Introduction
Heart failure (HF) is now one of the most common
causes of cardiovascular disease, with increasing morbidity and
mortality rates (1), and HF
occurrence is closely related to the long-term survival of patients
with acute coronary syndrome (2).
Chronic HF (CHF) is a common complication of several heart
diseases, characterized by impaired ventricular filling and
ejection (3). Diabetes,
hypertension, coronary heart disease, and chronic renal diseases
are all risk factors for CHF (4).
Doxorubicin (DOX) is an anthracycline initially extracted and
identified from Streptomyces pneumonia (5). The highly effective anticancer drug
DOX causes progressive cardiac remodeling due to myocardial damage
during the early stages of treatment and leads to cardiomyopathy in
the later periods (6), involving
the excessive generation of reactive oxygen species (ROS),
mitochondrial dysfunction, and cardiomyocyte death, frequently
resulting in hospitalization (7,8).
Additionally, cardiac cell injury, left ventricular dysfunction,
and congestive HF associated with a high dose of DOX treatment
further worsen a patient's quality of life (9). DOX-induced cardiotoxicity is a side
effect when used to treat CHF.
Ferroptosis is a relatively newly discovered type of
programmed cell death characterized by iron overload-induced
production of large amounts of ROS and lipid peroxide flocking
(10). Excessive death of
myocardial cells can lead to various cardiovascular diseases and
even develop into HF, so developing ideal treatment plans based on
pathogenesis is of great significance for cardiopathy (11). Cardiac remodeling is widely
accepted as the primary mechanism underlying the progression of HF
(12). Cardiac remodeling causes
myocardial fibrosis and hypertrophy, resulting in cardiac size,
shape, and functional changes, eventually leading to cardiac
systolic or diastolic dysfunction (13). Current studies have found that the
mechanisms related to ferroptosis, including iron homeostasis
imbalance, GSH deficiency, oxidative stress, cardiac stimulation,
and mitochondrial dysfunction, play a role in cardiac remodeling
(14). Substantial evidence has
shown a close relationship between HF pathogenesis and the
mechanism of ferroptosis, as the mechanism of HF and ferroptosis
has been studied (15). Iron
homeostasis is a complex process regulated by multiple iron
metabolism proteins; iron homeostasis imbalances can lead to iron
overload and ferroptosis. Iron homeostasis plays an important role
in maintaining normal heart physiological function, which is
susceptible to iron overload, resulting in oxidative stress, and
promoting the development of HF (16). Furthermore, the activation center
of GPX4 is an important component of cysteine and prevents
ferroptosis caused by lipid ROS, implying that GPX4-related
ferroptosis may be important in CHF.
Echinacoside (ECH) is a natural bioactive compound
isolated from the natural herbs Cistanche and
Echinacea. ECH has several biological and pharmacological
activities, including anti-apoptotic, neuroprotective,
hepatoprotective, immunomodulatory, anti-aging, anti-diabetic, and
bone formation-promoting properties (17,18).
However, there are no studies assessing the cardioprotective
effects of ECH in CHF to the best of our knowledge, and the
potential underlying mechanisms remain elusive. Thus, the aim of
the present study was to investigate the effects of ECH in GPX4
inhibition-induced ferroptosis of cardiomyocytes in vivo and
in vitro.
Materials and methods
Cell culture and treatment
Rat H9c2 cells were obtained from The Cell Bank of
Type Culture Collection of The Chinese Academy of Sciences. Cells
were cultured in DMEM supplemented with 10% FBS, 2 mM l-glutamine,
and 1% penicillin/streptomycin (all from Thermo Fisher Scientific,
Inc.) at 37˚C in a humidified incubator supplied with 5%
CO2 air. DOX (2 µM, MilliporeSigma, cat. no. 25316-40-9)
was used to create the model of using H9c2 cells pretreated for 24
h with or without 5, 10, or 20 µM ECH (purity: ≥99.85%;
MedChemExpress, cat. no. HY-N0020). Otherwise, 35 µM erastin was
used to induce ferroptosis in H9c2 cells pretreated for 24 h with
or without 10 µM ECH.
CCK-8 assay
The CCK-8 assay (Signalway Antibody LLC) was used to
detect cell proliferation. Cells were incubated with CCK-8 solution
for 1 h after 0 and 24 h in culture. Cell proliferation was
measured using a microplate reader, and optical densities at 450 nm
were assessed. Each experiment was repeated three times.
Knockdown of Gpx4
Short hairpin-interfering RNAs (shRNA) targeting rat
Gpx4 (shGpx4-1: 5'-GGTTTGACATGTACAGCAA-3', site: 377-398;
shGpx4-2, 5'-GAAGTAATCAAGAAATCAA-3', site: 332-353; and shGpx4-3
5'-GGATGAAAGTCCAGCCCAA-3', site: 437-458) and scramble shRNA
(5'-GGACGAGCTGTACAAGTAA-3') were produced by General Bio Co., Ltd.
and assembled into lentiviral plasmids (pLKO.1). Recombinant
plasmids were expressed in 293T cells in the presence of psPAX2 and
pMD2G using Lipofectamine® 2000 (Thermo Fisher
Scientific, Inc.) as per the manufacturer's instructions to produce
transducer plasmids. After 48 h, cells were transduced with the
pLKO.1-scramble shRNA (shNC) as a negative control.
Measurement of biochemical
indices
The Fe2+ concentration in H9c2 cells was
determined using an Iron Assay kit (cat. no. ab83366, Abcam), and
the LDH (cat. no. A020-1-1), GSH (cat. no. A006-2-1), and MDA
levels (cat. no. A003-1-1) in H9c2 cells or serum samples were
determined using commercial biochemical kits (Jiancheng
Bioengineering Institute). All procedures were performed in
accordance with the manufacturer's protocols.
Lipid peroxidation assessment using
C11-BODIPY
A total of 10 µM C11-BODIPY (Thermo Fisher
Scientific, Inc.) was added to the cell suspension
(1x106 cells/ml), and the suspension was incubated in
the dark at 37˚C for 30 min. Cells were then washed with PBS, and
the fluorescence intensity of the dye C11-BODIPY was measured using
an Accuri™ C6 flow cytometer (BD Biosciences, Inc.).
Data were analyzed using FlowJo version 7.6.1 (FlowJo LLC).
