Introduction
Congenital heart disease is hypothesized to be the
most prevalent type of birth anomaly in humans, occurring in ~1/100
live newborns and 10/100 early miscarriages worldwide (1,2). If
minor cardiovascular developmental abnormalities are included, such
as aortic bicuspid valve, which represents the most frequent
congenital heart deformity with a prevalence of ~1% in the general
pediatric population (3), the
total prevalence of congenital heart defects is up to 5% in live
newborns (4). As a global
pediatric concern, congenital heart defects comprise a wide
spectrum of cardiovascular developmental defects, which are
categorized into >25 distinct clinical subtypes, including
tetralogy of Fallot (TOF) (1).
Although certain minor congenital heart defects spontaneously
resolve, severe congenital heart disease may lead to poor health
and quality of life (5-8),
diminished physical exercise capacity (9-13),
impaired neurodevelopment (the most prevalent extracardiac
manifestation in patients with a congenital heart defect) and brain
damage (14-18),
thromboembolic complications (19-21),
acute renal injury and chronic kidney disease (22-24),
hepatic dysfunction (25),
pulmonary arterial hypertension (26-28),
infective endocarditis (29-31),
congestive cardiac failure (32-34),
miscellaneous cardiac dysrhythmia (35-37)
and cardiovascular demise (38-40).
Improvement has been made in cardiovascular surgery and
transcatheter interventional treatment, which has allowed >90%
of children with congenital heart defects to survive to adulthood;
adults living with various congenital heart defects outnumber
children affected by congenital heart defects (41-43).
However, despite the lifespan of these survivors being markedly
prolonged, the long-term prognostic effects are suboptimal because
of complications, including cerebrovascular infarction, chronic
renal dysfunction, hypertension, myocardial fibrosis, congestive
cardiac failure, cardiac arrhythmias and death (44,45).
Therefore, congenital heart disease has resulted in strikingly
increased morbidity, mortality and socioeconomic burden, which
underscores the need for defining the causes of congenital heart
disease (1).
In vertebrates, embryonic cardiac organogenesis
arises from complicated biological processes that involve cellular
commitment, differentiation, proliferation, apoptosis and migration
(46); both
non-inheritable/environmental predisposing factors and heritable
abnormal components may interrupt the finely controlled process,
leading to congenital heart disease (2,47-51).
Environmental precipitating factors may contribute to ~10% of
congenital heart disease cases, although their underlying
mechanisms are largely unclear (2). Non-inheritable factors predisposing
congenital heart disease encompass maternal viral infection, folate
deficiency, early-onset pre-eclampsia, obesity, diabetes mellitus,
autoimmune imbalance and maternal consumption of alcohol, tobacco
and medications as well as exposure to toxicants and air pollutants
during gestation (47,52,53).
However, ever-mounting evidence demonstrates that heritable
pathogenic determinants are the leading cause of congenital heart
disease (2,51). At present, in addition to copy
number variations (loss or gain) and aneuploidies, mutations in
>100 genes have been identified as responsible for congenital
heart disease (2,51,54-75).
Nevertheless, the definitive genetic components for congenital
heart disease are identified in only a minority of patients
(2,51,54-75),
which highlights the genetic heterogeneity of congenital heart
disease and makes it essential that new congenital heart
disease-causing mutations or genes are investigated.
Recent aggregating evidence has underscored the key
roles of some nuclear transcriptional factors in regulating proper
cardiovascular morphogenesis, including the
guanine-adenine-thymine-adenine (GATA) family of transcriptional
factors (2,51,76).
At present, six members of the GATA family have been categorized
fundamentally into a cardiac subfamily (GATA4/5/6) and a
hematopoietic subfamily (GATA1/2/3) (76). GATA4 and GATA6, as
well as GATA5, are among the first genes expressed
abundantly in the embryonic heart with a partially overlapping mode
of expression spectrum, and these three cardiogenic GATA factors
regulate cardiac organogenesis (76). In addition, germline mutations in
all three cardiogenic GATA genes (GATA4/5/6) are associated
with various forms of congenital heart disease, including TOF
(77-81),
the most prevalent type of cyanotic birth defect with an estimated
prevalence of 3/10,000 in live newborns (46). Furthermore, somatic mutations in
both GATA6 and GATA5 are causally related to TOF
(46,82), which implies that somatic mutations
in GATA4 may also play a role in TOF.
Materials and methods
Human research individuals
The present human case-control study adhered to
ethical standards outlined in the Declaration of Helsinki (2013).
The protocol was approved by The Medical Ethics Committee of Tongji
Hospital [approval no. LL(H)-09-07, Shanghai, China]. Informed
consent was signed by each individual's legal guardian prior to
recruitment. A total of 62 patients with sporadic TOF (33 male
cases and 29 female cases) who underwent cardiac surgery were
recruited from the Tongji Hospital (Shanghai, China) between March
2009 and October 2022. The age range of patients was 6-12 months,
with a mean age of 0.91 years (~11 months) at the time of surgical
treatment. TOF was diagnosed by echocardiographic images and
validated by cardiologist direct view during surgery. The inclusion
criteria for the patients included a diagnosis of sporadic TOF,
available heart tissue and peripheral blood samples as well as
clinical data, and informed consent. The exclusion criteria
included a positive familial history of congenital heart disease, a
known monogenic mutation or pathogenic copy number variation
responsible for TOF, and presence of acquired risk factors
predisposing to congenital heart disease. Cases with definite
anomalous chromosomes or syndromic cardiac deformations, such as
Marfan, Char, DiGeorge, Alagille, Noonan, Holt-Oram and Turner's
syndrome, were also excluded. Controls comprised 68 patients with
rheumatic heart disorder who underwent cardiac valve displacement
(36 male and 32 female cases) and 216 healthy subjects (115 male
and 101 female subjects). The age range and location and date range
of recruitment for the control subjects were the same as those for
the patients with TOF. In terms of echocardiograms, no control
patients presented with cardiovascular developmental deformation.
All the study subjects were unrelated and enrolled from the Chinese
population of the Han race.
Sample preparation and DNA
extraction
A section of heart tissue was routinely resected
from the right ventricular outflow tract of patients with TOF
during cardiac surgery. The right outflow tract tissue from TOF
repair was collected and cleared of blood contaminants with sterile
normal saline, then stored in a -80˚C refrigerator. The peripheral
blood samples from the patients with TOF were collected (2 ml for
each patient). The cardiac tissue from the heart valves and venous
blood specimens of cases who underwent cardiac valve displacement
because of rheumatic heart disorder, as well as venous blood
specimens of healthy subjects, were collected as control specimens.
Somatic genomic DNA was isolated from cardiac tissue samples using
the DNeasy Blood & Tissue Kit (cat. no. 69504; Qiagen, Inc.)
following the manufacturer's instructions. Purification of genomic
DNA from blood leucocytes was performed using the
Wizard® Genomic DNA Purification Kit (cat. no. A1125;
Promega Corporation) according to the manufacturer's
instructions.
Genetic investigation
The oligonucleotide primers applied to amplify
coding exons and splicing donors/acceptors of the GATA4 gene
via PCR, as well as the reaction mixtures and conditions for the
PCR, were as previously described (83). Briefly, the HotStar Taq DNA
Polymerase (cat. no. 203205; Qiagen, Inc.) was used according to
the manufacturer's instructions. The primers to amplify the whole
coding regions of GATA4 by PCR were as follows: Exon 2 (part
a) forward, 5'-GATCTTCGCGACAGTTCCTC-3' and reverse,
5'-GTCCCCGGGAAGGAGAAG-3' (amplicon size, 458 bp); exon 2 (part b)
forward, 5'-GCTGGGCCTGTCCTACCT-3' and reverse,
5'-AAAAACAAGAGGCCCTCGAC-3' (amplicon size, 554 bp); exon 3 forward,
5'-GGGCTGAAGTCAGAGTGAGG-3' and reverse, 5'-GATGCACACCCTCAAGTTCC-3'
(amplicon size, 437 bp); exon 4 forward, 5'-GAGATCTCATGCAGGGTCGT-3'
and reverse, 5'-GCCCCTTCCAAATCTAAGTC-3' (amplicon size, 390 bp);
exon 5 forward, 5'-TCTTTCTCGCTGAGTTCCAG-3' and reverse,
5'-GGGATGTCCGATGCTGTC-3' (amplicon size, 379 bp); exon 6 forward
5'-GCCATCCCTGTGAGAACTGT-3' and reverse, 5'-GAGGGTAGCTCACTGCTTGC-3'
(amplicon size, 444 bp) and exon 7 forward,
5'-AAGTGCTCCTTGGTCCCTTC-3' and reverse, 5'-TTCCCCTAACCAGATTGTCG-3'
(amplicon size, 479 bp). The PCR-amplified products were fragmented
by electrophoresis on 1.3% agarose gel and isolated with the
QIAquick Gel Extraction Kit (cat. no. 28704; Qiagen, Inc.). The
amplicons were sequenced and analyzed as previously described
(83). For each GATA4
variation detected, databases such as gnomAD (gnomad-sg.org/) and SNP (ncbi.nlm.nih.gov/SNP) were consulted to evaluate its
novelty. Additionally, once a GATA4 mutation was identified,
it would be deposited in a genetics database (https://databases.lovd.nl/shared/genes/GATA4).
