Introduction
Acute pancreatitis (AP) is characterized by
pancreatic acinar cell death and persistent local or systemic
inflammation. The incidence of the disease is increasing and the
mortality is high (1). Currently,
the complex pathogenesis of AP is still unclear. Studies have
confirmed that mitochondrial Ca2+ overload is associated
with acinar cell death in AP (2,3).
However, most of the current studies regarding the pathogenesis of
AP primarily focus on acinar cells, while only a few studies focus
on pancreatic ductal epithelial cells (PDECs), which is another
important cell type closely related to AP.
PDECs play a key role in maintaining pancreatic
homeostasis and avoiding substantial pancreatic inflammation
(4-6).
Studies have shown that transient high pressure, intracellular
Ca2+ overload, and cytoskeletal changes of PDECs can
aggravate AP (7-9).
In addition, PDECs are an important component of the pancreatic
ductal mucosal barrier (PDMB) and play an important role in
preventing abnormal activation of trypsinogen and the reflux of
digestive enzymes to the pancreas (5,10).
Furthermore, impaired ductal fluid secretion and a lower
intraluminal pH lead to zymogen activation and acinar cell
dysfunction (11,12). Therefore, the decrease of PDMB
function caused by PDECs damage may be the main cause of pancreatic
injury and AP occurrence. Studying PDECs will help to uncover the
underlying mechanisms of the development of AP.
A previous study from our group has identified
intracellular Ca2+ overload in PDECs in AP (9). As one of the most important
intracellular signal transduction molecules, Ca2+ is
involved in almost every physiological activity within cells,
including mitophagy. Mitophagy is the selective autophagy of
mitochondria whereby dysfunctional mitochondria are transported to
lysosomes for degradation (13)
and it has a crucial role in mitochondrial quality and quantity
control as well as cell survival. Mitochondrial Ca2+
overload is related to the mitochondrial structure, mitochondrial
reactive oxygen species, mitochondrial membrane potential (MMP),
and ATP production, which all contribute to the initiation of
mitophagy (14).
Recent studies have shown that mitophagy within
pancreatic acinar cells plays an important role in the pathogenesis
and development of AP. This includes both PTEN-induced putative
kinase 1 (PINK1)/Parkin-activated mitophagy and vacuole membrane
protein 1-related mitophagy (15,16).
However, whether similar mechanisms exist in PDECs remains to be
elucidated. Intracellular Ca2+ overload in acinar cells
leads to mitochondrial depolarization through the opening of the
mitochondrial permeability transition pore (17) and these damaged mitochondria can be
removed by mitophagy. However, the relationship between
mitochondrial Ca2+ overload and mitophagy in PDECs
during AP has not been investigated.
The mitochondrial calcium uniporter (MCU), as a
highly selective Ca2+ uptake channel, transports
Ca2+ into the mitochondrial matrix along an
electrochemical gradient (18). As
such, the MCU plays a crucial role in mitochondrial Ca2+
homeostasis and mitochondrial energy metabolism. There has been
convincing evidence to suggest a close relationship between the MCU
and mitophagy. Guan et al (19) found that upregulation of the MCU
inhibited mitochondrial fusion and mitophagy through the
calpain/optic atrophy type 1 signaling pathway, leading to
myocardial ischemia-reperfusion injury. Chen et al (20) found that proliferation stimulated
by apelin-13 in vascular smooth muscle cells was regulated by
mitophagy that was caused by MCU-associated mitochondrial
Ca2+ uptake. The MCU may be a key protein that links
mitochondrial Ca2+ overload and mitophagy. A previous
study by our group indicated that MCU expression and mitochondrial
Ca2+ were increased in the rat model of AP (21). However, the exact mechanism by
which MCU regulates mitophagy through mitochondrial Ca2+
regulation in AP remains to be elucidated.
Caerulein (CAE), an analog of cholecystokinin, has
been widely used in vitro and in vivo to establish
models of AP (22). Our previous
studies found that the change in protein expression of autophagy
marker, the apoptosis and pyroptosis protein expression of NLPR3,
cleaved caspase-1 and cleaved caspase-3, the tight junction protein
expression of ZO-1 and occludin in HPDE6-C7 cells treated with CAE
in vitro were consistent with the rat model of AP (8,9,23,24).
In the present study, it was hypothesized that MCU-associated
mitochondrial Ca2+ overload promotes mitophagy by
regulating the PINK1/Parkin pathway in AP. To test this hypothesis,
CAE-treated HPDE6-C7 cells were used to imitate the pathological
changes of PDECs in AP. In addition, ruthenium red (RR) is an
inorganic polycationic dye that has been reported as an MCU
inhibitor (25). It was first used
to stain specific plant material and was subsequently found to be a
voltage-sensitive calcium channel inhibitor (26,27).
Therefore, RR was used as an MCU-specific inhibitor to assess the
effect of MCU on mitophagy in the present study.