Reverse transcription-quantitative
(RTq)PCR
Total RNA of different samples was extracted using
TRIzol® Reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Using a cDNA synthesis kit, RNA was reverse-transcribed into
cDNA using a PrimeScript™ kit (Takara Biotechnology Co.,
Ltd.) according to the manufacturer's protocol. For qPCR, the
thermocycling conditions were: 95˚C for 10 min; followed by 40
cycles of 95˚C for 15 sec and 60˚C for 45 sec, and Gapdh was
used as the loading control. The relative gene expression was
calculated using the 2-ΔΔCq method (19). Data are presented as the mean of
three replicates. Table I contains
the sequences of all the primers.
 | Table ISequences of the primers. |
Table I
Sequences of the primers.
| Gene | Forward | Reverse |
|---|
| Tfrc |
5'-GTTTCTGCCAGCCCCCTATT-3' |
5'-CACCTCTGCTGCTGTACGAA-3' |
| Slc11a2 |
5'-TCCCCATTCCTGAGGAGGAG-3' |
5'-ATCCGTGGGACCTTGGGATA-3' |
| Slc7a11 |
5'-TCGTCCTTTCAAGGTGCCTC-3' |
5'-AGAGTCTTCTGGTACAACTTCTAGT-3' |
| Gpx4 |
5'-ACGCCAAAGTCCTAGGAAGC-3' |
5'-CTGCGAATTCGTGCATGGAG-3' |
| Ptgs2 |
5'-CTCAGCCATGCAGCAAATCC-3' |
5'-GGGTGGGCTTCAGCAGTAAT-3' |
| Gapdh |
5'-GGAGTCTACTGGCGTCTTCAC-3' |
5'-ATGAGCCCTTCCACGATGC-3'. |
Western blot analysis
RIPA lysis buffer (JRDUN) was used to lyse the
samples, and the protein yield was determined using an enhanced BCA
protein assay kit (Thermo Fisher Scientific, Inc.). Total proteins
were separated by 10% SDS-PAGE and transferred to a nitrocellulose
membrane overnight before being blocked with 5% nonfat dry milk
overnight at 4˚C. Subsequently, membranes were incubated with one
of the primary antibodies overnight at 4˚C, followed by secondary
antibody anti-mouse IgG (cat. no. A0208; Beyotime Institute of
Biotechnology; 1:1,000) for 1 h at 37˚C. Signals were visualized
and densitometry analysis was performed using an enhanced
chemiluminescence system (Bio-Rad Laboratories, Inc.). The primary
antibodies were used: GPX4 (cat. no. DF6701; Affinity Biosciences
Ltd.; 1:1,000), PTGS2 (cat. no. AF7003; Affinity Biosciences Ltd.;
1:1,000), and GAPDH (cat. no. #5174s; Cell Signaling Technology,
Inc.; 1:30,000).
Rat model of CHF and treatments
Male Sprague-Dawley rats weighing 180-220 g were
purchased from HFK Bioscience Co. Ltd. All experimental procedures
were performed in accordance with the Guidelines for the
Institutional Animal Care and Use Committee of Shanghai Rat &
Mouse Biotech Co., Ltd. (Approval no. 202109) (18). All procedures followed
internationally recognized ethical standards. The rats were divided
into the following four groups (n=6 per group): normal group, DOX
group (rat model of CHF), DOX + E50 group (DOX + ECH 50 mg/kg), and
DOX + E100 group (DOX + ECH 100 mg/kg). A CHF rat model was induced
by DOX (intraperitoneal injection of 2.5 mg/kg, twice a week) as
previously described (7).
Furthermore, the rats were given ECH (50 or 100 mg/kg) by gavage
for 2 weeks. To monitor cardiac function, an echocardiogram was
performed, and hemodynamic variables were assessed (20). After treatment completion, the
animals were anesthetized with 2% sodium pentobarbital (50 mg/kg)
(B005, Jiancheng Bioengineering Institute) and sacrificed by
cervical dislocation, and their hearts were removed. Myocardial
tissues were fixed in 4% paraformaldehyde overnight at 4˚C,
dehydrated in ascending ethanol gradient and embedded in paraffin
for 2 h at room temperature and made into paraffin sections (5 µm).
Subsequently, the sections were placed in an oven and heated at
60˚C for 1 h, immersed in xylene solution at room temperature for
10 min, and then immersed in descending ethanol gradient at room
temperature for 5 min, followed by staining with hematoxylin and
eosin at room temperature for 5 and 3 min, respectively, using an
H&E staining kit (cat. no. C0105S; Beyotime Institute of
Biotechnology). After dehydrating in 100% alcohol at room
temperature for 5 min and mounting, tissues were observed under a
microscope. Blood samples were taken from the abdominal aorta to
detect brain natriuretic peptide (BNP) using a BNP ELISA Kit (cat.
no. E-EL-R0126c; Elabscience Biotechnology, Inc.).
Statistical analysis
GraphPad Prism version 8.4.2 (GraphPad Software,
Inc.) was used for statistical analysis. Data are presented as the
mean ± standard deviation. Comparisons between two groups were
performed using a Student's t-test, whereas an ANOVA followed by a
Tukey's post hoc test was used to compare multiple groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
ECH ameliorates DOX-mediated increases
in LDH and MDA levels and decreases in GSH levels in rat H9c2
cells
To determine the optimal concentration of ECH for
regulating DOX-induced rat H9c2 cell cytotoxicity, H9c2 cells were
cultured with ECH at various concentrations (5-20 µM) for 24 h, and
the cell proliferation activity was detected using a CCK-8 assay
kit (Fig. 1A). When 20 µM ECH was
used, cell proliferation activity decreased significantly and in a
dose-dependent manner. The MDA, GSH, and LDH levels were also
measured. The MDA and LDH levels were elevated by DOX, whereas ECH
significantly reduced this trend (Fig.
1B and C). Furthermore, when
compared to the control group, the GSH levels in the DOX group were
significantly lower, and an increase in this level was observed
after ECH treatment (Fig. 1D).
