Introduction
Chronic myeloid leukemia (CML) and Philadelphia
Chromosome (Ph)-positive acute lymphoblastic leukemia are caused by
expression of the oncogenic fusion protein Bcr-Abl, which is a
constitutively active tyrosine kinase (1). Malignant transformation of affected
cells by Bcr-Abl is mediated by a number of signaling mechanisms,
including pathways of PI3K/Akt, mitogen-activated protein kinases,
RhoA-Rac, and JAK/STAT, leading to dysregulation of cell survival,
proliferation, differentiation and metabolism (2,3).
Tyrosine kinase inhibitors (TKIs) such as imatinib and dasatinib,
which target the binding of Bcr-Abl with ATP, have been
successfully used in the treatment of CML. However, a major
challenge associated with the use of these drugs in the clinic is
the development of resistance (4).
It has been shown that primary or acquired resistance occurs in
>20% of CML patients undergoing imatinib treatment (5).
TKI resistance can be mediated by Bcr-Abl-dependent
and -independent mechanisms. Bcr-Abl-dependent resistance is
primarily caused by mutations (e.g. the well characterized T315I
mutation) in the kinase domain of Abl (4). By comparison, the mechanisms of
Bcr-Abl-independent resistance remain to be elucidated. It has been
recognized that compensatory activation of the Akt/mTOR pathway
and/or inactivation of the p53 gene are involved in this process
(6,7). In fact, Bcr-Abl-independent
resistance has particular clinical significance, because in the
presence of effective kinase inhibition, Bcr-Abl-independent
resistance may be a major contributor to the maintenance of minimal
residual disease and relapse of CML (4). Supporting this notion, evidence shows
that reactivation of the p53 pathway, either directly or
indirectly, may boost the eradication of CML leukemia stem cells,
which are thought to be TKI resistant (8-10).
Clinical studies have revealed that progression of CML into blastic
crisis is often associated with mutations or loss of the p53 gene
(11-13).
It is estimated that ≤30% of CML cases are associated with
inactivation of the p53 gene (14). Therefore, an intriguing question is
whether there is a means to enhance the therapeutic efficacy of
TKIs in the absence of functional p53.
Cancer cells are generally associated with a
phenotype of oncogene and/or non-oncogene addiction, which results
in a status of enhanced levels of various cellular stresses
(15). Emerging evidence suggests
that exploiting these stress pathways by stress overload might be
used to enhance the efficacy of current anti-cancer therapies
(16,17). In eukaryotic cells, the nucleolus
may act as a signaling hub involved in mediating cellular stress
responses (18,19), while inhibiting ribosomal (r)DNA
transcription or inhibiting rRNA processing induces a unique
cellular stress response termed nucleolar stress response (NSR)
(18,19). NSR can be induced by RNA polymerase
I (Pol I) inhibitors. Of the available small molecule Pol I
inhibitors, CX-5461 is the first-in-class selective Pol I inhibitor
with an IC50 value at the 100 nM range (20). Currently, CX-5461 has entered into
clinical studies to treat blood malignancies (21).
The conventional NSR pathway involves stabilization
and accumulation of the tumor suppressor p53, through disruption of
the association between p53 and the E3 ubiquitin ligase Mdm2,
leading to decreased p53 ubiquitination and degradation (18,19).
Indeed, the cancer killing activity of CX-5461 appears to be
largely attributable to this mechanism (22). Nonetheless, there is evidence
suggesting that induction of NSR may also lead to p53-independent
consequences (19). Currently, the
pharmacodynamic properties of combined treatment with CX-5461 and
TKI in CML cells remain to be explored. The present study,
therefore, tested whether non-cytotoxic concentrations of CX-5461
can potentiate the efficacy of imatinib to induce apoptosis.
Especially, in order to clarify whether CX-5461 can enhance the
activity of imatinib in a setting of p53 loss-of-function, study
was performed in the human CML cell line K562, which is
p53-deficient (23-26).
Materials and methods
Reagents
CX-5461 and BMH-21 were obtained from Selleck
Chemicals. Imatinib mesylate was from Cayman Chemical Company. The
compounds were initially dissolved in DMSO and a series of 200X
stock solutions were prepared according to proposed final
concentrations. Drug treatment was performed by adding 5 µl of the
stock solution in each ml of culture medium. DMSO was dissolved
1:200 in the culture medium as vehicle control. Primary antibodies
used were: Anti-Kif1b (cat. no. A6638; ABclonal Biotech Co., Ltd. )
and anti-Rbfox2 (cat. no. A05389-1; Wuhan Boster Biological
Technology, Ltd. ). The dilution ratios of anti-Kif1b and
anti-Rbfox2 used in western blotting experiments were 1:500 and
1:1,000 respectively. The secondary antibody used was horseradish
peroxidase-conjugated goat anti-rabbit IgG (cat. no. SA00001-2;
Wuhan Sanying Biotechnology; 1:5,000).
Cell cultures
K562 (p53-/Ph+) and NALM-6
(p53+/Ph-) cells were originally obtained
from American Type Culture Collection. The THP-1 cell line
(p53-/Ph-) was obtained from National
Collection of Authenticated Cell Cultures. All of the cells were
cultured in RPMI 1640 medium supplemented with 10% fetal bovine
serum and antibiotics (all from Thermo Fisher Scientific, Inc.), in
a humidified environment with 5% CO2 at 37˚C. Untreated
cells were maintained at a density of below 1x106
cells/ml. Medium renewal was performed every 2 to 3 days.
Cell proliferation and viability
assay
The number of viable cells was assessed using a
colorimetric Enhanced Cell Counting Kit-8 (cat. no. C0042; Beyotime
Institute of Biotechnology) according to the manufacturer's
instructions. Briefly, 100 µl aliquots of cell suspension were
transferred to a 96-well plate, and 10 µl of the CCK-8 solution was
added to each well. After mixing, the cells were placed back to the
cell incubator and further incubated for 2 h at 37˚C. Cell number
was assessed by measuring absorbance at 450 nm using a microplate
reader (EMax Plus; Molecular Devices, LLC).
Flow cytometry
Cell apoptosis was assessed using a FITC-Annexin V
Apoptosis Detection kit (BD Biosciences). Cells were washed with
cold PBS and resuspended in 1X Binding Buffer at a concentration of
1x106 cells/ml. Then 100 µl aliquot of the cell
suspension was transferred to a 5-ml test tube, and working
solutions (5 µl each) of Annexin V and propidium iodide (PI),
prepared according to the manufacturer's protocols, were added.
After 15 min of incubation at room temperature in the dark, 400 µl
of the Binding Buffer was added to each tube. Flow cytometry
analysis was performed using a BD Accuri C6 Plus machine (BD
Biosciences). The following controls were used to set up the
quadrants: Unstained cells, cells stained with Annexin only, and
cells stained only with PI. Cells in both early stage
(Annexin+/PI−) and late stage
(Annexin+/PI+) apoptosis were equally counted
as apoptotic cells. Data were analyzed with WinMDI (version 2.9;
The Scripps Institute; http://www.cyto.purdue.edu/flowcyt/software/Winmdi.htm)
and FlowJo (version 10; FlowJo LLC) software.
Transcriptome sequencing
Cells were divided into three treatment groups (with
3 independent biological replications in each group): control,
imatinib alone, and imatinib + CX-5461. Total RNA was extracted
using TRIzol® (Thermo Fisher Scientific, Inc.) following
the manufacturer's instructions. RNA integrity was confirmed with
an Agilent Bioanalyzer 2100 (Agilent Technologies, Inc.). Qualified
total RNA was further purified by RNAClean XP Kit (Beckman Coulter,
Inc.) and RNase-Free DNase Set (Qiagen GmbH). RNA concentration was
determined using a NanoDrop ND-2000 spectrophotometer (Thermo
Fisher Scientific, Inc.). Library construction was performed using
VAHTS Stranded mRNA-seq Library Prep Kit for Illumina (Vazyme
Biotech Co., Ltd.). RNA sequencing was performed using a NextSeq
Illumina550 platform (Illumina, Inc.) in a paired-end manner. RNA
processing, library construction and sequencing services were
provided by Shanghai Biotechnology Corporation. The complete
dataset of the raw count values and fragments per kilo base per
million mapped reads (FPKM) values are provided in Table SI. Differentially expressed genes
were defined by the false discovery rate (q value) <0.05
and fold change >2. The reproducibility of the sequencing assay
was confirmed by the degree of correlation between biological
duplicate samples (see ENCODE Guidelines and Best Practices for
RNA-Seq; https://www.encodeproject.org/about/experiment-guidelines)
(27).
Reverse transcription-quantitative
(RT-q) PCR
Around 5x105 cells were homogenized in
TRIzol® (Thermo Fisher Scientific, Inc.) and total RNA
was isolated according to the manufacturer's instructions. The RNA
concentration was determined using a NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific, Inc.). cDNA was
synthesized from 50 ng of total RNA using the PrimeScript RT-PCR
Kit (cat. no. RR037A; Takara Bio, Inc.) according to the
manufacturer's instructions. Reverse transcription was carried out
using 100 µM of random 6mers, under a thermocycler condition of
37˚C for 15 min followed by 85˚C for 5 sec. The real-time PCR
reaction was carried out using the UltraSYBR Mixture kit (cat. no.