Materials and methods
Cell culture
Normal human pancreatic ductal epithelial cells
(HPDE6-C7) were obtained from WHELAB (https://www.whelab.com/pro/580.html; cat. no. C1248;
passage 3) and cultured in Dulbecco's Modified Eagle's Medium
(DMEM; cat. no. C11995500BT; Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (cat. no. S711-001S;
Shanghai Shuangru Biotechnology Co., Ltd.), 100 U/ml penicillin,
and 0.1 mg/ml streptomycin (cat. no. P1400; Beijing Solarbio
Science & Technology Co., Ltd.). Cells were placed in an
incubator with sterile humidified air with 5% CO2 at
37˚C. Cells were treated at the same time with CAE (100 nmol/l;
cat. no. EY5113; Shanghai Amquar Biological Technology Co., Ltd.)
and RR (10 µmol/l; cat. no. 00541; MilliporeSigma) for 24 h. CAE
and RR were both dissolved in phosphate-buffered saline for the
experiments. Cells were divided into four groups according to the
type of treatment: CAE, RR, CAE+RR, and control.
Flow cytometry
Following treatment with RR (1, 5, 10, 50 and 100
µmol/l) for 24 h, the rate of apoptosis in HPDE6-C7 cells was
detected using the annexin V-FITC/PI kit [cat. no. AT101;
Multisciences (Lianke) Biotech Co., Ltd.], following the
manufacturer's instructions. In brief, after digestion with an
accutase solution, the cells were resuspended in binding buffer,
then incubated with 5 µl of annexin V-FITC and 10 µl of PI for 5
min at 25˚C in an incubator. Cell apoptosis rates were calculated
as the percentage of early + late apoptotic cells and measured by
flow cytometry (FACSVerse, BD Biosciences) and scatter diagrams
were visualized using FlowJo software version 10.8.1 (FlowJo
LLC).
Cell Counting Kit (CCK-8) assay
HPDE6-C7 cells were plated in a 96-well plate and
cultured for 6 h. After treatment with RR (1, 5, 10, 50 and 100
µmol/l) for 24 h at 37˚C, the cell medium was replaced with a
mixture of fresh medium (90%) and CCK-8 reagent (10%). Cells were
incubated for 1 h at 37˚C in the dark, and the absorbance was
measured at 450 nm.
Western blotting
Protein was isolated from HPDEE6-C7 cells using RIPA
lysis buffer (cat. no. R0020; Beijing Solarbio Science &
Technology Co., Ltd.) supplemented with 1 mmol/l PMSF, and the
protein concentration was measured by a BCA Protein Assay Kit (cat.
no. P0012; Beyotime Institute of Biotechnology). Equivalent amounts
of protein (50 µg/well) were separated by 10 or 15% SDS-PAGE. The
gels were electroblotted onto PVDF membranes (162-0219; Bio-Rad
Laboratories, Inc.). Next, membranes were placed in nonfat milk
(cat. no. P0216; Beyotime Institute of Biotechnology) for 40 min at
room temperature and incubated at 4˚C for 16-20 h with primary
antibodies, including: MCU (cat. no. 14997S; Cell Signaling
Technology, Inc.; 1:1,000), LC3 (cat. no. ab48394; Abcam; 1:1,000),
SQSTM1/p62 (cat. no. ab109012; Abcam; 1:15,000), and translocase of
the outer mitochondrial membrane complex subunit 20 (cat. no.
TOMM20; ab186735; Abcam; 1:2,000), PINK1 (cat. no. ab300623; Abcam;
1:1,000), Parkin (cat. no. 14060-1-AP; Proteintech Group, Inc.;
1:1,000), β-Actin (cat. no. 4970S; Cell Signaling Technology, Inc.;
1:1,000), and GAPDH (cat. no. ab9485; Abcam; 1:2,500). The
following day, the membranes were incubated with the DyLight
800-conjugated secondary anti-rabbit antibody for 1 h at room
temperature (cat. no. 5151; Cell Signaling Technology, Inc.;
1:10,000). Proteins were visualized using the Odyssey infrared
imaging system (LI-COR, Inc.), and the gray value of the protein
band was analyzed using ImageJ software (version 1.53e; National
Institutes of Health).
Transmission electron microscopy
(TEM)
HPDE6-C7 cells were immersed in electron microscope
fixation fluid for 2.5 h at 4˚C for fixation and stained in 1%
osmium acid for 2 h at room temperature. After dehydration, cells
were embedded in epoxy resin at 37˚C overnight. Epoxy resin samples
were heated at 60˚C for 48 h, and slices (80 nm) were obtained
using an ultramicrotome (EM UC7; Leica Microsystems GmbH). After
counterstaining sequentially with 2% uranium acetate and 2.6% lead
citrate for 8 min at room temperature, images were observed by TEM
(HT7700; Hitachi High-Technologies Corporation).
Measurement of mitochondrial membrane
potentials (MMP)
MMP changes in HPDE6-C7 cells were measured with an
MMP assay kit and JC-1 (cat. no. C2006; Beyotime Institute of
Biotechnology). Briefly, cells were loaded with the JC-1 working
solution (X1) at 37˚C for 20 min. After washing with pre-cooled
staining buffer, cells were immediately observed using fluorescence
microscopy (Olympus Corporation). Fluorescent images were analyzed
using ImageJ software (version 1.53e; National Institutes of
Health).