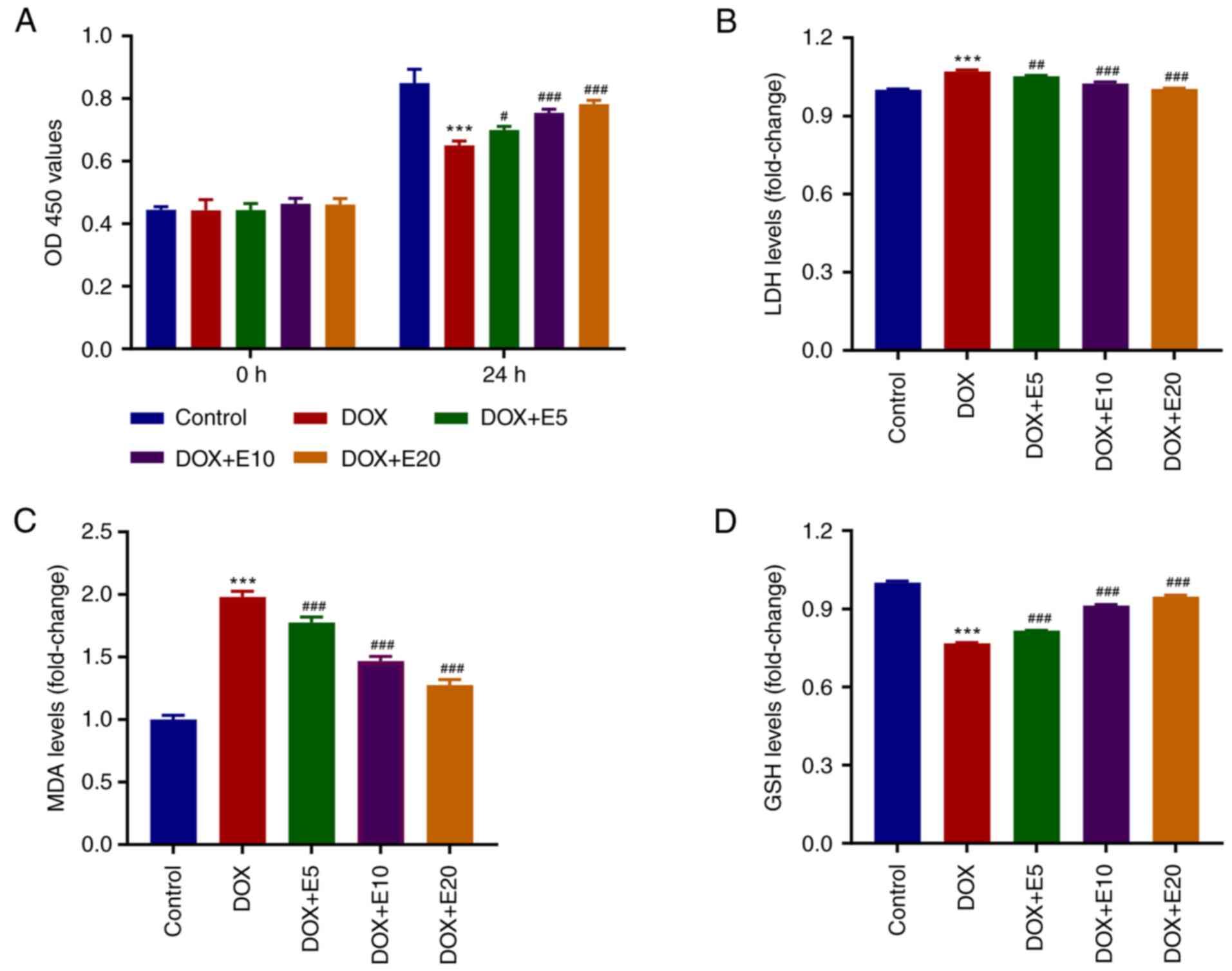 | Figure 1ECH ameliorates DOX-mediated
increases in LDH and MDA levels and decreases in GSH levels in rat
H9c2 cells. Rat H9c2 cells were treated with 2 µM DOX and 5, 10, or
20 µM ECH, and the (A) cell proliferation, (B) LDH, (C) MDA, and
(D) GSH levels were measured. ***P<0.001 vs. control;
#P<0.05, ##P<0.01,
###P<0.001 vs. DOX. ECH, Echinacoside; DOX,
doxorubicin; LDH, lactate dehydrogenase; MDA, malondialdehyde; GSH,
glutathione. |
ECH ameliorates DOX-induced
ferroptosis in rat H9c2 cells
The mechanisms related to ferroptosis, such as iron
homeostasis imbalance, GSH deficiency, oxidative stress, cardiac
stimulation, and mitochondrial dysfunction, play a role in cardiac
remodeling (14). Moreover, our
previous study found that DOX caused cardiac injury by activating
cardiomyocyte ferroptosis (7).
Therefore, the effects of ECH on DOX-induced Fe2+ and
lipid ROS in H9c2 cells were assessed. Interestingly, it was found
that DOX exposure increased the Fe2+ levels in H9c2
cells by threefold when compared to the control group. However, ECH
significantly reduced this elevation in a dose-dependent manner
(from 5-20 µM; Fig. 2A). When H9c2
cells were treated with DOX, they produced significantly more lipid
ROS as compared to the control group. Meanwhile, lipid ROS levels
in the ECH groups were significantly reduced in a dose-dependent
manner (Fig. 2B and C). GPX4 is currently recognized as a
central inhibitor of ferroptosis, and its activity depends on
glutathione produced by cystine-glutamate antitransporter solute
carrier family seven member 11 (SLC7A11) activation, which is
considered an anti-ferroptosis molecule (21). Additionally, transferrin receptor
protein 1 (TFRC) is the key receptor mediating the cellular uptake
of iron, and solute carrier family 11 member 2 (SLC11A2) is
involved in the release of irons from acidified endosomes into the
cytoplasm (22).
Prostaglandin-endoperoxide synthase 2 (PTGS2) plays dual functions
as both a cyclooxygenase and peroxidase, and is known to be induced
by various ferroptosis inducers (22). Thus, the mRNA levels of hub
ferroptosis-related genes Tfrc, Slc11a2,
Slc7a11, Gpx4, and Ptgs2 in cells were
assessed. DOX exposure significantly increased the mRNA expression
levels of Tfrc, Slc11a2, Slc7a11, and
Ptgs2, while decreasing the mRNA expression levels of
Gpx4. However, ECH significantly increased the mRNA
expression levels of Gpx4 and decreased the Ptgs2
mRNA expression levels in DOX-treated rat H9c2 cells in a
dose-dependent manner (Fig. 2D).
These two proteins were therefore further examined by western
blotting. DOX decreased GPX4 protein expression while increasing
PTGS2 protein expression, and this was inhibited by ECH (Fig. 2E).
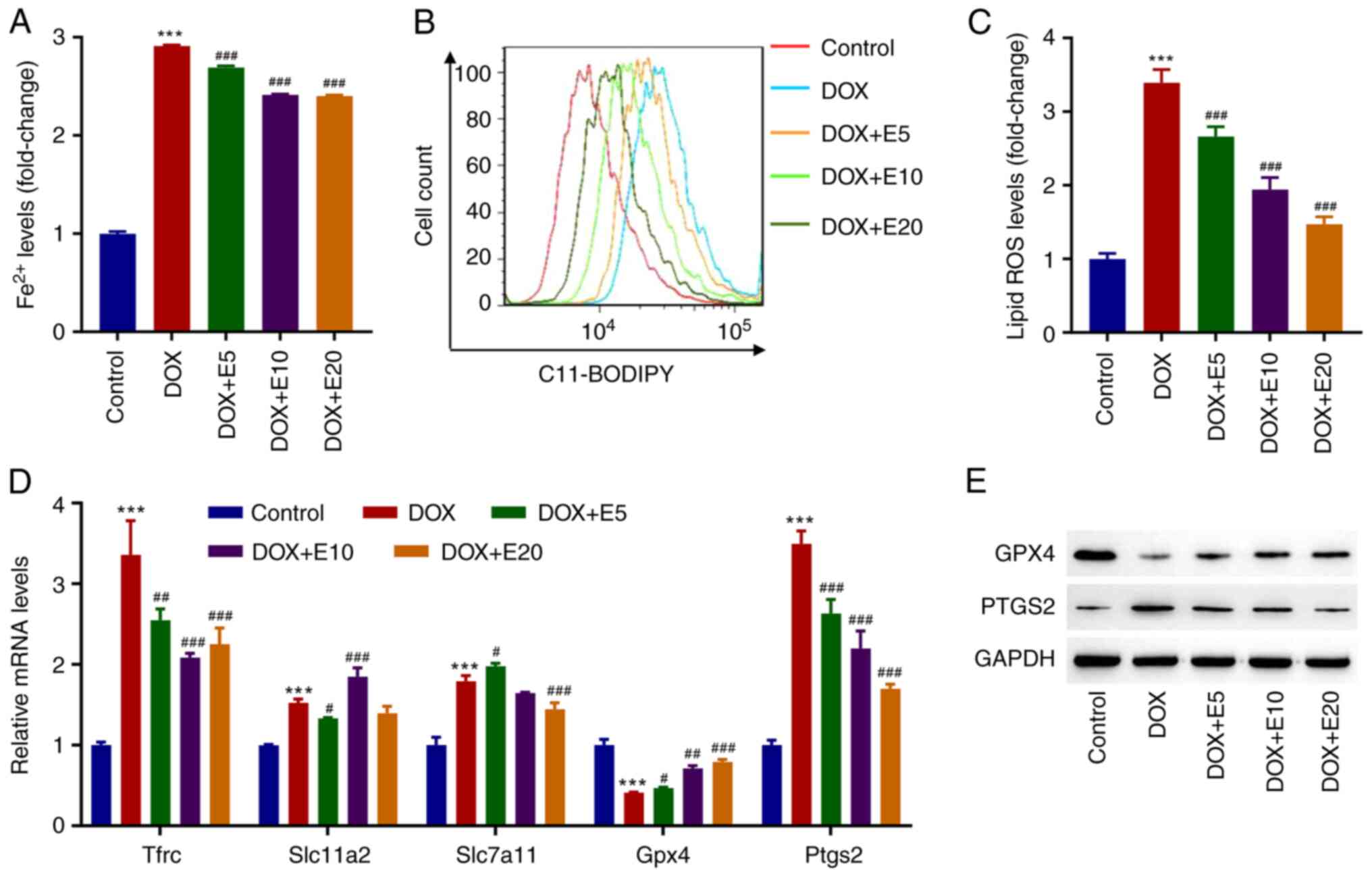 | Figure 2ECH reduced DOX-induced ferroptosis
in rat H9c2 cells. Rat H9c2 cells were treated with 2 µM DOX and 5,
10, or 20 µM ECH, and the (A) Fe2+ concentrations, (B
and C) lipid ROS levels, and (D) mRNA levels of Tfrc,
Slc11a2, Slc7a11, Gpx4 and Ptgs2, and
(E) protein expression of GPX4 and PTGS2 were measured.
