1. Introduction
According to the International Agency for Research
on Cancer GLOBOCAN Cancer Statistics for 2020, prostate cancer
(PCa) is the second most prevalent malignancy in men worldwide
(1). The detection of biomarkers
for PCa may influence clinical decision-making, guide low-risk
patients to avoid unnecessary biopsies and overtreatment, and
design the best strategy for patients with high-risk diseases
(2,3). The treatment for PCa includes
androgen deprivation therapy (ADT), radiation therapy (RT),
chemotherapy, newly emerging immunotherapies, and surgery. However,
numerous patients cannot be cured after treatment and are prone to
develop fatal metastatic Castration-Resistant PCa (mCRPC) (4). Currently, ~10 million men worldwide
have the condition, of which ~700,000 are affected (5). It is well known that the occurrence
and progression of PCa are androgen-dependent, and profiles of the
PCa transcriptome and genome have identified chromosomal
rearrangements and copy number increases or decreases, including
androgen receptor (AR) amplification (6). Previous studies on PCa have mainly
focused on AR mutations or related pathways, and ADT was once the
most effective clinical treatment strategy. However, once patients
enter a state of castration resistance, their cancer progression is
difficult to control (7).
Mutations in mCRPC have received increasing attention in recent
years, with the highest mutation rate occurring in the DNA damage
response (DDR)-associated BRCA2 gene (8,9).
Studies have further found that mCRPC patients with mutations in
the BRCA2 gene are effectively treated with the PARP inhibitor
(PARPi) olaparib. In another study, PARPi was found to interact
with the E3 ubiquitin-protein ligase TRIP12(10), indicating an emerging crosstalk
between the DDR and ubiquitination in PCa.
Numerous studies have shown that the
ubiquitin-proteasome system (UPS) is essential for maintaining
homeostasis in vivo by controlling a wide range of cellular
functions. UPS dysfunction contributes to various human diseases,
especially cancer and neurodegenerative disorders (11,12),
Currently, the UPS is considered to be a very promising target for
cancer therapy and is receiving increasing attention. Ubiquitin can
be used not only as a signal molecule mediating proteasome
degradation but also as a signal molecule for DNA repair,
transcription factor activation and other biological processes
(13). Generally, ubiquitin binds
covalently to substrates via various enzymes, modifying or
degrading them to control various cellular processes. In addition
to mediating proteasomal degradation of substrates, ubiquitin can
also mediate non-degradative ubiquitination of substrates, which is
often associated with the regulation of kinase activity. However,
owing to the diversity and complexity of the UPS, the mechanisms of
its physiological and pathophysiological actions are not fully
understood. As the UPS is involved in a wide range of cellular
functional activities, the role of the UPS in the DDR pathway was
investigated in the present review, with a particular focus on
PCa-related DDR.
2. DDR
It is estimated that a large number of cells in the
human organism suffer tens of thousands of DNA lesions per day
(14). Most lesions (75%) are DNA
single-strand breaks (SSBs) that can be caused by oxidative damage
during the process of metabolism or base hydrolysis. In addition,
double-strand breaks (DSBs) form when two SSBs occur in close
proximity or when the DNA replication apparatus encounters a
single-strand break or other lesions, which are less frequent but
challenging to repair and highly toxic (15). These lesions can prevent genome
replication and transcription; if not repaired or repaired
improperly, they can lead to mutations or broader genomic
aberrations, threatening the viability of the cell or organism
(16). Given the potentially
destructive effects of genomic instability, cells have evolved a
complex array of interlocking mechanisms to maintain their genomic
integrity. The type and frequency of DNA lesions match the
complexity of the mechanisms that counteract these threats to
genomic integrity. These mechanisms are collectively referred to as
DDR (17). In general, the DDR
pathway consists of a similar set of closely coordinated processes,
first detecting DNA damage, then recruiting a set of DNA repair
factors at the site of the damage, and finally physically repairing
the damage (14). DDR is a series
of distinct but functionally intertwined pathways that depend on
the type of DNA damage.
For example, base excision repair (BER) can rectify
small base lesions caused by oxidation, deamination and alkylation,
which do not significantly distort the DNA helix structure. When
oxidative and alkylation damage occurs, the resulting base mutation
is recognized by DNA glycosylase and excises the bases at the
damaged site. A series of enzymes are used to complete the chain
incision, cut the chain treatment to achieve DNA synthesis, fill
gaps and connections, and complete BER (18). Nucleotide excision repair (NER) is
used to repair damage to large segments of DNA in two ways: the
global-genome NER (GG-NER) and the transcription-coupled NER
(TC-NER), including lesions caused by solar radiation. GG-NER can
correct DNA damage that occurs throughout the whole genome, whereas
TC-NER specifically acts on DNA damage of the transcribed strand of
transcriptionally active genes. DNA mismatch repair (MMR) is a
highly conserved genomic pathway that can rectify DNA replication
error, limit chromatin rearrangement, and mediate multiple types of
DNA damage (19,20). The main mechanism of DSBs repair
plays a crucial role in inhibiting genomic instability. There are
two main mechanisms for DSBs repair in mammalian cells: Homologous
recombination (HR) and non-homologous end joining (NHEJ) (21). The damaged site is identified by
ATM/ATR, and BRCA1 is activated, which can recruit exonuclidenase
MRE11 to excise a sequence near the damage of the two chains. The
single chain was complementary to other homologous chains in the
vicinity under the action of the recombinant enzyme RAD51, which is
equivalent to DNA synthesis using the homologous chain as a
template. After the synthesis of the original double strand can be
partially complementary, this strand breaks off from the homologous
chain, binds to another strand that was originally complementary to
it, and uses it as a template for DNA synthesis by DNA polymerase.
Finally, DNA ligase forms phosphate bonds to obtain a lossless DNA
double strand, thus completing the HR (22). HR is an important process that is
necessary to repair DNA DSBs, restart folding replication forks,
and rearrange parental chromosome genetic information during
meiosis (23).
The DDR pathway is complex and convoluted, and it is
worth noting the DDR core components interact with cell cycle
checkpoints and chromosome segregation mechanisms (17). These interactions allow DNA repair
before mitosis and ensure the delivery of the correct complement of
genetic material to daughter cells, which is essential for
maintaining genomic stability (17). A large proportion of patients with
PCa have DDR-associated gene alterations, and 19% of the 333 PCa
patients' samples from The Cancer Genome Atlas had deleterious
aberrations in the DDR-associated gene (24). The American Association for Cancer
Research PCa Study Group identified 23% of DDR-associated gene
alterations in 150 metastatic biopsies (25). Common aberrant DDR genes in PCa
include BRCA1/2, ATM, CDK12, FANCD2 and RAD51C. Among these,
BRCA2 is the most commonly altered DDR-associated gene that
results in aggression and poorer prognosis of PCa (26).
DDR in cancer
Maintaining genomic integrity and stability is
crucial for intracellular DDR, and any disruption of this
kinase-based signaling pathway can lead to the development of
various diseases, especially cancer. A study has shown that one of
the most common features of human tumors is genomic instability,
which facilitates the development of driver mutations and expansion
of tumor heterogeneity (27).
Cytotoxic chemotherapy and radiation have long been the main
treatments for tumors, and they cause severe DNA damage in
proliferating cancer cells. However, tumor cells are often altered
in the DDR-associated pathway, leading to genomic instability that
can promote tumorigenesis and cancer cell growth such as driver
mutations (28).
Although DDR defects in most cancers are unknown, a
correlation between specific DDR dysfunction and tumor phenotype
has been demonstrated in some cancers. For example, ~10% of breast
cancer (BC) cases have been reported to be associated with germline
defects in DDR-associated gene BRCA1/2 and a small
percentage of mutations in the genes encoding CHK2 and
RAD51(29). The expression of
DNA-PKC was reduced in 57% of patients with early BC (30), and in >10% of aggressive BC
samples, the CDK12 gene was amplified or mutated (31). These findings have sparked
extensive research and provided support for the use of DDR-targeted
agents, such as PARPi, for treating BC.
DDR dysfunction has also been found in colorectal
cancer (CRCA), and brain metastasis (BM) is a rare but fatal
complication of CRCA. Patients with BM exhibit elevated mutational
features of HR defects and MMR defects compared with primary CRCA
(32). The importance of DDR in
CRCA is supported by elevated levels of BM-specific mutations and
microsatellite instability (MSI) in DDR-associated genes. MSI is
observed in sporadic CRCA and familial hereditary non-polyposis
CRCA, which is associated with loss-of-function mutations in MMR
genes, such as MSH2 and MLH1(33).
In fact, MSI of CRCA is not only associated with DDR dysfunction
but also with UPS-associated aberrations. A previous study has
revealed that MSH2 acts as a critical DNA MMR protein and also
functions as an E3 ligase that mediates MSH2 ubiquitination and
degradation (34).
In conclusion, during tumorigenesis, DDR components
are frequently dysfunctional, DNA damage cannot be efficiently
repaired, and cells continue to have intact DNA damage during the
cell cycle, which increases the chance of mutation occurrence. DDR
disorders eventually lead to the occurrence and progression of
cancer (17).
Although the treatment of PCa has progressed
considerably in the past decades with the widespread use of ADT, AR
antagonists, and androgen synthesis inhibitors, drug resistance
often develops and progresses to mCRPC due to the amplification and
overexpression of AR genes, AR mutations, and splice variants
(28). In the case of mCRPC, AR
function is reactivated and previous treatment options fail; new
treatment strategies become the hope of patients, and DDR-related
treatment strategies become particularly important. An increasing
number of DDR-targeted drugs have rapidly spread to inhibitors of
several members of the DDR pathway, including PARP, ATM, ATR, CHK1,
CHK2, WEE1 and DNA-PK (35). Some
of these are under clinical study, especially with PARPi olaparib
and niraparib (36).