Construction of expression
vectors
The expression vectors of GATA-binding protein 4
(GATA4)-pSSRa, T-box transcription factor 5 (TBX5)-pcDNA3.1 and K2
homeobox 5 (NKX2.5)-pEFSA, which express human GATA4, TBX5 and
NKX2.5, respectively, reporter vector of atrial natriuretic peptide
(ANP)-luciferase (Luc), where the ANP promoter drives the
expression of firefly luciferase, and the reporter plasmid of
myosin heavy chain 6 (MYH6)-luciferase (Luc), where the promoter of
MYH6 (expressing myosin heavy chain 6) drives the expression
of firefly luciferase, were generated as previously described
(84). Expression vectors of
GATA4-pSSRa and NKX2.5-pEFSA as well as the reporter vector ANP-Luc
were provided by Dr Ichiro Shiojima at The Department of
Cardiovascular Science and Medicine of Chiba University (Chiba,
Japan). The mutant-type GATA4-pSSRa plasmid harboring the
c.708T>G (p.Tyr236*) mutation was created via site-directed
mutagenesis using the GeneArt Site-Directed Mutagenesis System
(Invitrogen; Thermo Fisher Scientific, Inc.) and an overlapping
pair of primers containing the target mutation (forward,
5'-TGGGACGGGTCACTAGCTGTGCAACGCCTGC-3' and reverse,
5'-GCAGGCGTTGCACAGCTAGTGACCCGTCCCA-3') and was validated via
PCR-sequencing assay performed as aforementioned. The primers used
for site-directed mutagenesis are located in the cDNA of human
GATA4 (Fig. S1).
Cellular transient transfection with
vectors and reporter activity assay
COS-7 cells (an African green monkey kidney
fibroblast-like cell line) from the Cell Bank of Chinese Academy of
Sciences were maintained as previously described (84). COS-7 cells plated onto a 24-well
plate at an initial density of 1x105 cells/well were
grown in Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc.) containing 10% fetal bovine serum (Invitrogen;
Thermo Fisher Scientific, Inc.) and 1% penicillin/streptomycin
(Thermo Fisher Scientific, Inc.) at 37˚C with 5% CO2.
COS-7 cells at ~80% confluency were transiently transfected with
the aforementioned expression vectors using
Lipofectamineâ 3000 (Thermo Fisher Scientific, Inc.), as
described previously (84). As an
internal control, the vector pGL4.75 (Promega Corporation), which
expresses Renilla luciferase, was used for normalized
transfection efficiency. A total of 1.0 µg wild-type GATA4-pSSRa
was used to mimic the human physiological status, 1.0 µg
Tyr236*-mutant GATA4-pSSRa was used to mimic pathogenic status of
patients harboring the homozygous mutation and 0.5 µg wild-type
GATA4-pSSRa + Tyr236*-mutant GATA4-pSSRa was used to mimic the
pathogenic status of patients harboring the heterozygous mutation.
Additionally, 0.5 µg wild-type GATA4-pSSRa + empty pcDNA3.1 was
compared with 0.5 µg wild-type GATA4-pSSRa + Tyr236*-mutant
GATA4-pSSRa to determine whether the Tyr236*-mutant GATA4 exerted a
dominant-negative effect on the wild-type GATA4. For each
transfection, three independent replicates were performed. Cells
were collected 48 h after transfection and lysed. The lysate was
used to assess dual-luciferase activity under a microplate
luminometer (Promega Corporation) with the
Dual-Luciferase® Reporter Assay System (cat. no. E1910;
Promega Corporation) according to the manufacturer's instructions.
The activity of the MYH6 or ANP promoter was
expressed as a relative value of firefly luciferase activity
divided by Renilla luciferase activity. The results were
representative of three independent experiments in triplicate.
Statistical analysis
Analyses of categorical data (such as demographic
data, including ethnicity, sex and family history) between two
groups were performed by χ2 or Fisher's exact test. For
the quantitative parameters given as mean ± standard deviation
(such as age and the MYH6 or ANP promoter activity),
Student's unpaired t-test was applied to perform comparisons
between two groups. For comparisons between ≥3 groups, one-way
ANOVA followed by Tukey's post hoc test was applied. Statistical
analysis was performed employing SPSS version 16.0 (IBM Corp.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Clinical data of patients
The present research included 62 non-familial cases
affected with TOF who underwent cardiac surgery, 68 sporadic cases
who underwent cardiac valve displacement because of rheumatic heart
disorder and 216 healthy patients as controls. All research
subjects, who were of Han race, had no known family history of
congenital heart defect and had no identified environmental factors
contributing to congenital heart disease, such as maternal disease,
medication and exposure to ionizing radiation, chemicals and toxins
during pregnancy. There was no significant difference in the ages
(t=-0.104976, P=0.9165) between the case group of 62 patients with
TOF (with an average of 0.91±0.59) and the control group of 216
healthy individuals (with an average of 0.92±0.68). The baseline
phenotypical data of the 62 non-familial cases with sporadic TOF
are summarized in Table I.
 | Table IBaseline phenotypical data of 62
unrelated patients with sporadic TOF. |
Table I
Baseline phenotypical data of 62
unrelated patients with sporadic TOF.
| Variable | Value |
|---|
| Male, n (%) | 33 (53.23) |
| Age at time of
surgery, years | 0.91±0.59 |
| Age at time of
recruitment, years | 0.87±0.62 |
| Family history of
TOF, n (%) | 0 (0.00) |
| Form of TOF, n
(%) | |
|
Isolated | 30 (48.39) |
|
Bicuspid
pulmonary valve | 8 (12.90) |
|
Patent
ductus arteriosus | 6 (9.68) |
|
Atrial
septal defect | 5 (8.06) |
|
Persistent
left superior vena cava | 4 (6.45) |
|
Anomalous
pulmonary venous connection | 2 (3.23) |
|
Partial
common atrioventricular canal | 2 (3.23) |
|
≥2 other
cardiovascular defects | 5 (8.06) |
| Dysrhythmia, n
(%) | |
|
Atrioventricular
block | 4 (6.45) |
|
Supraventricular
tachycardia | 2 (3.23) |
|
Atrial
fibrillation | 1 (1.61) |
| Surgical repair, n
(%) | 100 (100.00) |
Discovery of a somatic GATA4 mutation
causative for TOF
Sequencing analysis of the GATA4 gene was
performed with the genomic DNA isolated from the diseased cardiac
tissue (the resected right ventricular outflow tract muscle to
release right ventricular outflow tract obstruction) of 62
non-familial patients with TOF and the heart valve tissues of 68
patients with rheumatic heart disorder, as well as the blood
leucocytes of all the 346 research participants. A heterozygous
GATA4 mutation, NM_002052.5: c.708T>G; p.(Tyr236*), was
discovered in the pathological myocardial tissue from an
11-month-old male patient with TOF. The sequencing chromatograms
illustrating the detected GATA4 mutation (G/T) as well as
its corresponding control counterpart (T/T) are exhibited in
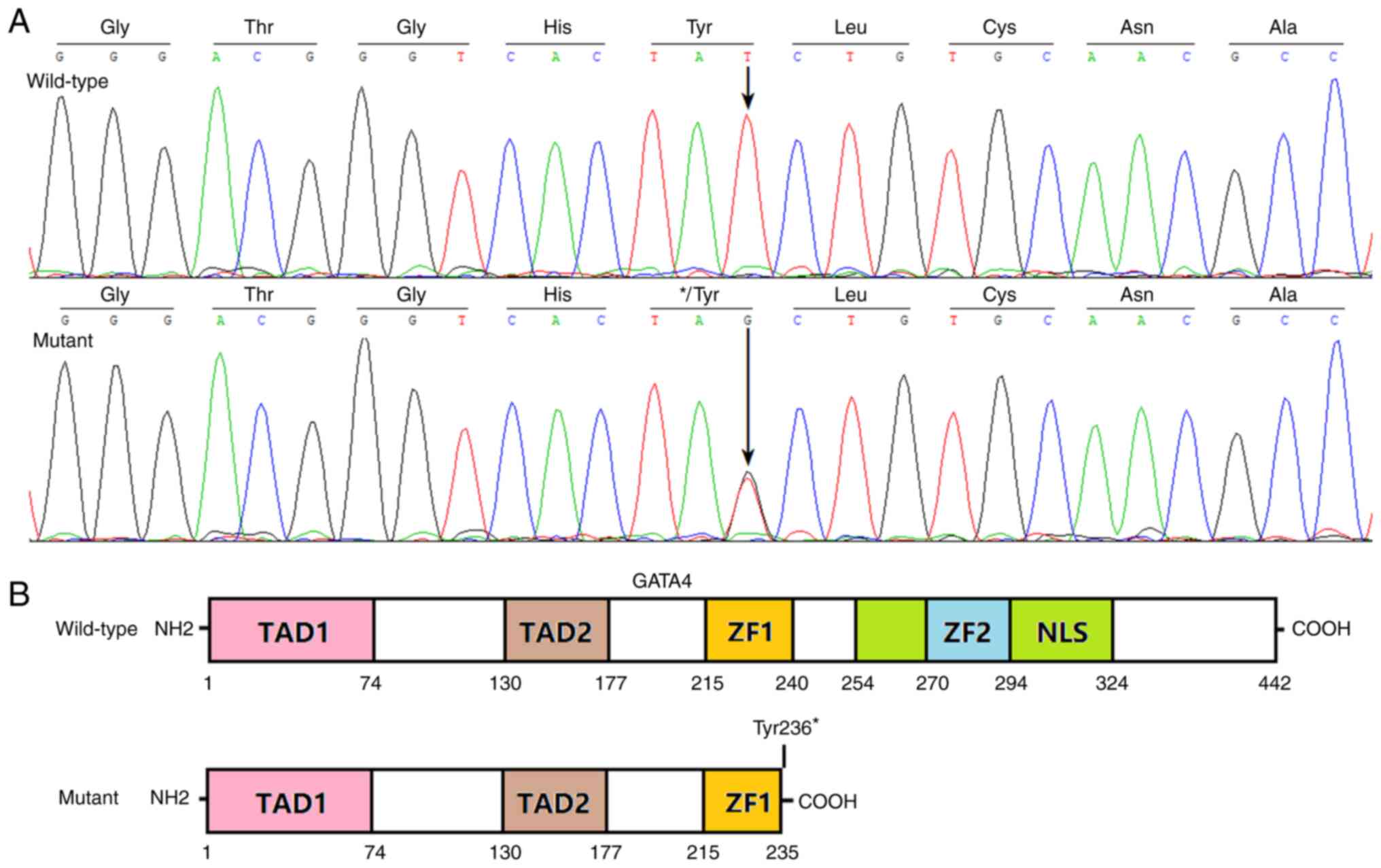
Fig. 1A. The schematic diagrams
delineating the key structural domains of wild-type GATA4 and
Tyr236*-mutant GATA4 are presented in Fig. 1B. The discovered heterozygous
GATA4 mutation was not detected in the heart valve tissue
samples from 68 cases with rheumatic heart disorder or blood cells
of all 346 patients and was not released in the SNP and gnomAD
databases (accessed August 2023).