***P<0.001 vs. control; #P<0.05,
##P<0.01, ###P<0.001 vs. DOX. ECH,
Echinacoside; DOX, doxorubicin; ROS, reactive oxygen species;
Tfrc, transferrin receptor protein 1; Slc11a2, solute
carrier family 11 member 2; Slc7a11, solute carrier family
seven member 11; Gpx4, glutathione peroxidase 4;
Ptgs2, prostaglandin-endoperoxide synthase 2. |
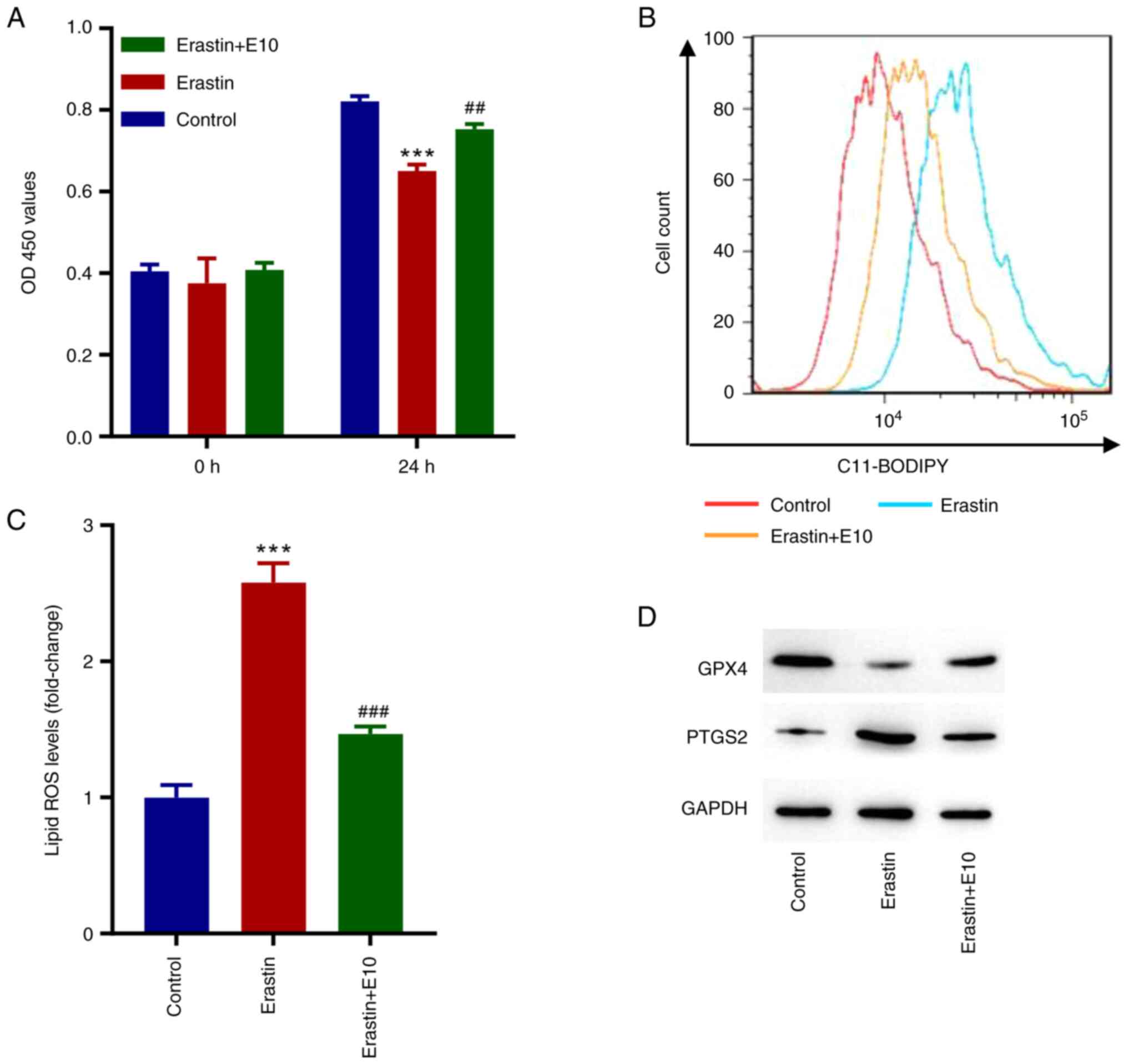
ECH inhibits erastin-induced
ferroptosis in rat H9c2 cells
To further examine the role of ECH in ferroptosis in
rat H9c2 cells, the ferroptosis inducer erastin was used. As
illustrated in Fig. 3A, cell
proliferation decreased significantly in erastin-treated cells,
whereas ECH significantly increased cell proliferation after 24 h.
Moreover, erastin significantly increased the lipid ROS levels, and
ECH reversed this (Fig. 3B and
C). When compared to the control
group, erastin exposure reduced GPX4 expression while increasing
PTGS2 expression, but ECH could reverse these effects (Fig. 3D).