DDR-associated genes mutation in
PCa
The incidence of germline mutations in the
DDR-associated genes ranges from 11-33% in patients with metastatic
PCa and was significantly higher than that in patients with
localized PCa (8,9). DDR pathway impairment can be detected
in a considerable proportion of cases, is more common in mCRPC, and
is highly enriched in metastatic PCa (37). There is a wide range of DDR
deficiencies in PCa, such as TMPRSS2-ERG gene fusion,
Speckle-type BTB/POZ protein (SPOP) mutation, and loss of
PTEN or CHD1, which are all related to DDR-related
phenotypic damage. Functional defects in the DDR pathway may lead
to sensitivity to genotoxic treatment programs, such as
radiotherapy and chemotherapy. This can be further strengthened by
molecular-targeting drugs to block the alternative DDR pathway
(37). Alterations in the DNA
damage repair pathway have recently been regarded as the main
hallmark of PCa. Next-generation sequencing studies identified that
~10% of primary tumors and 25% of metastases from PCa have DDR
defects, of which BRCA2 mutation in BER pathway is
considered to be the most common events (25).
In a landmark study, the most frequent aberrations
in metastatic PCa patients were found to be BRCA2 (5.3%),
followed by CHEK2 (1.9%), ATM (1.6%), BRCA1
(0.9%) and RAD51 (0.4%) (9). Another multi-institutional
comprehensive clinical sequencing analysis found positive
DDR-associated gene aberrations in 23% of 150 mCRPC biopsies.
BRCA2 was mutated in 13% of samples, followed by ATM
(7.3%), MSH2 (2%), BRCA1, FANCA, MLH1 and
RAD51 (0.3%) (8).
DDR-associated gene mutations usually increase during tumor
progression (38). For example,
CDK12, which plays an essential role in transcriptional regulation
and genomic stability, is mutated in 1-2% of localized PCa and 4-7%
of mCRPC (39). CDK12
double allele inactivation mutations define a distinct subtype of
advanced mCRPC. CDK12 deletion is associated with genomic
instability and localized tandem replication, leading to increased
gene fusion and significant differential gene expression,
especially in genes involved in cell cycle and DNA replication
(39). Tandem duplication has also
been described as an AR enhancer, possibly associated with disease
progression in androgen pathway inhibitors (40).
PCa is a clinically heterogeneous disease that
exhibits different responses to RT or chemotherapy, leading to
different clinical outcomes. Several studies have investigated the
prognostic role of BRCA2 (BRCA2 is often considered a central
mediator of HR repair of DSBs) aberrations in patients with
localized PCa and mCRPC receiving standard therapy (41). It is involved in initiating
homology search, strand invasion, strand exchange, and limiting
replication stress, and is a central regulator of genomic stability
(42). In a retrospective study,
BRCA2 mutations were associated with higher Gleason scores,
lymph node involvement, metastatic disease at diagnosis and T3/4
stage (26). In addition,
BRCA2 mutation is an independent prognostic factor
associated with a poorer prognosis. In localized PCa, 5-year
cancer-specific survival and metastasis-free survival were
significantly shorter in BRCA2 mutation carriers than in
non-carriers (82 and 96%; 77 and 93%, respectively) (26). Disruption of BRCA2 leads to
defects in HR, resulting in a lack of sensitivity to DNA-damaging
agents that induce DSBs and replication fork stall (43). In conclusion, among PCa-associated
DDR defects, BRCA2 mutations show relevant clinical
significance by correlating with the poor clinical features of
primary tumors and poor prognosis in patients with mCRPC.
Studies have shown that individuals with a reduced
NER ability have an increased risk of PCa. In addition to the
BRCA2, some of the established PCa-susceptibility genes
include RNASEL, ELAC2, MSR1, AR, CYP17 and SRD5A2
(44). Germline mutations and
polymorphisms of DDR genes [including BRCA1, 8-oxoguanine
DNA glycosylase (OGG1), XRCC1, CHEK2 and
ADPRT] are associated with PCa risk (45,46).
A previous study assessing NER polymorphisms and PCa risk revealed
that the combined variant genotypes of ERCC2/XPD D312N in
NER and XRCC1 R399G in BER significantly increased the risk
of PCa tumorigenesis (47). NER
and other repair pathways play essential roles in PCa risk
(44).
A total of ~10-23% of PCa patients show high-level
MSI associated with MMR gene mutations and corresponding altered
MMR protein (48,49). Although the reduction or deletion
of MSH2 protein expression may be associated with an increased risk
of PCa tumorigenesis, it also appears to correspond to a
hormone-sensitive phenotype. Compared with patients with moderate
to strong MSH2 expression, the prognosis of patients with reduced
or missing MSH2 expression is relatively improved (50). Interestingly, in patients with PCa,
elevated PMS2 expression, a component of the post-replicative DNA
MMR, also appears to be negatively correlated with prognosis
(48,50).
3. Effect of the UPS on the DDR pathway in
PCa
The UPS is an essential component of DNA damage
recognition and repair. The UPS plays an indispensable role in the
recruitment and removal of proteins at DNA damage sites and in the
regulation of downstream effectors. In addition, the UPS can
participate in the arrangement and regulation of the assembly and
disassembly of DDR-associated proteins at DNA damage sites to
ensure the regulatory progress of DNA damage repair (51,52).
DNA damage triggers corresponding cellular responses depending on
the type of damage, ranging from cell cycle arrest to the
activation of specific DNA repair mechanisms (53). Regulatory proteins such as E3
ligases (MDM2, Siah2, and Pirh2 in PCa) carry out the corresponding
ubiquitination of p53, which determines the fate of cells, such as
survival or apoptosis (54,55).
SPOP mediates the non-degradative ubiquitination of HIPK2 and
activates downstream targets of DDR (11,56).
Ubiquitin-specific protease 14 (USP14) regulates recombinant ring
finger protein 168 (RNF168) and is involved in recruiting the DDR
effector protein TP53BP1(57).
HUWE1 induces non-degradative ubiquitination of KDM3A and enhances
the transcription of DDR-associated genes, including NBS1
and RNF8 in HR repair, and XRCC6 and PRKDC are
involved in NHEJ repair (58). The
deubiquitination function of USP14 reduces RNF168-induced γH2AFX
ubiquitination signaling, which enhances cell sensitivity to
ionizing radiation (IR). These UPS-associated proteins guarantee
the timely repair of DNA damage, maintain the integrity of the
genome, and prevent the development of a range of human diseases,
including cancers and premature aging (59). Thus, the UPS-related DDR signaling
pathway has been implicated in the occurrence and progression of
PCa, and the specific mechanism is explained in each pathway
(Fig. 1).
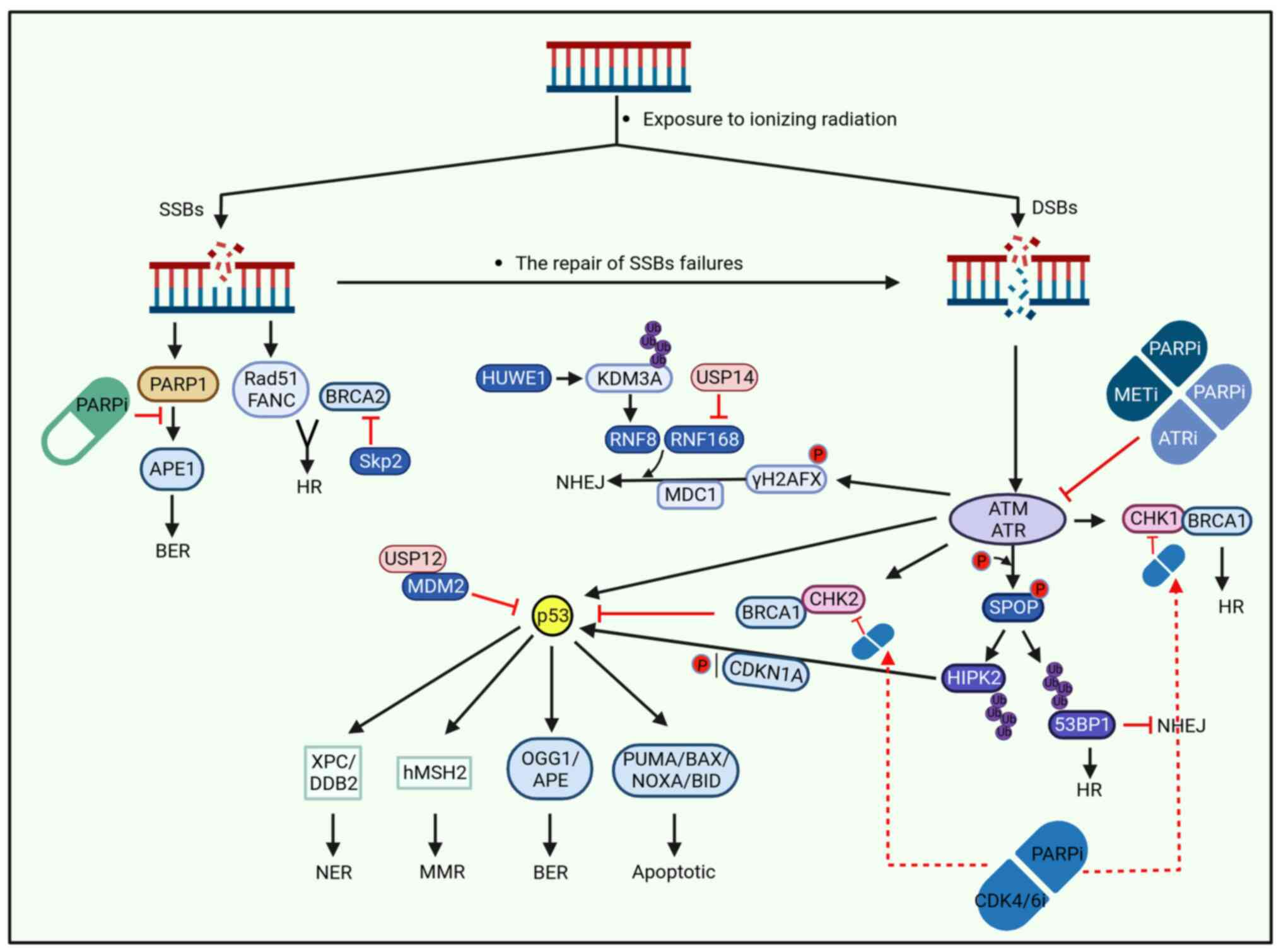 | Figure 1DDR is caused by exposure of DNA
double strands to ionizing radiation. Due to the different damage
degrees of ionizing radiation exposure, DNA exhibits DNA SSBs when
the damage was weak. When the damage is severe, DSBs appear. SSBs
are repaired by HR and BER, and if the repair fails, it can be
further developed into more severe DSBs. DSBs can be repaired in
various ways, including HR, NHEJ, BER, NER and MMR. The specific
repair process is complex and varied, and ubiquitin protease system
can participate in DDR by influencing the ubiquitination of related
proteins. Drugs can also affect the progression of DDR, with PARPi,
ATRi, METi and CDK4/6i drugs affecting multiple processes of DDR as
demonstrated in the figure. DDR, DNA damage response; SSBs, single
strand breaks; DSBs, double strand breaks; HR, homologous
recombination; NHEJ, non-homologous end joining; BER, base-excision
repair; NER, nucleotide excision repair; MMR, mismatch repair. |
SPOP-HIPK2/53BP1 in PCa
SPOP is a well-known component of the E3 ligase
complex and is frequently mutated in PCa. SPOP contains multiple
domains, including an N-terminal MATH domain, internal BTB
structure and C-terminal nuclear localization sequence, where the
MATH structural domain is essential for substrate recruitment
(60).