CW0957M; CWBio) according to the manufacturer's instructions in a
LightCycler 96 Instrument (Roche Diagnostics). The primer sequences
used in the study are listed in Table
SII. The reaction was carried out using 5 ng of cDNA in a 50-µl
reaction volume, containing 0.2 µM of primers. The following
thermocycler settings were applied: 95˚C for 10 min; 40 cycles of
95˚C for 16 sec and 60˚C for 1 min. Human GAPDH was used as the
housekeeping gene. Fold changes were determined using the
2-ΔΔCq method, where ΔCq=Cq target gene-Cq housekeeping
gene; ΔΔCq=ΔCq test sample-ΔCq calibrator (control) sample (see
‘Real-time PCR handbook’ at https://www.thermofisher.com/content/dam/LifeTech/global/Forms/PDF/real-time-pcr-handbook.pdf)
(28).
Small interfering (si)RNA
transfection
siRNA constructs targeting RBFOX2 (sense
sequence CCGGAGUUAUAUGCAGCAUTT; anti-sense AUGCUGCAUAUAACUCCGGTT)
and KIF1B-β (sense GCCAUCCUCUCCCUAAAUATT; anti-sense
UAUUUAGGGAGAGGAUGGCTT) were obtained from Shanghai GenePharma Co.,
Ltd. The RBFOX2 siRNA was designed using RBFOX2
transcript variant 1 mRNA (Reference Sequence accession:
NM_001031695.4) as the template, targeting the 54-nucleotide exon
which was conserved in transcript variants 1-18. The KIF1B-β
siRNA was designed using KIF1B transcript variant 1 mRNA
(Reference Sequence accession: NM_015074.3) as the template,
targeting the nucleotides 4108 to 4126. A universal non-targeting
siRNA was used as control (sense UUCUCCGAACGUGUCACGUTT; anti-sense
ACGUGACACGUUCGGAGAATT). K562 cells were re-suspended in
antibiotic-free Opti-MEM I Reduced Serum medium (cat. no. 31985-062
from Thermo Fisher Scientific, Inc.) at 1x106 cells per
ml, and transfected using Lipofectamine® RNAiMAX (cat.
no. 13778-075; Thermo Fisher Scientific, Inc.) with a final siRNA
concentration of 200 nM at 37˚C. Fresh medium was replenished after
6 h of incubation with siRNA, and the cells were used for
experimentation at 48 h after transfection.
Western blotting
Total proteins were extracted using RIPA lysis
buffer (cat. no. R0010; Beijing Solarbio Science & Technology
Co., Ltd.) containing Protein Phosphatase Inhibitors cocktail (cat.
no. P1260; Beijing Solarbio Science & Technology Co., Ltd.).
The protein concentration was determined using a BCA protein assay
kit (cat. no. P0010; Beyotime Institute of Biotechnology). Samples
were boiled in SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS,
10% glycerol, 5% 2-mercaptoethanol and 0.02% bromophenol blue).
Protein samples (25 µg per lane) were loaded onto precast 4-20%
gradient Bis-Tris PAGE gel (Dalian Meilun Biotechnology Co., Ltd.),
separated by electrophoresis, and transferred to Immobilon-NC
nitrocellulose membrane (Merck KGaA). The membrane was blocked with
5% non-fat milk for 2 h at room temperature, and then incubated
with primary antibodies overnight at 4˚C. After washing, the
membrane was probed with horseradish peroxidase-conjugated
secondary antibody (1:5,000) for 1-2 h at room temperature. The
bands were developed by enhanced chemiluminescence using SuperKine
West Femto Maximum Sensitivity Substrate (cat. no. BMU102-CN;
Abbkine Scientific Co., Ltd.), and visualized with a Tanon 3500 Gel
Imaging System (Tanon Science and Technology Co., Ltd.). Band
densitometry analysis was performed using ImageJ software (version
1.46r) (National Institutes of Health).
Caspase 3 activity assay
Caspase 3 activity was measured with a colorimetric
assay kit (cat. no. C1115; Beyotime Institute of Biotechnology),
which detected the cleavage of substrate (Ac-DEVD-p-nitroanilide)
to the yellow-colored product p-nitroaniline, according to the
manufacturer's protocol. Briefly, 4x105 cells were
collected by centrifugation (600 x g for 5 min), washed with PBS,
and incubated in 200 µl of the lysis buffer supplied on ice for 15
min with vortexing. The sample was then centrifuged at 16,000 x g
4˚C for 15 min. The supernatant was transferred to a test tube,
mixed with 0.2 mM substrate, and incubated at 37˚C overnight. The
absorbance at 405 nm was measured using a SUNRISE microplate reader
(Tecan Group, Ltd.).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. Data analysis was performed with unpaired t-test or
one-way analysis of variance followed by post hoc Tukey's test as
appropriate. All of the n values represented the number of
independent biological replicates. P<0.05 was considered to
indicate a statistically significant difference.
Results
CX-5461 potentiates imatinib-induced
cytotoxicity and apoptosis
In order to determine the potential synergy between
imatinib and CX-5461, the combination subthresholding approach was
adopted, an effect-based method which detects additivity by showing
that combination of non-effective doses of two drugs yields a
significant effect (29).
Following this principle, the synergy is determined by showing that
neither [drug A] nor [drug B] has a significant effect
individually, while the effect of [drug A]/[drug B] combination is
significant. According to Picard et al (30), the effective plasma concentration
of imatinib in humans is between 300-3,000 nM. Based on this
information, 100 nM was selected as the reference concentration for
imatinib in the following experiments. The rationale for using 100
nM of imatinib was that this concentration represented a
non-effective one for single treatment, thereby allowing the
present study to test drug synergy with the subthresholding
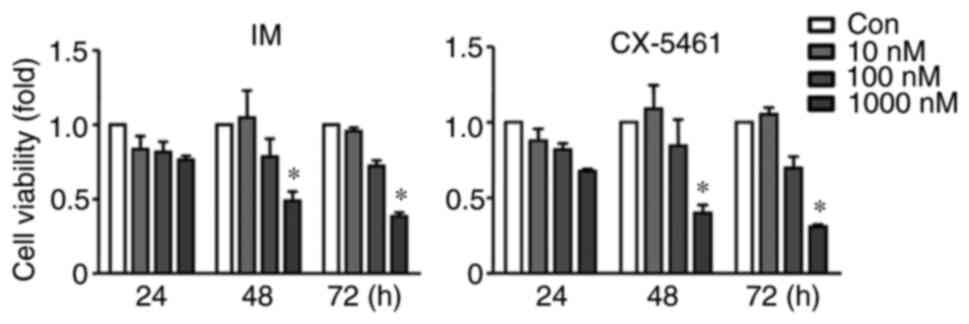
approach. First, it was confirmed that at a concentration of 100
nM, neither imatinib nor CX-5461 individually had any significant
effect on the viability of K562 cells after 48 h of treatment
(Fig. 1). In comparison, under the
same experimental conditions, combined treatment with CX-5461 and
imatinib, both at 100 nM (for 48 h), significantly decreased the
cell viability (Fig. 2A). To
precisely define the nature of the synergistic effect of CX-5461 on
imatinib-induced cell toxicity, concentration-response analysis was
performed in the absence or presence of 100 nM CX-5461. As shown in
Fig. 2B, CX-5461 increased the
Emax (maximum effect) of imatinib (83.4±3.1 vs. 70.8±1.9%, P=0.026,
unpaired t-test, n=3), but had no significant effect on the
EC50 value (concentration producing 50% of the maximum
effect; 0.23±0.02 vs. 0.20±0.009 mM; P=0.215). Furthermore,
treatment with imatinib/CX-5461 combination (for 48 h) increased
the number of apoptotic cells as compared with CX-5461 or imatinib
mono-treatment (Fig. 2C).