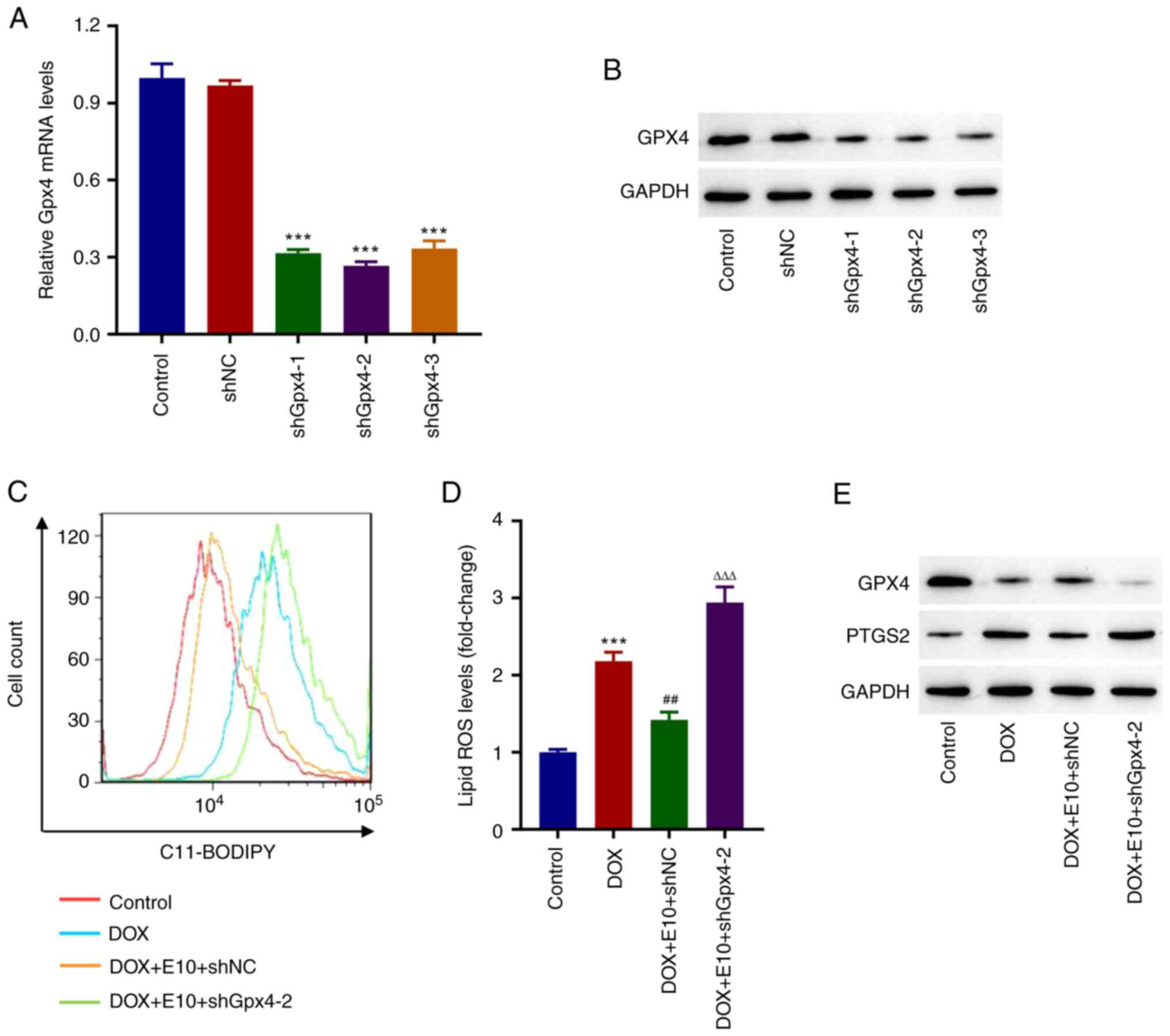
Knockdown of Gpx4 inhibits the
protective effects of ECH on DOX-induced accumulation of lipid ROS
in rat H9c2 cells
shRNA targeting of Gpx4 was used to
investigate the effects of GPX4 on the protective effects of ECH on
DOX-induced ferroptosis in rat H9c2 cells. Western blot analysis
revealed that using specific Gpx4 shRNAs (Fig. 4A and B) significantly reduced Gpx4
expression when compared to the shNC group. Furthermore, ECH
significantly reduced the DOX-induced increases in lipid ROS
levels, whereas shGpx4 reversed this effect (Fig. 4C and D). Additionally, Western blotting data
showed that DOX inhibited GPX4 expression while increasing PTGS2
expression; however, this trend was reversed by the knockdown of
Gpx4 (Fig. 4E). These
findings showed that knockdown of Gpx4 inhibited the
protective effects of ECH on DOX-induced accumulation of lipid ROS
in rat H9c2 cells.
ECH ameliorates DOX-induced CHF in
rats
DOX was used to establish a CHF rat model to assess
the effects of ECH on CHF in vivo. DOX reduced the left
ventricular ejection fraction, fractional shortening, end-systolic
pressure, and heart rate while increasing left ventricular
end-diastolic pressure (Table
II). Conversely, ECH may improve DOX-induced cardiac function.
Tissue sections from the control group exhibited a normal
morphology, whereas those from DOX-treated rats exhibited
myocardial disorganization, increased intracellular spaces, and
cytoplasmic vacuolization (Fig.
5A). Moreover, eosinophilic cells with pyknotic nuclei and loss
of myofibrils were observed among cardiomyocytes in the DOX-treated
rats (Fig. 5A). However, ECH
administration resulted in a considerable improvement in the
histopathology as compared to that observed in the DOX group; the
histological appearance of high-dose ECH group was similar to that
of the control group (Fig. 5A).
Given that BNP is an important indicator of ventricular function,
the BNP levels in the CHF rat model were measured by ELISA. The
findings revealed that, when compared to the control group, DOX
exposure significantly increased the BNP levels, whereas ECH
exposure decreased the BNP levels in a dose-dependent manner
(Fig. 5B). LDH is an enzyme found
in myocardial cell damage. DOX significantly increased the LDH
levels compared with the control group, whereas ECH administration
reversed this effect (Fig. 5C). Of
note, the effect of ECH on the protein expression levels of GPX4
and PTGS2 matched the in vitro findings, which showed that
ECH administration reversed the effects in the CHF rat model
(Fig. 5D).