Multiple studies have confirmed that SPOP plays a
tumor suppressor role in PCa by targeting a variety of proteins,
but PCa-associated SPOP mutants often exhibit loss of function and
negative dominant function, impairing tumor suppressor function and
promoting the occurrence and progression of PCa. For example, SPOP
induces ubiquitination and degradation of AR, repressing
AR-mediated gene transcription and PCa cell growth. However,
PCa-associated SPOP mutants abrogate this inhibition (61).
Previously, as a critical tumor suppressor in PCa,
the relationship between SPOP and DDR has attracted attention. On
the one hand, SPOP is associated with several proteins involved in
transcription, mRNA splicing and export, including BRCA2, ATR, CHK1
and RAD51, promoting DDR and transcriptional expression of
replication factors (62). By
contrast, SPOP is phosphorylated by ATM kinase at Ser119 after DNA
damage, which enhances SPOP binding to homologous interacting
protein kinase 2 (HIPK2) and leads to the non-degradative
ubiquitination of HIPK2(11).
Furthermore, SPOP-53BP1 interaction is enhanced in response to DNA
damage by ATM-dependent phosphorylation of SPOP-Ser119(56).
HIPK2 is a DNA damage-responsive kinase that
activates downstream targets including p53(63). HIPK2 phosphorylates p53 at Ser46,
which activates apoptotic target genes such as PUMA, BAX,
NOXA and BID in response to lethal DNA damage (64). When damage is mild, HIPK2 mediates
p53 recruitment to the cyclin-dependent kinase inhibitor 1A
(CDKN1A) promoter site, thus inducing cell cycle arrest followed by
DNA repair (63). HIPK2
contributes to the DDR by regulating cell cycle arrest and
apoptosis, thereby preventing mutations, genomic instability and
carcinogenesis. In addition, it was found that SPOP-mediated
ubiquitination of HIPK2 increases its phosphorylation activity on
HP1γ, which further leads to the uncoupling of phosphorylated HP1γ
from trimethylated H3K9me3 heterochromatin and the initiation of
DNA damage repair (11). HHowever,
PCa-associated SPOP mutants, such as SPOPS119A/N
are defective in SPOP-HIPK2 interaction, which may lead to DDR
abnormalities and genomic instability. 53BP1 is a DDR-associated
protein that plays a role in the DSBs repair pathway selection.
53BP1 promotes NHEJ repair by facilitating long-range end joining
of broken DNA and restricts HR by inhibiting DNA end resection.
SPOP induces non-degradative polyubiquitination of 53BP1 and
extracts 53BP1 from chromatin, which promotes DNA repair by more
accurate HR over error-prone NHEJ. However, PCa-associated
SPOPS119N promoted 53BP1 retention at DSBs sites and
impaired DNA end excision. The lack of HR selection causes genomic
instability in SPOPS119N cells (56).
These studies suggested that mutations in the
PCa-associated SPOP-Ser119 locus impair the DDR, contributing to
the genomic instability of PCa. PCa patients with SPOP-Ser119
mutations performed more sensitively to RT and chemotherapy, which
may guide the clinical treatment of PCa patients with SPOP
mutations.
USP12-MDM2-p53 regulates DDR in
PCa
USP12 is similar to other USP family members, and
contains a conserved catalytic cysteine/histidine structural domain
(65). A previous study has found
that USP12 can directly target AR, and induce its deubiquitination
and stabilization, controlling the AR-AKT signaling network. The
aberrant activity of AKT signaling is one of the most common
features of mCRPC (66). MDM2 is a
nuclear-localized E3 ligase consisting of a p53-binding domain,
acid domain, zinc finger domain and ring finger domain. MDM2
targets p53 for proteasomal degradation to MDM2 to inhibit
p53-mediated cell cycle checkpoint activation and DDR, thus
promoting the tumorigenesis of PCa.
USP12 was previously identified as a deubiquitinase
of histones H2A/H2B, Notch, PH domain leucine-rich repeat protein
phosphatase 1 and AR (67).
Previously, it was found that USP12 not only facilitates PCa
progression by regulating AR but also regulates MDM2
deubiquitination, which in turn controls the protein level of p53
in PCa (65).
In addition to transmitting apoptotic signals, p53
can facilitate the clearance of DNA lesions by enhancing several
DNA repair pathways (53). p53 is
involved in NER through transcriptional upregulation of xeroderma
pigmentosum complementation group C and damage-specific DNA binding
protein 2, both critical damage recognition factors required to
initiate global genomic NER (68).
However, it has also been implicated in the transcriptional control
of the MMR component human muts homolog 2 during DNA damage
(69). In addition to
transcriptional regulation, p53 interacts directly with the
critical BER enzymes, OGG1 and apurinic/apyrimidinic endonuclease,
to enhance their activity, thereby increasing the efficiency of DNA
lesion excision to regulate BER (53,70).
However, a high USP12 protein level in mCRPC
correlates with a poor prognosis in PCa (53). USP12 stabilizes MDM2 and positively
correlates with MDM2, leading to p53 degradation (53). Reduced abundance of p53 impairs its
function, leading to DDR disruption and genomic instability. Thus,
MDM2 inhibition may represent an attractive and feasible strategy
for treating PCa. MDM2 inhibitors can increase the sensitivity of
PCa to IR and ADT, thereby improving treatment outcomes.
Skp2-BRCA2 regulates DDR in PCa
Skp2, a member of the F-box protein family, is a
substrate recognition component of the SCFSkp2 E3 ligase
complex and plays a role in phosphorylation-dependent
ubiquitination. It recognizes phosphorylated cyclin-dependent
kinase inhibitor 1 B (also known as p27 or KIP1), mainly in the
S-phase, and is overexpressed in several cancers including BC,
gastric cancer and PCa (71,72).
Overexpression of Skp2 and decreased p27 abundance
are often associated with aggressive PCa (73,74).
Skp2, RB transcriptional corepressor 1 and p53 can trigger the
degradation of p27, and overexpression of Skp2 mediates the
ubiquitination and degradation of p27 and blocks cell cycle arrest
and apoptosis in PCa (73). Skp2
inhibitors would lead to the accumulation of p27, activate the
p27-E2F1-p73 axis, and induce apoptosis to inhibit PCa (74,75).
In addition, Skp2 can activate AR directly or indirectly via
TRAF6-EZH2/H3K27me3 to contribute to PCa progression (76,77).
Skp2 downregulates TRAF6-mediated lysine demethylase 5B (KDM5B)
ubiquitination, inhibits H3K4me3 stabilization, and promotes mCRPC
migration (78).
Previously, with more attention paid to
DDR-associated studies in PCa, the effect of Skp2 on BRCA2 has been
gradually discovered (79). In
PCa, abnormal upregulation of Skp2 leads to hydrolytic degradation,
reduced abundance and impairment of BRCA2 protein-related
functions. On the one hand, impairment of BRCA2 forms a complex
with Rad51/FANC, which participates in HR repair of SSBs and
coordinates the function of HR repair of DSBs (80). However, the functions of BRCA2 in
stabilizing DNA replication forks, centrosome replication and
transcriptional regulation are impaired. In addition, BRCA2
deficiency is closely associated with migratory behavior and tumor
growth during PCa development, while Skp2 overexpression impairs
DDR and promotes PCa progression (79,80).
