Introduction
Acute myocardial infarction (AMI), which has been
reported to be a main cause of death and disability worldwide, is a
severe cardiovascular disease that results from the acute loss of
viable myocardium due to hypoxia (1). Restoring blood flow to the ischemic
area, namely reperfusion therapy, is the most common treatment for
AMI, but it can sometimes abnormally aggravate myocardial injury,
myocardial cell death and cause additional cardiac dysfunction in
clinical practice in a process known as myocardial
ischemia/reperfusion (I/R) injury (2). As a complex pathophysiological
process during hypoxia and subsequent reperfusion, myocardial I/R
has been reported to be promoted by oxidative stress radicals,
inflammation and apoptosis in previous studies (3-5).
However, the exact molecular mechanism remains unknown. Therefore,
clarifying the mechanisms underlying the progression of myocardial
I/R injury is necessary for identifying new therapeutic strategies
and medicines.
Chemokine-like factor 1 (CKLF1) is a member of the
CKLF protein family that has multiple biological functions,
including chemotactic activities, inducing cell growth in multiple
organs, and regulating vascular smooth muscle cell migration and
proliferation after vascular injury (6-8).
An increasing number of studies has validated the relationship
between CKLF1 and ischemic disease. For example, CKLF1 aggravates
early focal cerebral ischemic injury by regulating the polarization
of microglia/macrophages to M1 type (9). Conversely, by promoting energy
metabolism and inhibiting apoptosis, CKLF1 knockdown improves focal
cerebral ischemia (10).
Furthermore, IMM-H004 can downregulate the expression of CKLF1 to
restrain activation of the NOD-like receptor family, pyrin domain
containing 3 (NLRP3) inflammasome and the subsequent inflammatory
reaction, ultimately protecting the ischemic brain (11). In addition, CKLF1 has been reported
to be upregulated during hepatic I/R, and CKLF1 inhibition can
reduce neutrophil infiltration and decrease the inflammatory
response to alleviate hepatic I/R injury (12). However, the role of CKLF1 in
myocardial I/R injury has rarely been studied (13,14).
The JASPAR database (https://jaspar.genereg.net/) indicated the putative
forkhead box protein C1 (FOXC1)-binding site on the CKLF1 promoter.
A previous study suggested that FOXC1 is a hypoxia-activated
transcription factor that can promote lung cancer cell growth
(15). Of note, FOXC1 can be
induced to promote the inflammatory response and cell damage during
myocardial ischemia (16).
Therefore, the present study focused on whether CKLF1 could be
transcriptionally activated by the transcription factor FOXC1 to
participate in the progression of myocardial I/R.
In the present study, the hypoxia/reoxygenation
(H/R)-induced H9c2 rat cardiomyoblast cell injury model was
established to mimic myocardial I/R injury in vitro with the
aim of investigating the effects of CKLF1 and FOXC1 on H/R injury
from the perspectives of oxidative stress, inflammation and
apoptosis. The present findings may provide information on the
mechanisms underlying myocardial I/R and could aid the development
of future therapies.
Materials and methods
Cell culture and H/R induction
The rat cardiomyoblast cell line H9c2 was obtained
from The Cell Bank of Type Culture Collection of The Chinese
Academy of Sciences. The cells were placed in a constant
temperature incubator at 37˚C and 5% CO2, and were
maintained in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (HyClone; Cytiva). To
simulate myocardial I/R in vitro, H9c2 cells at 80%
confluence were transferred to serum- and glucose-free DMEM, and
were cultivated in a hypoxic atmosphere of 0.1% O2, 95%
N2 and 5% CO2 for 6 h. Subsequently, the
cells were maintained in the incubator with normal DMEM under
normoxic conditions in the presence of 95% air and 5%
CO2 for 12 h for reoxygenation (17,18).
H9c2 cells in the control group were cultured under normal culture
conditions.
Transfection
For transfection, CKLF1 small interfering RNAs
(siRNAs) (si-CKLF1#1 sense, 5'-GTCTTGACAAGACAATGAGATCT-3',
antisense, 5'-AGATCTCATTGTCTTGTCAAGAC-3'; si-CKLF1#2 sense,
5'-GCCTTTGCTTGATGTTATCAACT-3', antisense,
5'-AGTTGATAACATCAAGCAAAGGC-3'), negative control siRNA (si-NC
sense, 5'-CCTTATGTACGTTGATTCAGTACAA-3', antisense
5'-TTGTACTGAATCAACGTACATAAGG-3'), FOXC1 pcDNA3.1 plasmid (Oe-FOXC1)
and empty vector (Oe-NC) were synthesized by Shanghai GeneChem Co.,
Ltd. H9c2 cells at the logarithmic phase were seeded in a 6-well
plate (1x105 cells/well) and were incubated at 37˚C
until they reached 80% confluence. Subsequently, 100 nM of
recombinants were transfected into H9c2 cells at 37˚C for 48 h
using Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. After
48 h, cells were collected and further experiments were performed.
For H9c2 cells both subjected to H/R stimulation and
co-transfection with si-CKLF1 and Oe-FOXC1, cells were
pre-transfected for 48 h and then received H/R stimulation.
Cell viability assay
H9c2 cells were plated into a 96-well plate at a
density of 1x104 cells/well. Following H/R stimulation
and indicated transfection, 10 µl Cell Counting Kit-8 (CCK-8)
solution (Sangon Biotech Co., Ltd.) was added to each well and
incubated for 3 h at 37˚C. The absorbance was detected at a
wavelength of 450 nm using a microplate reader (BioTek Instruments,
Inc.) to evaluate the viability of H9c2 cells.
Flow cytometric analysis
The apoptotic rate of H9c2 cells was determined
following H/R stimulation and indicated transfection using an
Annexin V-fluorescein isothiocyanate (FITC) kit (Shanghai GeneChem
Co., Ltd.) according to the manufacturer's instructions. H9c2 cells
were obtained and resuspended in 500 µl binding buffer.
Subsequently, 100 µl cell suspension was transferred to a tube, to
which 5 µl Annexin V-FITC and 10 µl propidium iodide was added. The
mixture was incubated at room temperature for 5 min in dark.
Finally, treated cells were analyzed using a flow cytometer
(FACSCalibur; BD Biosciences) and Flowjo vX.0.7 software (FlowJo
LLC) was used. Apoptotic cells were counted and expressed as a
percentage of the total cell count.
Measurement of caspase 3 activity
Caspase 3 activity was measured using a caspase 3
colorimetric assay kit (cat. no. C1115; Beyotime Institute of
Biotechnology) according to the manufacturer's guidelines. H9c2
cells were lysed in RIPA lysis buffer (cat. no. P0013B; Beyotime
Institute of Biotechnology) and the lysates were centrifuged at
12,000 x g for 10 min at 4˚C. Cell lysates were then incubated with
caspase 3 substrate (Ac-DEVD-pNA) for 2 h at 37˚C in the dark. The
relative fluorescence was detected at 450 nm using a fluorescence
plate reader.
Evaluation of intracellular reactive
oxygen species (ROS)
The measurement of intracellular ROS production was
performed using the ROS assay kit (cat. no. S0033S; Beyotime
Institute of Biotechnology) using the fluorescent probe
2,7-dichlorodihydrofluorescein diacetate (DCFH-DA). Following
transfection and/or H/R treatment, H9c2 cells were incubated with
10 µmol/l DCFH-DA for 20 min at 37˚C in the dark. After washing
three times with PBS, the fluorescence intensity of cells in each
group was detected using a fluorescence spectrophotometer with 488
nm excitation and 525 nm emission filters.
Detection of oxidative stress
indicators
After H/R stimulation and indicated transfection,
H9c2 cells were centrifuged at 1,500 x g for 10 min at 4˚C. The
activities of superoxide dismutase (SOD) and glutathione peroxidase
(GSH-Px), and the content of malondialdehyde (MDA) in the
supernatant of H9c2 cells were tested using SOD assay kits (cat.
no. S0086; Beyotime Institute of Biotechnology), GSH-Px assay kits
(cat. no. A005-1-2; Nanjing Jiancheng Bioengineering Institute) and
MDA assay kits (cat. no. A003-1-2; Nanjing Jiancheng Bioengineering
Institute), respectively, according to the manufacturer's
protocols. The optical density was measured using a microplate
reader (BioTek Instruments, Inc.).