Functional insufficiency of
Tyr236*-mutant GATA4
In the cultured COS-7 cells transiently transfected
with various expression vectors, wild-type GATA4 (GATA4) and
Tyr236*-mutant GATA4 (Tyr236*) transcriptionally activated
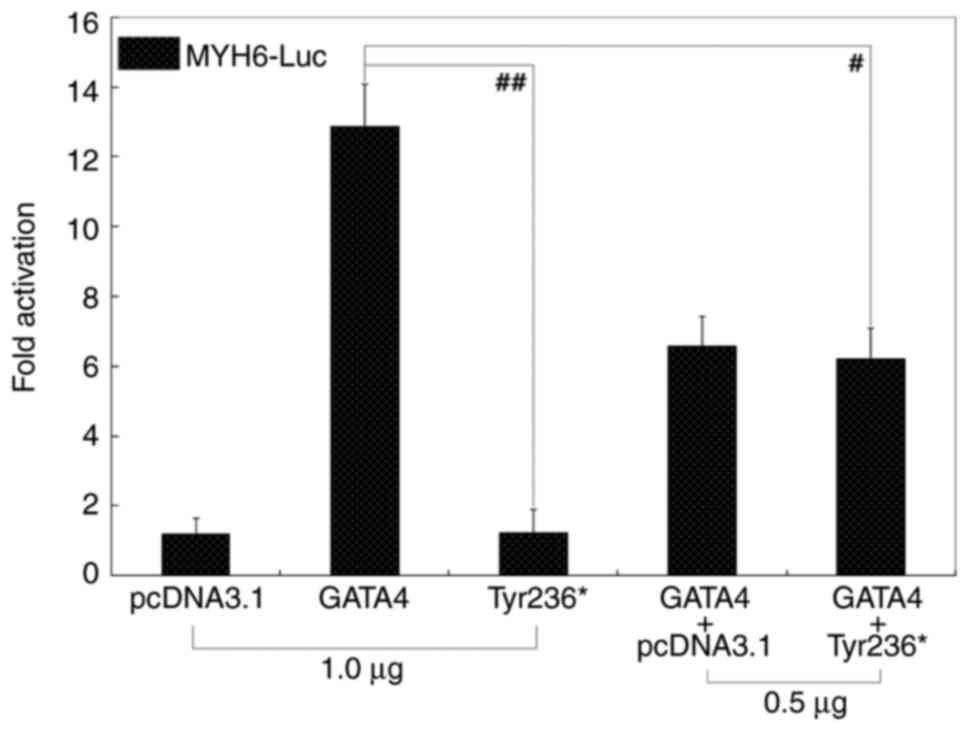
MYH6 by ~13-fold and ~1-fold, respectively (t=14.6834;
P=0.00013; Fig. 2). When Tyr236*
and GATA4 were co-expressed, transactivation on MYH6 was
~6-fold (t=7.69231; P=0.00154). Wild-type GATA4 retained its
activity in the presence of Tyr236*-mutant GATA4, indicating no
significant dominant-negative effect for this GATA4 mutation.
Similar results were obtained when the comparison of multiple
groups (among all the control and experimental groups) was
performed (P=6.555x10-8; F=94.859). Specifically,
multiple comparisons were conducted between pcDNA3.1 and GATA4
(t=11.6767, P<0.00001), pcDNA3.1 and Tyr236* (t=0.03, P=1.0),
pcDNA3.1 and pcDNA3.1 + GATA4 (t=5.3767, P=0.00013), pcDNA3.1 and
pcDNA3.1 + Tyr236* (t=5.01, P=0.00023), GATA4 and Tyr236*
(t=11.6467, P<0.00001), GATA4 and pcDNA3.1 + GATA4 (t=6.3,
P=0.00003), GATA4 and GATA4 + Tyr236* (t=6.6667, P=0.00002),
Tyr236* and GATA4 + pcDNA3.1 (t=5.3467, P=0.00013), Tyr236* and
GATA4 + Tyr236* (t=4.98, P=0.00024) and GATA4 + pcDNA3.1 and GATA4
+ Tyr236* (t=0.3667, P=0.98237).
Synergistic transactivation
dysfunction of Tyr236*-mutant GATA4 with NKX2.5 or TBX5
Cultivated COS-7 cells transiently transfected with
multiple expression vectors, GATA4 and Tyr236* transcriptionally
activated ANP by ~7-fold and ~2-fold, respectively
(t=9.7248, P=0.00063; Fig. 3). In the presence of NKX2.5, GATA4
and Tyr236* transactivated ANP by ~32- and ~11-fold,
respectively (t=13.4306, P=0.00018); while in the
presence of TBX5, GATA4 and Tyr236* transactivated ANP by
~38- and ~15-fold, respectively (t=12.4266,
P=0.00024). Additionally, similar results were obtained when
the comparisons of multiple groups were conducted
[P=2.249x10-11 (F=220.56) for the synergy of GATA4 with
NKX2.5 and P=2.852x10-11 (F=211.89) for the synergy of
GATA4 with TBX5]. Specifically, multiple comparisons were conducted
between pcDNA3.1 (-) and GATA4 (t=5.92, P=0.00123), pcDNA3.1 and
Tyr236* (t=0.04, P=1.0), pcDNA3.1 and NKX2.5 (t=9.92, P=0.00001),
pcDNA3.1 and GATA4 + NKX2.5 (t=30.02, P<0.00001), pcDNA3.1 and
Tyr236* + NKX2.5 (t=9.5533, P=0.00001), GATA4 and Tyr236* (t=5.88,
P=0.00130), GATA4 and NKX2.5 (t=4.0, P=0.02408), GATA4 and GATA4 +
NKX2.5 (t=24.1, P<0.00001), GATA4 and Tyr236* + NKX2.5
(t=3.6333, P=0.04329), Tyr236* and NKX2.5 (t=9.88, P=0.00001),
Tyr236* and GATA4 + NKX2.5 (t=29.98, P<0.00001), Tyr236* and
Tyr236* + NKX2.5 (t=9.5133, P=0.00001), NKX2.5 and GATA4 + NKX2.5
(t=20.1, P<0.00001), NKX2.5 and Tyr236* + NKX2.5 (t=0.3667,
P=0.99914), GATA4 + NKX2.5 and Tyr236* + NKX2.5 (t=20.4667,
P<0.00001); pcDNA3.1 and TBX5 (t=14.7533, P<0.00001),
pcDNA3.1 and GATA4 + TBX5 (t=36.72, P<0.00001), pcDNA3.1 and
Tyr236* + TBX5 (t=13.52, P<0.00001), GATA4 and TBX5 (t=8.8333,
P=0.00028), GATA4 and GATA4 + TBX5 (t=30.8, P<0.00001), GATA4
and Tyr236* + TBX5 (t=7.6, P=0.00109), Tyr236* and TBX5 (t=14.7133,
P<0.00001), Tyr236* and GATA4 + TBX5 (t=36.68, P<0.00001),
Tyr236* and Tyr236* + TBX5 (t=13.48, P<0.00001), TBX5 and GATA4
+ TBX5 (t=21.9667, P<0.00001), TBX5 and Tyr236* + TBX5
(t=1.2333, P=0.93282) and GATA4 + TBX5 and Tyr236* + TBX5 (t=23.2,
P<0.00001).