These studies suggested that Skp2 is at least a key
regulator determining BRCA2 abundance in PCa cell lines and that
novel inhibitors targeting Skp2 activity or specifically
counteracting Skp2 BRCA2 interaction, providing a new idea of
therapeutic treatment for PCa patients (79,80).
USP14-RNF168 regulates DDR in PCa
USP14 is a deubiquitinating enzyme that interacts
with the proteasome to regulate substrate ubiquitination (81). It contains an N-terminal
ubiquitin-like (UBL) domain and C-terminal USP domain (81). The UBL domain regulates proteasome
activity, whereas the USP domain displays deubiquitinase activity.
USP14 protects the substrate from degradation by removing ubiquitin
chains through the cooperative function of these two domains
(81).
Previous studies have confirmed that USP14
overexpression accelerates PCa cell proliferation by
deubiquitinating and inhibiting AR degradation in
androgen-responsive PCa cells (82). Moreover, it can enhance the
stability of the cancer-associated AR mutant protein AR-V7 by
deubiquitination modification, promoting the progression of PCa
(83,84). USP14-mediated deubiquitination of
activating transcription factor 2 upregulates its abundance, which
functions as an oncogenic transcription factor in PCa, thereby
leading to the proliferation of PCa cells (85).
Apart from affecting AR stability to influence the
occurrence and progression of PCa, it has been reported that UPS14
also affects RNF168-associated ubiquitination signaling, playing a
role in NHEJ repair of DDR in PCa (57).
Sharma et al (57) examined the role of autophagy in
regulating the DDR in response to IR using PCa cell lines as a
model system and observed that RNF168 protein levels were reduced,
and DSBs were not repaired when PCa autophagy-deficient cells were
damaged by continuous IR. IR is a canonical DNA-damaging method,
and the DDR network recognizes induced DSBs, which subsequently
recruit ATM kinase, thus triggering downstream phosphorylation of
histone H2AFX at Ser139. Phosphorylated H2AFX (γH2AFX) recruits E3
ligases RNF8 and RNF168 via MDC1, which initiates γH2AFX
ubiquitination required for recruiting DDR factors, such as
TP53BP1, to DSBs sites to coordinate NHEJ repair (57). The researchers further found that
USP14 directly interacted with RNF168 and affected its associated
ubiquitination process, and RNF168-mediated ubiquitination
decreased in the presence of USP14(57). In summary, USP14 negatively
regulates DDR signaling, inhibits NHEJ repair, and enhances
cellular sensitivity to IR by suppressing RNF168-induced γH2AFX
ubiquitination.
Notably, a previous study has suggested that
nuclear-localized p62 is a major factor regulating RNF168 in tumor
cells, such as HeLa cells from cervical cancer and HCT116 cells
from CRCA (86). However, in more
advanced PCa, p62 is mainly located in the cytoplasm and is barely
detectable in the nucleus (57).
Additionally, USP14 was confirmed to be a novel autophagic
substrate that accumulated in PCa autophagy-deficient cells, and
p62 interacted with USP14 to regulate its autophagic degradation
(57). Therefore, regulation of
RNF168 by USP14 may be an effective mechanism to stabilize NHEJ to
avoid genomic deranging in PCa cells that lack nuclear p62(57).
This finding has significant implications in guiding
PCa treatment. Firstly, the detection of UPS14 can be used to
predict radiosensitivity (57).
Second, autophagy signaling is usually enhanced in advanced PCa,
and the application of autophagy inhibitors or p62 inhibitors to
regulate the abundance of UPS14 to inhibit DDR signaling may
enhance IR sensitivity.
HUWE1-KDM3A regulates DDR in PCa
HUWE1 is an evolutionarily conserved E3 ligase
belonging to the HECT family. HUWE1 contains a HECT domain, UBA
domain, Bcl-2 homology region 3 domain and UBM1 domain (87). The C-terminal HECT domain is the
primary domain that acts as an E3 ligase that mediates
ubiquitination and subsequent proteasomal degradation of substrates
(87). HUWE1 is a crucial
regulator of DDR transcription, autophagy and apoptosis (88,89).
HUWE1 may play different roles in different cancers
as it is upregulated as an oncogene in non-small cell lung cancer
(NSCLC) and downregulated as a tumor suppressor in colon
adenocarcinoma (COAD) (90,91).
In NSCLC, HUWE1 directly binds to and degrades the tumor suppressor
p53, and an increase in HUWE1 expression is significantly
associated with a worse prognosis in patients with NSCLC (91). The inactivation of endogenous HUWE1
is essential for p53 stability, and the HUWE1-p53 axis may be a
potential target for NSCLC therapy (91). HUWE1 is a critical COAD suppressor
that destabilizes MYC-MIZ1 and prevents DNA damage accumulation and
tumor initiation (90). Notably,
there are studies indicating a relationship between HUWE1 and DDR
in PCa. KDM3A, a histone demethylase, has been reported to be
overexpressed and play a tumor-promoting role in PCa. It was found
that HUWE1 induced the non-degradative ubiquitination of KDM3A and
enhanced its transcription of DDR-associated genes, including
NBS1 and RNF8 involved in DSBs HR repair, and
XRCC6 and PRKDC involved in NHEJ repair. However, PCa
cells expressing the KDM3AK918R mutant, which cannot be
ubiquitinated by HUWE1, exhibit DSBs repair defects and sensitivity
to genotoxic stress (92).
The aforementioned study suggested that KDM3A
modification by HUWE1 is an important event affecting
DDR-associated gene expression and DSBs repair in PCa. This
interference with non-degradative ubiquitination of KDM3A by HUWE1
may be a means of regulating DSBs repair, which may improve DSBs
repair and the response to RT in advanced PCa.
4. Treatment for DDR defects in PCa
Currently, DDR-associated treatments for patients
with PCa are mainly focused on PARPi, ATR inhibitors (ATRi) and
platinum-based chemotherapy (7).
Among the currently approved PCa regimens, PARPi and platinum-based
chemotherapy are effective in other cancer types associated with
BRCA1/2 alterations, and several PARPi have been clinically
studied in patients with mCRPC. Other DDR inhibitor targets, such
as ATM, ATR, CHK1, CHK2 and WEE1, have been
extensively studied (35,41). PARPi combined with RT is commonly
used to treat CRCA and glioblastoma (93,94).
Currently, the combination of PARPi is often used in clinical
practice for the treatment of mCRPC. The currently ongoing clinical
practice is summarized in Table I.
In the present review, the selection of these agents for improved
treatment was discussed.
 | Table IOngoing clinical trials assessing the
role of PARPi in metastatic castration-resistant prostate
cancer. |
Table I
Ongoing clinical trials assessing the
role of PARPi in metastatic castration-resistant prostate
cancer.
| NCT number | Phase | PARPi | Interventions | Primary
endpoint |
|---|
| NCT05171816 | III | Olaparib | Drug: Olaparib,
Abiraterone acetate | rPFS |
| NCT03732820 | III | | Drug: Olaparib,
Abiraterone acetate | rPFS |
| NCT05457257 | IV | | Drug: Olaparib,
Enzalutamide, Abiraterone acetate, Prednisone | rPFS |
| NCT03874884 | I | | Drug: Olaparib.
Combination Product: 177Lu-PSMA | DLT, MTD, RP2D |
| NCT02987543 | III | | Drug: Olaparib,
Enzalutamide, Abiraterone acetate | rPFS |
| NCT04556617 | I/II | | Drug: Olaparib,
PLX2853, Abiraterone acetate, Prednisone | DLT |
| NCT01972217 | II | | Drug: Olaparib,
Placebo, Abiraterone, Prednisone | AEs, DLT, rPFS |
| NCT03012321 | II | | Drug: Olaparib,
Abiraterone acetate, Prednisone | PFS |
| NCT03834519 | III | | Drug: Olaparib,
Abiraterone acetate, Prednisone, Enzalutamide. Biological:
Pembrolizumab | OS, rPFS |
| NCT05005728 | II | | Combination
Product: XmAb20717 + Olaparib. Combination Product: XmAb20717 +
Carboplatin + Cabazitaxel. Biological: XmAb20717 monotherapy | AEs |
| NCT02861573 | I/II | | Drug: Olaparib,
Docetaxel, Prednisone, Enzalutamide, Ebiraterone acetate,
Lenvatinib, Carboplatin, Etoposide. Biological: Pembrolizumab,
Pembrolizumab/Vibostolimab coformulation | PSA, AEs, ORR |
| NCT05262608 | II | | Drug: Olaparib | ORR |
| NCT03317392 | I/II | | Drug: Olaparib.