ELISA
After H/R stimulation and indicated transfection,
H9c2 cells were centrifuged at 2.000 x g for 5 min at 4˚C. The
levels of tumor necrosis factor-α (TNF-α), interleukin (IL)-6 and
IL-1β in the culture supernatant were detected using ELISA kits for
TNF-α (cat. no. F3768; Shanghai Westang Biotechnology Co., Ltd.),
IL-6 (cat. no. F3743; Shanghai Westang Biotechnology Co., Ltd.) and
IL-1β (cat. no. F3739; Shanghai Westang Biotechnology Co., Ltd.).
All experimental procedures were conducted in accordance with the
manufacturer's guidelines.
Reverse transcription-quantitative PCR
(RT-qPCR)
After H/R stimulation and indicated transfection,
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) was used to isolate total RNA from H9c2 cells according to
the manufacturer's recommendations. cDNA was synthesized from total
RNA using the Prime Script RT Reagent Kit with gDNA Eraser (Takara
Biotechnology Ltd.) according to the manufacturer's instructions.
qPCR was performed using SYBR Green master mix (Vazyme Biotech Co.,
Ltd.) and detected on an ABI Prism 7500 Sequence Detector (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The following
thermocycling conditions were used for qPCR: 95˚C for 10 min;
followed by 40 cycles of 95˚C for 10 sec and 60˚C for 60 sec. The
following primer pairs were used for qPCR: CKLF1 forward,
5'-GTTGAAGTTGTTGCGCGAGT-3' and reverse, 5'-ATACGGTTCAGGGGCTTGTG-3';
FOXC1 forward, 5'-CTATCCAGAATGCCCCGGAC-3' and reverse,
5'-CGTACCGTTCTCCGTCTTGA-3'; GAPDH forward,
5'-GCATCTTCTTGTGCAGTGCC-3' and reverse, 5'-GATGGTGATGGGTTTCCCGT-3'.
GAPDH was used as an internal reference and the 2-ΔΔCq
method (19) was used for the
calculation of relative mRNA expression levels.
Western blot analysis
After H/R stimulation and indicated transfection,
H9c2 cells were lysed in RIPA lysis buffer and the lysates were
centrifuged at 12,000 x g for 10 min at 4˚C to obtain proteins. The
protein concentration was determined using the bicinchoninic acid
method. Equal amounts of protein (40 µg) were separated by SDS-PAGE
on 10% gels and were then transferred to PVDF membranes. After
blocking in 5% non-fat milk for 1 h at room temperature, the
membranes were probed with primary antibodies overnight at 4˚C,
followed by incubation with a HRP-conjugated secondary antibody
(1:5,000; cat. no. 7074P2; Cell Signaling Technology, Inc.) at room
temperature for 1 h. Immunoreactive proteins were visualized using
an enhanced chemiluminescence kit (Thermo Fisher Scientific, Inc.)
and gray intensity analysis was performed using ImageJ 1.8.0
software (National Institutes of Health). The expression levels of
specific protein were normalized to GAPDH. The primary antibodies
used in the present study were as follows: CKLF1 (1:1,000; cat. no.
ab180512), Bcl-2 (1:2,000; cat. no. ab196495), NLRP3 (1:1,000; cat.
no. ab263899), IL-18 (1:1,000; cat. no. ab191860) and IL-1β
(1:1,000; cat. no. ab254360) (all from Abcam); gasdermin D
N-terminal domain (GSDMD-N; 1:200; cat. no. DF13758; Affinity
Biosciences); FOXC1 (1:1,000; cat. no. 8758S), Bax (1:1,000; cat.
no. 14796S), cleaved PARP (1:1,000; cat. no. 94885S), PARP
(1:1,000; cat. no. 9532T), caspase 1 (1:1,000; cat. no. 83383S) and
GAPDH (1:1,000; cat. no. 5174T) (all from Cell Signaling
Technology, Inc.).
Dual-luciferase reporter assay
Lipofectamine 3000 was used to co-transfect H9c2
cells with pGL3 vectors (Promega Corporation) containing the
wild-type (WT) CKLF1 promoter sequence or the corresponding mutant
CKLF1 promoter sequence, and Oe-FOXC1 or Oe-NC. A total of 48 h
post-transfection, the firefly and Renilla luciferase
activities were detected using a dual luciferase reporter assay
system (Promega Corporation). The results obtained were normalized
to Renilla luciferase activity.
Chromatin immunoprecipitation (ChIP)
assay
The binding of FOXC1 to the CKLF1 promoter was
validated using a ChIP assay kit (Beyotime Institute of
Biotechnology). H9c2 cells were cross-linked with 1% formaldehyde
solution for 10 min at 37˚C. Subsequently, the cross-linking
reaction was quenched with 1.1 ml glycine solution (10X). DNA
fragments were extracted by sonication using a 10 sec on and 10 sec
off mode for 12 cycles at 4˚C and immunoprecipitated overnight at
4˚C with anti-FOXC1 (cat. no. ab227977; 1:50; Abcam) or negative
control IgG antibodies (cat. no. 3423; 1:20; Cell Signaling
Technology). The DNA isolated through ChIP reactions was evaluated
using PCR as aforementioned.
Statistical analysis
GraphPad Prism 8.0 (Dotmatics) was used for
statistical analysis. All experimental data are presented as the
mean ± standard deviation from three experiments. The statistical
significance between two groups was analyzed using an unpaired
Student's t-test. One-way analysis of variance followed by Tukey's
post-hoc test was performed to compare the data of multiple groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
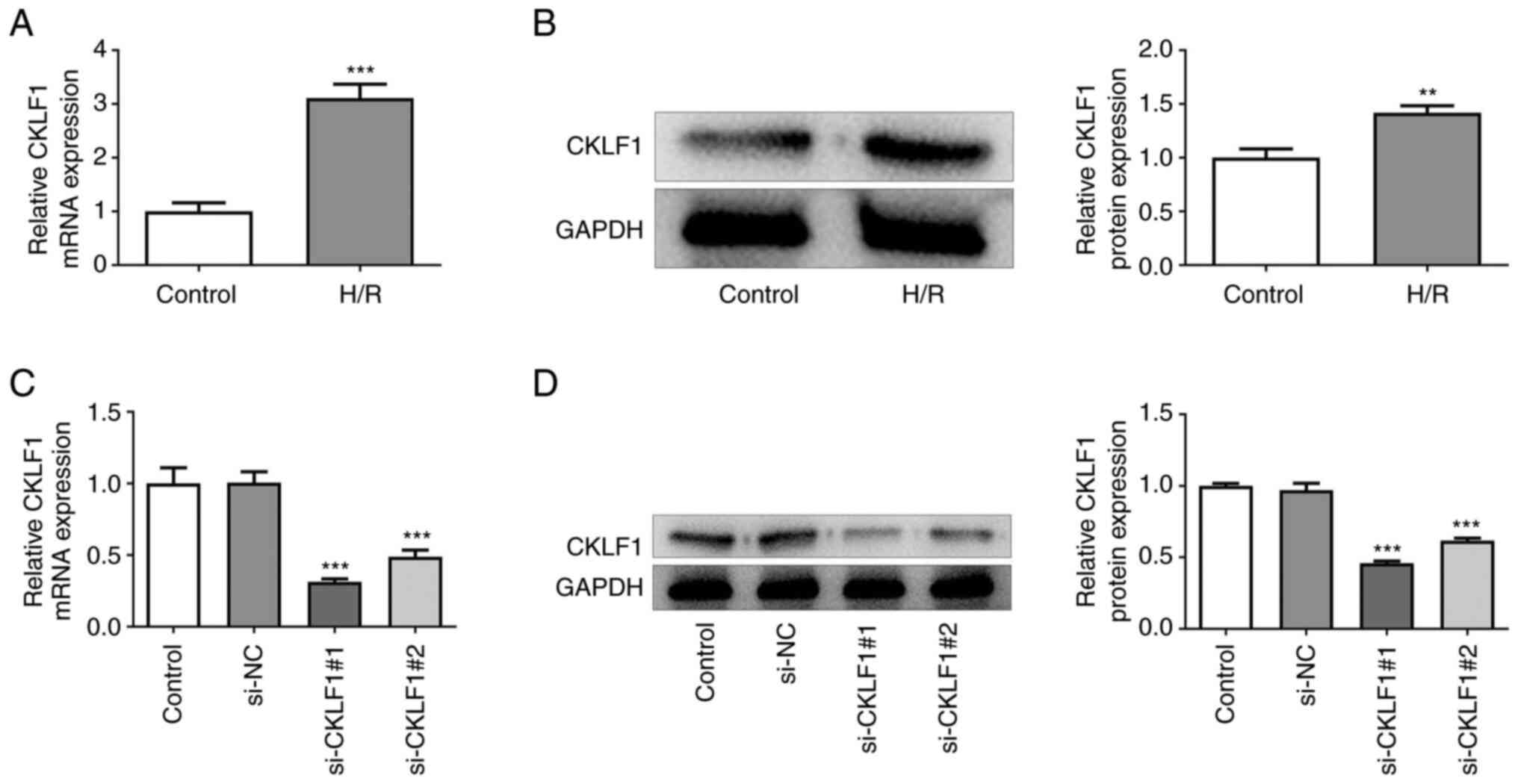
CKLF1 is significantly upregulated in
H/R-induced H9c2 cells
H9c2 cells exposed to H/R were used to establish an
in vitro model of myocardial I/R injury. The results of
RT-qPCR and western blotting indicated that H/R stimulation
significantly upregulated the expression levels of CKLF1 compared
with those in the control group (Fig.