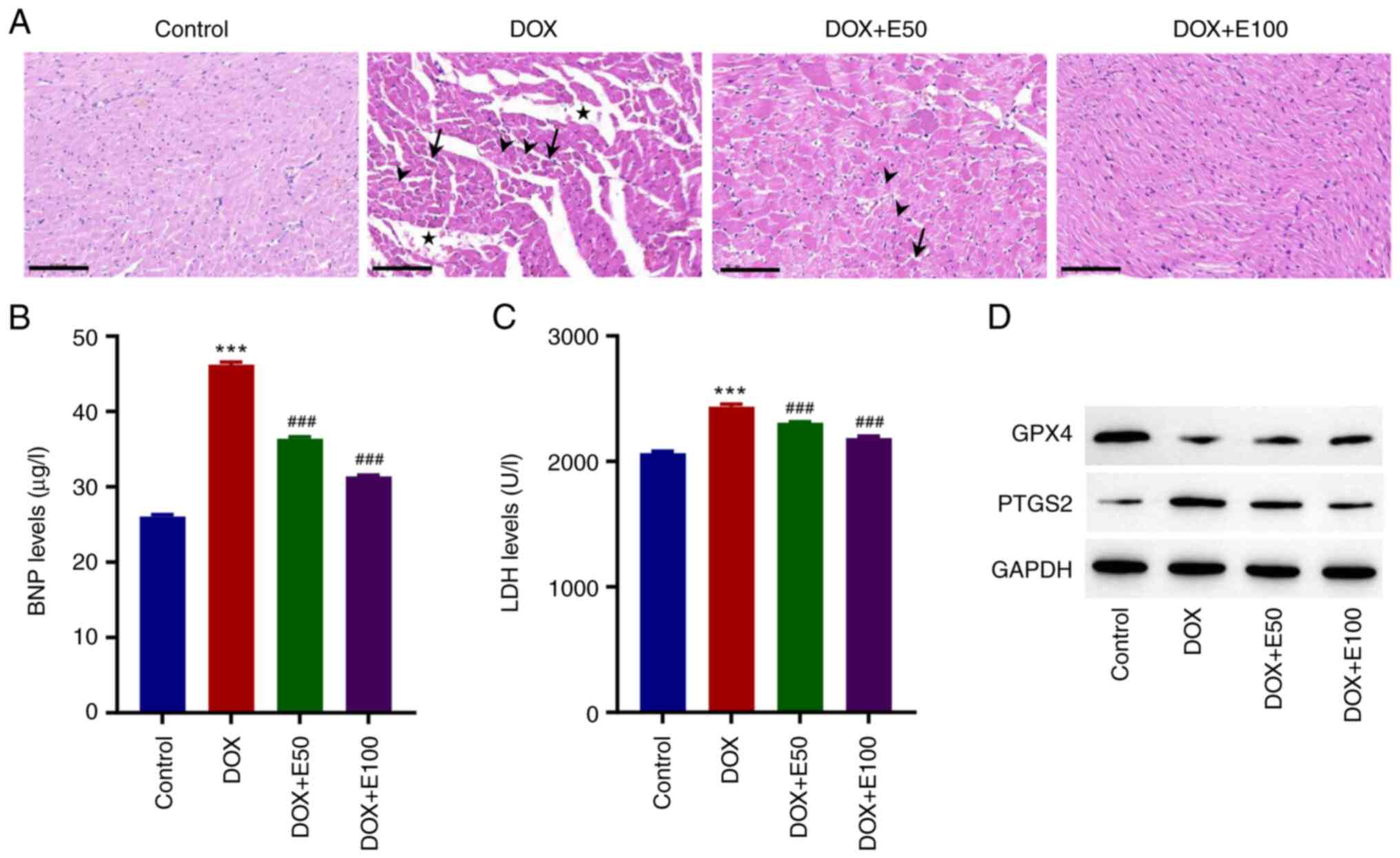 | Figure 5ECH reduced the DOX-induced cardiac
injury in rat. (A) Images of hematoxylin and eosin-stained
cardiomyocytes. Scale bar, 100 µm. Serum (B) BNP and (C) LDH
levels. (D) Protein expression levels of GPX4 and PTGS2. Stars,
intracellular spaces; arrowheads, myofibril loss; black arrows,
eosinophilia and pyknotic nuclei. ***P<0.001 vs.
control; ###P<0.001 vs. DOX. ECH, Echinacoside; DOX,
doxorubicin; Gpx4, glutathione peroxidase 4; PTGS2,
prostaglandin-endoperoxide synthase 2; BNP, brain natriuretic
peptide; LDH, lactate dehydrogenase. |
| Table IICardiac parameters in the rat models
of heart failure. |
Table II
Cardiac parameters in the rat models
of heart failure.
| | Echocardiographic
data, % | Left ventricular
end pressure, mmHg | |
|---|
| Group | Ejection
fraction | Fractional
shortening | Systolic | Diastolic | Heart rate,
beats/min |
|---|
| Control | 85.33±1.72 | 47.33±2.08 | 102.89±0.59 | 7.53±0.78 | 370.00±9.42 |
| DOX |
60.54±4.45a |
26.74±2.78a |
71.98±1.54a |
12.57±0.21a |
298.17±9.15a |
| DOX+E50 |
73.25±3.37c |
35.66±2.67c |
79.31±1.75c |
11.78±0.56b | 308.17±11.05 |
| DOX+E100 |
83.17±1.45c |
44.83±1.64c |
94.63±6.19c |
10.72±0.40c |
338.00±5.83c |
Discussion
HF is a complicated clinical syndrome characterized
by cardiac systolic and diastolic dysfunction (23). Despite numerous clinical and
experimental studies on the treatment of CHF, a satisfactory
treatment regimen to prevent HF progression is lacking (24). Hence, it is critical to find a safe
and effective therapeutic to treat HF. ECH has been studied for its
potential to protect against HF (25). The effect and molecular mechanism
of HF, however, are unknown.
In the present study, ECH protected DOX-treated H9c2
cells from oxidative stress and ferroptosis. Furthermore, ECH
alleviated DOX-induced cardiac injury by regulating GPX4
inhibition-induced ferroptosis. A growing body of evidence suggests
that oxidative stress is a direct or indirect pathophysiological
pathway in the HF process (26).
Excess ROS production causes cellular protein, lipid, and DNA
damage, and the physiological antioxidant defense system cannot
eliminate it, resulting in cell necrosis and apoptosis (27). MDA is formed as the result of lipid
oxidation by oxygen-free radicals, causing cytotoxicity and
worsening cell membrane damage, and is commonly used to reflect the
degree of oxidative stress (28,29).
However, an excess of ROS leads to an excess of lipid peroxide
conversion in HF development. Meanwhile, MDA production increases,
activating a feedforward loop mechanism, and oxidative stress
increases, resulting in further deterioration of myocardial
systolic and diastolic functions (30). DOX was found to increase the
production of inflammatory cytokines during cardiotoxicity and
oxidative stress (31,32). When patients develop HF, the plasma
BNP levels are markedly increased, and the degree of increase is
related to the degree of ventricular dilatation, which is widely
used as an HF biomarker, and is involved in ventricular
dysfunction; that is, the content of BNP is closely associated with
HF severity (33,34). In the present study, the effects of
ECH on DOX-induced BNP in plasma for the alleviation of HF symptoms
were investigated. The results showed that ECH effectively
alleviated cardiac injury in vivo and in vitro.
As more evidence emerged that myocardial cell
ferroptosis is associated with myocardial ischemia-reperfusion
injury, which plays a role in HF development, it was shown that
ferroptosis inhibition could improve myocardial injury and HF
(35-38).
Ferroptosis is caused by an imbalance in the production and
clearance of intracellular lipid peroxides (39). Ferroptosis involves lipid, GSH, and
iron metabolisms, and mitochondrial dysfunction. When cells
experience iron overload, the ROS levels increase, resulting in
intracellular lipid peroxide accumulation, and iron overload in
cardiomyocytes results in oxidative stress and mitochondrial
damage, eventually leading to systolic and diastolic dysfunction
and myocardial fibrosis (40,41).
Iron overload damages the heart by causing oxidative stress,
promoting HF development (42).