Other: Laboratory Biomarker Analysis, Quality-of-Life Assessment
Radiation: Radium Ra 223 Dichloride | rPFS |
| NCT02893917 | II | | Drug: Olaparib,
Cediranib | rPFS |
| NCT03787680 | II | | Drug: Olaparib,
AZD6738 | ORR |
| NCT03568656 | I/II | | Drug: Olaparib,
CCS1477, Abiraterone acetate, Enzalutamide, Darolutamide,
Atezolizumab | AEs |
| NCT04038502 | II | | Drug: Olaparib,
Carboplatin | PFS |
| NCT05252390 | I/II | | Drug: Olaparib,
NUV-868, Enzalutamide | RP2D |
| NCT03903835 | III | Niraparib | Drug: Niraparib
plus Abiraterone acetate plus Prednisone, Enzalutamide Oral
Capsule, Abiraterone Oral Tablet, Carboplatin, Docetaxel Injectable
Solution, Radium Chloride Ra-223 | PFS |
| NCT02854436 | II | | Drug:
Niraparib | ORR |
| NCT03431350 | I/II | | Drug: Niraparib,
Cetrelimab, Abiraterone acetate, Prednisone | ORR, AEs, RR |
| NCT02924766 | I | | Drug: Niraparib,
Apalutamide, Abiraterone acetate, Prednisone | RP2D |
| NCT03748641 | III | | Drug: Niraparib,
Abiraterone acetate, Prednisone, Placebo, New Formulation of
Niraparib and Abiraterone acetate | rPFS |
| NCT04179396 | I | Rucaparib | Drug: Rucaparib,
Enzalutamide, Abiraterone | AEs, SAEs |
| NCT04253262 | I/II | | Drug: Rucaparib,
Copanlisib | MTD |
| NCT02952534 | II | | Drug:
Rucaparib | ORR |
| NCT02975934 | III | | Drug: Rucaparib,
Abiraterone acetate or Enzalutamide or Docetaxel | rPFS |
| NCT03442556 | II | | Drug: Rucaparib,
Rucaparib Camsylate, Carboplatin, Docetaxel. Other: Laboratory
Biomarker Analysis | rPFS |
| NCT03338790 | II | | Drug: Rucaparib,
Docetaxel, Enzalutamide, Prednisone Biological: Nivolumab | ORR, RR |
| NCT04455750 | III | | Drug: Rucaparib
camsylate, Enzalutamide, Placebo, Leuprolide acetate,Goserelin
acetate, Degarelix. Other: Quality-of-Life Assessment. Other:
Questionnaire Administration | rPFS, OS |
| NCT04676334 | III | | Drug:
Rucaparib | AEs, SAEs |
| NCT05425862 | I | Talazoparib | Drug: Talazoparib,
Pidnarulex | MTD |
| NCT04846478 | I | | Drug: Talazoparib,
Tazemetostat | DLT, AEs |
| NCT04703920 | I | | Drug: Talazoparib,
Belinostat | DLT |
| NCT03148795 | II | | Drug:
Talazoparib | ORR |
| NCT04052204 | I/II | | Drug: Talazoparib,
Avelumab, Bempegaldesleukin, Enzalutamide | DLT |
| NCT03395197 | III | | Drug: Talazoparib
with enzalutamide, Placebo with enzalutamide | MTD, rPFS |
| NCT04824937 | II | | Drug: Talazoparib,
Telaglenastat | ORR |
| NCT04019327 | I/II | | Drug: Talazoparib,
Temozolomide | AEs, ORR |
| NCT03330405 | I/II | | Drug: Talazoparib,
Avelumab | DLT, OR |
| NCT01576172 | II | Veliparib | Drug: Veliparib,
Abiraterone Acetate, Prednisone. Other: Laboratory Biomarker
Analysis | RR |
| NCT01085422 | I | | Drug: Veliparib,
Temozolomide | PSA test |
PARPi is the first class of drugs to enter the
clinic targeting DDR, and is a successful example of the concept of
selective targeting of cancer cells introduced by precision
medicine (36). PARPi causes the
conversion of SSBs gaps to DSBs by blocking BER, which can result
in BRCA1/2 aberrations and HR-deficient cell death (95). Notably, PARPi was first approved
for treating BC and OC with BRCA aberrations through the synthetic
killing effect of DDR-associated gene mutations and has been
further applied to PCa treatment (36). ADP ribosylation is involved in
various cellular processes including cell growth and
differentiation, transcriptional regulation and apoptosis. In
addition, ADP ribosylation plays a crucial role in DNA repair by
promoting DSBs repair via HR (96). PARPi takes advantage of genomic
instability induced by oxidative and replicative stress and defects
in DDR pathways to destabilize replication forks by entrapment of
PARP DNA and to induce cell death by mitotic disasters induced by
replication stress (36).
Olaparib, a representative drug, was the first PARPi
drug to enter a PCa clinical trial. One clinical study found that
the application of olaparib was associated with prolonged
progression-free survival (PFS), improved response measures and
patient-reported endpoints compared with patients with mCRPC who
received ADT, such as enzalutamide or abiraterone, while still
experiencing disease progression (97). The aforementioned study suggested
that the clinical benefit of olaparib is promising and that its
combination with PARPi is a hot topic in current research. For
example, the combination of olaparib and the CDK4/6 inhibitor
(CDK4/6i) drugs palbociclib or abemaciclib for mCRPC and
neuroendocrine PCa has been demonstrated to synergistically inhibit
the p-RB-E2F1 signaling axis at the transcriptional and
post-translational levels, leading to the disruption of cell cycle
progression and inhibition of E2F1 gene targets (CHK1 and CHK2),
including genes involved in DDR signaling damage repair
CDK1(98). The combination of
PARPi and CDK4/6i not only inhibits the growth of PCa cells but
also promotes apoptosis, giving greater play to the ability of
PARPi to induce cell death.
Additionally, the proto-oncogene
mesenchymal-epithelial transition (MET) is highly expressed in
human mCRPC tissues, and MET is critical for tumor cell growth,
proliferation, migration and invasion. A trial combining PARPi
olaparib with the MET inhibitor (METi) crizotinib found that
olaparib and crizotinib jointly downregulated the ATM/ATR signaling
pathway. Drugs enhance the antitumor effects of olaparib-induced
DU145 and PC3 in PCa cells by inhibiting the phosphoinositide
3-kinase/protein kinase B (PI3K/AKT) pathway to induce apoptosis,
increase mCRPC sensitivity to PARPi, and provide a new combination
treatment option for Mcrpc (99).
In addition to PARP, the targeting of other
DDR-related proteins is an attractive therapeutic strategy. ATR, a
DDR kinase, plays a key role in preventing excessive genomic
instability in tumors. ATR is responsible for sensing replication
stress and sending it to the S and G2/M checkpoints to promote
repair. When the DNA damage load is high enough, loss or inhibition
of ATR can lead to genomic instability or cell death (100). A recent study found a new type of
ATRi induction that differs from PARPi, ATRi, through abrogation of
the ATR-CHK1-CDK1 regulated G2-M cell cycle checkpoint, which leads
to cell death and activation of cGAS-STING signaling (101). Moreover, in contrast to PARPi,
ATRi-induced abrogation of ATR-CHK1 signaling and activation of
CDK1 results in the activation of the CDK1-SPOP axis, which leads
to destabilization and degradation of PD-L1 in PCa cells. This
difference in mechanisms provides new opportunities for combination
therapy with ATRi and PARPi (101).
The main ATRi currently entering clinical oncology
studies are Berzosertib, Ceralasertib, RP-3500, ART-0380, ATRN-119,
M-4344, M-1774, M-6620 and Elimusertib. The ATRi drugs currently
entering clinical studies in PCa have been summarized in Table II.
| Table IIOngoing clinical trials assessing the
role of ATR inhibitors in prostate cancer. |
Table II
Ongoing clinical trials assessing the
role of ATR inhibitors in prostate cancer.
| NCT number | Phase | ATR
iinhibitors | Interventions | Primary
endpoint |
|---|
| NCT03787680 | II | Ceralasertib | Drug: Ceralasertib,
Olaparib | CR, PR |
| NCT03682289 | II | | Drug: Ceralasertib.
Drug: Olaparib. Drug: Durvalumab | ORR |
| NCT04564027 | II | | Drug:
Ceralasertib | ORR |
| NCT03517969 | II | Berzosertib | Drug: Berzosertib,
Carboplatin, Docetaxel. Other: Laboratory Biomarker Analysis | PAS test, PFS,
rPFS |
| NCT04267939 | I | Elimusertib | Drug: Elimusertib,
Niraparib | TEAEs, TESAEs, MTD,
DLT |
ATM is the most commonly mutated
DDR-associated gene for PCa, except for BRCA1/2. Previous
research data suggested that patients with ATM mutations may
be less likely to benefit from PARPi treatment than patients with
BRCA1/2 alterations, and PCa patients with harmful
ATM mutations are more likely to benefit from ATRi
treatment. This may be a manifestation of the different mechanisms
of action of ATRi and PARPi.
As aforementioned, platinum-based chemotherapy is a
popular research topic for mCRPC treatment. A study evaluated the
response of mCRPC patients with multiple DDR-associated gene
mutations, including BRCA1/2, ATM, PALB2, FANCA and
CDK12 to platinum-based chemotherapy and found that a
subgroup of patients with DDR-associated gene alterations may
benefit from platinum-based chemotherapy (102). DDR aberration carriers exhibited
improved response to platinum-based chemotherapy, indicating that
DDR status deserves further validation as a potential biomarker for
patient selection (102,103).
5. Discussion
The role of DDR in cancer has received increasing
attention in recent years, and numerous clinical trials of
DDR-related drugs are underway; however, they remain very limited
in terms of clinical application. The application of DDR-related
drugs is limited by the presence of specific genetic aberrations.
For example, PCa cells with BRCA1/2 mutations are more
sensitive to PARPi, PCa cells with ATM aberrations are more
sensitive to ATRi drugs, and cells without the corresponding
aberrations are less sensitive. DDR-related drugs often have
limited effects owing to their high drug specificity. Therefore,
effectively blocking the various escape routes of cancer cells is
key to deciphering the limitations of these drugs.