1A and B). Subsequently, CKLF1
was silenced by transfection with siRNAs targeting CKLF1.
Transfection with si-CKLF1#1 and si-CKLF1#2 led to significantly
reduced CKLF1 expression when compared with the si-NC group
(Fig. 1C and D). It is worthwhile to mention that
si-CKLF1#1 was selected for subsequent experiments due to its
better knockdown efficacy.
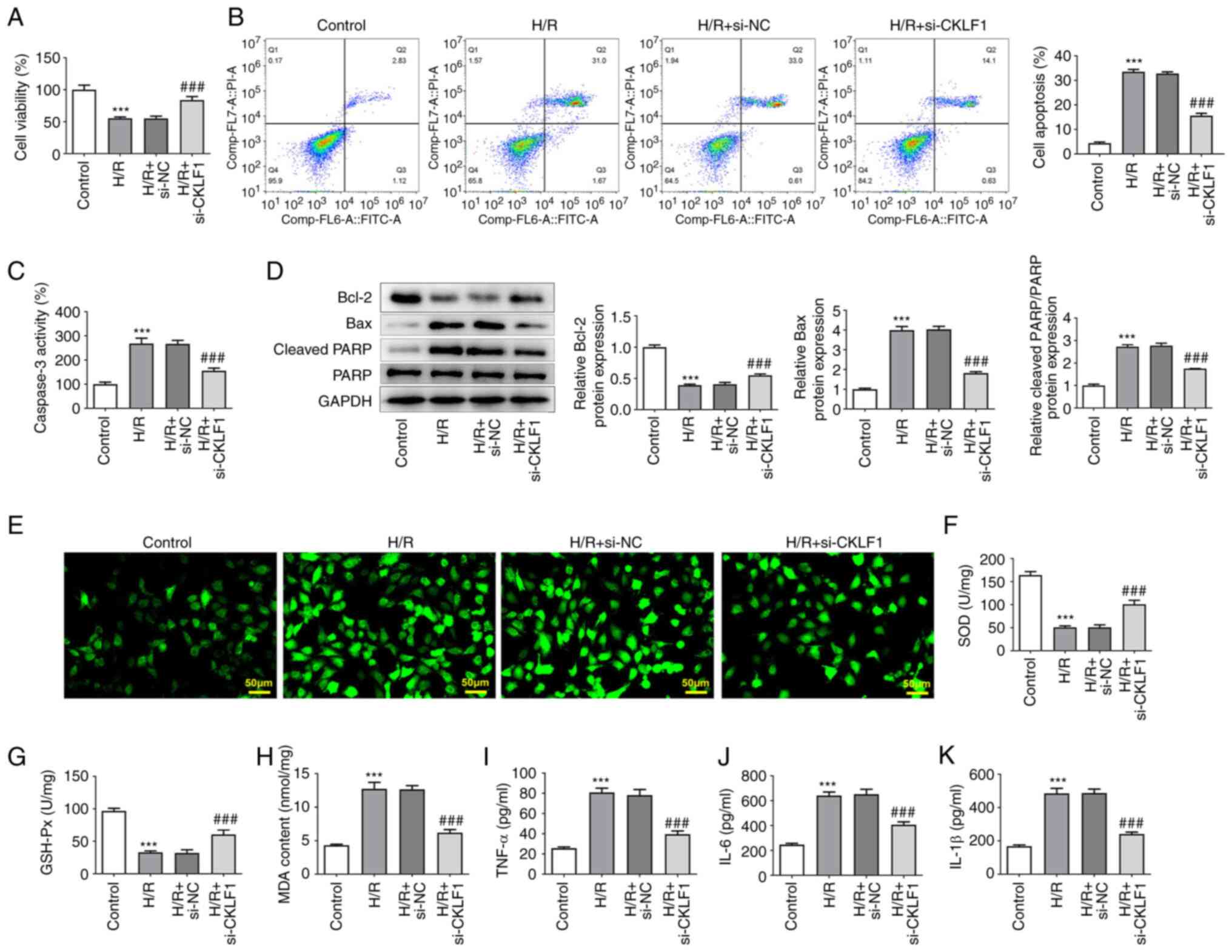
CKLF1 knockdown inhibits the
apoptosis, oxidative stress and inflammation of H/R-induced H9c2
cells
Cell viability was evaluated using the CCK-8 assay
in the presence or absence of H/R and si-CKLF1. As shown in
Fig. 2A, H/R stimulation
significantly decreased the viability of H9c2 cells when compared
with the control group; however, this was reversed by CKLF1
silencing. In addition, the percentage of apoptotic H9c2 cells was
significantly elevated following H/R induction, whereas CKLF1
knockdown partially alleviated the H/R-triggered increase in
apoptosis (Fig. 2B). Furthermore,
H9c2 cells under H/R conditions exhibited enhanced caspase 3
activity, upregulated Bax and cleaved PARP expression, and
downregulated Bcl-2 expression compared with that in the control
group (Fig. 2C and D). However, CKLF1 knockdown attenuated
the effects of H/R on the levels of caspase 3, Bax, cleaved PARP
and Bcl-2. Subsequently, the levels of intracellular ROS were
evaluated using DCFH-DA as a fluorescent probe in H9c2 cells
exposed to H/R conditions. Markedly enhanced fluorescence intensity
was observed in the H/R group compared with that in the control
group (Fig. 2E). By contrast,
CKLF1 silencing reduced the fluorescence intensity induced by H/R
exposure. Furthermore, compared with that in the control group, H/R
stimulation promoted the oxidative stress and inflammation of H9c2
cells, as evidenced by decreased SOD and GSH-Px activities, and
increased MDA, TNF-α, IL-6 and IL-1β contents (Fig. 2F-K). Conversely, CKLF1 knockdown
relieved the effects of H/R on the aforementioned oxidative stress
and inflammation-related factors. These findings indicated that
interference with CKLF1 inhibited the apoptosis, oxidative stress
and inflammation of H/R-induced H9c2 cells.