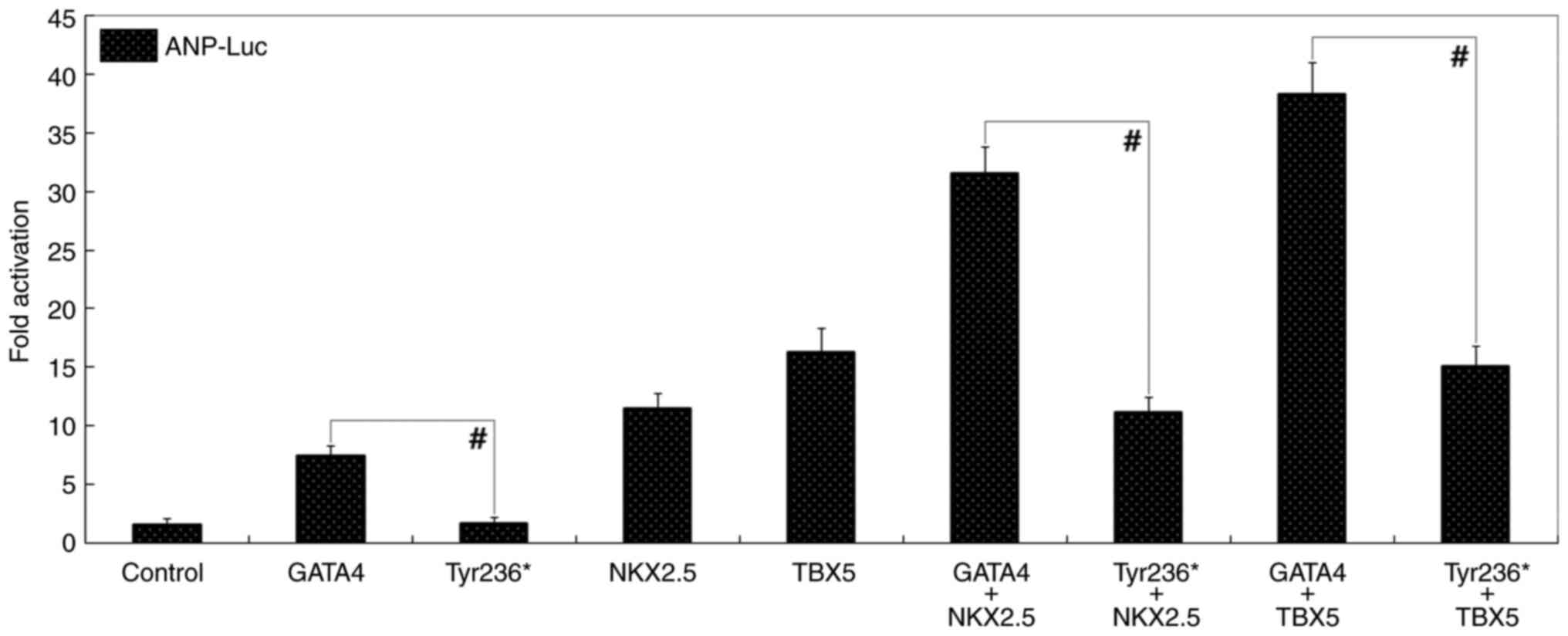 | Figure 3Lost synergistic transactivation
between Tyr236*-mutant GATA4 and NKX2.5 or TBX5. In cultured COS-7
cells overexpressing various interest proteins (Tyr236*-mutant
GATA4, wild-type GATA4, NKX2.5, TBX5, firefly luciferase and
Renilla luciferase), dual-luciferase activity measurement of
the synergistic activation of ANP by GATA4 in combination
with NKX2.5 or TBX5 showed that synergy was disrupted by the
Tyr236* mutation. #P<0.001. Luc, luciferase; GATA4,
GATA-binding protein 4; NKX2.5, NK2 homeobox 5; ANP, atrial
natriuretic peptide; TBX5, T-box transcription factor 5. |
Discussion
In the present study, through sequencing analysis a
new GATA4 mutation in a heterozygous status,
NM_002052.5:c.708T>G;p.(Tyr236*), was found in diseased heart
tissue derived from one male patient out of 62 non-familial
patients with sporadic TOF. The mutant allele was not detected in
the diseased heart tissues of 68 cases with rheumatic heart
disorder or in the blood cells of all the 346 research subjects,
encompassing 216 healthy participants matched for ethnicity and
sex, suggesting the identified mutation was somatic in origin. This
mutation in GATA4 was absent from gnomAD and SNP databases.
Quantitative reporter gene measurements unveiled that
Tyr236*-mutant GATA4 was unable to trans-activate the key target
genes of MYH6 and ANP, singly or in synergy with
NKX2.5 or TBX5, two other TOF-causative genes (85-88).
ANP and MYH6 are well-characterized downstream target
genes of GATA4 and GATA4 loss-of-function mutations decrease the
transcription of ANP or MYH6 (89-91).
Additionally, GATA4, alone or in synergy with transcriptionally
cooperative partners such as NKX2.5 and TBX5, has been shown to
activate transcription of target genes such as ANP and
MYH6, highlighting the important role of physical and
functional interactions between GATA4 and NKX2.5 as well as TBX5 in
proper heart development (89,92,93).
Furthermore, multiple germline deleterious mutations in
GATA4 cause cardiac developmental deformations, including
bicuspid aortic valve, atrial septal defect, double-outlet right
ventricle, Ebstein's anomaly, ventricular septal defect and TOF
(77,78,94).
The present results strongly support that somatic GATA4
mutation is responsible for the molecular pathogenesis underpinning
TOF in the mutation carrier, although the mechanism by which the
somatic GATA4 mutation causes TOF remains to be
elucidated.
Although progress has been made in the discovery of
germline mutations contributing to occurrence of congenital heart
defects (2,51,54-75)
and the significant effects of somatic mutations on genesis and
progression of cancer and aging are well defined (95-97),
the roles of somatic mutations in the development of congenital
heart disease are unclear. Furthermore, depending on the type of
disease and class of mutation (insertion/deletion, single
nucleotide substitution, copy number variation, chromosomal
aberration and transposon-mediated mutation), somatic mutations may
be causative in 6-20% of patients and the frequency of gene
mutation in embryonic cells is not significantly different from
that in germline cells (98).
Given the intensive oxidative metabolism of cardiomyocytes,
increased oxidative DNA damage and/or decreased base excision
repair as well as defective mismatch repair of damaged DNA may lead
to somatic mutations in cardiomyocytes, and emerging evidence
indicates that non-inherited/acquired mutations involving somatic
cells are key in cardiovascular disorder (99,100). In agreement with this evidence,
the present sequencing analysis of GATA4 on genomic DNA from
resected cardiac tissue along with peripheral blood leucocytes of a
patient with TOF identified a somatic mutation responsible for TOF,
suggesting that TOF could be partially due to cardiac somatic
mutations and somatic mosaicism may be an alternative molecular
mechanism of TOF.
The prevalence of somatic GATA4 variations in
patients suffering from congenital heart disease undergoing cardiac
surgery has been examined. Salazar et al (101) analyzed the GATA4 gene in
fresh-frozen pathological heart tissues as well as corresponding
non-diseased tissue obtained from 62 patients with sporadic
congenital heart disease (35 cases with cardiac septal defects and
27 cases presented with other heart deformities), and detected six
rare variants as well as two frequent polymorphisms in GATA4
in both the cardiac and the corresponding normal tissues,
indicating that they were constitutional variations rather than
somatically derived mutations. Wang et al (102) performed a sequencing assay of
GATA4 derived from muscle tissue of the right ventricular
outflow tract as well as peripheral venous blood leucocytes of 38
patients with isolated TOF undergoing routine cardiac surgery and
identified a previously reported GATA4 mutation
(p.Pro407Gln) in an affected child, both in the diseased heart
tissue and in blood lymphocytes, implying that a germline
GATA4 mutation contributes to non-syndromic TOF. Cheng et
al (103) sequenced
GATA4 on DNA samples obtained from cardiac tissue and
peripheral blood leucocytes of 20 patients undergoing surgery for
ventricular septal defects; seven novel variations in a
heterozygous status were observed in the heart tissues but none in
the blood leucocytes of patients or in the control samples of 500
healthy individuals, indicating that they are of somatic origin.
Esposito et al (104)
utilized freshly frozen cardiac tissue samples of right ventricular
myocardium and matched blood samples from nine cases undergoing
surgical treatment for TOF and 24 patients with left heart
hypoplasia to evaluate the incidence of somatic GATA4
mutations in heart tissue by direct sequencing analysis; no somatic
or germline mutations were identified. Yin et al (105) performed direct PCR-sequencing
analysis of GATA4 on genomic DNA purified from heart tissue
and peripheral blood cells of 98 cases with sporadic congenital
heart disease and found two well-known SNPs (rs3729856 and
rs56166237) in GATA4 in both heart tissue and blood samples,
indicating a role of germline GATA4 variations in
development of congenital heart disease. Given these conflicting
reports on the contribution of somatic mutations to congenital
heart disease, the finding of a somatic mutation of GATA4 in
a case of TOF is rare and may depend on various factors such as
analytical methods, ethnicity and environmental factors. More
in-depth investigations with larger samples sizes from individuals
of different ethnicities are required to determine the genetic
contribution of somatic mosaicism to pathogenesis of congenital
heart defects.
A number of germline GATA4 mutations have
been causally implicated in distinct forms of congenital heart
disease, including TOF. Nemer et al (94) screened exon 2 of GATA4 in 26
patients with TOF and 94 cases with other types of congenital heart
defect and identified a novel heterozygous GATA4 mutation,
namely NM_002052.5: c.648C>G; p.(Asp216Glu), in two of 26
patients with TOF. Asp216Glu-mutant GATA4 decreases transactivation
of a downstream target gene, ANP, although this mutation has
no effect on the binding affinity of GATA4 to its target gene
promoter DNA or the physical and functional interaction of GATA4
with zinc finger protein FOG family member 2. Yang et al
(77) sequenced GATA4 in 52
probands with TOF with a positive family history and found three
novel heterozygous mutations, namely p.Ala9Pro, p.Leu51Val and
p.Asn285Ser, in three TOF families. Functional analysis indicated
that all three GATA4 mutants had markedly reduced DNA-binding
ability and significantly diminished transcriptional activity.