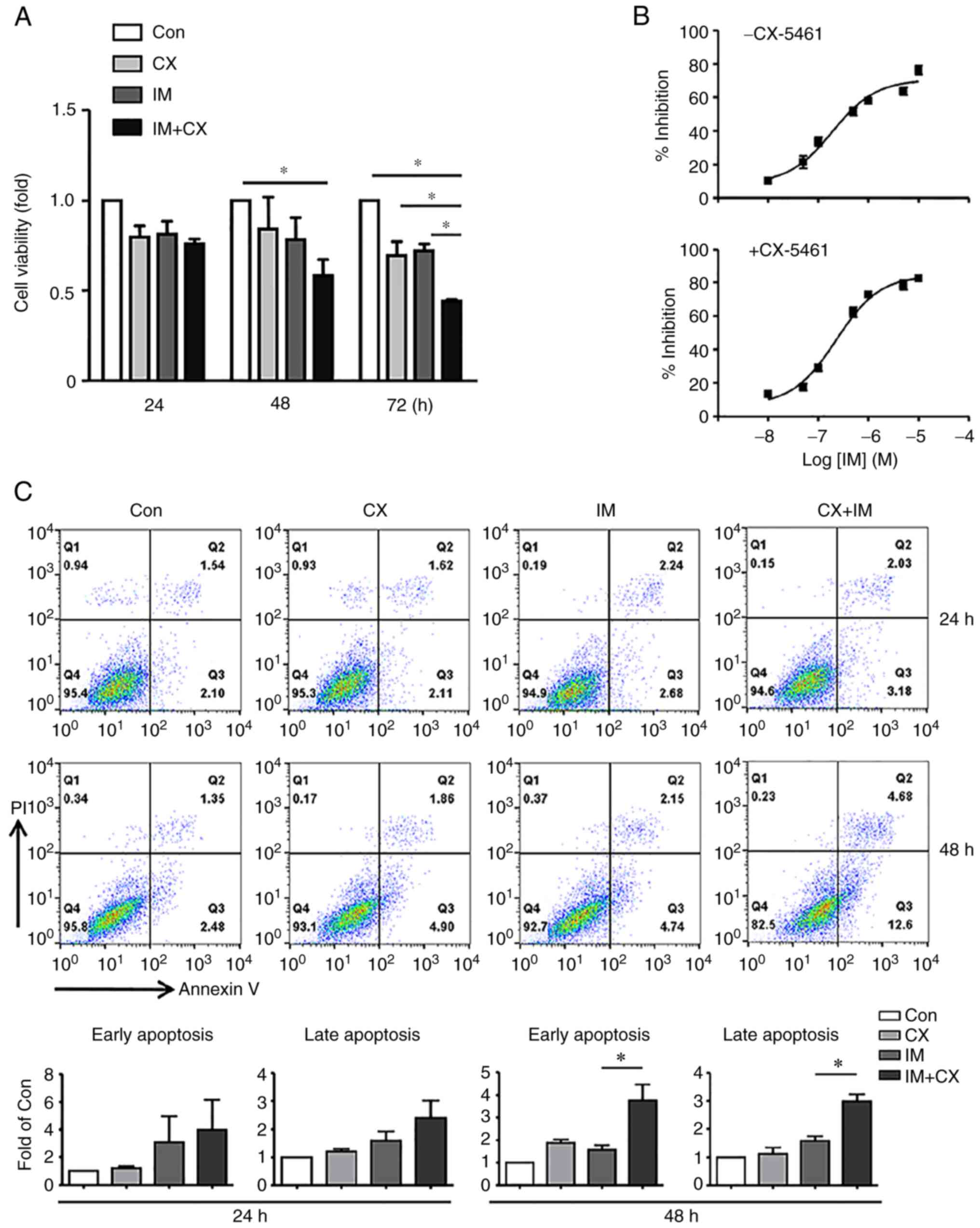 | Figure 2Synergistic effects of CX on
IM-induced cytotoxicity and apoptosis in K562 cells. (A) Effects of
CX (100 nM), IM (100 nM), and their combination on cell viability.
(B) Concentration-response curves of imatinib-induced inhibition of
cell viability in the absence or presence of CX (100 nM, treatment
for 48 h). (C) Representative flow cytometry results and
quantitative data showing the effects of CX (100 nM), IM (100 nM)
and their combination on cell apoptosis. Early-stage apoptosis and
late-stage apoptosis were shown separately. Data were expressed as
mean ± standard error of the mean. *P<0.05, one-way
analysis of variance, n=3 (for panel A), 3 (for panel B), 4
(for the 24 h experiments in panel C), and 3 (for the 48 h
experiments in panel C). CX, CX-5461; IM, imatinib; Con,
control. |
BMH-21 does not mimic the actions of
CX-5461
To clarify whether the synergistic action of CX-5461
with imatinib was due to inhibition of rDNA transcription, the
cells were treated with another unrelated Pol I inhibitor BMH-21.
It was found that treatment with BMH-21 at 1 µM for 48 h moderately
reduced the cell viability (Fig.
S1A). At this concentration, however, a synergistic effect of
BMH-21 on imatinib-induced apoptosis was not observed (Fig. S1B). These data indicated that the
synergistic effect of CX-5461 with imatinib was unlikely to be a
direct result of the reduced rDNA transcription.
Imatinib/CX-5461 synergy is not
related to changed expressions of various apoptosis regulators
To clarify whether the synergistic effect of CX-5461
on imatinib-induced apoptosis was related to changed expressions of
essential apoptosis-regulating factors, the mRNA levels of
FAS, BAX, BIM, BCL-2 and BCL-XL
genes were examined. As shown in Fig.
3, CX-5461 exhibited no significant effects on the expression
of these genes either in the absence or presence of imatinib
co-treatment.
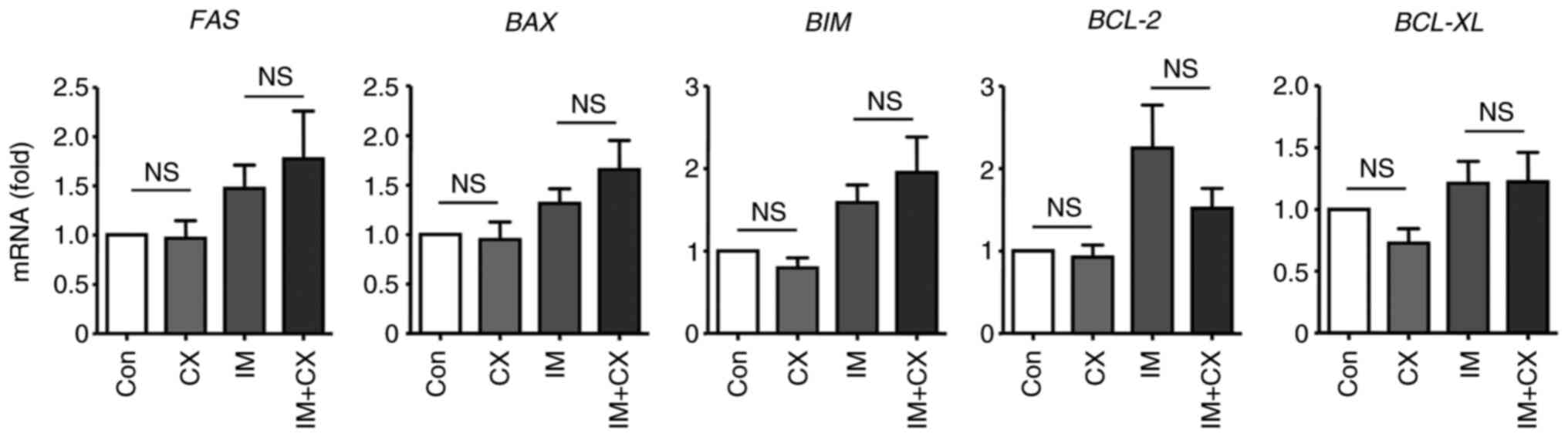 | Figure 3Reverse transcription-quantitative
PCR results showing that CX (100 nM for 48 h), in the absence or
presence of IM (100 nM), had no effects on the expression of
various apoptosis-related genes in K562 cells. Data were expressed
as mean ± standard error of the mean, n=10 for FAS, 8
for BAX, 9 for BIM, 10 for BCL-2 and 10 for
BCL-XL. NS, non-significant; CX, CX-5461; IM, imatinib; Con,
control. |
Transcriptome sequencing analysis of
the effects of imatinib/CX-5461 in K562 cells
To further explore the possible mechanism underlying
the synergy between CX-5461 and imatinib, genome-wide RNA
sequencing experiments were performed in cells which were
untreated, treated with imatinib alone, and treated with
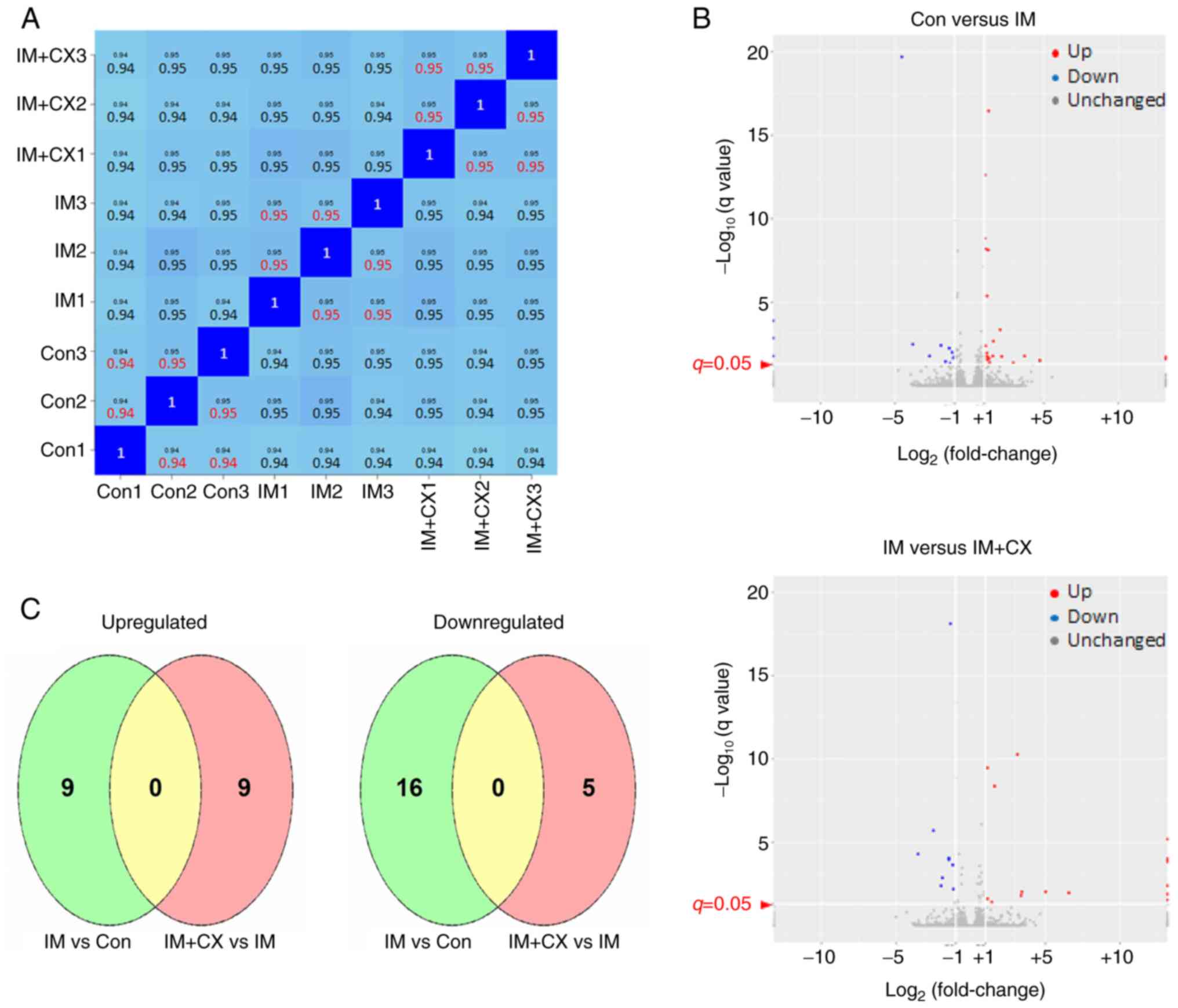
imatinib/CX-5461 combination respectively. Technically, the
reproducibility of the assay was confirmed by the high degree of
correlation (coefficient >0.9) between biological duplicate
samples (27). The specific
correlation coefficient values between sample pairs are shown in
Fig. 4A. Consistent with the
sub-efficacious concentration of imatinib used in this experiment,
it was shown that only a small number of gene expressions were
altered by imatinib treatment alone (Fig. 4B). Specifically, nine genes were
upregulated and 16 genes were downregulated significantly by
imatinib. Similarly, it was demonstrated that in the presence of
imatinib, only a minor group of genes were responsive to CX-5461
co-treatment (Fig. 4B and Table I). Specifically, nine genes were
upregulated and five genes were downregulated significantly. As the
transcription factor p53 has a central role in mediating the