There is no doubt that targeted DDR is an important
clinical strategy for the PCa patients treatment, but previous
research is also a solid basis for improving the efficiency of PCa
treatment. Therefore, a combination of drugs with different
mechanisms of action is key to PCa treatment. As shown in Table I, some clinical trials have already
tested the combined application of PARPi and ADT, which may be a
promising approach. Several trials have suggested an increase in
radiographic PFS of 5.6 months in the abiraterone combined with
olaparib group compared with the ADT drug abiraterone alone group
in some patients with mCRPC, but there was also a more severe
incidence of adverse events (AEs) (104). Therefore, phase III clinical
trials (NCT03732820) are ongoing to assess the feasibility of
abiraterone in combination with olaparib as a first-line agent for
mCRPC. Future combination applications could not be limited to ADT.
ATRi, epidermal growth factor receptor, vascular endothelial growth
factor (VEGF) and immunotherapy are also important research
directions for PCa treatment, but their combination applications
are much less frequent in PCa than in BC and OC. For example,
combining PARPi and ATRi overcomes PARPi and platinum resistance in
an OC model and significantly improves patient survival (105). Combination immunotherapy of PARPi
with immune checkpoint inhibitors for mCRPC has been poorly
studied; although previous studies suggested improved overall
survival, the high incidence of AEs cannot be ignored, and further
exploration of effective combination immunotherapy strategies with
few adverse effects is warranted (106). VEGF pathway inhibition enhanced
the efficacy of PARPi in OC and reduced growth and survival in OC
models, irrespective of HR repair mutation status (107).
Overall, existing research provides support for new
drug combination therapies with molecular mechanisms that offer
more opportunities for the treatment of patients with advanced PCa.
Of course, research needs to overcome the current limitations and
build on existing studies to more thoughtfully apply drug
combinations to further contribute to the treatment and prognosis
of PCa.
Acknowledgements
The authors would like to thank Dr Tian Tang (Ningbo
University, Ningbo, China) for providing help and advice in the
creation of this manuscript.
Funding
Funding: This research was funded by The National Natural
Science Foundation of China (grant no. 32270821), Natural Science
Foundation of Ningbo (grant no. 2021J065), the Youth Science and
technology innovation leader of Ningbo (grant no. 2023QL052) and
the K.C. Wong Magna Fund in Ningbo University.
Availability of data and materials
Not applicable.
Authors' contributions
YL drafted the manuscript. XJ and YL made
substantial contributions to interpretation, drafting the study,
and revising it critically for important intellectual content. XJ
and YL were the major contributors to the manuscript. Both authors
have read and approved the final manuscript. Data authentication is
not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Colombet M, Soerjomataram I,
Parkin DM, Pineros M, Znaor A and Bray F: Cancer statistics for the
year 2020: An overview. Int J Cancer: Apr 5, 2021 (Epub ahead of
print). doi: 10.1002/ijc.33588.
|
|
2
|
Álvarez Múgica M and Jalón Monzón A:
Tissue biomarkers in prostate cancer. Arch Esp Urol. 75:185–194.
2022.PubMed/NCBI(In Spanish).
|
|
3
|
Plata Bello A, Tamayo Jover MA, Gutierrez
Nicolas F, Acosta López S, Concepción Masip T and Plata Bello J:
Biomarkers for characterization and therapeutic orientation in
castration-resistant prostate cancer. Arch Esp Urol. 75:195–202.
2022.PubMed/NCBI(In English, Spanish).
|
|
4
|
Evans AJ: Treatment effects in prostate
cancer. Mod Pathol. 31 (Suppl 1):S110–S121. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sandhu S, Moore CM, Chiong E, Beltran H,
Bristow RG and Williams SG: Prostate cancer. Lancet. 398:1075–1090.
2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Grasso CS, Wu YM, Robinson DR, Cao X,
Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC,
et al: The mutational landscape of lethal castration-resistant
prostate cancer. Nature. 487:239–243. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Teo MY, Rathkopf DE and Kantoff P:
Treatment of advanced prostate cancer. Annu Rev Med. 70:479–499.
2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Mateo J, Carreira S, Sandhu S, Miranda S,
Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A,
Tunariu N, et al: DNA-repair defects and olaparib in metastatic
prostate cancer. N Engl J Med. 373:1697–1708. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Pritchard CC, Mateo J, Walsh MF, De Sarkar
N, Abida W, Beltran H, Garofalo A, Gulati R, Carreira S, Eeles R,
et al: Inherited DNA-Repair gene mutations in men with metastatic
prostate cancer. N Engl J Med. 375:443–453. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Gatti M, Imhof R, Huang Q, Baudis M and
Altmeyer M: The ubiquitin ligase TRIP12 limits PARP1 trapping and
constrains PARP inhibitor efficiency. Cell Rep.
32(107985)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Jin X, Qing S, Li Q, Zhuang H, Shen L, Li
J, Qi H, Lin T, Lin Z, Wang J, et al: Prostate cancer-associated
SPOP mutations lead to genomic instability through disruption of
the SPOP-HIPK2 axis. Nucleic Acids Res. 49:6788–6803.
2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang H, Cao X, Wang J, Li Q, Zhao Y and
Jin X: LZTR1: A promising adaptor of the CUL3 family. Oncol Lett.
22(564)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zhai F, Li J, Ye M and Jin X: The
functions and effects of CUL3-E3 ligases mediated non-degradative
ubiquitination. Gene. 832(146562)2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lindahl T and Barnes DE: Repair of
endogenous DNA damage. Cold Spring Harb Symp Quant Biol.
65:127–133. 2000.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Tubbs A and Nussenzweig A: Endogenous DNA
damage as a source of genomic instability in cancer. Cell.
168:644–656. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078.
2009.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lord CJ and Ashworth A: The DNA damage
response and cancer therapy. Nature. 481:287–294. 2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chou WC, Wang HC, Wong FH, Ding SL, Wu PE,
Shieh SY and Shen CY: Chk2-dependent phosphorylation of XRCC1 in
the DNA damage response promotes base excision repair. EMBO J.
27:3140–3150. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Iyer RR and Pluciennik A: DNA Mismatch
repair and its role in Huntington's disease. J Huntingtons Dis.
10:75–94. 2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li Z, Pearlman AH and Hsieh P: DNA
mismatch repair and the DNA damage response. DNA Repair (Amst).
38:94–101. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Scully R, Panday A, Elango R and Willis
NA: DNA double-strand Break repair-pathway choice in somatic
mammalian cells. Nat Rev Mol Cell Biol. 20:698–714. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
van Wilpe S, Tolmeijer SH, Koornstra RHT,
de Vries IJM, Gerritsen WR, Ligtenberg M and Mehra N: Homologous
recombination repair deficiency and implications for tumor
immunogenicity. Cancers (Basel). 13(2249)2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Huselid E and Bunting SF: The regulation
of homologous recombination by helicases. Genes (Basel).
11(498)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
The Molecular Taxonomy of Primary Prostate
Cancer. Cell. 163:1011–1025. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Robinson D, Van Allen EM, Wu YM, Schultz
N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC,
Attard GM, et al: Integrative clinical genomics of advanced
prostate cancer. Cell. 161:1215–1228. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Castro E, Goh C, Olmos D, Saunders E,
Leongamornlert D, Tymrakiewicz M, Mahmud N, Dadaev T, Govindasami
K, Guy M, et al: Germline BRCA mutations are associated with higher
risk of nodal involvement, distant metastasis, and poor survival
outcomes in prostate cancer. J Clin Oncol. 31:1748–1757.
2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wengner AM, Scholz A and Haendler B:
Targeting DNA damage response in prostate and breast cancer. Int J
Mol Sci. 21(8273)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Santana Dos Santos E, Lallemand F,
Petitalot A, Caputo SM and Rouleau E: HRness in breast and ovarian
cancers. Int J Mol Sci. 21(3850)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Söderlund Leifler K, Queseth S, Fornander
T and Askmalm MS: Low expression of Ku70/80, but high expression of
DNA-PKcs, predict good response to radiotherapy in early breast
cancer. Int J Oncol. 37:1547–1554. 2010.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Lui GYL, Grandori C and Kemp CJ: CDK12: An
emerging therapeutic target for cancer. J Clin Pathol. 71:957–962.
2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Sun J, Wang C, Zhang Y, Xu L, Fang W, Zhu
Y, Zheng Y, Chen X, Xie X, Hu X, et al: Genomic signatures reveal
DNA damage response deficiency in colorectal cancer brain
metastases. Nat Commun. 10(3190)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Jiricny J: The multifaceted
mismatch-repair system. Nat Rev Mol Cell Biol. 7:335–346.
2006.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhang M, Xiang S, Joo HY, Wang L, Williams
KA, Liu W, Hu C, Tong D, Haakenson J, Wang C, et al: HDAC6
deacetylates and ubiquitinates MSH2 to maintain proper levels of
MutSα. Mol Cell. 55:31–46. 2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Staniszewska M, Iking J, Lückerath K,
Hadaschik B, Herrmann K, Ferdinandus J and Fendler WP: Drug and
molecular radiotherapy combinations for metastatic castration
resistant prostate cancer. Nucl Med Biol. 96-97:101–111.
2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Slade D: PARP and PARG inhibitors in
cancer treatment. Genes Dev. 34:360–394. 2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Burdak-Rothkamm S, Mansour WY and Rothkamm
K: DNA damage repair deficiency in prostate cancer. Trends Cancer.