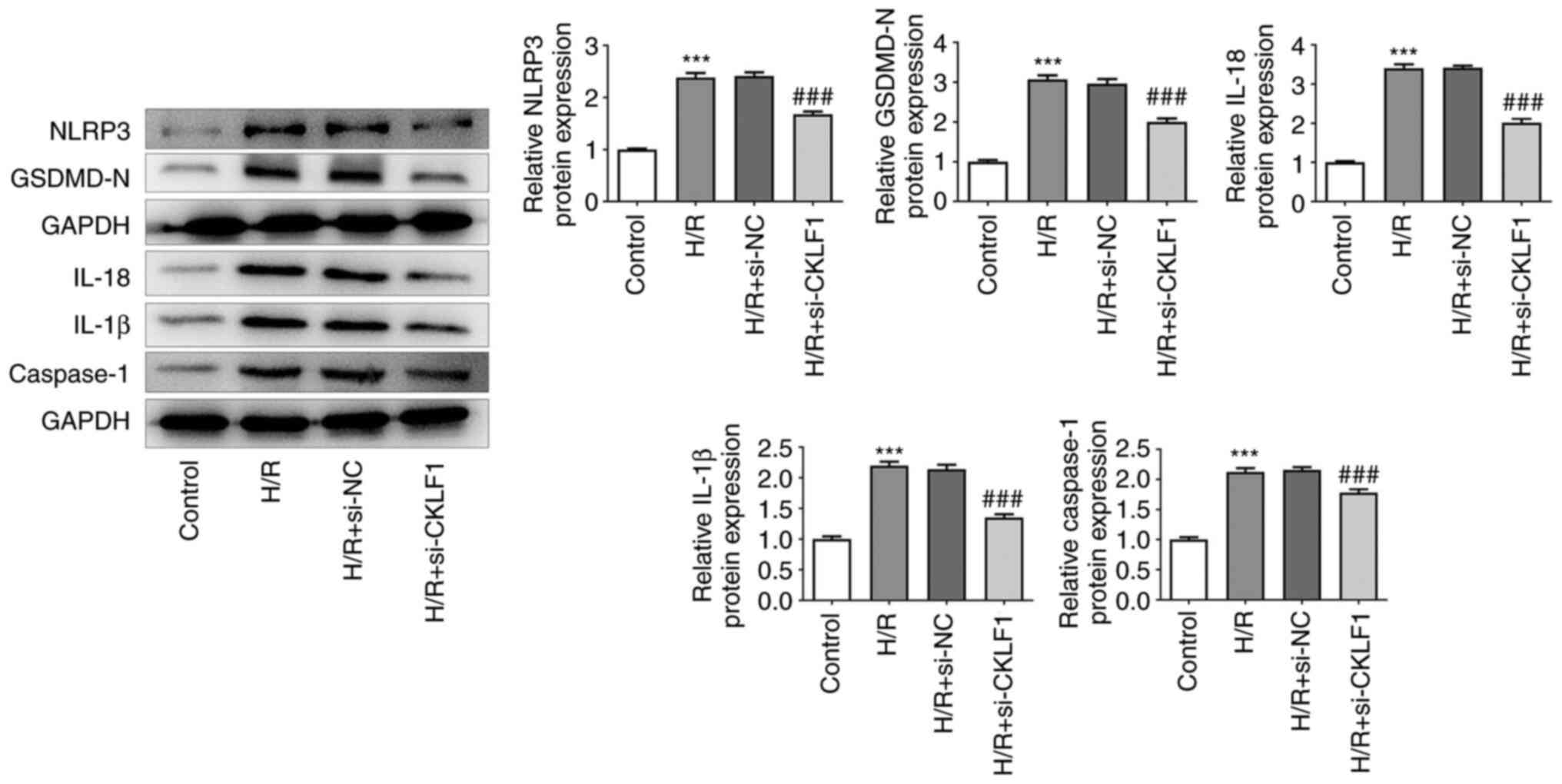
 | Figure 2CKLF1 knockdown inhibits the
apoptosis, oxidative stress and inflammation of H/R-induced H9c2
cells. (A) Cell viability was assessed by Cell Counting Kit-8
assay. (B) Apoptotic rate was measured by flow cytometric analysis.
(C) Caspase 3 activity was detected using a caspase 3 colorimetric
assay kit. (D) Western blotting was used to evaluate the expression
levels of apoptosis-related proteins. (E) Intracellular reactive
oxygen species were evaluated using 2,7-dichlorodihydrofluorescein
diacetate as a fluorescent probe in H9c2 cells exposed to H/R
conditions. The activities of (F) SOD and (G) GSH-Px, and the
levels of (H) MDA, (I) TNF-α, (J) IL-6 and (K) IL-1β were examined
using commercially available kits. ***P<0.001 vs.
control; ###P<0.001 vs. H/R + si-NC. CKLF1,
chemokine-like factor 1; GSH-Px, glutathione peroxidase; H/R,
hypoxia/reoxygenation; IL, interleukin; MDA, malondialdehyde; NC,
negative control; si, small interfering; SOD, superoxide dismutase;
TNF-α, tumor necrosis factor-α. |
CKLF1 knockdown suppresses NLRP3
inflammasome activation in H/R-induced H9c2 cells
The results of western blotting indicated that H/R
led to the activation of NLRP3 inflammasome signaling by
upregulating NLRP3, GSDMD-N, IL-18, IL-1β and caspase 1 expression
(Fig. 3). Notably, CKLF1 knockdown
contributed to the inactivation of NLRP3 inflammasome signaling
compared with the H/R + si-NC group. These results suggested that
CKLF1 silencing suppressed the oxidative stress, inflammation and
NLRP3 inflammasome activation in H/R-induced H9c2 cells.
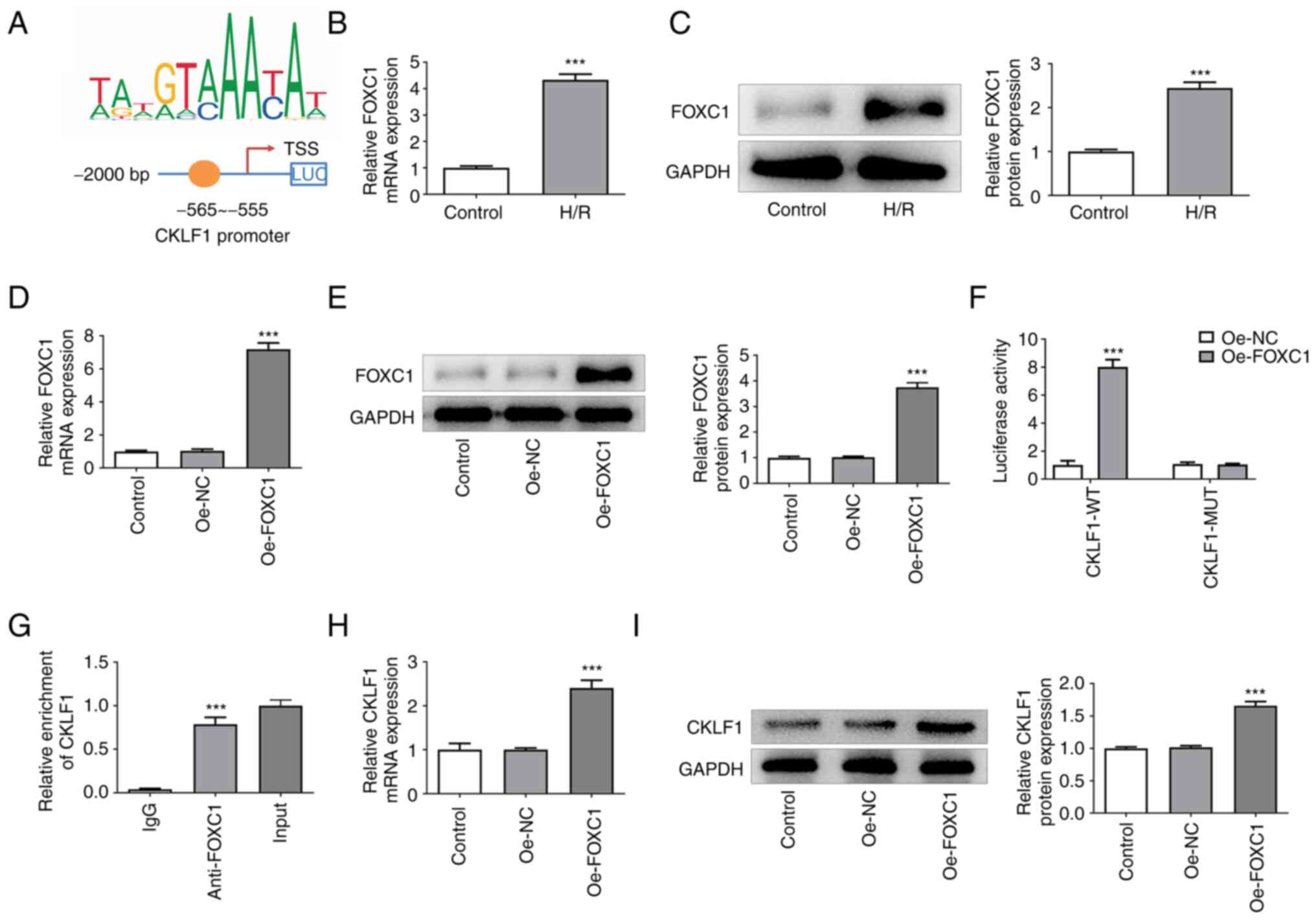
CKLF1 is transcriptionally activated
by FOXC1 in H9c2 cells
The putative FOXC1-binding site on the CKLF1
promoter was identified using the JASPAR database (Fig. 4A). As shown in Fig. 4B and C, H9c2 cells under H/R conditions
exhibited higher FOXC1 expression than that in the control group.