cellular effects of Pol I inhibition-induced NSR (18,19),
in K562 cells (without functional p53) the CX-5461-responsive genes
did not contain typical p53 targets (see Table I). Moreover, it was shown that
under the present experimental settings, CX-5461-responsive genes
in K562 cells were totally distinct from those responsive to
imatinib (Fig. 4C). Among the
differentially expressed genes responsive to CX-5461, special
attention was paid to the RBFOX2 gene (downregulated), which
encodes a RNA splicing factor (31-33).
Rbfox2 mediates the splicing of KIF1B mRNA, while reduced
Rbfox2 expression has been shown to facilitate the preferential
expression of the KIF1B-β isoform (over KIF1B-α),
which has a pro-apoptotic role in solid tumor cells (34-40).
 | Table IGenes regulated by CX-5461 in the
presence of IM. |
Table I
Genes regulated by CX-5461 in the
presence of IM.
| Ensembl ID | Gene name | Fold change | P-value | q-value | Change in
regulation vs. IM alone |
|---|
|
ENSG00000180354 | MTURN | 0.21 |
8.35x10-29 |
2.11x10-24 | Down |
|
ENSG00000166532 | RIMKLB | 2.56 |
9.02x10-23 |
7.62x10-19 | Up |
|
ENSG00000167842 | MIS12 | 0.46 |
8.11x10-14 |
3.42x10-10 | Down |
|
ENSG00000100320 | RBFOX2 | 0.33 |
1.35x10-12 |
4.28x10-9 | Down |
|
ENSG00000198932 | GPRASP1 | 5.65 |
7.84x10-10 |
1.99x10-6 | Up |
|
ENSG00000102780 | DGKH | 2.76 |
4.90x10-8 |
8.87x10-5 | Up |
|
ENSG00000131779 | PEX11B | 2.76 |
7.00x10-8 |
1.11x10-4 | Up |
|
ENSG00000125841 | NRSN2 | 2.30 |
1.64x10-7 |
2.31x10-4 | Up |
|
ENSG00000116731 | PRDM2 | 3.71 |
1.29x10-6 |
1.37x10-3 | Up |
|
ENSG00000112242 | E2F3 | 2.23 |
7.98x10-6 |
6.52x10-3 | Up |
|
ENSG00000158985 |
CDC42SE2 | 0.46 |
4.00x10-5 |
2.47x10-2 | Down |
|
ENSG00000138942 | RNF185 | 0.37 |
6.93x10-5 |
3.99x10-2 | Down |
|
ENSG00000102524 |
TNFSF13B | 2.05 |
8.39x10-5 |
4.52x10-2 | Up |
|
ENSG00000171574 | ZNF584 | 2.01 |
8.39x10-5 |
4.52x10-2 | UP |
Imatinib/CX-5461 combination modulates
KIF1B expression in K562 cells
The present study examined whether changed
expression of KIF1B was involved in the pro-apoptotic effect
of imatinib/CX-5461 combination. Using qPCR, it was found that
CX-5461 treatment significantly upregulated the expression levels
of both KIF1B-α and KIF1B-β in the presence of
imatinib (Fig. 5A); however,
CX-5461 alone had no significant effect on KIF1B expression
in the absence of imatinib (Fig.
S2). Similarly, imatinib alone did not significantly affect the
expression of KIF1B (Fig.
S2). To further confirm the finding that imatinib/CX-5461 could
upregulate the expression of KIF1B, two additional PCR
primers were designed targeting the N-terminal part of
Kif1b, which was 100% identical for Kif1b-α and Kif1b-β. As shown
in Fig. 5B, the mRNA level of
total KIF1B was significantly increased in
imatinib/CX-5461-treated cells. Moreover, it was confirmed that
imatinib/CX-5461 significantly increased the protein level of total
Kif1b as compared with the imatinib mono-treatment (Fig. 5B). Remarkably, the present study
did not detect any significant change in the KIF1B-α to
KIF1B-β ratio following imatinib/CX-5461 treatment (Fig. 5A), which seemed to be in contrast
to the reported effect of Rbfox2(38). To clarify this question, gene
silencing for RBFOX2 was performed (Fig. 5C), and it was demonstrated that
knocking down the RBFOX2 expression in K562 cells did not
change the expressions of KIF1B-α or KIF1B-β, nor did
it affect the ratio between the two isoforms (Fig. 5D). These results suggest that there
is no association between Rbfox2 and KIF1B mRNA splicing in
K562 cells under the present experimental settings.