6:974–984. 2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zhang W, van Gent DC, Incrocci L, van
Weerden WM and Nonnekens J: Role of the DNA damage response in
prostate cancer formation, progression and treatment. Prostate
Cancer Prostatic Dis. 23:24–37. 2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Wu YM, Cieślik M, Lonigro RJ, Vats P,
Reimers MA, Cao X, Ning Y, Wang L, Kunju LP, de Sarkar N, et al:
Inactivation of CDK12 delineates a distinct immunogenic class of
advanced prostate cancer. Cell. 173:1770–1782.e14. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Viswanathan SR, Ha G, Hoff AM, Wala JA,
Carrot-Zhang J, Whelan CW, Haradhvala NJ, Freeman SS, Reed SC,
Rhoades J, et al: Structural alterations driving
castration-resistant prostate cancer revealed by linked-read genome
sequencing. Cell. 174:433–447.e19. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Nombela P, Lozano R, Aytes A, Mateo J,
Olmos D and Castro E: BRCA2 and other DDR genes in prostate cancer.
Cancers (Basel). 11(352)2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Fradet-Turcotte A, Sitz J, Grapton D and
Orthwein A: BRCA2 functions: From DNA repair to replication fork
stabilization. Endocr Relat Cancer. 23:T1–T17. 2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Mesman RLS, Calleja F, Hendriks G, Morolli
B, Misovic B, Devilee P, van Asperen CJ, Vrieling H and Vreeswijk
MPG: The functional impact of variants of uncertain significance in
BRCA2. Genet Med. 21:293–302. 2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Lockett KL, Snowhite IV and Hu JJ:
Nucleotide-excision repair and prostate cancer risk. Cancer Lett.
220:125–135. 2005.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Gayther SA, de Foy KA, Harrington P,
Pharoah P, Dunsmuir WD, Edwards SM, Gillett C, Ardern-Jones A,
Dearnaley DP, Easton DF, et al: The frequency of germ-line
mutations in the breast cancer predisposition genes BRCA1 and BRCA2
in familial prostate cancer. The Cancer Research Campaign/British
Prostate Group United Kingdom Familial Prostate Cancer Study
Collaborators. Cancer Res. 60:4513–4518. 2000.PubMed/NCBI
|
|
46
|
Dong X, Wang L, Taniguchi K, Wang X,
Cunningham JM, McDonnell SK, Qian C, Marks AF, Slager SL, Peterson
BJ, et al: Mutations in CHEK2 associated with prostate cancer risk.
Am J Hum Genet. 72:270–280. 2003.PubMed/NCBI View
Article : Google Scholar
|
|
47
|
Rybicki BA, Conti DV, Moreira A, Cicek M,
Casey G and Witte JS: DNA repair gene XRCC1 and XPD polymorphisms
and risk of prostate cancer. Cancer Epidemiol Biomarkers Prev.
13:23–29. 2004.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Norris AM, Woodruff RD, D'Agostino RB Jr,
Clodfelter JE and Scarpinato KD: Elevated levels of the mismatch
repair protein PMS2 are associated with prostate cancer. Prostate.
67:214–225. 2007.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Sun X, Chen C, Vessella RL and Dong JT:
Microsatellite instability and mismatch repair target gene
mutations in cell lines and xenografts of prostate cancer.
Prostate. 66:660–666. 2006.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Martin L, Coffey M, Lawler M, Hollywood D
and Marignol L: DNA mismatch repair and the transition to hormone
independence in breast and prostate cancer. Cancer Lett.
291:142–149. 2010.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Schwertman P, Bekker-Jensen S and Mailand
N: Regulation of DNA double-strand break repair by ubiquitin and
ubiquitin-like modifiers. Nat Rev Mol Cell Biol. 17:379–394.
2016.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Brinkmann K, Schell M, Hoppe T and Kashkar
H: Regulation of the DNA damage response by ubiquitin conjugation.
Front Genet. 6(98)2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Ou HL and Schumacher B: DNA damage
responses and p53 in the aging process. Blood. 131:488–495.
2018.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Li J and Kurokawa M: Regulation of MDM2
stability after DNA damage. J Cell Physiol. 230:2318–2327.
2015.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Chan P, Möller A, Liu MC, Sceneay JE, Wong
CS, Waddell N, Huang KT, Dobrovic A, Millar EK, O'Toole SA, et al:
The expression of the ubiquitin ligase SIAH2 (seven in absentia
homolog 2) is mediated through gene copy number in breast cancer
and is associated with a basal-like phenotype and p53 expression.
Breast Cancer Res. 13(R19)2011.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Wang D, Ma J, Botuyan MV, Cui G, Yan Y,
Ding D, Zhou Y, Krueger EW, Pei J, Wu X, et al: ATM-phosphorylated
SPOP contributes to 53BP1 exclusion from chromatin during DNA
replication. Sci Adv. 7(eabd9208)2021.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Sharma A, Alswillah T, Singh K, Chatterjee
P, Willard B, Venere M, Summers MK and Almasan A: USP14 regulates
DNA damage repair by targeting RNF168-dependent ubiquitination.
Autophagy. 14:1976–1990. 2018.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Qu H, Liu H, Jin Y, Cui Z and Han G: HUWE1
upregulation has tumor suppressive effect in human prostate cancer
cell lines through c-Myc. Biomed Pharmacother. 106:309–315.
2018.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Gewirtz DA, Alotaibi M, Yakovlev VA and
Povirk LF: Tumor cell recovery from senescence induced by radiation
with PARP inhibition. Radiat Res. 186:327–332. 2016.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Wang Z, Song Y, Ye M, Dai X, Zhu X and Wei
W: The diverse roles of SPOP in prostate cancer and kidney cancer.
Nat Rev Urol. 17:339–350. 2020.PubMed/NCBI View Article : Google Scholar
|
|
61
|
An J, Wang C, Deng Y, Yu L and Huang H:
Destruction of full-length androgen receptor by wild-type SPOP, but
not prostate-cancer-associated mutants. Cell Rep. 6:657–669.
2014.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Hjorth-Jensen K, Maya-Mendoza A, Dalgaard
N, Sigurethsson JO, Bartek J, Iglesias-Gato D, Olsen JV and
Flores-Morales A: SPOP promotes transcriptional expression of DNA
repair and replication factors to prevent replication stress and
genomic instability. Nucleic Acids Res. 46:9484–9495.
2018.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Kuwano Y, Nishida K, Akaike Y, Kurokawa K,
Nishikawa T, Masuda K and Rokutan K: Homeodomain-interacting
protein Kinase-2: A critical regulator of the DNA damage response
and the Epigenome. Int J Mol Sci. 17(1638)2016.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008.PubMed/NCBI View Article : Google Scholar
|
|
65
|
McClurg UL, Chit N, Azizyan M, Edwards J,
Nabbi A, Riabowol KT, Nakjang S, McCracken SR and Robson CN:
Molecular mechanism of the TP53-MDM2-AR-AKT signalling network
regulation by USP12. Oncogene. 37:4679–4691. 2018.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Aron R, Pellegrini P, Green EW, Maddison
DC, Opoku-Nsiah K, Oliveira AO, Wong JS, Daub AC, Giorgini F and
Finkbeiner S: Publisher Correction: Deubiquitinase Usp12 functions
noncatalytically to induce autophagy and confer neuroprotection in
models of Huntington's disease. Nat Commun. 9(4333)2018.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Joo HY, Jones A, Yang C, Zhai L, Smith
ADt, Zhang Z, Chandrasekharan MB, Sun ZW, Renfrow MB, Wang Y, et
al: Regulation of histone H2A and H2B deubiquitination and Xenopus
development by USP12 and USP46. J Biol Chem. 286:7190–7201.
2011.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Adimoolam S and Ford JM: p53 and DNA
damage-inducible expression of the xeroderma pigmentosum group C
gene. Proc Natl Acad Sci USA. 99:12985–12990. 2002.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Scherer SJ, Maier SM, Seifert M,
Hanselmann RG, Zang KD, Muller-Hermelink HK, Angel P, Welter C and
Schartl M: p53 and c-Jun functionally synergize in the regulation
of the DNA repair gene hMSH2 in response to UV. J Biol Chem.
275:37469–37473. 2000.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Achanta G and Huang P: Role of p53 in
sensing oxidative DNA damage in response to reactive oxygen
species-generating agents. Cancer Res. 64:6233–6239.
2004.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Roe JS, Kim HR, Hwang IY, Cho EJ and Youn
HD: Von Hippel-Lindau protein promotes Skp2 destabilization on DNA
damage. Oncogene. 30:3127–3138. 2011.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Schulman BA, Carrano AC, Jeffrey PD, Bowen
Z, Kinnucan ER, Finnin MS, Elledge SJ, Harper JW, Pagano M and
Pavletich NP: Insights into SCF ubiquitin ligases from the
structure of the Skp1-Skp2 complex. Nature. 408:381–386.
2000.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Zhao H, Bauzon F, Fu H, Lu Z, Cui J,
Nakayama K, Nakayama KI, Locker J and Zhu L: Skp2 deletion unmasks
a p27 safeguard that blocks tumorigenesis in the absence of pRb and
p53 tumor suppressors. Cancer Cell. 24:645–659. 2013.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Zhao H, Lu Z, Bauzon F, Fu H, Cui J,
Locker J and Zhu L: p27T187A knockin identifies Skp2/Cks1 pocket
inhibitors for advanced prostate cancer. Oncogene. 36:60–70.