Moreover, Asn285Ser mutation prevented the functional interplay of
GATA4 with TBX5. Additionally, Dixit et al (78) screened GATA4 in 285 probands
with congenital heart defects and detected nine heterozygous
mutations (p.Pro407Gln, p.Trp228Arg, p.Ala8Asp, p.Ala75Ser,
p.Glu128Val, p.Thr355Ser, p.Ser358Thr, p.Ser133Cys and p.Ala9Thr)
in 22 unrelated patients with congenital heart disease. Notably,
GATA4 mutants were more commonly involved in TOF (45%) and
pulmonary stenosis (22.7%) regardless of the profusion of cardiac
septal defects in the research cohort. Biochemical measurements
showed that three of the nine GATA4 mutants, p.Trp228Arg,
p.Ser133Cys and p.Glu128Val, had impaired combinatorial synergy
with TBX5, NKX2.5 or serum response factor (SRF) and diminished
DNA-binding affinity. Here, no germline GATA4 mutations were
found except for one somatic GATA4 mutation, highlighting a
somatic mosaic basis of TOF in a minority of patients.
In humans, GATA4 is located at chromosome
8p23.1 and comprises seven exons, coding for a protein with 442
amino acids (77). GATA4, one of
the earliest genetic markers expressed in the developing heart, is
amply expressed in the embryonic heart; GATA4 transactivates
expression of multiple target genes in the cardiovascular system
during embryonic development, including genes that encode MYH6,
ANP, β myosin heavy chain, brain natriuretic factor, vascular
endothelial growth factor, cardiac troponin I and cardiac troponin
C, alone or synergistically with cofactors such as TBX5, NKX2.5,
GATA6, heart and neural crest derivatives expressed 2 and SRF,
which indicates the key role of GATA4 in embryogenic cardiac
organogenesis (77,106,107). In chick embryos, knockdown of
Gata4 by small interfering RNAs targeting Gata4 in
the cardiac mesodermal cells inhibits ability of bilateral cardiac
rudiments to migrate to the midline, resulting in development of
two isolated hearts at lateral locations, a deformity of cardia
bifida, due to the downregulated expression of N-cadherin (108). In mice, knockout of Gata4
causes embryonic lethality due to anomalous morphogenesis of the
heart tube, including TOF, endocardial cushion defect, cardiac
septal defect, right ventricular hypoplasia, double-outlet right
ventricle and cardiomyopathy (109-111).
In a transgenic murine model overexpressing Val217Gly-mutant GATA4,
embryonic death occurs, manifesting similar cardiovascular
developmental defects with those observed in humans carrying
GATA4 mutations (112). In
a knock-in mouse model expressing Gly295Ser-mutant GATA4,
homozygous mice manifested a single ventricular chamber, thin
ventricular myocardium and embryonic lethality while heterozygous
mice are viable, with minor structural aberrations of the atrial
septum and semilunar valve stenosis (113). Moreover, Gata4 is required
for normal cardiovascular morphogenesis in the xenopus, fly and
fish (114). Collectively, these
observations from experimental animals highlight the sensitivity of
the heart to GATA4 mutants during cardiac organogenesis,
suggesting that GATA4 exerts a pivotal role in the
developing heart and functionally defective GATA4
predisposes humans to numerous types of congenital heart disease,
including TOF.
Notably, in addition to a range of congenital heart
defects, germline GATA4 mutations cause dilated
cardiomyopathy and atrial fibrillation in humans (115,116). As indicated by the present
research findings and others (46,82),
a higher rate of gene mutations in heart tissue and peripheral
blood samples suggests a genetic contribution to dilated
cardiomyopathy and atrial fibrillation. Sequencing analysis of
GATA4 from resected cardiac tissue of patients with dilated
cardiomyopathy and atrial fibrillation may reveal cardiac somatic
mutations contributing to dilated cardiomyopathy and atrial
fibrillation.
There are some limitations to this investigation.
Firstly, the sample size of the study is relatively small, and
larger sample sizes may lead to the discovery of more pathogenic
mutations. Secondly, in this study, a pathogenic GATA4
mutation was identified through candidate gene analysis, hence it
cannot be ruled out that other genetic defects may also play a
pathogenic role. Whole exome or genome sequencing analysis can help
address this problem. Thirdly, the subcellular localization and
distribution of the mutated GATA4 protein, as well as the changes
in its ability to bind target gene promoters, remain to be
clarified. Finally, the pathogenicity of the GATA4 mutation
is still to be further explored at the level of genetically
modified animal models.
In conclusion, the present study identified a
somatic GATA4 loss-of-function mutation predisposing TOF,
which indicated that somatic mosaicism plays a prominent role in
the molecular pathogenesis of TOF in a minority of cases.
Supplementary Material
Locations of the primers used for
site-directed mutagenesis in the cDNA of human GATA4. The cDNA
sequences of the human GATA4 gene derived from the
Nucleotide database with an accession number of NM_002052.5
(https://www.ncbi.nlm.nih.gov/nuccore/NM_002052.5).
The red color marks the coding region and the green color marks the
wild-type forward primer. A rectangle marks the nucleotide where
the mutation occurs.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by The Basic Research
Project of Shanghai, China (grant no. 20JC1418800) and The Natural
Science Foundation of Shanghai, China (grant no. 18ZR1431000).
Availability of data and materials
All data generated or analysed during this study are
included in this published article. The GATA4 mutation,
NM_002052.5: c.708T>G; p.(Tyr236*), was deposited in a genetics
database (https://databases.lovd.nl/shared/genes/GATA4), having
an individual ID of 00436129 (phenotype ID: 0000326313; screening
ID: 0000437610; variant ID: 0000932923).
Authors' contributions
JW and YQY conceived the study and wrote the
manuscript. PA, YJL, RTH, XYL, JNG, CXY, YJX, JW and YQY performed
clinical research including collection and analysis of clinical
data. PA, YJL, JNG, CXY, JW and YQY performed genetic and
biochemical experiments. All authors have read and approved the
final manuscript. JW and YQY confirm the authenticity of all the
raw data.
Ethics approval and consent to
participate
The present study was approved by The Medical Ethics
Committee of Tongji Hospital [approval no. LL(H)-09-07; Shanghai,
China]. Informed consent was signed by the legal guardians of all
patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tsao CW, Aday AW, Almarzooq ZI, Anderson
CAM, Arora P, Avery CL, Baker-Smith CM, Beaton AZ, Boehme AK,
Buxton AE, et al: Heart disease and stroke statistics-2023 update:
A report from the American Heart Association. Circulation.
147:e93–e621. 2023.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Diab NS, Barish S, Dong W, Zhao S,
Allington G, Yu X, Kahle KT, Brueckner M and Jin SC: Molecular
genetics and complex inheritance of congenital heart disease. Genes
(Basel). 12(1020)2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Spaziani G, Girolami F, Arcieri L, Calabri
GB, Porcedda G, Di Filippo C, Surace FC, Pozzi M and Favilli S:
Bicuspid aortic valve in children and adolescents: A comprehensive
review. Diagnostics (Basel). 12(1751)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Martin LJ and Benson DW: Focused
strategies for defining the genetic architecture of congenital
heart defects. Genes (Basel). 12(827)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Brudy L, Meyer M, Oberhoffer R, Ewert P
and Müller J: Move more-be happier? Physical activity and
health-related quality of life in children with congenital heart
disease. Am Heart J. 241:68–73. 2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Moons P, Luyckx K, Thomet C, Budts W,
Enomoto J, Sluman MA, Lu CW, Jackson JL, Khairy P, Cook SC, et al:
Physical functioning, mental health, and quality of life in
different congenital heart defects: Comparative analysis in 3538
patients from 15 countries. Can J Cardiol. 37:215–223.
2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Freiberger A, Busse A, Ewert P,
Huntgeburth M, Kaemmerer H, Kohls N, Nagdyman N, Richter C, Röhrich
C, von Scheidt F, et al: Quality of life in adults with congenital
heart disease with and without pulmonary hypertension: A
comparative study. Cardiovasc Diagn Ther. 12:758–766.
2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ly R, Karsenty C, Amedro P, Cohen S,
Domanski O, Godart F, Radojevic J, Vaksmann G, Naccache N, Boubrit
A, et al: Health-Related quality of life and its association with
outcomes in adults with congenital heart disease and heart failure:
Insight From FRESH-ACHD Registry. J Am Heart Assoc.
12(e027819)2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Meyer M, Brudy L, Fuertes-Moure A, Hager
A, Oberhoffer-Fritz R, Ewert P and Müller J: E-Health exercise
intervention for pediatric patients with congenital heart disease:
A randomized controlled trial. J Pediatr. 233:163–168.
2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fritz C, Hock J, Oberhoffer R, Hager A,
Ewert P and Müller J: reduced parasympathetic activity in patients
with different types of congenital heart disease and associations
to exercise capacity. J Cardiopulm Rehabil Prev. 41:35–39.