Measurement of mitochondrial
Ca2+
Levels of mitochondrial Ca2+ in HPDE6-C7
cells were identified using Rhod-2 AM (cat. no. 40776ES50, Shanghai
Yeasen Biotechnology Co., Ltd.) and Mito-Tracker Green (MTG; cat.
no. C1048; Beyotime Institute of Biotechnology). After washing with
D-Hank's balanced salt solution (cat. no. H1046; Beijing Solarbio
Science & Technology Co., Ltd.), cells were loaded with the
Rhod-2 AM working solution (4 µmol/l) at 37˚C for 30 min.
Subsequently, cells were loaded with MTG (200 nmol/l) at 37˚C for
30 min and immediately observed with a fluorescence microscope
(Olympus Corporation). Fluorescent images were analyzed using
ImageJ software (version 1.53e; National Institutes of Health).
Fluorescence colocalization assay of
mitochondria and lysosomes
To visualize the formation of autolysosomes during
mitophagy, MTG and Lyso-Tracker Red (LTR; cat. no. C1046; Beyotime
Institute of Biotechnology) were used to label the mitochondria and
lysosomes, respectively. Cells were cultured in confocal dishes
(BS-15-GJM, Biosharp) and stained in medium containing 200 nmol/l
MTG for 30 min and 75 nmol/l LTR for 15 min at 37˚C. Confocal
fluorescent images were obtained using a laser scanning confocal
microscope (TCS SP8 STED; Leica Microsystems GmbH) with a x63 oil
immersion objective.
Immunofluorescence
At room temperature, HPDE6-C7 cells were fixed in 4%
paraformaldehyde for 15 min, then permeabilized with 0.1% Triton
X-100 for 5 min. After treatment with blocking buffer for 30 min,
cells were incubated at 4˚C overnight with primary antibodies,
including LC3 (cat. no. ab48394; Abcam; 1:1,000), Parkin (cat. no.
14060-1-AP; Proteintech Group, Inc.; 1:500), and TOMM20 (cat. no.
66777-1-Ig; Proteintech Group, Inc.; 1:500). Subsequently, cells
were incubated with Alexa Fluor 555 (cat. no. 4413S; Cell Signaling
Technology, Inc.; 1:1,000) and Alexa Fluor 488 (cat. no. 550036,
Chengdu Zen-Bioscience Co., Ltd.; 1:1,000)-conjugated secondary
antibodies for 1 h at room temperature in the dark. Fluorescent
images were obtained using a fluorescence microscope (Carl Zeiss
AG).
Statistical analysis
All experiments were repeated at least three times.
Data were presented as the mean ± standard deviation, and SPSS 26
software (IBM Corp.) was used to perform all statistical analyses.
Pearson's correlation coefficient (PCC) was determined as a
statistic for quantifying colocalization using the ImageJ plugin
Colocalization Finder. Student's t-test or one-way analysis
of variance followed by Tukey's test was used to determine the
statistical significance between indicated groups. P<0.05 was
considered to indicate a statistically significant difference.
Cartograms were generated using GraphPad Prism 9 software
(Dotmatics).
Results
RR inhibited MCU expression in
CAE-treated HPDE6-C7 cells
RR has been reported as a specific inhibitor of MCU
(28). To identify a working
nontoxic concentration of RR for the current study, the cytotoxic
effect of RR in HPDE6-C7 cells was evaluated using a CCK-8 assay
and flow cytometry. The results of the CCK-8 assay revealed
decreased cell viability with higher concentrations of RR (50 or
100 µmol/l; Fig. 1A). Flow
cytometry also confirmed that apoptosis was increased in cells
exposed to 50 or 100 µmol/l RR (Fig.
1B and C). Thus, the maximum
nontoxic concentration of RR (10 µmol/l) was used in subsequent
experiments. Additionally, western blotting revealed that CAE
increased the expression level of MCU and RR treatment restrained
MCU expression in CAE-treated cells (Fig. 1D and E).