2017.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Lin HK, Chen Z, Wang G, Nardella C, Lee
SW, Chan CH, Yang WL, Wang J, Egia A, Nakayama KI, et al: Skp2
targeting suppresses tumorigenesis by Arf-p53-independent cellular
senescence. Nature. 464:374–379. 2010.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Lu W, Liu S, Li B, Xie Y, Izban MG,
Ballard BR, Sathyanarayana SA, Adunyah SE, Matusik RJ, Chen Z, et
al: SKP2 loss destabilizes EZH2 by promoting TRAF6-mediated
ubiquitination to suppress prostate cancer. Oncogene. 36:1364–1373.
2017.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Li B, Lu W, Yang Q, Yu X, Matusik RJ and
Chen Z: Skp2 regulates androgen receptor through ubiquitin-mediated
degradation independent of Akt/mTOR pathways in prostate cancer.
Prostate. 74:421–432. 2014.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Lu W, Liu S, Li B, Xie Y, Adhiambo C, Yang
Q, Ballard BR, Nakayama KI, Matusik RJ and Chen Z: SKP2
inactivation suppresses prostate tumorigenesis by mediating JARID1B
ubiquitination. Oncotarget. 6:771–788. 2015.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Arbini AA, Greco M, Yao JL, Bourne P,
Marra E, Hsieh JT, di Sant'agnese PA and Moro L: Skp2
overexpression is associated with loss of BRCA2 protein in human
prostate cancer. Am J Pathol. 178:2367–2376. 2011.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Moynahan ME, Pierce AJ and Jasin M: BRCA2
is required for homology-directed repair of chromosomal breaks. Mol
Cell. 7:263–272. 2001.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Wang F, Ning S, Yu B and Wang Y: USP14:
Structure, function, and target inhibition. Front Pharmacol.
12(801328)2021.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Liao Y, Liu N, Hua X, Cai J, Xia X, Wang
X, Huang H and Liu J: Proteasome-associated deubiquitinase
ubiquitin-specific protease 14 regulates prostate cancer
proliferation by deubiquitinating and stabilizing androgen
receptor. Cell Death Dis. 8(e2585)2017.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Gao L, Zhang W, Zhang J, Liu J, Sun F, Liu
H, Hu J, Wang X, Wang X, Su P, et al: KIF15-mediated stabilization
of AR and AR-V7 contributes to Enzalutamide resistance in prostate
cancer. Cancer Res. 81:1026–1039. 2021.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Liu Y, Yu C, Shao Z, Xia X, Hu T, Kong W,
He X, Sun W, Deng Y and Huang H: Selective degradation of AR-V7 to
overcome castration resistance of prostate cancer. Cell Death Dis.
12(857)2021.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Geng L, Chen X, Zhang M and Luo Z:
Ubiquitin-specific protease 14 promotes prostate cancer progression
through deubiquitinating the transcriptional factor ATF2. Biochem
Biophys Res Commun. 524:16–21. 2020.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Wang Y, Zhang N, Zhang L, Li R, Fu W, Ma
K, Li X, Wang L, Wang J, Zhang H, et al: Autophagy regulates
chromatin Ubiquitination in DNA damage response through elimination
of SQSTM1/p62. Mol Cell. 63:34–48. 2016.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Sander B, Xu W, Eilers M, Popov N and
Lorenz S: A conformational switch regulates the ubiquitin ligase
HUWE1. ELife. 6(e21036)2017.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Zhong Q, Gao W, Du F and Wang X:
Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the
polyubiquitination of Mcl-1 and regulates apoptosis. Cell.
121:1085–1095. 2005.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Gong X, Du D, Deng Y, Zhou Y, Sun L and
Yuan S: The structure and regulation of the E3 ubiquitin ligase
HUWE1 and its biological functions in cancer. Invest New Drugs.
38:515–524. 2020.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Myant KB, Cammareri P, Hodder MC, Wills J,
Von Kriegsheim A, Győrffy B, Rashid M, Polo S, Maspero E, Vaughan
L, et al: HUWE1 is a critical colonic tumour suppressor gene that
prevents MYC signalling, DNA damage accumulation and tumour
initiation. EMBO Mol Med. 9:181–197. 2017.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Yang D, Cheng D, Tu Q, Yang H, Sun B, Yan
L, Dai H, Luo J, Mao B, Cao Y, et al: HUWE1 controls the
development of non-small cell lung cancer through down-regulation
of p53. Theranostics. 8:3517–3529. 2018.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Fan L, Xu S, Zhang F, Cui X, Fazli L,
Gleave M, Clark DJ, Yang A, Hussain A, Rassool F and Qi J: Histone
demethylase JMJD1A promotes expression of DNA repair factors and
radio-resistance of prostate cancer cells. Cell Death Dis.
11(214)2020.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Shen D, Luo J, Chen L, Ma W, Mao X, Zhang
Y, Zheng J, Wang Y, Wan J, Wang S, et al: PARPi treatment enhances
radiotherapy-induced ferroptosis and antitumor immune responses via
the cGAS signaling pathway in colorectal cancer. Cancer Lett.
550(215919)2022.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Lesueur P, Lequesne J, Grellard JM, Dugué
A, Coquan E, Brachet PE, Geffrelot J, Kao W, Emery E, Berro DH, et
al: Phase I/IIa study of concomitant radiotherapy with olaparib and
temozolomide in unresectable or partially resectable glioblastoma:
OLA-TMZ-RTE-01 trial protocol. BMC cancer. 19(198)2019.PubMed/NCBI View Article : Google Scholar
|
|
95
|
D'Andrea AD: Mechanisms of PARP inhibitor
sensitivity and resistance. DNA Repair (Amst). 71:172–176.
2018.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Messina C, Cattrini C, Soldato D, Vallome
G, Caffo O, Castro E, Olmos D, Boccardo F and Zanardi E: BRCA
mutations in prostate cancer: Prognostic and predictive
implications. J Oncol. 2020(4986365)2020.PubMed/NCBI View Article : Google Scholar
|
|
97
|
de Bono J, Mateo J, Fizazi K, Saad F,
Shore N, Sandhu S, Chi KN, Sartor O, Agarwal N, Olmos D, et al:
Olaparib for metastatic castration-resistant prostate cancer. N
Engl J Med. 382:2091–2102. 2020.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Wu C, Peng S, Pilie PG, Geng C, Park S,
Manyam GC, Lu Y, Yang G, Tang Z, Kondraganti S, et al: PARP and
CDK4/6 inhibitor combination therapy induces apoptosis and
suppresses neuroendocrine differentiation in prostate cancer. Mol
Cancer Ther. 20:1680–1691. 2021.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Zhou S, Dai Z, Wang L, Gao X, Yang L, Wang
Z, Wang Q and Liu Z: MET inhibition enhances PARP inhibitor
efficacy in castration-resistant prostate cancer by suppressing the
ATM/ATR and PI3K/AKT pathways. J Cell Mol Med. 25:11157–11169.
2021.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Enriquez-Rios V, Dumitrache LC, Downing
SM, Li Y, Brown EJ, Russell HR and McKinnon PJ: DNA-PKcs, ATM, and
ATR interplay maintains genome integrity during neurogenesis. J
Neurosci. 37:893–905. 2017.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Tang Z, Pilié PG, Geng C, Manyam GC, Yang
G, Park S, Wang D, Peng S, Wu C, Peng G, et al: ATR inhibition
induces CDK1-SPOP signaling and enhances Anti-PD-L1 cytotoxicity in
prostate cancer. Clin Cancer Res. 27:4898–4909. 2021.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Mota JM, Barnett E, Nauseef JT, Nguyen B,
Stopsack KH, Wibmer A, Flynn JR, Heller G, Danila DC, Rathkopf D,
et al: Platinum-Based chemotherapy in metastatic prostate cancer
with DNA repair gene alterations. JCO Precis Oncol. 4:355–366.
2020.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Schmid S, Omlin A, Higano C, Sweeney C,
Martinez Chanza N, Mehra N, Kuppen MCP, Beltran H, Conteduca V,
Vargas Pivato de Almeida D, et al: Activity of Platinum-Based
chemotherapy in patients with advanced prostate cancer with and
without DNA repair gene aberrations. JAMA Netw Open.
3(e2021692)2020.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Su YX, Yu CF, Xue P, Li LL, Xiao KM, Chu
XL and Zhu SJ: Research progress on the treatment of advanced
prostate cancer with Olaparib. Neoplasma. 68:1132–1138.
2021.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Kim H, Xu H, George E, Hallberg D, Kumar
S, Jagannathan V, Medvedev S, Kinose Y, Devins K, Verma P, et al:
Combining PARP with ATR inhibition overcomes PARP inhibitor and
platinum resistance in ovarian cancer models. Nat Commun.
11(3726)2020.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Karzai F, VanderWeele D, Madan RA, Owens
H, Cordes LM, Hankin A, Couvillon A, Nichols E, Bilusic M, Beshiri
ML, et al: Activity of durvalumab plus olaparib in metastatic
castration-resistant prostate cancer in men with and without DNA
damage repair mutations. J Immunother Cancer. 6(141)2018.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Bizzaro F, Fuso Nerini I, Taylor MA,
Anastasia A, Russo M, Damia G, Guffanti F, Guana F, Ostano P,
Minoli L, et al: VEGF pathway inhibition potentiates PARP inhibitor
efficacy in ovarian cancer independent of BRCA status. J Hematol
Oncol. 14(186)2021.PubMed/NCBI View Article : Google Scholar
|