Significantly elevated FOXC1 expression was observed after Oe-FOXC1
transfection (Fig. 4D and E). Moreover, FOXC1 overexpression
significantly increased the luciferase activity in the CKLF1-WT
group when compared with the Oe-NC group (Fig. 4F). ChIP assay also demonstrated
that FOXC1 could bind with the CKLF1 promoter region (Fig. 4G). Furthermore, enhanced CKLF1 mRNA
and protein expression levels were found in H9c2 cells in the
Oe-FOXC1 group compared with the Oe-NC group (Fig. 4H and I). These results suggested that FOXC1
could transcriptionally activate CKLF1 and upregulate CKLF1
expression in H9c2 cells.
FOXC1 overexpression alleviates the
inhibitory effects of CKLF1 knockdown on the apoptosis, oxidative
stress and inflammation of H/R-induced H9c2 cells
FOXC1 was overexpressed in H9c2 cells to further
explore whether FOXC1 transcriptionally activated CKLF1 to regulate
H/R-induced damage. As shown in Fig.
5A, compared with in the H/R + si-CKLF1 + Oe-NC group, FOXC1
overexpression increased the apoptotic rate of H9c2 cells. In
addition, FOXC1 overexpression elevated caspase 3 activity in H9c2
cells exposed to H/R; however, there was no significant difference
when compared with the Oe-NC group (Fig. 5B). Furthermore, Bcl-2 expression
was significantly decreased, and Bax and cleaved PARP expression
was significantly increased in the H/R + si-CKLF1 + Oe-FOXC1 group
when compared with the H/R + si-CKLF1 + Oe-NC group (Fig. 5C). As shown in Fig. 5D-G, FOXC1 overexpression in H9c2
cells transfected with si-CKLF1 under H/R conditions exhibited
elevated oxidative stress compared with that in the H/R + si-CKLF1
+ Oe-NC group, as evidenced by the enhanced fluorescence intensity,
increased MDA content, and decreased SOD and GSH-Px activities.
Furthermore, the levels of inflammatory factors, including TNF-α,
IL-6 and IL-1β, were significantly elevated in the H/R + si-CKLF1 +
Oe-FOXC1 group compared with those in the H/R + si-CKLF1 + Oe-NC
group (Fig. 5H-J). These findings
indicated that FOXC1 overexpression partially reversed the
inhibitory effects of CKLF1 knockdown on the apoptosis, oxidative
stress and inflammation of H/R-induced H9c2 cells.
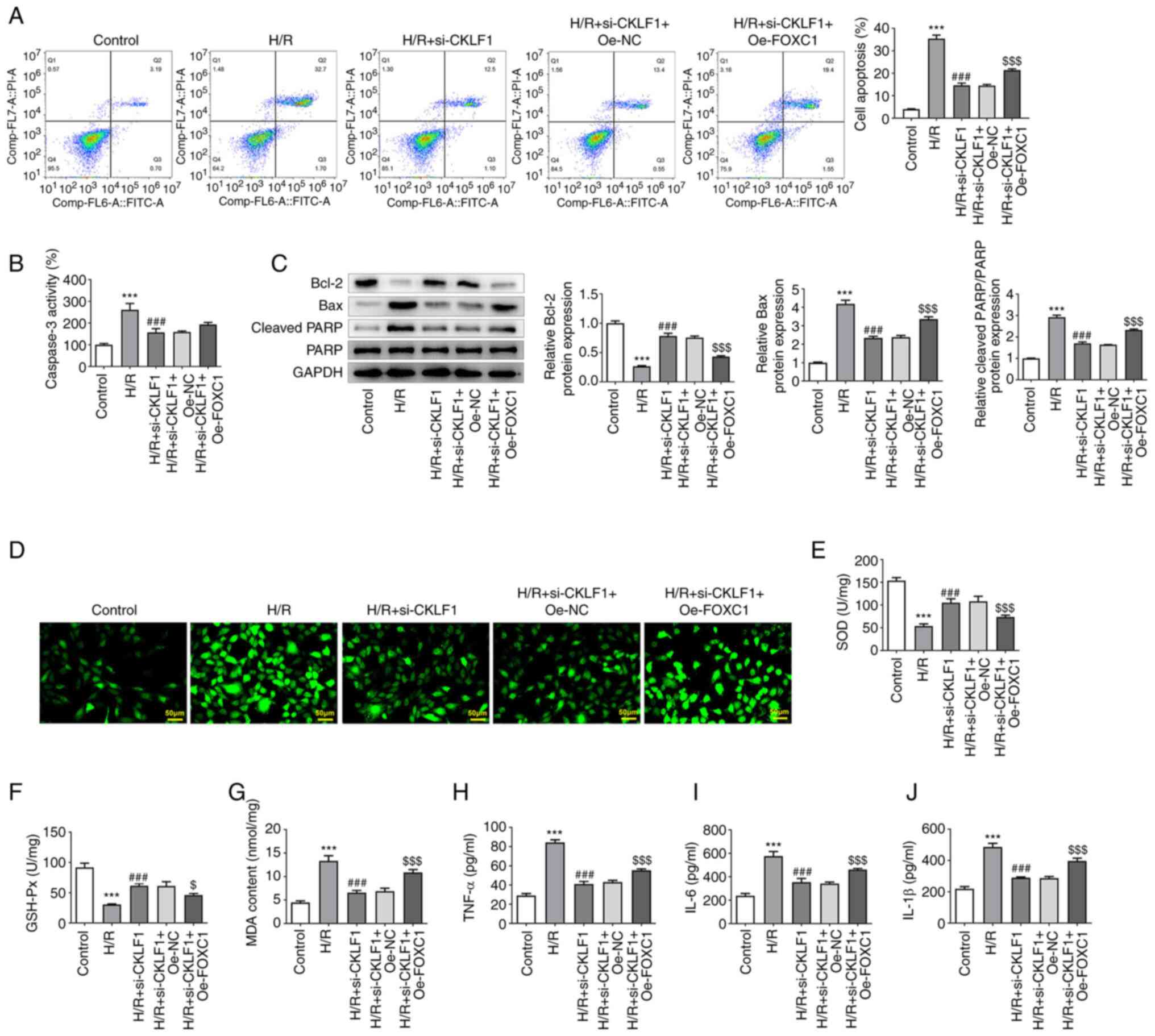 | Figure 5Oe-FOXC1 alleviates the inhibitory
effects of CKLF1 knockdown on the apoptosis, oxidative stress and
inflammation of H/R-induced H9c2 cells. (A) Cell apoptosis was
measured using flow cytometry. (B) Caspase 3 activity was detected
using a caspase 3 colorimetric assay kit. (C) Western blotting was
used to evaluate the expression of apoptosis-related proteins. (D)
Intracellular reactive oxygen species were evaluated using
2,7-dichlorodihydrofluorescein diacetate as a fluorescent probe.