According to research, inhibiting GSH synthesis increases cardiac
lipid peroxidation, which inhibits myocardial contractility
(43). One of the
ferroptosis-related proteins is GPX4, and selenium deficiency is a
risk factor for HF development. By lowering the GPX4 activity,
selenium deficiency promotes CHF progression. PTGS2 participated in
the prostaglandin biosynthesis process, which catalyzes lipid
oxidation and is involved in ferroptosis (44). Furthermore, it was discovered that
ECH administration decreased PTGS2 expression while increasing GPX4
expression in DOX-treated rats and H9c2 cells. As a regulator of
ferroptosis, GPX4 induces ferroptosis by inhibiting its substrate
glutathione or glutathione components (45). When cardiomyocytes produce a large
amount of ROS, the cell's antioxidant activity is reduced, and
lipid ROS are produced, inhibiting GSH absorption, and the GSH
levels are associated with HF severity (46). GSH deficiency causes cysteine
deficiency, lowering the GPX4 activity and resulting in
ferroptosis. Furthermore, Gpx4 knockdown inhibited the
protective effects of ECH on DOX-induced accumulation of lipid ROS
in rat H9c2 cells, indicating that ECH could protect DOX-induced
cardiac injury by affecting ferroptosis hub proteins. These
important findings provide a foundation for the clinical treatment
of CHF. Owing to the limited literature reports on the involvement
of ferroptosis in CHF, the present study may be considered for
developing a potential therapy for the disease based on targeting
of ferroptosis.
In conclusion, ECH ameliorated DOX-induced cardiac
injury by regulating GPX4 inhibition-induced ferroptosis. Thus, ECH
may serve as a promising and important therapy for the treatment of
CHF injury.
Acknowledgements
Not applicable.
Funding
Funding: The present study was financially supported by the
Shanghai Pudong Commission of Health and Family Planning (grant no.
PW2020D-10), Shanghai Municipal Commission of Health and Family
Planning (grant no. 202040188), Medical Disciplinary Development
Project of Pudong New Area Health System (grant no. PWYgy2021-10)
and Multiplication plan for traditional Chinese medicine specialty
brand building (grant no. PDZY-2021-0310).
Availability of data and material
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YM, SZ, and JZ conceived and designed the study. YM
wrote, reviewed, and revised the manuscript. XY, NJ, CL and JZ
acquired, analyzed, and interpreted the data, and performed the
statistical analysis. YM, SZ, and JZ confirm the authenticity of
all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All experimental procedures were performed in
accordance with the Guidelines for the Institutional Animal Care
and Use Committee of Shanghai Rat & Mouse Biotech Co., Ltd.
(approval no. 202109).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sola S, Mir MQS, Lerakis S, Tandon N and
Khan BV: Atorvastatin improves left ventricular systolic function
and serum markers of inflammation in nonischemic heart failure. J
Am Coll Cardiol. 47:332–337. 2006.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wen W, Zhang Z, She J, Bai X, Wu Y, Gao L,
Zhou J and Yuan Z: The predictive values of white blood cell
indices (lymphocyte and eosinophilic granulocyte) for heart failure
in acute coronary syndrome patients following percutaneous coronary
intervention: A prospective cohort study. Clin Interv Aging.
18:951–962. 2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Khan H, Anker SD, Januzzi JL Jr, McGuire
DK, Sattar N, Woerle HJ and Butler J: Heart failure epidemiology in
patients with diabetes mellitus without coronary heart disease. J
Card Fail. 25:78–86. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hunt SA, Abraham WT, Chin MH, Feldman AM,
Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K,
et al: ACC/AHA 2005 guideline update for the diagnosis and
management of chronic heart failure in the adult: A report of the
American college of cardiology/American heart association task
force on practice guidelines (writing committee to update the 2001
guidelines for the evaluation and management of heart failure):
Developed in collaboration with the American college of chest
physicians and the international society for heart and lung
transplantation: Endorsed by the heart rhythm society. Circulation.
112:e154–e235. 2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Long K, Zhao Z, Chen J, Zhi L, Wang C,
Liao D, Wang M and Gao P: Yang-xin-xue keli exerts therapeutic
effects via regulating mitochondrial homeostasis and function in
doxorubicin-induced rat heart failure. Front Pharmacol.
13(931453)2022.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ermis N, Ulutas Z, Ozhan O, Yildiz A,
Vardi N, Colak C and Parlakpinar H: Angiotensin II type 2 receptor
agonist treatment of doxorubicin induced heart failure. Biotech
Histochem. 98:326–335. 2023.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhuang S, Ma Y, Zeng Y, Lu C, Yang F,
Jiang N, Ge J, Ju H, Zhong C, Wang J, et al: METTL14 promotes
doxorubicin-induced cardiomyocyte ferroptosis by regulating the
KCNQ1OT1-miR-7-5p-TFRC axis. Cell Biol Toxicol. 39:1015–1035.
2023.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Liao HH, Ding W, Zhang N, Zhou ZY, Ling Z,
Li WJ, Chen S and Tang QZ: Activation of AMPKα2 attenuated
doxorubicin-induced cardiotoxicity via inhibiting lipid
peroxidation associated ferroptosis. Free Radic Biol Med.
205:275–290. 2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Syamprasad NP, Jain S, Rajdev B, Panda SR,
Gangasani JK, Challa VS, Vaidya JR, Kundu GC and Naidu VGM: AKR1B1
inhibition using NARI-29-an epalrestat analogue-alleviates
doxorubicin-induced cardiotoxicity via modulating
calcium/CaMKII/MuRF-1 axis. Chem Biol Interact.
381(110566)2023.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Yang J, Ma S, Xu R, Wei Y, Zhang J, Zuo T,
Wang Z, Deng H, Yang N and Shen Q: Smart biomimetic metal organic
frameworks based on ROS-ferroptosis-glycolysis regulation for
enhanced tumor chemo-immunotherapy. J Control Release. 334:21–33.
2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wang R, Chen X, Li X and Wang K: Molecular
therapy of cardiac ischemia-reperfusion injury based on
mitochondria and ferroptosis. J Mol Med (Berl): Jul 28, 2023 (Epub
ahead of print).
|
|
12
|
Kumarswamy R and Thum T: Non-coding RNAs
in cardiac remodeling and heart failure. Circ Res. 113:676–689.
2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Matsumoto T, Wada A, Tsutamoto T, Ohnishi
M, Isono T and Kinoshita M: Chymase inhibition prevents cardiac
fibrosis and improves diastolic dysfunction in the progression of
heart failure. Circulation. 107:2555–2558. 2003.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zy A, Gd A, Xu CA, Qin X, Xu H, Zeng B,
Ren J, Zheng Q and Wang S: Beclin1 haploinsufficiency rescues low
ambient temperature-induced cardiac remodeling and contractile
dysfunction through inhibition of ferroptosis and mitochondrial
injury. Metabolism. 113(154397)2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Chen X, Xu S, Zhao C and Liu B: Role of
TLR4/NADPH oxidase 4 pathway in promoting cell death through
autophagy and ferroptosis during heart failure. Biochem Biophys Res
Commun. 516:37–43. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang H, Zhabyeyev P, Wang S and Oudit GY:
Role of iron metabolism in heart failure: From iron deficiency to
iron overload. Biochim Biophys Acta Mol Basis Dis. 1865:1925–1937.
