kudo H and Takahashi H: NACP/alpha-synuclein-positive filamentous
inclusions in astrocytes and oligodendrocytes of Parkinson's
disease brains. Acta Neuropathol. 99:14–20. 2000.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Reyes JF, Rey NL, Bousset L, Melki R,
Brundin P and Angot E: Alpha-synuclein transfers from neurons to
oligodendrocytes. Glia. 62:387–398. 2014.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Kisos H, Pukaß K, Ben-Hur T,
Richter-Landsberg C and Sharon R: Increased neuronal α-synuclein
pathology associates with its accumulation in oligodendrocytes in
mice modeling α-synucleinopathies. PLoS One.
7(e46817)2012.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Ihse E, Yamakado H, van Wijk XM, Lawrence
R, Esko JD and Masliah E: Cellular internalization of
alpha-synuclein aggregates by cell surface heparan sulfate depends
on aggregate conformation and cell type. Sci Rep.
7(9008)2017.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Stefanova N, Reindl M, Neumann M, Kahle
PJ, Poewe W and Wenning GK: Microglial activation mediates
neurodegeneration related to oligodendroglial
alpha-synucleinopathy: Implications for multiple system atrophy.
Mov Disord. 22:2196–2203. 2007.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Raffaele S, Boccazzi M and Fumagalli M:
Oligodendrocyte dysfunction in amyotrophic lateral sclerosis:
Mechanisms and therapeutic perspectives. Cells.
10(565)2021.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Ratnam NM, Sonnemann HM, Frederico SC,
Chen H, Hutchinson MND, Dowdy T, Reid CM, Jung J, Zhang W, Song H,
et al: Reversing epigenetic gene silencing to overcome immune
evasion in CNS malignancies. Front Oncol. 11(719091)2021.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Tait AS, Butts CL and Sternberg EM: The
role of glucocorticoids and progestins in inflammatory, autoimmune,
and infectious disease. J Leukoc Biol. 84:924–931. 2008.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Enzmann G, Kargaran S and Engelhardt B:
Ischemia-reperfusion injury in stroke: Impact of the brain barriers
and brain immune privilege on neutrophil function. Ther Adv Neurol
Disord. 11(1756286418794184)2018.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Chandra G, Roy A, Rangasamy SB and Pahan
K: Induction of adaptive immunity leads to nigrostriatal disease
progression in MPTP mouse model of Parkinson's disease. J Immunol.
198:4312–4326. 2017.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Earls RH, Menees KB, Chung J, Barber J,
Gutekunst CA, Hazim MG and Lee JK: Intrastriatal injection of
preformed alpha-synuclein fibrils alters central and peripheral
immune cell profiles in non-transgenic mice. J Neuroinflammation.
16(250)2019.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Sun C, Jia G, Wang X, Wang Y and Liu Y:
Immunoproteasome is up-regulated in rotenone-induced Parkinson's
disease rat model. Neurosci Lett. 738(135360)2020.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Ferreira SA and Romero-Ramos M: Microglia
response during Parkinson's Disease: Alpha-synuclein intervention.
Front Cell Neurosci. 12(247)2018.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Kannarkat GT, Cook DA, Lee JK, Chang J,
Chung J, Sandy E, Paul KC, Ritz B, Bronstein J, Factor SA, et al:
Common genetic variant association with altered HLA expression,
synergy with pyrethroid exposure, and risk for Parkinson's disease:
An observational and case-control study. NPJ Parkinsons Dis.
1(15002)2015.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Galiano-Landeira J, Torra A, Vila M and
Bové J: CD8 T cell nigral infiltration precedes synucleinopathy in
early stages of Parkinson's disease. Brain. 143:3717–3733.