2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sheng SP, Feinberg JL, Bostrom JA, Tang Y,
Sweeney G, Pierre A, Katz ES, Whiteson JH, Haas F, Dodson JA and
Halpern DG: Adherence and exercise capacity improvements of
patients with adult congenital heart disease participating in
cardiac rehabilitation. J Am Heart Assoc.
11(e023896)2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Masood IR, Detterich J, Cerrone D,
Lewinter K, Shah P, Kato R and Sabati A: Reduced forced vital
capacity and the number of chest wall surgeries are associated with
decreased exercise capacity in children with congenital heart
disease. Pediatr Cardiol. 43:54–61. 2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Willinger L, Hock J, Hager A,
Oberhoffer-Fritz R, Ewert P and Müller J: Heart-Focused anxiety is
prevalent in adults with congenital heart disease and associated
with reduced exercise capacity. J Cardiopulm Rehabil Prev.
43:277–281. 2023.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sadhwani A, Wypij D, Rofeberg V, Gholipour
A, Mittleman M, Rohde J, Velasco-Annis C, Calderon J, Friedman KG,
Tworetzky W, et al: Fetal brain volume predicts neurodevelopment in
congenital heart disease. Circulation. 145:1108–1119.
2022.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Parekh SA, Cox SM, Barkovich AJ, Chau V,
Steurer MA, Xu D, Miller SP, McQuillen PS and Peyvandi S: The
effect of size and asymmetry at birth on brain injury and
neurodevelopmental outcomes in congenital heart disease. Pediatr
Cardiol. 43:868–877. 2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Peyvandi S and Rollins C: Fetal brain
development in congenital heart disease. Can J Cardiol. 39:115–122.
2023.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Brossard-Racine M and Panigrahy A:
Structural brain alterations and their associations with function
in children, adolescents, and young adults with congenital heart
disease. Can J Cardiol. 39:123–132. 2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Cromb D, Bonthrone AF, Maggioni A, Cawley
P, Dimitrova R, Kelly CJ, Cordero-Grande L, Carney O, Egloff A,
Hughes E, et al: Individual assessment of perioperative brain
growth trajectories in infants with congenital heart disease:
Correlation with clinical and surgical risk factors. J Am Heart
Assoc. 12(e028565)2023.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Karsenty C, Waldmann V, Mulder B, Hascoet
S and Ladouceur M: Thromboembolic complications in adult congenital
heart disease: the knowns and the unknowns. Clin Res Cardiol.
10:1380–1391. 2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Giang KW, Fedchenko M, Dellborg M,
Eriksson P and Mandalenakis Z: Burden of ischemic stroke in
patients with congenital heart disease: A nationwide, case-control
study. J Am Heart Assoc. 10(e020939)2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yeh HR, Kim EH, Yu JJ, Yun TJ, Ko TS and
Yum MS: Arterial ischemic stroke in children with congenital heart
diseases. Pediatr Int. 64(e15200)2022.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kourelis G, Kanakis M, Samanidis G,
Tzannis K, Bobos D, Kousi T, Apostolopoulou S, Kakava F,
Kyriakoulis K, Bounta S, et al: Acute kidney injury predictors and
outcomes after cardiac surgery in children with congenital heart
disease: An observational cohort study. Diagnostics (Basel).
12(2397)2022.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Xie Y, Jiang W, Cao J and Xie H:
Dexmedetomidine attenuates acute kidney injury in children
undergoing congenital heart surgery with cardiopulmonary bypass by
inhibiting the TLR3/NF-κB signaling pathway. Am J Transl Res.
13:2763–2773. 2021.PubMed/NCBI
|
|
24
|
Gillesén M, Fedchenko M, Giang KW,
Dimopoulos K, Eriksson P, Dellborg M and Mandalenakis Z: Chronic
kidney disease in patients with congenital heart disease: A
nationwide, register-based cohort study. Eur Heart J Open.
2(oeac055)2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Reiter FP, Hadjamu NJ, Nagdyman N,
Zachoval R, Mayerle J, De Toni EN, Kaemmerer H and Denk G:
Congenital heart disease-associated liver disease: A narrative
review. Cardiovasc Diagn Ther. 11:577–590. 2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Rosenzweig EB and Krishnan U: Congenital
heart disease-associated pulmonary hypertension. Clin Chest Med.
42:9–18. 2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chiu SN, Lu CW, Lin MT, Chen CA, Wu MH and
Wang JK: Pulmonary hypertension in adult congenital heart disease
in Asia: A distinctive feature of complex congenital heart disease.
J Am Heart Assoc. 11(e022596)2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Lindberg L: Long-Term follow-up of
pediatric patients with severe postoperative pulmonary hypertension
after correction of congenital heart defects. Pediatr Cardiol.
43:827–836. 2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Snygg-Martin U, Giang KW, Dellborg M,
Robertson J and Mandalenakis Z: Cumulative incidence of infective
endocarditis in patients with congenital heart disease: A
nationwide, case-control study over nine decades. Clin Infect Dis.
73:1469–1475. 2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
van Melle JP, Roos-Hesselink JW, Bansal M,
Kamp O, Meshaal M, Pudich J, Luksic VR, Rodriguez-Alvarez R,
Sadeghpour A, Hanzevacki JS, et al: Infective endocarditis in adult
patients with congenital heart disease. Int J Cardiol. 370:178–185.
2023.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Havers-Borgersen E, Butt JH, Østergaard L,
Petersen JK, Torp-Pedersen C, Køber L and Fosbøl EL: Long-term
incidence of infective endocarditis among patients with congenital
heart disease. Am Heart J. 259:9–20. 2023.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Arnaert S, De Meester P, Troost E, Droogne
W, Van Aelst L, Van Cleemput J, Voros G, Gewillig M, Cools B, Moons
P, et al: Heart failure related to adult congenital heart disease:
prevalence, outcome and risk factors. ESC Heart Fail. 8:2940–2950.
2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Egbe AC, Miranda WR, Jain CC, Bonnichsen
CR, Anderson JH, Dearani JA, Warnes CA, Crestanello J and Connolly
HM: Incidence and outcomes of advanced heart failure in adults with
congenital heart disease. Circ Heart Fail.
15(e009675)2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lu CW, Wang JK, Yang HL, Kovacs AH, Luyckx
K, Ruperti-Repilado FJ, Van De Bruaene A, Enomoto J, Sluman MA,
Jackson JL, et al: Heart failure and patient-reported outcomes in
adults with congenital heart disease from 15 countries. J Am Heart
Assoc. 11(e024993)2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Fischer AJ, Enders D, Wasmer K, Marschall
U, Baumgartner H and Diller GP: Impact of specialized
electrophysiological care on the outcome of catheter ablation for
supraventricular tachycardias in adults with congenital heart
disease: Independent risk factors and gender aspects. Heart Rhythm.
18:1852–1859. 2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Casteigt B, Samuel M, Laplante L, Shohoudi
A, Apers S, Kovacs AH, Luyckx K, Thomet C, Budts W, Enomoto J, et
al: Atrial arrhythmias and patient-reported outcomes in adults with
congenital heart disease: An international study. Heart Rhythm.
18:793–800. 2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wasmer K, Eckardt L, Baumgartner H and
Köbe J: Therapy of supraventricular and ventricular arrhythmias in
adults with congenital heart disease-narrative review. Cardiovasc
Diagn Ther. 11:550–562. 2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Vehmeijer JT, Koyak Z, Leerink JM,
Zwinderman AH, Harris L, Peinado R, Oechslin EN, Robbers-Visser D,
Groenink M, Boekholdt SM, et al: Identification of patients at risk
of sudden cardiac death in congenital heart disease: The
PRospEctiVE study on implaNTable cardIOverter defibrillator therapy
and suddeN cardiac death in Adults with Congenital Heart Disease
(PREVENTION-ACHD). Heart Rhythm. 18:785–792. 2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Diller GP, Orwat S, Lammers AE, Radke RM,
De-Torres-Alba F, Schmidt R, Marschall U, Bauer UM, Enders D,
Bronstein L, et al: Lack of specialist care is associated with
increased morbidity and mortality in adult congenital heart
disease: A population-based study. Eur Heart J. 42:4241–4248.
2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Williams JL, Torok RD, D'Ottavio A, Spears
T, Chiswell K, Forestieri NE, Sang CJ, Paolillo JA, Walsh MJ,
Hoffman TM, et al: Causes of death in infants and children with
congenital heart disease. Pediatr Cardiol. 42:1308–1315.
2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Triedman JK and Newburger JW: Trends in
congenital heart disease: The next decade. Circulation.
133:2716–2733. 2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Bouma BJ and Mulder BJ: Changing landscape
of congenital heart disease. Circ Res. 120:908–922. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Rao PS and Agarwal A: Advances in the
diagnosis and management of congenital heart disease in children.
Children (Basel). 9(1056)2022.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Williams RG: Late causes of death after
congenital heart defects: A population-based study from finland. J
Am Coll Cardiol. 68:499–501. 2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Niwa K, Kaemmerer H and von Kodolitsch Y:
Current diagnosis and management of late complications in adult
congenital heart disease. Cardiovasc Diagn Ther. 11:478–480.
2021.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Huang RT, Xue S, Xu YJ, Zhou M and Yang
YQ: Somatic GATA5 mutations in sporadic tetralogy of Fallot. Int J
Mol Med. 33:1227–1235. 2014.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Boyd R, McMullen H, Beqaj H and Kalfa D:
Environmental exposures and congenital heart disease. Pediatrics.