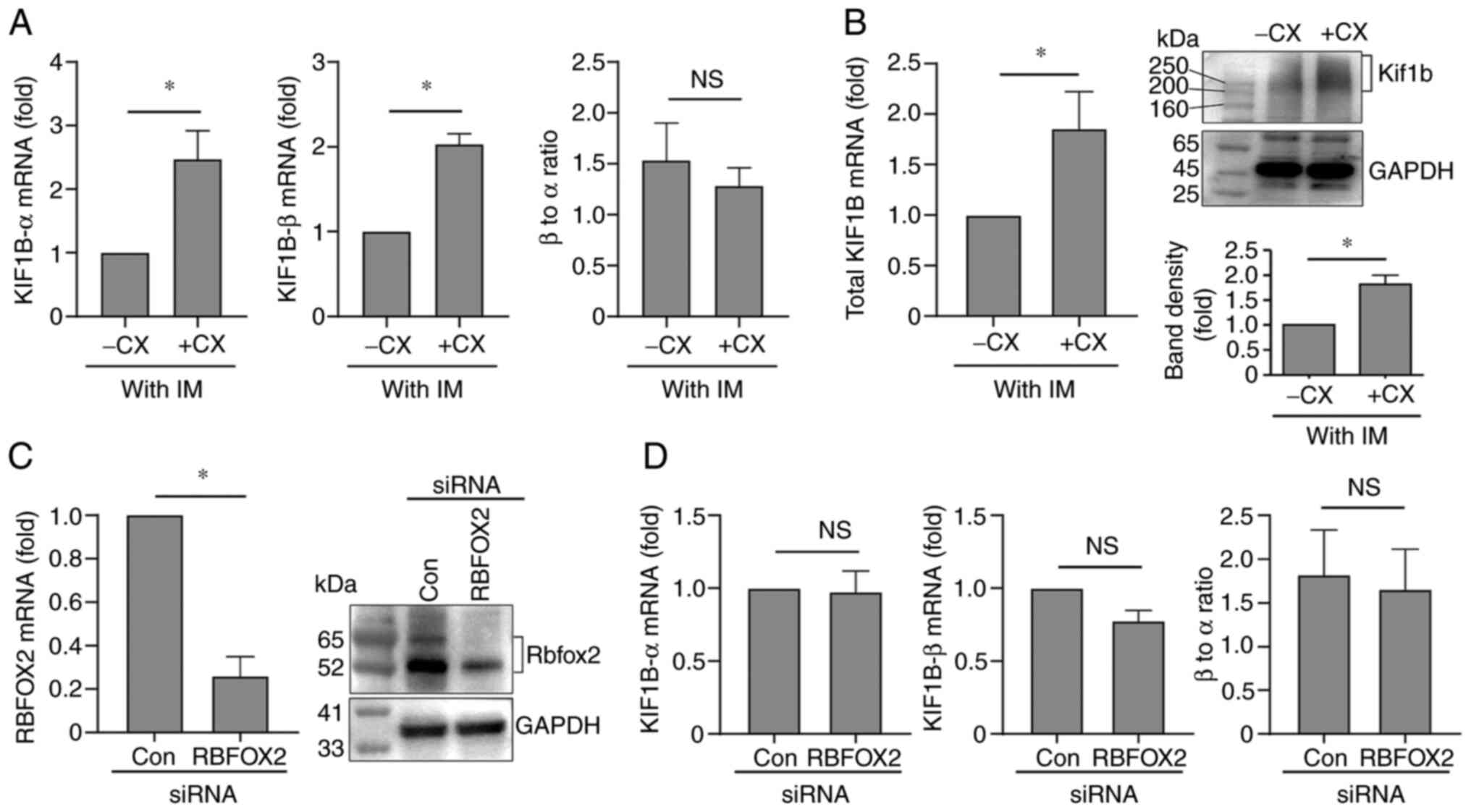 | Figure 5Regulation of the expression of
KIF1B isoforms in K562 cells. (A) Reverse
transcription-quantitative PCR results showing the effects of CX
(100 nM for 48 h in the presence of 100 nM IM) on the levels of
KIF1B-α, KIF1B-β, and their relative ratio. Con.
groups of untreated cells and cells treated with CX or IM
separately were not included in this experiment. Instead, for
results of the control experiments with untreated, CX-5461
alone-treated, and imatinib alone-treated cells, see Fig. S2. (B) Effects of IM/CX combination
on the mRNA (left panel) and protein (right panel) levels of total
KIF1B as compared with the IM mono-treatment. (C) Reverse
transcription-quantitative PCR (left panel) and western blotting
(right panel) results showing the gene silencing efficiency of the
RBFOX2 siRNA. (D) Effects of RBFOX2 gene silencing on
the levels of KIF1B-α, KIF1B-β, and their relative
ratio. Data were expressed as mean ± standard error of the mean.
*P<0.05, unpaired t-test, n=8 (A), 7 (left
panel of B), 3 (right panel of B), 5 (left panel of C), 2 (right
panel of C), 5 (left panel of D) and 4 (middle and right panels of
D). NS, non-significant; CX, CX-5461; CX, CX-5461; Con, control,
si, small interfering. |
KIF1B-β has a pro-apoptotic role in
K562 cells
As KIF1B-α was not implicated in modulating
cell apoptosis (34,40), it was then tested whether
KIF1B-β was involved in mediating the pro-apoptotic action
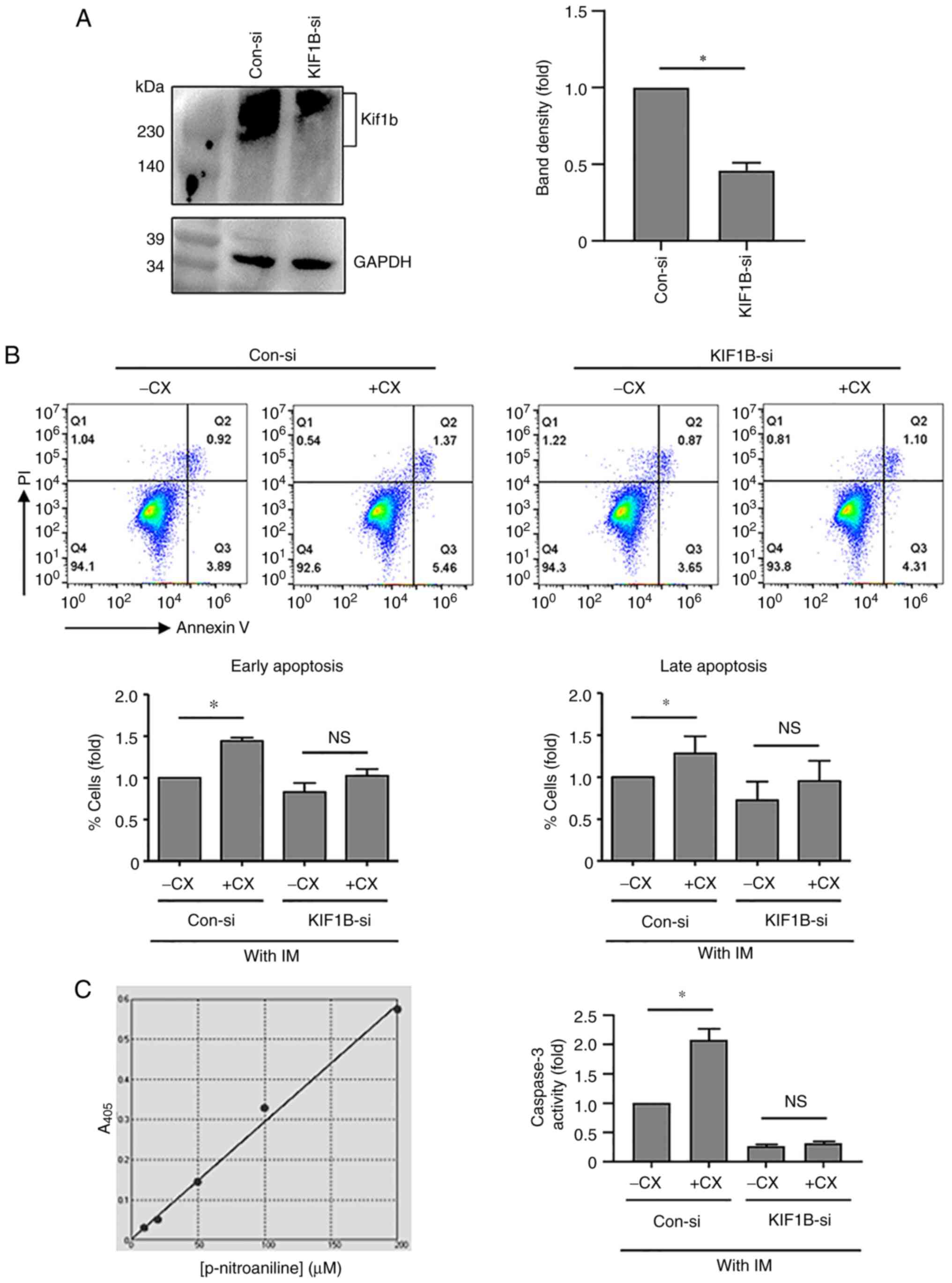
of imatinib/CX-5461 combination in K562 cells. For this purpose,
K562 cells were transfected with KIF1B-β siRNA, of which the
gene silencing effect was confirmed by western blotting (Fig. 6A). Flow cytometry assays,
demonstrated that in cells transfected with the control siRNA,
CX-5461 in the presence of imatinib increased the apoptotic
response; by contrast, in cells transfected with KIF1B-β
siRNA, the pro-apoptotic effect of CX-5461 was blunted (Fig. 6B). To further corroborate the above
flow cytometry results, caspase 3 activity assay was performed in
K562 cells. In cells transfected with the control siRNA,
imatinib/CX-5461 increased the level of caspase 3 activation as
compared with imatinib mono-treatment, whereas in cells transfected
with KIF1B-β siRNA, the effect of CX-5461 was diminished
(Fig. 6C).
Regulation of KIF1B-β expression by
imatinib/CX-5461 in other cell lines
The effects of imatinib/CX-5461 on KIF1B
expression in NALM-6 (human acute lymphoblastic leukemia cell line;
p53+/Ph-) and THP-1 (human monocytic leukemia
cell line; p53-/Ph-) cells was also tested.
In NALM-6 cells, it was shown that only imatinib/CX-5461
combination significantly increased the expression of
KIF1B-α and KIF1B-β (Fig. S3), whereas the effects of imatinib
or CX-5461 alone did not reach a statistical significance, although
the moderate increases induced by CX-5461 were significant using
unpaired t-test. In THP-1 cells, however, imatinib and CX-5461,
either alone or in combination, had no significant effects on the
expression of KIF1B isoforms (Fig. S3). The partial effect of imatinib
in NALM-6 cells (lacking Bcr-Abl) may be due to the off-target
activities of the drug (41).
Discussion
Selective targeting of Pol I-dependent rDNA
transcription is a conceptually novel strategy to treat cancers.
Being the first selective Pol I inhibitor, CX-5461 shows
therapeutic effects on a spectrum of solid and blood cancers in
both pre-clinical and clinical investigations (20,21,42).