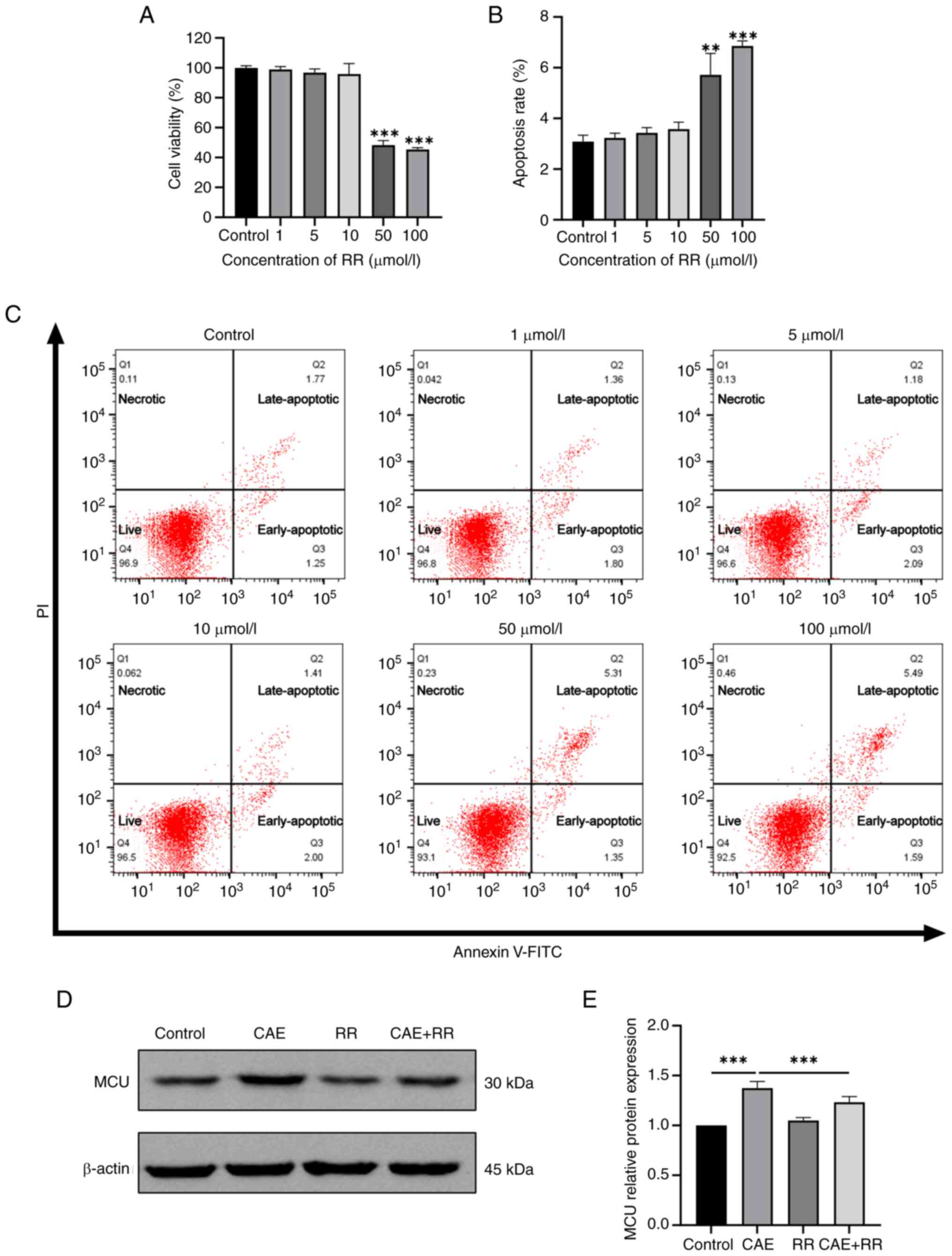 | Figure 1Effects of RR on the viability and
apoptosis of HPDE6-C7 cells, and MCU expression in CAE-treated
HPDE6-C7 cells. HPDE6-C7 cells were treated with RR (1, 5, 10, 50
and 100 µmol/l) for 24 h. (A) CCK-8 assay was used to detect cell
viability. (B and C) Cell apoptosis was detected by flow cytometry.
Q1: Necrotic cells, Q2: Late-apoptotic cells, Q3: Early-apoptotic
cells, Q4: Live cells. HPDE6-C7 cells were treated with 10 µmol/l
RR and 100 nmol/l CAE for 24 h. (D) Western blot and (E)
semiquantitative analysis of MCU expression. Data were presented as
the mean ± standard deviation. **P<0.01,
***P<0.001 vs. control. All experiments were repeated
at least three times. RR, ruthenium red; MCU, mitochondrial calcium
uniporter; CAE, caerulein. |
Inhibition of MCU attenuated
CAE-induced mitochondrial Ca2+ accumulation and MMP
decline in HPDE6-C7 cells
The effect of CAE on mitochondrial Ca2+
levels and MMP in HPDE6-C7 cells was explored using fluorescence
microscopy. The results of PCC indicated that the Rhod-2-loaded
Ca2+ were strongly correlated with the Mito-Tracker
(Fig. 2C). Therefore, it can be
speculated that the Rhod-2-loaded Ca2+ may come from
mitochondria. The results indicated that treatment of HPDE6-C7
cells with CAE overtly increased mitochondrial Ca2+
accumulation (Fig. 2A and B). Furthermore, cells were loaded with
JC-1 to measure the MMP. The red/green fluorescence density was
decreased in CAE-treated cells, indicating that treatment with CAE
induced MMP depolarization (Fig.
2D and E). Notably, RR
effectively attenuated mitochondrial Ca2+ accumulation
and restored MMP in CAE-treated cells. These data demonstrated that
CAE promotes MCU-associated mitochondrial Ca2+ overload
and MMP decline.