The activities of (E) SOD and (F) GSH-Px, and the levels of (G)
MDA, (H) TNF-α, (I) IL-6 and (J) IL-1β were examined using
commercially available kits. ***P<0.001 vs. control;
###P<0.001 vs. H/R; $P<0.05,
$$$P<0.001 vs. H/R + si-CKLF1 + Oe-NC. CKLF1,
chemokine-like factor 1; GSH-Px, glutathione peroxidase; FOXC1,
forkhead box protein C1; H/R, hypoxia/reoxygenation; IL,
interleukin; MDA, malondialdehyde; NC, negative control; OE,
overexpression; si, small interfering; SOD, superoxide dismutase;
TNF-α, tumor necrosis factor-α. |
FOXC1 overexpression reverses the
effects of CKLF1 knockdown on H/R-induced H9c2 cell damage via
NLRP3 inflammasome signaling
Post-transfection with Oe-FOXC1 and si-CKLF1 in H9c2
cells exposed to H/R, the expression levels of proteins associated
with NLRP3 inflammasome signaling were assessed by western
blotting. As shown in Fig. 6,
FOXC1 overexpression in H9c2 cells with CKLF1 knockdown led to
markedly increased expression levels of NLRP3, GSDMD-N, IL-18,
IL-1β and caspase 1. Taken together, these findings indicated that
CKLF1 silencing, potentially mediated by FOXC1 downregulation,
suppressed H/R-induced H9c2 cell damage by inhibiting NLRP3
inflammasome activation.
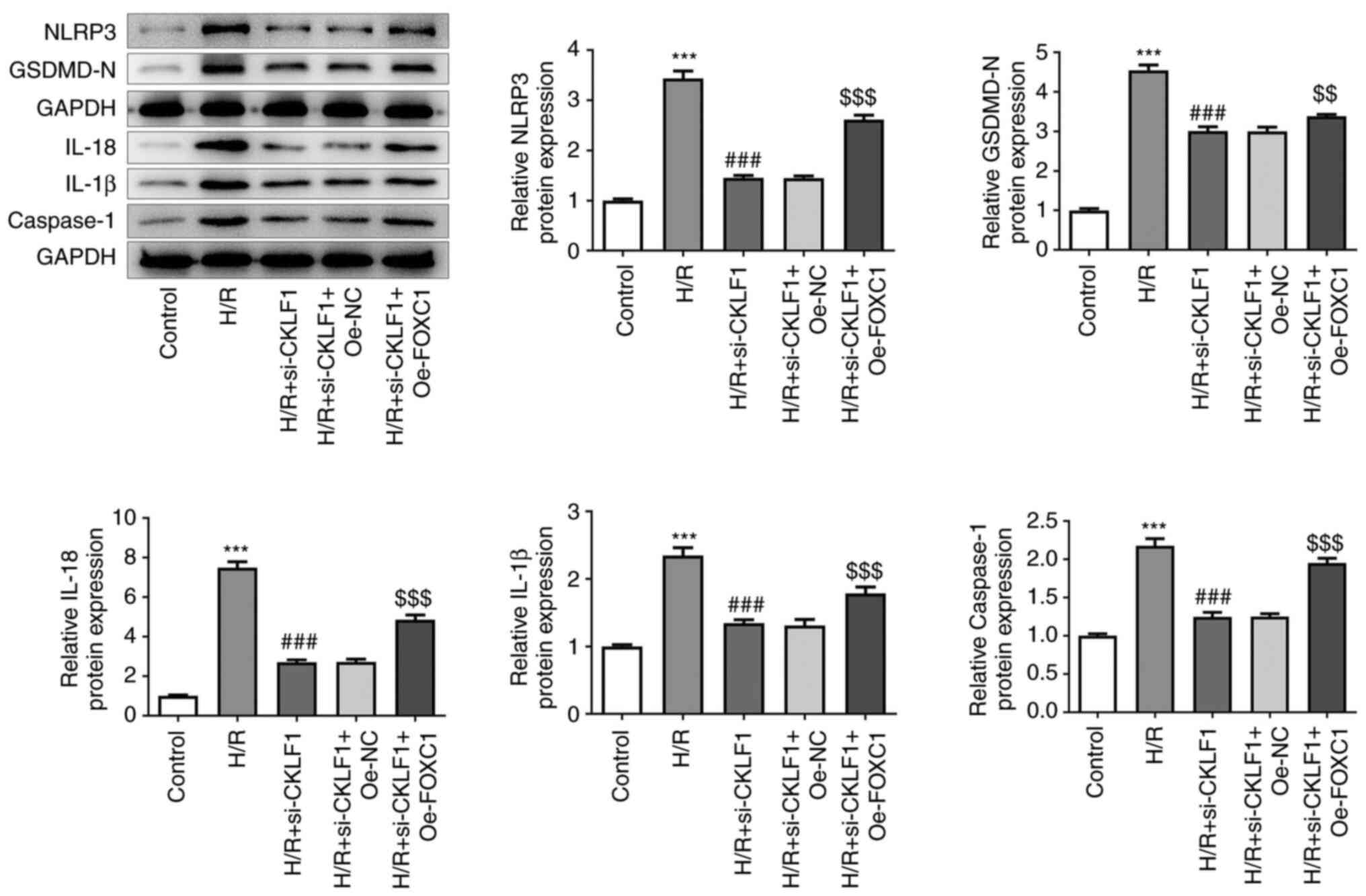 | Figure 6Oe-FOXC1 reverses the effects of
CKLF1 knockdown on H/R-induced H9c2 cell damage via NLRP3
inflammasome signaling. The expression levels of proteins
associated with NLRP3 inflammasome signaling were assessed by
western blotting post-transfection with Oe-FOXC1 and si-CKLF1 in
H9c2 cells exposed to H/R. ***P<0.001 vs. control;
###P<0.001 vs. H/R; $$P<0.01,
$$$P<0.001 vs. H/R + si-CKLF1 + Oe-NC. CKLF1,
chemokine-like factor 1; FOXC1, forkhead box protein C1; GSDMD-N,
gasdermin D N-terminal domain; H/R, hypoxia/reoxygenation; IL,
interleukin; NC, negative control; NLRP3, NOD-like receptor family,
pyrin domain containing 3; OE, overexpression; si, small
interfering. |
Discussion
Excessive apoptosis has long been considered a
promoting factor for the pathological progression of myocardial I/R
injury, and intervening in myocardial apoptosis is considered a
promising approach in the prevention and reduction of myocardial
I/R injury (20,21). Bcl-2 and Bax are two important
proteins in the Bcl-2 protein family, which serve anti-apoptotic
and pro-apoptotic roles in intrinsic apoptosis, respectively
(22). PARP has a crucial role in
DNA damage repair and loses its activity once PARP cleavage occurs,
thus accelerating the apoptotic process (23). CKLF1, a novel chemokine discovered
in 2001, is implicated in neuronal apoptosis following cerebral
I/R, and CKLF1 inhibition has been shown to reduce Bax expression
and elevate Bcl-2 expression in brain tissues (10). Furthermore, by attenuating
CKLF1-mediated inflammation and apoptosis, hydroxytyrosol has been
reported to exert protective effects on cisplatin-induced
nephrotoxicity (24). In the
present study, to the best of our knowledge, the role of CKLF1 in
H/R-induced cardiomyocyte damage was explored for the first time.
The results demonstrated that CKLF1 was highly expressed in
H/R-stimulated H9c2 cells, and CKLF1 knockdown decreased the
apoptotic rate of cells, accompanied by upregulated Bcl-2
expression, and downregulated Bax and cleaved PARP expression.
Cardiomyocytes are more susceptible to free radical
damage because they contain fewer antioxidants and antioxidant
enzymes, such as SOD and GSH-Px (25,26).