2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhang D, Li H and Wang JB: Echinacoside
inhibits amyloid fibrillization of HEWL and protects against
Aβ-induced neurotoxicity. Int J Biol Macromol. 72:243–253.
2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yang X, Li F, Yang Y, Shen J, Zou R, Zhu
P, Zhang C, Yang Z and Li P: Efficacy and safety of echinacoside in
a rat osteopenia model. Evid Based Complement Alternat Med.
2013(926928)2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Yao L, Gui M, Li J, Lu B, Wang J, Zhou X
and Fu D: Shengxian decoction decreases doxorubicin-induced cardiac
apoptosis by regulating the TREM1/NF-κB signaling pathway. Mol Med
Rep. 23(219)2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chen H, Wang L, Liu J, Wan Z, Zhou L, Liao
H and Wan R: LncRNA ITGB2-AS1 promotes cisplatin resistance of
non-small cell lung cancer by inhibiting ferroptosis via activating
the FOSL2/NAMPT axis. Cancer Biol Ther. 24(2223377)2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Yuan H, Xia P, Sun X, Ma J, Xu X, Fu C,
Zhou H, Guan Y, Li Z, Zhao S, et al: Photothermal nanozymatic
nanoparticles induce ferroptosis and apoptosis through tumor
microenvironment manipulation for cancer therapy. Small.
18(e2202161)2022.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Aurigemma GP, Gottdiener JS, Shemanski L,
Gardin J and Kitzman D: Predictive value of systolic and diastolic
function for incident congestive heart failure in the elderly: The
cardiovascular health study. J Am Coll Cardiol. 37:1042–1048.
2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Bonsu KO, Owusu IK, Buabeng KO, Reidpath
DD and Kadirvelu A: Review of novel therapeutic targets for
improving heart failure treatment based on experimental and
clinical studies. Ther Clin Risk Manag. 12:887–906. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ni Y, Zhang J, Zhu W, Duan Y, Bai H and
Luan C: Echinacoside inhibited cardiomyocyte pyroptosis and
improved heart function of HF rats induced by isoproterenol via
suppressing NADPH/ROS/ER stress. J Cell Mol Med. 26:5414–5425.
2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Landmesser U, Spiekermann S, Dikalov S,
Tatge H, Wilke R, Kohler C, Harrison DG, Hornig B and Drexler H:
Vascular oxidative stress and endothelial dysfunction in patients
with chronic heart failure: Role of xanthine-oxidase and
extracellular superoxide dismutase. Circulation. 106:3073–3078.
2002.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Xiang XY, Yang XC, Su J, Kang JS, Wu Y,
Xue YN, Dong YT and Sun LK: Inhibition of autophagic flux by ROS
promotes apoptosis during DTT-induced ER/oxidative stress in HeLa
cells. Oncol Rep. 35:3471–3479. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ergüç A, Karakuş F, Arzuk E, Mutlu N and
Orhan H: Role of oxidative stress and reactive metabolites in
cytotoxicity & mitotoxicity of clozapine, diclofenac and
nifedipine in CHO-K1 cells in vitro. Endocr Metab Immune Disord
Drug Targets: Apr 19, 2023 (Epub ahead of print).
|
|
29
|
Granieri MC, Rocca C, De Bartolo A,
Nettore IC, Rago V, Romeo N, Ceramella J, Mariconda A, Macchia PE,
Ungaro P, et al: Quercetin and its derivative counteract
palmitate-dependent lipotoxicity by inhibiting oxidative stress and
inflammation in cardiomyocytes. Int J Environ Res Public Health.
20(3492)2023.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Turk R, Juretić D, Geres D, Svetina A,
Turk N and Flegar-Mestrić Z: Influence of oxidative stress and
metabolic adaptation on PON1 activity and MDA level in transition
dairy cows. Anim Reprod Sci. 108:98–106. 2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ichihara S, Yamada Y, Kawai Y, Osawa T,
Furuhashi K, Duan Z and Ichihara G: Roles of oxidative stress and
Akt signaling in doxorubicin cardiotoxicity. Biochem Biophys Res
Commun. 359:27–33. 2007.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Birari L, Wagh S, Patil KR, Mahajan UB,
Unger B, Belemkar S, Goyal SN, Ojha S and Patil CR: Aloin
alleviates doxorubicin-induced cardiotoxicity in rats by abrogating
oxidative stress and pro-inflammatory cytokines. Cancer Chemother
Pharmacol. 86:419–426. 2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Gaggin HK and Januzzi JL Jr: Biomarkers
and diagnostics in heart failure. Biochim Biophys Acta.
1832:2442–2450. 2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Salah K, Stienen S, Pinto YM, Eurlings LW,
Metra M, Bayes-Genis A, Verdiani V, Tijssen JGP and Kok WE:
Prognosis and NT-proBNP in heart failure patients with preserved
versus reduced ejection fraction. Heart. 105:1182–1189.
2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Liu B, Zhao C, Li H, Chen X, Ding Y and Xu
S: Puerarin protects against heart failure induced by pressure
overload through mitigation of ferroptosis. Biochem Biophys Res
Commun. 497:233–240. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Conrad M and Proneth B: Broken hearts:
Iron overload, ferroptosis and cardiomyopathy. Cell Res.
29:263–264. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Fang X, Cai Z, Wang H, Han D, Cheng Q,
Zhang P, Gao F, Yu Y, Song Z, Wu Q, et al: Loss of cardiac ferritin
H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ
Res. 127:486–501. 2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Miess H, Dankworth B, Gouw AM, Rosenfeldt
M, Schmitz W, Jiang M, Saunders B, Howell M, Downward J, Felsher
DW, et al: The glutathione redox system is essential to prevent
ferroptosis caused by impaired lipid metabolism in clear cell renal
cell carcinoma. Oncogene. 37:5435–5450. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Wu T, Liang X, Liu X, Li Y, Wang Y, Kong L
and Tang M: Induction of ferroptosis in response to graphene
quantum dots through mitochondrial oxidative stress in microglia.
Part Fibre Toxicol. 17(30)2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Paterek A, Mackiewicz U and Mączewski M:
Iron and the heart: A paradigm shift from systemic to cardiomyocyte
abnormalities. J Cell Physiol. 234:21613–21629. 2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Cheng CF and Lian WS: Prooxidant
mechanisms in iron overload cardiomyopathy. Biomed Res Int.
2013(740573)2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Chen L, Yin Z, Qin X, Zhu X, Chen X, Ding
G, Sun D, Wu NN, Fei J, Bi Y, et al: CD74 ablation rescues type 2
diabetes mellitus-induced cardiac remodeling and contractile
dysfunction through pyroptosis-evoked regulation of ferroptosis.
Pharmacol Res. 176:2022.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Wu J, Minikes AM, Gao M, Bian H, Li Y,
Stockwell BR, Chen ZN and Jiang X: Intercellular interaction
dictates cancer cell ferroptosis via NF2-YAP signalling. Nature.
572:402–406. 2019.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ren J, Privratsky JR, Yang X, Dong F and
Carlson EC: Metallothionein alleviates glutathione
depletion-induced oxidative cardiomyopathy in murine hearts. Crit
Care Med. 36:2106–2116. 2008.PubMed/NCBI View Article : Google Scholar
|