At present, the precise molecular mechanisms underlying the
pharmacological effects of CX-5461 are still under debate (20,43-46).
Indeed, most of the evidence suggests that the CX-5461 effects are
mainly attributable to the induction of a non-canonical DNA damage
response, rather than direct impairment of the ribosome biogenesis
(42). The effects of CX-5461 may
be either p53-dependent or p53-independent, depending on the
specific cellular context (42).
The major finding from the present study is that CX-5461 at a
non-cytotoxic concentration can potentiate the pro-apoptotic effect
of imatinib in K562 cells in the absence of functional p53, via an
unrecognized pathway, i.e., upregulation of the KIF1B-β
expression. This pharmacological property of CX-5461 had not been
reported previously, to the best of the authors' knowledge.
Bcr-Abl-targeting TKIs including imatinib have significantly
improved the prognosis of CML patients; however, the occurrence of
adverse events remains to be a critical issue in the clinic for
long-term TKI treatments (47).
Moreover, p53 loss-of-function may compromise the anti-leukemic
efficacy of imatinib (7) and may
contribute to the evolution from chronic phase CML to blast crisis
(48). Therefore, the synergistic
interaction between CX-5461 and imatinib may be of potential value
in the clinical management of refractory CML.
Following the initial success of CX-5461 single
therapy in cancer treatment, emerging evidence from several studies
has indicated that combined treatment with CX-5461 and other
chemotherapeutic agents may achieve enhanced effectiveness as
compared with single therapies. For example, CX-5461 has been shown
to have synergistic effects in conjunction with the DNA
topoisomerase 1 inhibitor topotecan (49), the mammalian target of rapamycin
complex (mTORC1/2) inhibitor INK128(50), the mTORC1 inhibitor everolimus
(51), the poly (ADP-ribose)
polymerase inhibitor talazoparib (52) and the pan-PIM kinase inhibitor
CX-6258(53), in different cancer
cells/models. In addition, it has been demonstrated that CX-5461,
at a concentration not sufficient to inhibit rDNA transcription,
sensitizes tumor cells to irradiation (54). In this regard, the results of the
present study have provided new information about a synergistic
effect of combining CX-5461 with imatinib. Given the favorable
safety profile of CX-5461 as revealed by a Phase I clinical trial
(55), it is probable that
imatinib/CX-5461 combination may represent a useful strategy to
alleviate the adverse effects caused by TKIs through dose reduction
(47).
Current evidence suggests that there is not a
unified mechanism to explain the synergy between CX-5461 and other
inhibitors, depending on the molecular target involved and the
status of p53 gene (49-53).
The RNA sequencing test in K562 cells identified only a small
number of differentially expressed genes; these genes included no
typical p53 targets. These data are consistent with the notion that
p53 is the predominant transcription factor mediating the cellular
effects of Pol I inhibition-induced NSR (18,19).
The weak genomic response to CX-5461 treatment also explains the
strong correlation between inter-group samples (Fig. 4A). It was noted that CX-5461
downregulated the expression of RBFOX2, which encodes a
RNA-binding protein with important roles in regulating pre-mRNA
splicing (31,32). It has been shown that Rbfox2 is
involved in mediating the alternative splicing events in B lymphoma
cells (33). Moreover, there is
evidence suggesting that Rbfox2 regulates the alternative splicing
of the mRNA of kinesin family member 1B (KIF1B), resulting in the
expression of two splicing variants, KIF1B-α and KIF1B-β (34,35).
It has been well documented that KIF1B-β has potent pro-apoptotic
functions (34-39).
Notably, a study demonstrated that reduction in Rbfox2 expression
resulted in the preferential expression of KIF1B-β (40). However, the present study failed to
detect significant alterations in the ratio between two isoforms in
K562 cells, either in response to imatinib/CX-5461 treatment or to
RBFOX2 gene silencing, suggesting that Rbfox2 protein was
unlikely to have a major role in mediating KIF1B mRNA
splicing in CML cells. Instead, it was observed that CX-5461
significantly upregulated the levels of both KIF1B-α and
KIF1B-β. This unanticipated finding is notable because there
is evidence suggesting that mutations in the KIF1B gene may
be associated with the pathogenesis of acute lymphoblastic leukemia
(56). Hence, the KIF1B
gene is likely to have important functional roles in leukemic
cells. Consistent with previous findings of the pro-apoptotic
property of KIF1B-β in solid tumor cells, the present study
confirmed that KIF1B gene silencing (presumably reducing
both KIF1B-α and KIF1B-β) in K562 cells blunted the
pro-apoptotic effect of imatinib/CX-5461. Based on these
observations, it is hypothesized that the synergistic pro-apoptotic
effect of imatinib/CX-5461 in K562 cells is mediated by
upregulating the expression of KIF1B-β.
Currently it is not understood how CX-5461 regulates
KIF1B expression. In a previous study by Ochiai et al
(57), it was shown that in
neuroblastoma cells, the transcription factor N-Myc could repress
the expression of KIF1B-β by upregulating the expression of
Bmi1, a component of the Polycomb transcriptional repressor
complexes. Notably, N-Myc is overexpressed in ~50% of T-cell acute
lymphoblastic leukemia patients and is implicated in promoting the
survival of leukemic cells (58).
Moreover, there is evidence indicating that CX-5461 may
downregulate the expression of N-MYC gene, thereby
suppressing the tumor growth (59). Taking these lines of data together,
it is suggested that CX-5461 may possibly stimulate KIF1B
expression in K562 cells by downregulating the expression of
N-MYC (57). This
hypothesis warrants further study. The inconsistent effects of
imatinib/CX-5461 in K562, NALM-6 and THP-1 cells indicate that the
regulation of KIF1B expression is not a ubiquitous
phenomenon in leukemic cells and should be examined in a cell
type-specific manner in future studies. On the other hand, the
similar effects in K562 (Ph+) and NALM-6
(Ph-) cells suggest that imatinib/CX-5461-induced
upregulation of KIF1B does not strictly depend on the
presence of Bcr-Abl. This phenomenon may be explained by the fact
that imatinib has limited target specificity; in addition to
Bcr-Abl, other tyrosine kinases such as platelet-derived growth
factor receptor and c-kit can also be inhibited by imatinib
(41). The relative independence
on Bcr-Abl and p53 of the action of imatinib/CX-5461 combination
may have two advantages: i) This strategy may be useful in CML
patients with TKI resistance caused by p53 loss-of-function
(6,7); ii) the combined treatment with
imatinib/CX-5461 is likely to be efficacious in CML cells with
resistance-causing mutations in Bcr-Abl (4). K562 cells represent the prototypical
cell culture model of CML; however, a limitation of the present
study was that no experimentation in additional Ph+ CML
cell lines as well as hypothetically sensitive Ph-
non-CML cell lines was conducted. The BCR-ABL translocation
in CML cells may occur via a number of different junction points.
In addition, CML is a disease of hematopoietic stem cells and, as a
result, can give rise to multiple lineages of tumor cells (60). Therefore, it would be of scientific
interest to further test the actions of imatinib/CX-5461
combination in additional Ph+ CML cell lines such as
LAMA-84 and CML-T1(60).
In summary, the present study demonstrated that the
selective Pol I inhibitor CX-5461, at a non-cytotoxic
concentration, potentiates the pro-apoptotic effect of imatinib in
the CML cell line K562 in a p53-independent manner. This effect of
CX-5461 is mainly mediated by upregulating the expression of
KIF1B-β, which is pro-apoptotic in K562 cells. This
pharmacological property of CX-5461 may be of potential clinical
value in the treatment of refractory CML.
Supplementary Material
Effects of BMH-21 on cell viability
and IM-induced apoptosis in K562 cells. (A) Concentration-dependent
effects of BMH-21 (treatment for 48 h) on cell viability. (B) Flow
cytometry results showing that BMH-21 (1 μM) did not modify
IM (100 nM)-induced apoptosis (example from two independent
experiments). Data were expressed as mean ± standard error of the
mean. *P<0.05, one-way analysis of variance,
n=3. IM, imatinib; Con, control.
Reverse transcription-quantitative PCR
results showing that (A) CX (100 nM) or (B) IM (100 nM) had no
significant effects on the expression of KIF1B-α or
KIF1B-β in K562 cells. Data were expressed as mean ±
standard error of the mean. Unpaired t-test, n=8 for panel A
and n=7 for panel B). NS, no significance; CX, CX-5461; IM,
imatinib.
Reverse transcription-quantitative PCR
results showing the effects of CX (100 nM for 48 h), IM (100 nM for
48 h), and their combination on the expression of KIF1B
isoforms in (A) NALM 6 and (B) THP 1 cells. Data were expressed as
mean ± standard error of the mean. *P<0.05, one-way
ANOVA, n=4 (for panel A) and 5 (for panel B). CX, CX-5461;
IM, imatinib; ANOVA, one-way analysis of variance.
Complete dataset of the raw count
values and FPKM values from RNA sequencing.
Primer sequences used for reverse
transcription-quantitative PCR.
Acknowledgements
The authors would like to thank Ms. Ye Chen and Ms.
Guopin Pan (Department of Physiology and Pathophysiology, School of
Basic Medical Sciences, Shandong University, Jinan, China) for
providing technical assistance.