MDA is the crucial indicator of membrane lipid peroxidation.
Excessive ROS can cause extensive oxidative damage to
cardiomyocytes, leading to loss of cell viability and myocardial
stunning (27). Numerous studies
have shown that ROS production is a hallmark of I/R injury, and
myocardial injury caused by H/R is closely associated with ROS
production and oxidative stress in cardiomyocytes (28,29).
Inhibition of oxidative stress and reduction of ROS production can
effectively alleviate myocardial I/R injury and protect the
myocardium (30). In addition,
H/R-injured cardiomyocytes undergo processes, such as inflammatory
reactions, which lead to the overproduction and excessive release
of inflammatory factors, including TNF-α, IL-1β and IL-6(31). Attenuating CKLF1-mediated
inflammation, oxidative stress and apoptosis has been shown to
exert protective effects on cisplatin-induced nephrotoxicity
(24). Furthermore, CKLF1 is
highly expressed during hepatic I/R, and CKLF1 inhibition
attenuates neutrophil infiltration and reduces the inflammatory
response to improve hepatic I/R injury (12). A previous study reported that
IMM-H004 downregulates the levels of CKLF1 in adult and aged rats
to inhibit the inflammatory response, and further protects from
cerebral ischemia injury (32).
The results of the present study suggested that knockdown of CKLF1
improved H/R-triggered oxidative stress and inflammation in H9c2
cells by decreasing the contents of ROS, MDA, TNF-α, IL-1β and
IL-6, and increasing the activities of SOD and GSH-Px.
Using the JASPAR database, FOXC1 was identified as a
putative transcription factor that can bind to the CKLF1 promoter.
FOXC1 serves a significant role in the development of the heart and
cardiovascular system, and abnormal FOXC1 function has been shown
to be closely related to congenital heart disease and myocardial
ischemia (16,33). Notably, FOXC1 expression can be
induced by hypoxia and it can subsequently initiate inflammatory
responses and cell injury (16).
During myocardial I/R, FOXC1 has been reported to transcriptionally
activate ELAVL1 to induce myocardial injury (34). Notably, suppressing FOXC1
expression exerts protective effects on human osteoarthritic
synovial fibroblasts by inhibiting inflammation (35). Furthermore, endogenous ROS
production in the early differentiation state of human cells
inhibits endodermal differentiation via transient FOXC1
upregulation (36). In the present
study, CKLF1 was identified as a novel target of FOXC1 via
luciferase reporter and ChIP assays. FOXC1 directly bound to the
CKLF1 promoter region to upregulate CKLF1 transcription and
expression.
The NLRP3 inflammasome refers to a group of
multimeric protein complexes consisting of NLRP3 and caspase 1, and
is known to be associated with multiple cellular functions,
including inflammation and apoptosis (37). Caspase 1 is the promoter of the
NLRP3 inflammasome, which induces the production of mature IL-1β
and IL-18(38). Caspase 1 can also
cleave GSDMD and generate GSDMD-N oligomers within the cell
membrane, leading to disruption of the cell membrane integrity and
promoting the release of cellular contents (39). H/R-induced upregulation of ROS
production increases NLRP3 and caspase 1 expression, and promotes
the release of pro-inflammatory cytokines (IL-18 and IL-1β) in rat
cardiomyocytes (40). An
increasing number of studies has validated that suppression of the
NLRP3 inflammasome can reduce myocardial infarct size and limit the
inflammatory response following myocardial I/R in mice (41-43).
Therefore, inhibition of NLRP3 inflammasome activation may help to
mitigate myocardial I/R injury, and the NLRP3 inflammasome may thus
be an important target for the prevention and treatment of
myocardial I/R injury. Notably, IMM-H004 can downregulate the
expression of CKLF1, thereby restraining activation of the NLRP3
inflammasome and subsequent inflammatory reaction, ultimately
protecting the ischemic brain (11). Consistently, the present data
showed that H/R led to the activation of NLRP3 inflammasome
signaling, as evidenced by upregulated expression levels of NLRP3,
GSDMD-N, IL-18, IL-1β and caspase 1 in H9c2 cells. CKLF1 silencing
inhibited activation of the NLRP3 inflammasome, which was restored
by FOXC1 overexpression.
In conclusion, to the best of our knowledge, this is
the first report showing that CKLF1 expression is significantly
increased in H/R-induced cardiomyocytes and CKLF1 knockdown plays
inhibitory effects on H/R-triggered oxidative stress and
inflammation. Mechanistically, CKLF1 could be transcriptionally
activated by FOXC1 to aggravate H/R-induced damage of H9c2 cells by
regulating NLRP3 inflammasome activation. These findings may
provide new insights into the pathogenesis and treatment of
myocardial I/R injury.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JP designed the study and drafted the manuscript. YJ
analyzed the data, searched the literature and revised the
manuscript. YJ and JP performed the experiments. JP and YJ confirm
the authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Benjamin EJ, Virani SS, Callaway CW,
Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling
FN, Deo R, et al: Heart disease and stroke statistics-2018 Update:
A report from the american heart association. Circulation.
137:e67–e492. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Jennings RB: Historical perspective on the
pathology of myocardial ischemia/reperfusion injury. Circ Res.
113:428–438. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Gonzalez-Montero J, Brito R, Gajardo AI
and Rodrigo R: Myocardial reperfusion injury and oxidative stress:
Therapeutic opportunities. World J Cardiol. 10:74–86.
2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Ischemia/Reperfusion. Compr Physiol. 7:113–170.
2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Mokhtari-Zaer A, Marefati N, Atkin SL,
Butler AE and Sahebkar A: The protective role of curcumin in
myocardial ischemia-reperfusion injury. J Cell Physiol.
234:214–222. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Han W, Lou Y, Tang J, Zhang Y, Chen Y, Li
Y, Gu W, Huang J, Gui L, Tang Y, et al: Molecular cloning and
characterization of chemokine-like factor 1 (CKLF1), a novel human
cytokine with unique structure and potential chemotactic activity.
Biochem J. 357:127–135. 2001.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Tan Y, Wang Y, Li L, Xia J, Peng S and He
Y: Chemokine-like factor 1-derived C-terminal peptides induce the
proliferation of dermal microvascular endothelial cells in
psoriasis. PLoS One. 10(e0125073)2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhang T, Zhang X, Yu W, Chen J, Li Q, Jiao
Y, He P and Shen C: Effects of chemokine-like factor 1 on vascular
smooth muscle cell migration and proliferation in vascular
inflammation. Atherosclerosis. 226:49–57. 2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chen C, Chu SF, Ai QD, Zhang Z, Guan FF,
Wang SS, Dong YX, Zhu J, Jian WX and Chen NH: CKLF1 aggravates
focal cerebral ischemia injury at early stage partly by modulating
Microglia/Macrophage toward M1 polarization through CCR4. Cell Mol
Neurobiol. 39:651–669. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kong LL, Wang ZY, Hu JF, Yuan YH, Han N,
Li H and Chen NH: Inhibition of chemokine-like factor 1 protects
against focal cerebral ischemia through the promotion of energy
metabolism and anti-apoptotic effect. Neurochem Int. 76:91–98.
2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ai QD, Chen C, Chu S, Zhang Z, Luo Y, Guan
F, Lin M, Liu D, Wang S and Chen N: IMM-H004 therapy for permanent
focal ischemic cerebral injury via CKLF1/CCR4-mediated NLRP3
inflammasome activation. Transl Res. 212:36–53. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Li FF, Zhou X, Chu SF and Chen NH:
Inhibition of CKLF1 ameliorates hepatic ischemia-reperfusion injury
via MAPK pathway. Cytokine. 141(155429)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yang B, Hong T, Liu QZ, Feng XR, Gong YJ,
Bu DF, Li XM, Xue L, Zhao CY and Huo Y: Effects of in vivo transfer
of human chemokine-like factor 1 gene on cardiac function after
acute myocardial infarction in rats. Beijing Da Xue Xue Bao Yi Xue
Ban. 41:144–147. 2009.PubMed/NCBI(In Chinese).
|
|
14
|
Feng XR, Hong T, Gong YJ, Bu DF, Yuan JY,
Xue L, Zhao CY and Huo Y: In vivo transfer of human chemokine-like
factor 1 gene increases peripheral blood CD34+ stem cells after
myocardial infarction in rats. Beijing Da Xue Xue Bao Yi Xue Ban.