2020.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Rostami J, Fotaki G, Sirois J, Mzezewa R,
Bergström J, Essand M, Healy L and Erlandsson A: Astrocytes have
the capacity to act as antigen-presenting cells in the Parkinson's
disease brain. J Neuroinflammation. 17(119)2020.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Sulzer D, Alcalay RN, Garretti F, Cote L,
Kanter E, Agin-Liebes J, Liong C, McMurtrey C, Hildebrand WH, Mao
X, et al: T cells from patients with Parkinson's disease recognize
α-synuclein peptides. Nature. 546:656–661. 2017.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Lindestam Arlehamn CS, Dhanwani R, Pham J,
Kuan R, Frazier A, Rezende Dutra J, Phillips E, Mallal S, Roederer
M, Marder KS, et al: α-Synuclein-specific T cell reactivity is
associated with preclinical and early Parkinson's disease. Nat
Commun. 11(1875)2020.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Kwon JH, Kim M, Um S, Lee HJ, Bae YK, Choi
SJ, Hwang HH, Oh W and Jin HJ: Senescence-associated secretory
phenotype suppression mediated by small-sized mesenchymal stem
cells delays cellular senescence through TLR2 and TLR5 signaling.
Cells. 10(63)2021.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Xu Z, Qu A, Zhang H, Wang W, Hao C, Lu M,
Shi B, Xu L, Sun M, Xu C and Kuang H: Photoinduced elimination of
senescent microglia cells in vivo by chiral gold nanoparticles.
Chem Sci. 13:6642–6654. 2022.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Badanjak K, Fixemer S, Smajić S, Skupin A
and Grünewald A: The contribution of microglia to neuroinflammation
in Parkinson's disease. Int J Mol Sci. 22(4676)2021.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Chaib S, Tchkonia T and Kirkland JL:
Cellular senescence and senolytics: The path to the clinic. Nat
Med. 28:1556–1568. 2022.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Choi I, Zhang Y, Seegobin SP, Pruvost M,
Wang Q, Purtell K, Zhang B and Yue Z: Microglia clear
neuron-released α-synuclein via selective autophagy and prevent
neurodegeneration. Nat Commun. 11(1386)2020.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Menon R, Behnia F, Polettini J, Saade GR,
Campisi J and Velarde M: Placental membrane aging and HMGB1
signaling associated with human parturition. Aging (Albany NY).
8:216–230. 2016.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Wang AS, Nakamizo S, Ishida Y, Klassen G,
Chong P, Wada A, Lim JSY, Wright GD, Kabashima K and Dreesen O:
Identification and quantification of senescent cell types by lamin
B1 and HMGB1 in Actinic keratosis lesions. J Dermatol Sci.
105:61–64. 2022.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Johmura Y, Yamanaka T, Omori S, Wang TW,
Sugiura Y, Matsumoto M, Suzuki N, Kumamoto S, Yamaguchi K,
Hatakeyama S, et al: Senolysis by glutaminolysis inhibition
ameliorates various age-associated disorders. Science. 371:265–270.
2021.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Enokido Y, Yoshitake A, Ito H and Okazawa
H: Age-dependent change of HMGB1 and DNA double-strand break
accumulation in mouse brain. Biochem Biophys Res Commun.
376:128–133. 2008.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Mandke P and Vasquez KM: Interactions of
high mobility group box protein 1 (HMGB1) with nucleic acids:
Implications in DNA repair and immune responses. DNA Repair (Amst).
83(102701)2019.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Gaikwad S, Puangmalai N, Bittar A,
Montalbano M, Garcia S, McAllen S, Bhatt N, Sonawane M, Sengupta U
and Kayed R: Tau oligomer induced HMGB1 release contributes to
cellular senescence and neuropathology linked to Alzheimer's
disease and frontotemporal dementia. Cell Rep.
36(109419)2021.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Yang JH, Petty CA, Dixon-McDougall T,
Lopez MV, Tyshkovskiy A, Maybury-Lewis S, Tian X, Ibrahim N, Chen
Z, Griffin PT, et al: Chemically induced reprogramming to reverse
cellular aging. Aging (Albany NY). 15:5966–5989. 2023.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Sarkar TJ, Quarta M, Mukherjee S, Colville
A, Paine P, Doan L, Tran CM, Chu CR, Horvath S, Qi LS, et al:
Transient non-integrative expression of nuclear reprogramming
factors promotes multifaceted amelioration of aging in human cells.
Nat Commun. 11(1545)2020.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Browder KC, Reddy P, Yamamoto M, Haghani
A, Guillen IG, Sahu S, Wang C, Luque Y, Prieto J, Shi L, et al: In
vivo partial reprogramming alters age-associated molecular changes
during physiological aging in mice. Nat Aging. 2:243–253.
2022.PubMed/NCBI View Article : Google Scholar
|