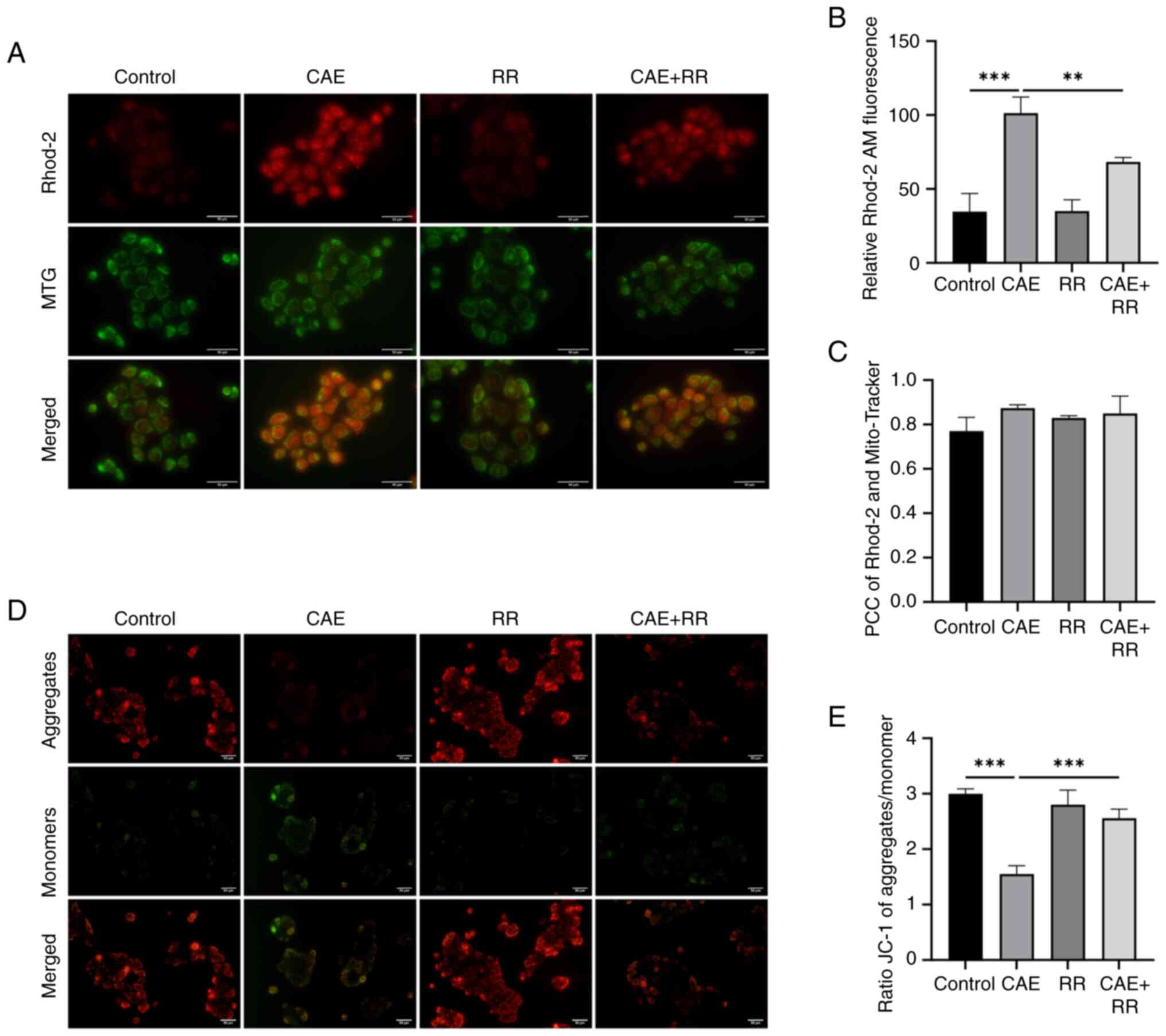 | Figure 2RR treatment inhibited mitochondrial
Ca2+ accumulation and reduced MMP decline in HPDE6-C7
cells. HPDE6-C7 cells were treated with 10 µmol/l RR and 100 nmol/l
CAE for 24 h. (A and B) Changes in mitochondrial Ca2+
were detected by labeling with Rhod-2 AM and MTG and observed under
a fluorescence microscope (magnification, x400; scale bar, 50 µm).
(C) PCC was used to calculate the correlation between Rhod-2 AM and
MTG. No correlation: PCC=0; weak: PCC <0.4; moderate: 0.4 ≤PCC
<0.7; strong: 0.7 ≤PCC<1; complete or perfect: PCC=1. (D and
E) Changes in MMP were detected by staining with JC-1 and
observation under a fluorescence microscope (magnification, x200;
scale bar, 50 µm). Data were presented as the mean ± standard
deviation. **P<0.01, ***P<0.001 vs.
control. All experiments were repeated at least three times. RR,
ruthenium red; MMP, mitochondrial membrane potential; CAE,
caerulein; MTG, Mito-Tracker Green; PCC, Pearson's correlation
coefficient. |
Inhibition of MCU attenuated
CAE-induced mitophagy in HPDE6-C7 cells
The effect of MCU on mitophagy in AP was
investigated by western blotting, immunofluorescence, fluorescence
probes and TEM. The results of western blotting indicated that CAE
improved the LC3-II/I ratio and reduced the expression of p62 and
TOMM20. In turn, the LC3-II/I ratio was reduced and the expression
of p62 and TOMM20 were increased by RR treatment in CAE-treated
cells (Fig. 3A and B). In addition, the results of the
immunofluorescence assays showed that the colocalization of LC3 and
TOMM20 was increased in the CAE-treated cells, but this increased
colocalization was decreased by treating the cells with RR
(Fig. 3C and D). Moreover, the fluorescence images
showed that the colocalization of mitochondria and lysosomes was
increased in CAE-treated cells, which indicate an increased fusion
of mitophagosomes and lysosomes. However, RR treatment reduced the
overlap of mitochondrial and lysosomal staining (Fig. 4A and B). Furthermore, autophagosomes during
mitophagy and impaired mitochondria were also detected by TEM and
RR treatment reduced the formation of mitophagosomes and damaged
mitochondria (Fig. 4C). These
results demonstrated that elevated mitophagy in CAE-treated cells
can be reduced by RR treatment.