Funding
Funding: This study was partially supported by research grants
from National Natural Science Foundation of China (grant nos.
82070265 and 82070382/82371574), Shandong Science and Technology
Development Project (grant no. 2017G006041), and Program of Taishan
Scholars Programme (grant no. 20190979).
Availability of data and materials
The original RNA sequencing data (in fastq format)
are freely accessible at Mendeley Data repository with the
following URL links: i) https://data.mendeley.com/datasets/n6jjyjbb76/1
(R1 data for Con-1/2/3 and IM-1 samples); ii) https://data.mendeley.com/datasets/b77ncvnb9d/1 (R1
data for IM-2/3 and IM+CX-1/2 samples); iii) https://data.mendeley.com/datasets/xfbrfhtt6p/1
(R1 data for IM+CX-3 sample and R2 data for Con-1/2/3 samples); iv)
https://data.mendeley.com/datasets/576tbt8m4b/1
(R2 data for IM-1/2/3 and IM+CX-1 samples); v) https://data.mendeley.com/datasets/ft27d96y63/1 (R2
data for IM+CX-2/3 samples).
Note: R1 and R2 represented the two files generated
by paired-end sequencing (i.e., 5' to 3' sequencing and 3' to 5'
sequencing respectively) for each sample.
The complete list of the raw count values and FPKM
values are presented in Table SI.
The datasets used and/or analyzed during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
CD was involved in experimentation, data
collection, and data analysis. JW was involved in experimentation
and data collection. XC was involved in experimental design and
data interpretation. BD was involved in analysis of the genomic
data, creating the illustrations, and manuscript revision. HG was
involved in data interpretation and project supervision. MC was
involved in experimental design, data interpretation, and project
supervision. FJ was involved in study conception, data
interpretation, manuscript writing, revision and approval. All
authors read and approved the final manuscript. CD and FJ confirm
the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Authors' information
ORCID: Professor Fan Jiang
(0000-0001-9466-2192)
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Butturini A, Arlinghaus RB and Gale RP:
BCR/ABL and leukemia. Leuk Res. 20:523–529. 1996.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kurzrock R, Kantarjian HM, Druker BJ and
Talpaz M: Philadelphia chromosome-positive leukemias: From basic
mechanisms to molecular therapeutics. Ann Intern Med. 138:819–830.
2003.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Maru Y: Molecular biology of chronic
myeloid leukemia. Cancer Sci. 103:1601–1610. 2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Patel AB, O'Hare T and Deininger MW:
Mechanisms of resistance to ABL kinase inhibition in chronic
myeloid leukemia and the development of next generation ABL kinase
inhibitors. Hematol Oncol Clin North Am. 31:589–612.
2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
de Lavallade H, Apperley JF, Khorashad JS,
Milojkovic D, Reid AG, Bua M, Szydlo R, Olavarria E, Kaeda J,
Goldman JM and Marin D: Imatinib for newly diagnosed patients with
chronic myeloid leukemia: Incidence of sustained responses in an
intention-to-treat analysis. J Clin Oncol. 26:3358–3363.
2008.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Burchert A, Wang Y, Cai D, von Bubnoff N,
Paschka P, Müller-Brüsselbach S, Ottmann OG, Duyster J, Hochhaus A
and Neubauer A: Compensatory PI3-kinase/Akt/mTor activation
regulates imatinib resistance development. Leukemia. 19:1774–1782.
2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wendel HG, de Stanchina E, Cepero E, Ray
S, Emig M, Fridman JS, Veach DR, Bornmann WG, Clarkson B, McCombie
WR, et al: Loss of p53 impedes the antileukemic response to BCR-ABL
inhibition. Proc Natl Acad Sci USA. 103:7444–7449. 2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Li L, Wang L, Wang Z, Ho Y, McDonald T,
Holyoake TL, Chen W and Bhatia R: Activation of p53 by SIRT1
inhibition enhances elimination of CML leukemia stem cells in
combination with imatinib. Cancer Cell. 21:266–281. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Peterson LF, Lo MC, Liu Y, Giannola D,
Mitrikeska E, Donato NJ, Johnson CN, Wang S, Mercer J and Talpaz M:
Induction of p53 suppresses chronic myeloid leukemia. Leuk
Lymphoma. 58:1–14. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Abraham SA, Hopcroft LE, Carrick E, Drotar
ME, Dunn K, Williamson AJ, Korfi K, Baquero P, Park LE, Scott MT,
et al: Dual targeting of p53 and c-MYC selectively eliminates
leukaemic stem cells. Nature. 534:341–346. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Guinn BA, Smith M, Padua RA, Burnett A and
Mills K: Changing p53 mutations with the evolution of chronic
myeloid leukaemia from the chronic phase to blast crisis. Leuk Res.
19:519–525. 1995.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Marasca R, Longo G, Luppi M, Barozzi P and
Torelli G: Double P53 point mutation in extramedullary blast crisis
of chronic myelogenous leukemia. Leuk Lymphoma. 16:171–175.
1994.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Stuppia L, Calabrese G, Peila R,
Guanciali-Franchi P, Morizio E, Spadano A and Palka G: p53 loss and
point mutations are associated with suppression of apoptosis and
progression of CML into myeloid blastic crisis. Cancer Genet
Cytogenet. 98:28–35. 1997.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mitelman F: The cytogenetic scenario of
chronic myeloid leukemia. Leuk Lymphoma. 11 (Suppl 1):S11–S15.
1993.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Luo J, Solimini NL and Elledge SJ:
Principles of cancer therapy: Oncogene and non-oncogene addiction.
Cell. 136:823–837. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Janczar S, Nautiyal J, Xiao Y, Curry E,
Sun M, Zanini E, Paige AJ and Gabra H: WWOX sensitises ovarian
cancer cells to paclitaxel via modulation of the ER stress
response. Cell Death Dis. 8(e2955)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Nguyen HG, Conn CS, Kye Y, Xue L, Forester
CM, Cowan JE, Hsieh AC, Cunningham JT, Truillet C, Tameire F, et
al: Development of a stress response therapy targeting aggressive
prostate cancer. Sci Transl Med. 10(eaar2036)2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Boulon S, Westman BJ, Hutten S, Boisvert
FM and Lamond AI: The nucleolus under stress. Mol Cell. 40:216–227.
2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
James A, Wang Y, Raje H, Rosby R and
DiMario P: Nucleolar stress with and without p53. Nucleus.
5:402–426. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Drygin D, Lin A, Bliesath J, Ho CB,
O'Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK,
Siddiqui-Jain A, et al: Targeting RNA polymerase I with an oral
small molecule CX-5461 inhibits ribosomal RNA synthesis and solid
tumor growth. Cancer Res. 71:1418–1430. 2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hein N, Cameron DP, Hannan KM, Nguyen NN,
Fong CY, Sornkom J, Wall M, Pavy M, Cullinane C, Diesch J, et al:
Inhibition of Pol I transcription treats murine and human AML by
targeting the leukemia-initiating cell population. Blood.
129:2882–2895. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Bywater MJ, Poortinga G, Sanij E, Hein N,
Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K, et al:
Inhibition of RNA polymerase I as a therapeutic strategy to promote
cancer-specific activation of p53. Cancer Cell. 22:51–65.
2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Law JC, Ritke MK, Yalowich JC, Leder GH
and Ferrell RE: Mutational inactivation of the p53 gene in the
human erythroid leukemic K562 cell line. Leuk Res. 17:1045–1050.
1993.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Di Bacco AM and Cotter TG: p53 expression
in K562 cells is associated with caspase-mediated cleavage of c-ABL
and BCR-ABL protein kinases. Br J Haematol. 117:588–597.
2002.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Bi S, Hughes T, Bungey J, Chase A, de
Fabritiis P and Goldman JM: p53 in chronic myeloid leukemia cell
lines. Leukemia. 6:839–842. 1992.PubMed/NCBI
|
|
26
|
Chylicki K, Ehinger M, Svedberg H, Bergh
G, Olsson I and Gullberg U: p53-mediated differentiation of the
erythroleukemia cell line K562. Cell Growth Differ. 11:315–324.
2000.PubMed/NCBI
|
|
27
|
ENCODE Guidelines and Best Practices for
RNA-Seq. https://www.encodeproject.org/about/experiment-guidelines.
|
|
28
|
Real-time PCR handbook. https://www.thermofisher.com/content/dam/LifeTech/global/Forms/PDF/real-time-pcr-handbook.pdf.
|
|
29
|
Foucquier J and Guedj M: Analysis of drug
combinations: Current methodological landscape. Pharmacol Res
Perspect. 3(e00149)2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Picard S, Titier K, Etienne G, Teilhet E,
Ducint D, Bernard MA, Lassalle R, Marit G, Reiffers J, Begaud B, et
al: Trough imatinib plasma levels are associated with both
cytogenetic and molecular responses to standard-dose imatinib in
chronic myeloid leukemia. Blood. 109:3496–3499. 2007.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Arya AD, Wilson DI, Baralle D and Raponi
M: RBFOX2 protein domains and cellular activities. Biochem Soc
Trans. 42:1180–1183. 2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Braeutigam C, Rago L, Rolke A, Waldmeier
L, Christofori G and Winter J: The RNA-binding protein Rbfox2: An
essential regulator of EMT-driven alternative splicing and a
mediator of cellular invasion. Oncogene. 33:1082–1092.