149(e2021052151)2022.PubMed/NCBI View Article : Google Scholar
|
|
48
|
García-Flores E, Rodríguez-Pérez JM,
Borgonio-Cuadra VM, Vargas-Alarcón G, Calderón-Colmenero J,
Sandoval JP, García-Montes JA, Espinoza-Gutiérrez VM, Reyes-García
JG, Cazarín-Santos BG, et al: DNA Methylation Levels of the TBX5
gene promoter are associated with congenital septal defects in
mexican paediatric patients. Biology (Basel). 11(96)2022.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Zhou J, Xiong Y, Dong X, Wang H, Qian Y,
Ma D and Li X: Genome-wide methylation analysis reveals
differentially methylated CpG sites and altered expression of heart
development-associated genes in fetuses with cardiac defects. Exp
Ther Med. 22(1032)2021.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Hu C, Huang S, Wu F and Ding H:
MicroRNA-219-5p participates in cyanotic congenital heart disease
progression by regulating cardiomyocyte apoptosis. Exp Ther Med.
21(36)2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Choudhury TZ and Garg V: Molecular genetic
mechanisms of congenital heart disease. Curr Opin Genet Dev.
75(101949)2022.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Sharma V, Goessling LS, Brar AK, Joshi CS,
Mysorekar IU and Eghtesady P: Coxsackievirus B3 infection early in
pregnancy induces congenital heart defects through suppression of
fetal cardiomyocyte proliferation. J Am Heart Assoc.
10(e017995)2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Han X, Wang B, Jin D, Liu K, Wang H, Chen
L and Zu Y: Precise dose of folic acid supplementation is essential
for embryonic heart development in zebrafish. Biology (Basel).
11(28)2021.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Wang C, Lv H, Ling X, Li H, Diao F, Dai J,
Du J, Chen T, Xi Q, Zhao Y, et al: Association of assisted
reproductive technology, germline de novo mutations and congenital
heart defects in a prospective birth cohort study. Cell Res.
31:919–928. 2021.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Lahrouchi N, Postma AV, Salazar CM, De
Laughter DM, Tjong F, Piherová L, Bowling FZ, Zimmerman D, Lodder
EM, Ta-Shma A, et al: Biallelic loss-of-function variants in PLD1
cause congenital right-sided cardiac valve defects and neonatal
cardiomyopathy. J Clin Invest. 131(e142148)2021.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Roifman M, Chung BHY, Reid DM, Teitelbaum
R, Martin N, Nield LE, Thompson M, Shannon P and Chitayat D:
Heterozygous NOTCH1 deletion associated with variable congenital
heart defects. Clin Genet. 99:836–841. 2021.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Ekure EN, Adeyemo A, Liu H, Sokunbi O,
Kalu N, Martinez AF, Owosela B, Tekendo-Ngongang C, Addissie YA,
Olusegun-Joseph A, et al: Exome sequencing and congenital heart
disease in sub-saharan Africa. Circ Genom Precis Med.
14(e003108)2021.PubMed/NCBI View Article : Google Scholar
|
|
58
|
van Walree ES, Dombrowsky G, Jansen IE,
Mirkov MU, Zwart R, Ilgun A, Guo D, Clur SB, Amin AS, Savage JE, et
al: Germline variants in HEY2 functional domains lead to congenital
heart defects and thoracic aortic aneurysms. Gene Med. 23:103–110.
2021.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Fu F, Li R, Lei TY, Wang D, Yang X, Han J,
Pan M, Zhen L, Li J, Li FT, et al: Compound heterozygous mutation
of the ASXL3 gene causes autosomal recessive congenital heart
disease. Hum Genet. 140:333–348. 2021.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Zhao L, Jiang WF, Yang CX, Qiao Q, Xu YJ,
Shi HY, Qiu XB, Wu SH and Yang YQ: SOX17 loss-of-function variation
underlying familial congenital heart disease. Eur J Med Genet.
64(104211)2021.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Shi HY, Xie MS, Yang CX, Huang RT, Xue S,
Liu XY, Xu YJ and Yang YQ: Identification of SOX18 as a new gene
predisposing to congenital heart disease. Diagnostics (Basel).
12(1917)2022.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Huang RT, Guo YH, Yang CX, Gu JN, Qiu XB,
Shi HY, Xu YJ, Xue S and Yang YQ: SOX7 loss-of-function variation
as a cause of familial congenital heart disease. Am J Transl Res.
14:1672–1684. 2022.PubMed/NCBI
|
|
63
|
Abhinav P, Zhang GF, Zhao CM, Xu YJ, Wang
J and Yang YQ: A novel KLF13 mutation underlying congenital patent
ductus arteriosus and ventricular septal defect, as well as
bicuspid aortic valve. Exp Ther Med. 23(311)2022.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Paszkowska A, Piekutowska-Abramczuk D,
Ciara E, Mirecka-Rola A, Brzezinska M, Wicher D, Kostrzewa G,
Sarnecki J and Ziółkowska L: Clinical presentation of left
ventricular noncompaction cardiomyopathy and bradycardia in three
families carrying HCN4 pathogenic variants. Genes (Basel).
13(477)2022.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Ke ZP, Zhang GF, Guo YH, Sun YM, Wang J,
Li N, Qiu XB, Xu YJ and Yang YQ: A novel PRRX1 loss-of-function
variation contributing to familial atrial fibrillation and
congenital patent ductus arteriosus. Genet Mol Biol.
45(e20210378)2022.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Debiec RM, Hamby SE, Jones PD, Safwan K,
Sosin M, Hetherington SL, Sprigings D, Sharman D, Lee K,
Salahshouri P, et al: Contribution of NOTCH1 genetic variants to
bicuspid aortic valve and other congenital lesions. Heart.
108:1114–1120. 2022.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Wang Z, Qiao XH, Xu YJ, Liu XY, Huang RT,
Xue S, Qiu HY and Yang YQ: SMAD1 Loss-of-Function variant
responsible for congenital heart disease. Biomed Res Int.
2022(9916325)2022.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Meerschaut I, Steyaert W, Bové T, François
K, Martens T, De Groote K, De Wilde H, Muiño Mosquera L, Panzer J,
Vandekerckhove K, et al: Exploring the mutational landscape of
isolated congenital heart defects: An exome sequencing study using
cardiac DNA. Genes (Basel). 13(1214)2022.PubMed/NCBI View Article : Google Scholar
|
|
69
|
De Ita M, Gaytán-Cervantes J, Cisneros B,
Araujo MA, Huicochea-Montiel JC, Cárdenas-Conejo A, Lazo-Cárdenas
CC, Ramírez-Portillo CI, Feria-Kaiser C, Peregrino-Bejarano L, et
al: Clustering of genetic anomalies of cilia outer dynein arm and
central apparatus in patients with transposition of the great
arteries. Genes (Basel). 13(1662)2022.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Okashah S, Vasudeva D, El Jerbi A,
Khodjet-El-Khil H, Al-Shafai M, Syed N, Kambouris M, Udassi S,
Saraiva LR, Al-Saloos H, et al: Investigation of genetic causes in
patients with congenital heart disease in qatar: Findings from the
Sidra Cardiac Registry. Genes (Basel). 13(1369)2022.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Azab B, Aburizeg D, Ji W, Jeffries L,
Isbeih NJ, Al-Akily AS, Mohammad H, Osba YA, Shahin MA, Dardas Z,
et al: TBX5 variant with the novel phenotype of mixed-type total
anomalous pulmonary venous return in Holt-Oram Syndrome and
variable intrafamilial heart defects. Mol Med Rep.
25(210)2022.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Li YJ, Wang J, Ye WG, Liu XY, Li L, Qiu
XB, Chen H, Xu YJ, Yang YQ, Bai D and Huang RT: Discovery of GJC1
(Cx45) as a new gene underlying congenital heart disease and
arrhythmias. Biology (Basel). 12(346)2023.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Wang H, Xiao F, Qian Y, Wu B, Dong X, Lu
Y, Cheng G, Wang L, Yan K, Yang L, et al: Genetic architecture in
neonatal intensive care unit patients with congenital heart
defects: a retrospective study from the China Neonatal Genomes
Project. J Med Genet. 60:247–253. 2023.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Wang Y, Xu YJ, Yang CX, Huang RT, Xue S,
Yuan F and Yang YQ: SMAD4 loss-of-function mutation predisposes to
congenital heart disease. Eur J Med Genet.
66(104677)2023.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Deng Q, Wang X, Gao J, Xia X, Wang Y,
Zhang Y and Chen Y: Growth restriction and congenital heart disease
caused by a novel TAB2 mutation: A case report. Exp Ther Med.
25(258)2023.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Afouda BA: Towards understanding the
gene-specific roles of GATA factors in heart development: Does
GATA4 lead the way? Int J Mol Sci. 23(5255)2022.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Yang YQ, Gharibeh L, Li RG, Xin YF, Wang
J, Liu ZM, Qiu XB, Xu YJ, Xu L, Qu XK, et al: GATA4
loss-of-function mutations underlie familial tetralogy of fallot.
Hum Mutat. 34:1662–1671. 2013.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Dixit R, Narasimhan C, Balekundri VI,
Agrawal D, Kumar A and Mohapatra B: Functionally significant, novel
GATA4 variants are frequently associated with Tetralogy of Fallot.