38:592–596. 2006.PubMed/NCBI(In Chinese).
|
|
15
|
Lin YJ, Shyu WC, Chang CW, Wang CC, Wu CP,
Lee HT, Chen LJ and Hsieh CH: Tumor hypoxia regulates forkhead box
C1 to promote lung cancer progression. Theranostics. 7:1177–1191.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang SP, Yang RH, Shang J, Gao T, Wang R,
Peng XD, Miao X, Pan L, Yuan WJ, Lin L and Hu QK: FOXC1
up-regulates the expression of toll-like receptors in myocardial
ischaemia. J Cell Mol Med. 23:7566–7580. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhao B, Li GP, Peng JJ, Ren LH, Lei LC, Ye
HM, Wang ZY and Zhao S: Schizandrin B attenuates
hypoxia/reoxygenation injury in H9c2 cells by activating the
AMPK/Nrf2 signaling pathway. Exp Ther Med. 21(220)2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Xu J, Huang J, He X, Hu M, Su S and Liu P:
Myosin 1b Participated in the Modulation of
Hypoxia/Reoxygenation-Caused H9c2 Cell Apoptosis and Autophagy.
Anal Cell Pathol (Amst). 2022(5187304)2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Del Re DP, Amgalan D, Linkermann A, Liu Q
and Kitsis RN: Fundamental mechanisms of regulated cell death and
implications for heart disease. Physiol Rev. 99:1765–1817.
2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Badalzadeh R, Mokhtari B and Yavari R:
Contribution of apoptosis in myocardial reperfusion injury and loss
of cardioprotection in diabetes mellitus. J Physiol Sci.
65:201–215. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhang Q, Dang YY, Luo X, Fu JJ, Zou ZC,
Jia XJ, Zheng GD and Li CW: Kazinol B protects H9c2 cardiomyocytes
from hypoxia/reoxygenation-induced cardiac injury by modulating the
AKT/AMPK/Nrf2 signalling pathway. Pharm Biol. 61:362–371.
2023.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhang Y, Wang Y, Ma Z, Liang Q, Tang X,
Tan H, Xiao C and Gao Y: Ginsenoside Rb1 inhibits
Doxorubicin-Triggered H9C2 cell apoptosis via aryl hydrocarbon
receptor. Biomol Ther (Seoul). 25:202–212. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chen C, Ai Q and Wei Y: Hydroxytyrosol
protects against cisplatin-induced nephrotoxicity via attenuating
CKLF1 mediated inflammation, and inhibiting oxidative stress and
apoptosis. Int Immunopharmacol. 96(107805)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chen YE, Yang H, Pang HB and Shang FQ:
Circ-CBFB exacerbates hypoxia/reoxygenation-triggered cardiomyocyte
injury via regulating miR-495-3p in a VDAC1-dependent manner. J
Biochem Mol Toxicol. 36(e23189)2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yang H, Wang C, Zhang L, Lv J and Ni H:
Rutin alleviates hypoxia/reoxygenation-induced injury in myocardial
cells by up-regulating SIRT1 expression. Chem Biol Interact.
297:44–49. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Radak Z, Zhao Z, Goto S and Koltai E:
Age-associated neurodegeneration and oxidative damage to lipids,
proteins and DNA. Mol Aspects Med. 32:305–315. 2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Cadenas S: ROS and redox signaling in
myocardial ischemia-reperfusion injury and cardioprotection. Free
Radic Biol Med. 117:76–89. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhao Q, Liu Z, Huang B, Yuan Y, Liu X,
Zhang H, Qiu F, Zhang Y, Li Y, Miao H, et al: PEDF improves cardiac
function in rats subjected to myocardial ischemia/reperfusion
injury by inhibiting ROS generation via PEDF-R. Int J Mol Med.
41:3243–3252. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhou T, Chuang CC and Zuo L: Molecular
characterization of reactive oxygen species in myocardial
Ischemia-Reperfusion Injury. Biomed Res Int.
2015(864946)2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Algoet M, Janssens S, Himmelreich U, Gsell
W, Pusovnik M, Van den Eynde J and Oosterlinck W: Myocardial
ischemia-reperfusion injury and the influence of inflammation.
Trends Cardiovasc Med. 33:357–366. 2023.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ai Q, Chen C, Chu S, Luo Y, Zhang Z, Zhang
S, Yang P, Gao Y, Zhang X and Chen N: IMM-H004 protects against
cerebral ischemia injury and cardiopulmonary complications via
CKLF1 mediated inflammation pathway in adult and aged rats. Int J
Mol Sci. 20(1661)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lambers E, Arnone B, Fatima A, Qin G,
Wasserstrom JA and Kume T: Foxc1 regulates early cardiomyogenesis
and functional properties of embryonic stem cell derived
cardiomyocytes. Stem Cells. 34:1487–1500. 2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Chen HY, Xiao ZZ, Ling X, Xu RN, Zhu P and
Zheng SY: ELAVL1 is transcriptionally activated by FOXC1 and
promotes ferroptosis in myocardial ischemia/reperfusion injury by
regulating autophagy. Mol Med. 27(14)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
He X and Deng L: miR-204-5p inhibits
inflammation of synovial fibroblasts in osteoarthritis by
suppressing FOXC1. J Orthop Sci. 27:921–928. 2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Oka S, Tsuzuki T, Hidaka M, Ohno M,
Nakatsu Y and Sekiguchi M: Endogenous ROS production in early
differentiation state suppresses endoderm differentiation via
transient FOXC1 expression. Cell Death Discov.
8(150)2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Fouad AA, Abdel-Aziz AM and Hamouda AAH:
Diacerein downregulates NLRP3/Caspase-1/IL-1beta and IL-6/STAT3
pathways of inflammation and apoptosis in a rat model of cadmium
testicular toxicity. Biol Trace Elem Res. 195:499–505.
2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Bian Y, Li X, Pang P, Hu XL, Yu ST, Liu
YN, Li X, Wang N, Wang JH, Xiao W, et al: Kanglexin, a novel
anthraquinone compound, protects against myocardial ischemic injury
in mice by suppressing NLRP3 and pyroptosis. Acta Pharmacol Sin.
41:319–326. 2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Shi H, Gao Y, Dong Z, Yang J, Gao R, Li X,
Zhang S, Ma L, Sun X, Wang Z, et al: GSDMD-mediated cardiomyocyte
pyroptosis promotes Myocardial I/R injury. Circ Res. 129:383–396.
2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Qiu Z, He Y, Ming H, Lei S, Leng Y and Xia
ZY: Lipopolysaccharide (LPS) Aggravates High Glucose- and
Hypoxia/Reoxygenation-Induced Injury through Activating
ROS-Dependent NLRP3 Inflammasome-Mediated pyroptosis in H9C2
cardiomyocytes. J Diabetes Res. 2019(8151836)2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Toldo S and Abbate A: The NLRP3
inflammasome in acute myocardial infarction. Nat Rev Cardiol.
15:203–214. 2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wang Y, Yan X, Mi S, Li Z, Wang Y, Zhu H,
Sun X, Zhao B, Zhao C, Zou Y, et al: Naoxintong attenuates
Ischaemia/reperfusion Injury through inhibiting NLRP3 inflammasome
activation. J Cell Mol Med. 21:4–12. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Toldo S, Marchetti C, Mauro AG, Chojnacki
J, Mezzaroma E, Carbone S, Zhang S, Van Tassell B, Salloum FN and
Abbate A: Inhibition of the NLRP3 inflammasome limits the
inflammatory injury following myocardial ischemia-reperfusion in
the mouse. Int J Cardiol. 209:215–220. 2016.PubMed/NCBI View Article : Google Scholar
|