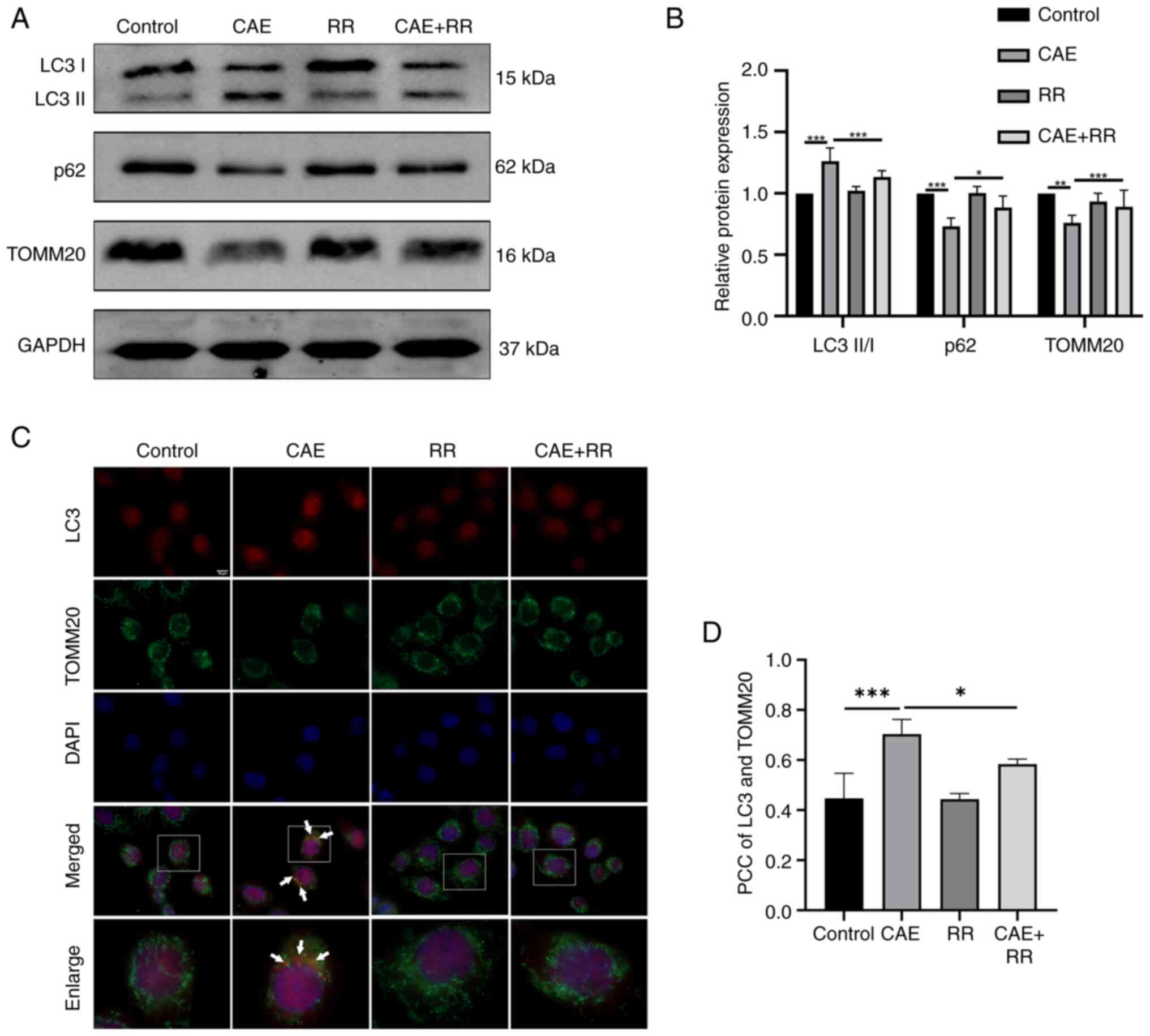 | Figure 3RR treatment inhibited CAE-induced
mitophagy in HPDE6-C7 cells. HPDE6-C7 cells were treated with 10
µmol/l RR and 100 nmol/l CAE for 24 h. (A and B) Expression of
microtubule-associated proteins 1A/1B LC3B, p62 and TOMM20 were
detected by western blotting. (C and D) TOMM20 (a mitochondrial
marker) colocalization with LC3 was observed by fluorescence
microscopy (magnification, x1,000; scale bar, 10 µm). The PCC was
used to calculate the colocalization between LC3 and TOMM20. No
correlation: PCC=0; weak: PCC <0.4; moderate: 0.4 ≤PCC <0.7;
strong: 0.7 ≤PCC<1; complete or perfect: PCC=1. Data were
presented as the mean ± standard deviation. *P<0.05,
**P<0.01, ***P<0.001 vs. control. All
experiments were repeated at least three times. RR, ruthenium red;
CAE, caerulein; TOMM20, translocase of the outer mitochondrial