2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Quentmeier H, Pommerenke C, Bernhart SH,
Dirks WG, Hauer V, Hoffmann S, Nagel S, Siebert R, Uphoff CC,
Zaborski M, et al: RBFOX2 and alternative splicing in B-cell
lymphoma. Blood Cancer J. 8(77)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Munirajan AK, Ando K, Mukai A, Takahashi
M, Suenaga Y, Ohira M, Koda T, Hirota T, Ozaki T and Nakagawara A:
KIF1Bbeta functions as a haploinsufficient tumor suppressor gene
mapped to chromosome 1p36.2 by inducing apoptotic cell death. J
Biol Chem. 283:24426–24434. 2008.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Schlisio S, Kenchappa RS, Vredeveld LC,
George RE, Stewart R, Greulich H, Shahriari K, Nguyen NV, Pigny P,
Dahia PL, et al: The kinesin KIF1Bbeta acts downstream from EglN3
to induce apoptosis and is a potential 1p36 tumor suppressor. Genes
Dev. 22:884–893. 2008.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Choo Z, Koh RY, Wallis K, Koh TJ, Kuick
CH, Sobrado V, Kenchappa RS, Loh AH, Soh SY, Schlisio S, et al:
XAF1 promotes neuroblastoma tumor suppression and is required for
KIF1Bβ-mediated apoptosis. Oncotarget. 7:34229–34239.
2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Li S, Fell SM, Surova O, Smedler E, Wallis
K, Chen ZX, Hellman U, Johnsen JI, Martinsson T, Kenchappa RS, et
al: The 1p36 tumor suppressor KIF 1Bβ is required for calcineurin
activation, controlling mitochondrial fission and apoptosis. Dev
Cell. 36:164–178. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Ando K, Yokochi T, Mukai A, Wei G, Li Y,
Kramer S, Ozaki T, Maehara Y and Nakagawara A: Tumor suppressor
KIF1Bβ regulates mitochondrial apoptosis in collaboration with
YME1L1. Mol Carcinog. 58:1134–1144. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Chen ZX, Wallis K, Fell SM, Sobrado VR,
Hemmer MC, Ramsköld D, Hellman U, Sandberg R, Kenchappa RS,
Martinson T, et al: RNA helicase A is a downstream mediator of
KIF1Bβ tumor-suppressor function in neuroblastoma. Cancer Discov.
4:434–451. 2014.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Gordon MA, Babbs B, Cochrane DR, Bitler BG
and Richer JK: The long non-coding RNA MALAT1 promotes ovarian
cancer progression by regulating RBFOX2-mediated alternative
splicing. Mol Carcinog. 58:196–205. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Wolf D, Tilg H, Rumpold H, Gastl G and
Wolf AM: The kinase inhibitor imatinib-an immunosuppressive drug?
Curr Cancer Drug Targets. 7:251–258. 2007.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Ferreira R, Schneekloth JS Jr, Panov KI,
Hannan KM and Hannan RD: Targeting the RNA polymerase I
transcription for cancer therapy comes of age. Cells.
9(266)2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Quin J, Chan KT, Devlin JR, Cameron DP,
Diesch J, Cullinane C, Ahern J, Khot A, Hein N, George AJ, et al:
Inhibition of RNA polymerase I transcription initiation by CX-5461
activates non-canonical ATM/ATR signaling. Oncotarget.
7:49800–49818. 2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Mars JC, Tremblay MG, Valere M, Sibai DS,
Sabourin-Felix M, Lessard F and Moss T: The chemotherapeutic agent
CX-5461 irreversibly blocks RNA polymerase I initiation and
promoter release to cause nucleolar disruption, DNA damage and cell
inviability. NAR Cancer. 2(zcaa032)2020.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Xu H, Di Antonio M, McKinney S, Mathew V,
Ho B, O'Neil NJ, Santos ND, Silvester J, Wei V, Garcia J, et al:
CX-5461 is a DNA G-quadruplex stabilizer with selective lethality
in BRCA1/2 deficient tumours. Nat Commun. 8(14432)2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Bruno PM, Lu M, Dennis KA, Inam H, Moore
CJ, Sheehe J, Elledge SJ, Hemann MT and Pritchard JR: The primary
mechanism of cytotoxicity of the chemotherapeutic agent CX-5461 is
topoisomerase II poisoning. Proc Natl Acad Sci USA. 117:4053–4060.
2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Lipton JH, Brümmendorf TH,
Gambacorti-Passerini C, Garcia-Gutiérrez V, Deininger MW and Cortes
JE: Long-term safety review of tyrosine kinase inhibitors in
chronic myeloid leukemia-What to look for when treatment-free
remission is not an option. Blood Rev. 56(100968)2022.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Honda H, Ushijima T, Wakazono K, Oda H,
Tanaka Y, Aizawa Si, Ishikawa T, Yazaki Y and Hirai H: Acquired
loss of p53 induces blastic transformation in
p210(bcr/abl)-expressing hematopoietic cells: A transgenic study
for blast crisis of human CML. Blood. 95:1144–1150. 2000.PubMed/NCBI
|
|
49
|
Yan S, Xuan J, Brajanovski N, Tancock MRC,
Madhamshettiwar PB, Simpson KJ, Ellis S, Kang J, Cullinane C,
Sheppard KE, et al: The RNA polymerase I transcription inhibitor
CX-5461 cooperates with topoisomerase 1 inhibition by enhancing the
DNA damage response in homologous recombination-proficient
high-grade serous ovarian cancer. Br J Cancer. 124:616–627.
2021.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Shi S, Luo H, Wang L, Li H, Liang Y, Xia
J, Wang Z, Cheng B, Huang L, Liao G and Xu B: Combined inhibition
of RNA polymerase I and mTORC1/2 synergize to combat oral squamous
cell carcinoma. Biomed Pharmacother. 133(110906)2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Devlin JR, Hannan KM, Hein N, Cullinane C,
Kusnadi E, Ng PY, George AJ, Shortt J, Bywater MJ, Poortinga G, et
al: Combination therapy targeting ribosome biogenesis and mRNA
translation synergistically extends survival in MYC-driven
lymphoma. Cancer Discov. 6:59–70. 2016.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Lawrence MG, Porter LH, Choo N, Pook D,
Grummet JP, Pezaro CJ, Sandhu S, Ramm S, Luu J, Bakshi A, et al:
CX-5461 sensitizes DNA damage repair-proficient castrate-resistant
prostate cancer to PARP inhibition. Mol Cancer Ther. 20:2140–2150.
2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Rebello RJ, Kusnadi E, Cameron DP, Pearson
HB, Lesmana A, Devlin JR, Drygin D, Clark AK, Porter L, Pedersen J,
et al: The dual inhibition of RNA Pol I transcription and PIM
kinase as a new therapeutic approach to treat advanced prostate
cancer. Clin Cancer Res. 22:5539–5552. 2016.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Lehman SL, Schwartz KR, Maheshwari S,
Camphausen K and Tofilon PJ: CX-5461 induces radiosensitization
through modification of the DNA damage response and not inhibition
of RNA polymerase I. Sci Rep. 12(4059)2022.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Khot A, Brajanovski N, Cameron DP, Hein N,
Maclachlan KH, Sanij E, Lim J, Soong J, Link E, Blombery P, et al:
First-in-Human RNA polymerase I transcription inhibitor CX-5461 in
patients with advanced hematologic cancers: Results of a phase I
dose-escalation study. Cancer Discov. 9:1036–1049. 2019.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Lindqvist CM, Nordlund J, Ekman D,
Johansson A, Moghadam BT, Raine A, Övernäs E, Dahlberg J, Wahlberg
P, Henriksson N, et al: The mutational landscape in pediatric acute
lymphoblastic leukemia deciphered by whole genome sequencing. Hum
Mutat. 36:118–128. 2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Ochiai H, Takenobu H, Nakagawa A,
Yamaguchi Y, Kimura M, Ohira M, Okimoto Y, Fujimura Y, Koseki H,
Kohno Y, et al: Bmi1 is a MYCN target gene that regulates
tumorigenesis through repression of KIF1Bbeta and TSLC1 in
neuroblastoma. Oncogene. 29:2681–2690. 2010.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Astolfi A, Vendemini F, Urbini M,
Melchionda F, Masetti R, Franzoni M, Libri V, Serravalle S, Togni
M, Paone G, et al: MYCN is a novel oncogenic target in pediatric
T-cell acute lymphoblastic leukemia. Oncotarget. 5:120–130.
2014.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Taylor JS, Zeki J, Ornell K, Coburn J,
Shimada H, Ikegaki N and Chiu B: Down-regulation of MYCN protein by
CX-5461 leads to neuroblastoma tumor growth suppression. J Pediatr
Surg. 54:1192–1197. 2019.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Wetzel R, Goss VL, Norris B, Popova L,
Melnick M and Smith BL: Evaluation of CML model cell lines and
imatinib mesylate response: Determinants of signaling profiles. J
Immunol Methods. 305:59–66. 2005.PubMed/NCBI View Article : Google Scholar
|