Hum Mutat. 39:1957–1972. 2018.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Wei D, Bao H, Liu XY, Zhou N, Wang Q, Li
RG, Xu YJ and Yang YQ: GATA5 loss-of-function mutations underlie
tetralogy of fallot. Int J Med Sci. 10:34–42. 2013.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Lin X, Huo Z, Liu X, Zhang Y, Li L, Zhao
H, Yan B, Liu Y, Yang Y and Chen YH: A novel GATA6 mutation in
patients with tetralogy of Fallot or atrial septal defect. J Hum
Genet. 55:662–667. 2010.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Wang J, Luo XJ, Xin YF, Liu Y, Liu ZM,
Wang Q, Li RG, Fang WY, Wang XZ and Yang YQ: Novel GATA6 mutations
associated with congenital ventricular septal defect or tetralogy
of fallot. DNA Cell Biol. 31:1610–1617. 2012.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Huang RT, Xue S, Xu YJ and Yang YQ:
Somatic mutations in the GATA6 gene underlie sporadic tetralogy of
Fallot. Int J Mol Med. 31:51–58. 2013.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Liu XY, Wang J, Zheng JH, Bai K, Liu ZM,
Wang XZ, Liu X, Fang WY and Yang YQ: Involvement of a novel GATA4
mutation in atrial septal defects. Int J Mol Med. 28:17–23.
2011.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Jiang WF, Xu YJ, Zhao CM, Wang XH, Qiu XB,
Liu X, Wu SH and Yang YQ: A novel TBX5 mutation predisposes to
familial cardiac septal defects and atrial fibrillation as well as
bicuspid aortic valve. Genet Mol Biol. 43(e20200142)2020.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Benson DW, Silberbach GM, Kavanaugh-McHugh
A, Cottrill C, Zhang Y, Riggs S, Smalls O, Johnson MC, Watson MS,
Seidman JG, et al: Mutations in the cardiac transcription factor
NKX2.5 affect diverse cardiac developmental pathways. J Clin
Invest. 104:1567–1573. 1999.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Goldmuntz E, Geiger E and Benson DW:
NKX2.5 mutations in patients with tetralogy of fallot. Circulation.
104:2565–2568. 2001.PubMed/NCBI View Article : Google Scholar
|
|
87
|
McElhinney DB, Geiger E, Blinder J, Benson
DW and Goldmuntz E: NKX2.5 mutations in patients with congenital
heart disease. J Am Coll Cardiol. 42:1650–1655. 2003.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Baban A, Postma AV, Marini M, Trocchio G,
Santilli A, Pelegrini M, Sirleto P, Lerone M, Albanese SB, Barnett
P, et al: Identification of TBX5 mutations in a series of 94
patients with Tetralogy of Fallot. Am J Med Genet A.
164A:3100–3107. 2014.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Amodio V, Tevy MF, Traina C, Ghosh TK and
Capovilla M: Transactivation in Drosophila of human enhancers by
human transcription factors involved in congenital heart diseases.
Dev Dyn. 241:190–199. 2012.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Charron F, Paradis P, Bronchain O, Nemer G
and Nemer M: Cooperative interaction between GATA-4 and GATA-6
regulates myocardial gene expression. Mol Cell Biol. 19:4355–4365.
1999.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Jiang Y and Evans T: The Xenopus
GATA-4/5/6 genes are associated with cardiac specification and can
regulate cardiac-specific transcription during embryogenesis. Dev
Biol. 174:258–270. 1996.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Garg V, Kathiriya IS, Barnes R,
Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS,
Hirayama-Yamada K, Joo K, et al: GATA4 mutations cause human
congenital heart defects and reveal an interaction with TBX5.
Nature. 424:443–447. 2003.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Durocher D, Charron F, Warren R, Schwartz
RJ and Nemer M: The cardiac transcription factors Nkx2-5 and GATA-4
are mutual cofactors. EMBO J. 16:5687–5696. 1997.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Nemer G, Fadlalah F, Usta J, Nemer M,
Dbaibo G, Obeid M and Bitar F: A novel mutation in the GATA4 gene
in patients with Tetralogy of Fallot. Hum Mutat. 27:293–294.
2006.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Martincorena I and Campbell PJ: Somatic
mutation in cancer and normal cells. Science. 349:1483–1489.
2015.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Maslov AY and Vijg J: Somatic mutation
burden in relation to aging and functional life span: Implications
for cellular reprogramming and rejuvenation. Curr Opin Genet Dev.
83(102132)2023.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Vijg J and Dong X: Pathogenic mechanisms
of somatic mutation and genome mosaicism in aging. Cell. 182:12–23.
2020.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Erickson RP: Somatic gene mutation and
human disease other than cancer: An update. Mutat Res. 705:96–106.
2010.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Walsh C, Choudhury S and Chen MH:
Landscape of somatic mutations in aging human heart muscle cells.
Nat Aging. 2:686–687. 2022.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Choudhury S, Huang AY, Kim J, Zhou Z,
Morillo K, Maury EA, Tsai JW, Miller MB, Lodato MA, Araten S, et
al: Somatic mutations in single human cardiomyocytes reveal
age-associated DNA damage and widespread oxidative genotoxicity.
Nat Aging. 2:714–725. 2022.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Salazar M, Consoli F, Villegas V, Caicedo
V, Maddaloni V, Daniele P, Caianiello G, Pachón S, Nuñez F,
Limongelli G, et al: Search of somatic GATA4 and NKX2.5 gene
mutations in sporadic septal heart defects. Eur J Med Genet.
54:306–309. 2011.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Wang J, Lu Y, Chen H, Yin M, Yu T and Fu
Q: Investigation of somatic NKX2-5, GATA4 and HAND1 mutations in
patients with tetralogy of Fallot. Pathology. 43:322–326.
2011.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Cheng C, Lin Y, Yang F, Wang W, Wu C, Qin
J, Shao X and Zhou L: Mutational screening of affected cardiac
tissues and peripheral blood cells identified novel somatic
mutations in GATA4 in patients with ventricular septal defect. J
Biomed Res. 25:425–430. 2011.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Esposito G, Butler TL, Blue GM, Cole AD,
Sholler GF, Kirk EP, Grossfeld P, Perryman BM, Harvey RP and Winlaw
DS: Somatic mutations in NKX2–5, GATA4, and HAND1 are not a common
cause of tetralogy of Fallot or hypoplastic left heart. Am J Med
Genet A. 155A:2416–2421. 2011.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Yin J, Qian J, Dai G, Wang C, Qin Y, Xu T,
Li Z, Zhang H and Yang S: Search of Somatic Mutations of NKX2-5 and
GATA4 Genes in Chinese patients with sporadic congenital heart
disease. Pediatr Cardiol. 40:17–22. 2019.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Heineke J, Auger-Messier M, Xu J, Oka T,
Sargent MA, York A, Klevitsky R, Vaikunth S, Duncan SA, Aronow BJ,
et al: Cardiomyocyte GATA4 functions as a stress-responsive
regulator of angiogenesis in the murine heart. J Clin Invest.
117:3198–3210. 2007.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Pikkarainen S, Tokola H, Kerkelä R and
Ruskoaho H: GATA transcription factors in the developing and adult
heart. Cardiovasc Res. 63:196–207. 2004.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Zhang H, Toyofuku T, Kamei J and Hori M:
GATA-4 regulates cardiac morphogenesis through transactivation of
the N-cadherin gene. Biochem Biophys Res Commun. 312:1033–1038.
2003.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Kuo CT, Morrisey EE, Anandappa R, Sigrist
K, Lu MM, Parmacek MS, Soudais C and Leiden JM: GATA4 transcription
factor is required for ventral morphogenesis and heart tube
formation. Genes Dev. 11:1048–1060. 1997.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Molkentin JD, Lin Q, Duncan SA and Olson
EN: Requirement of the transcription factor GATA4 for heart tube
formation and ventral morphogenesis. Genes Dev. 11:1061–1072.
1997.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Watt AJ, Battle MA, Li J and Duncan SA:
GATA4 is essential for formation of the proepicardium and regulates
cardiogenesis. Proc Natl Acad Sci USA. 101:12573–12578.
2004.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Crispino JD, Lodish MB, Thurberg BL,
Litovsky SH, Collins T, Molkentin JD and Orkin SH: Proper coronary
vascular development and heart morphogenesis depend on interaction
of GATA-4 with FOG cofactors. Genes Dev. 15:839–844.
2001.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Misra C, Sachan N, McNally CR, Koenig SN,
Nichols HA, Guggilam A, Lucchesi PA, Pu WT, Srivastava D and Garg
V: Congenital heart disease-causing Gata4 mutation displays
functional deficits in vivo. PLoS Genet. 8(e1002690)2012.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Epstein JA and Parmacek MS: Recent
advances in cardiac development with therapeutic implications for
adult cardiovascular disease. Circulation. 112:592–597.
2005.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Jiang JQ, Shen FF, Fang WY, Liu X and Yang
YQ: Novel GATA4 mutations in lone atrial fibrillation. Int J Mol
Med. 28:1025–1032. 2011.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Zhao L, Xu JH, Xu WJ, Yu H, Wang Q, Zheng
HZ, Jiang WF, Jiang JF and Yang YQ: A novel GATA4 loss-of-function
mutation responsible for familial dilated cardiomyopathy. Int J Mol
Med. 33:654–660. 2014.PubMed/NCBI View Article : Google Scholar
|