membrane complex subunit 20; PCC, Pearson's correlation
coefficient. |
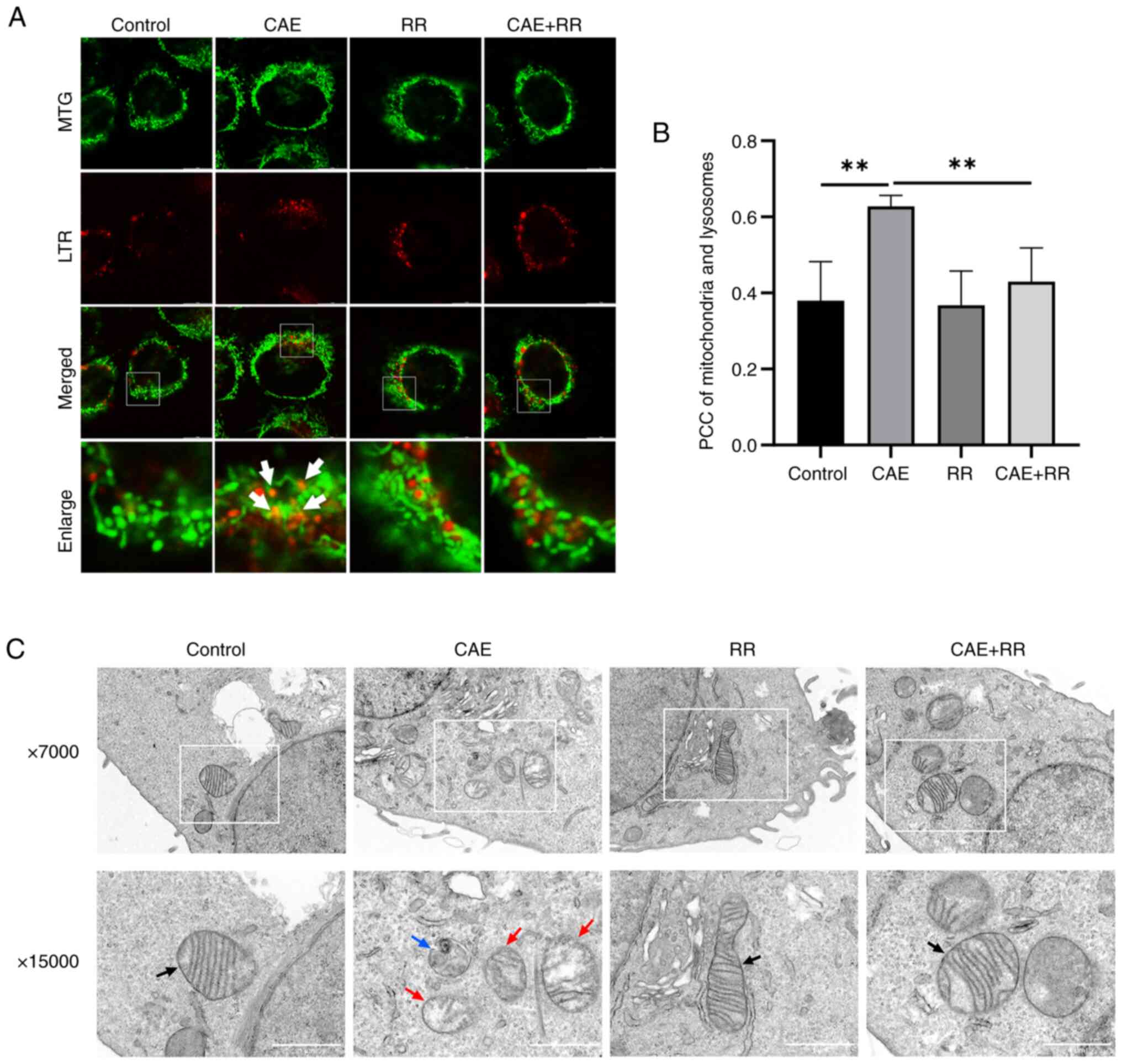 | Figure 4RR treatment reduced CAE-induced
mitophagosome formation in HPDE6-C7 cells. (A and B) Mitochondrial
colocalization with lysosomes was observed by fluorescence confocal
microscopy (magnification, x630; scale bar, 10 µm). The PCC was
used to calculate the colocalization between mitochondria and
lysosomes. No correlation: PCC=0; weak: PCC <0.4; moderate: 0.4
≤PCC <0.7; strong: 0.7 ≤PCC<1; complete or perfect: PCC=1.
(C) Mitophagy and damaged mitochondria in HPDE6-C7 cells were
observed by transmission electron microscopy. Black arrows indicate
normal mitochondria. Red arrows indicate damaged mitochondria and
the blue arrow indicates mitophagy (magnification, x15,000; scale
bar, 1.0 µm). Data were presented as the mean ± standard deviation.
**P<0.01 vs. control. All experiments were repeated
at least three times. RR, ruthenium red; CAE, caerulein; TOMM20,
translocase of the outer mitochondrial membrane complex subunit 20;
PCC, Pearson's correlation coefficient; MTG, Mito-Tracker Green;
LTR, Lyso-Tracker Red. |
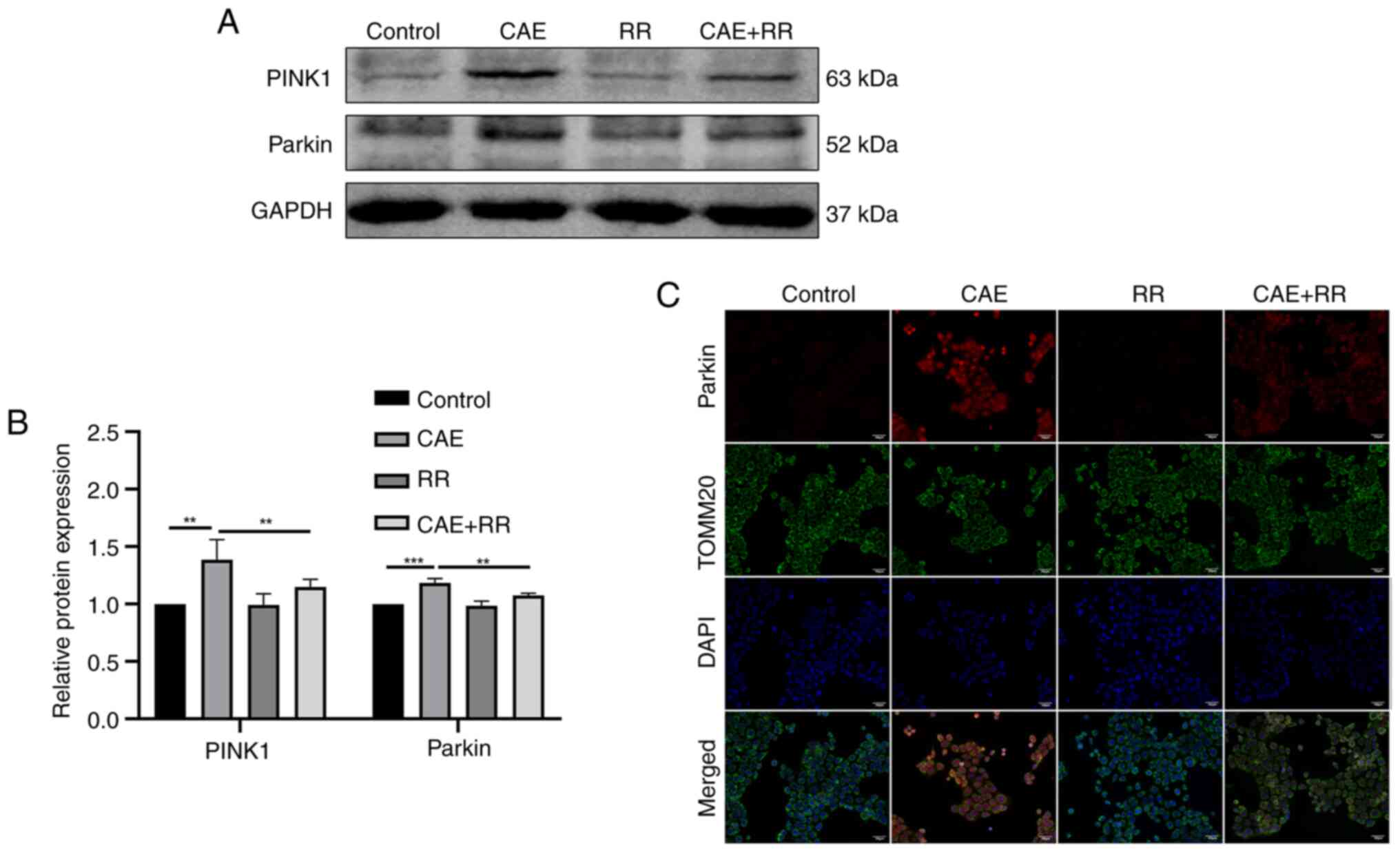
Inhibition of MCU attenuated
PINK1/Parkin pathway activation in HPDE6-C7 cells
The effect of MCU on the PINK1/Parkin pathway in AP
was investigated by western blotting and immunofluorescence. The
results of western blotting showed that CAE increased the
expression of PINK1 and Parkin, but this increased expression could
be reversed by treating the cells with RR (Fig. 5A and B). Moreover, the immunofluorescence of
Parkin and TOMM20 indicated that the expression of Parkin was
increased in CAE-treated cells, demonstrating that CAE promoted the
translocation of Parkin into the mitochondrial membrane. As
expected, this effect was alleviated by RR treatment (Fig. 5C).
Discussion
Previous studies have confirmed that mitophagy is
involved in the pathogenesis of AP (15,29,30).
In addition, MCU-associated mitochondrial Ca2+ overload
plays a key role in AP (31,32).
However, the effect of MCU on mitophagy in AP remains ambiguous.
The present study was the first to investigate the regulatory role
of MCU on mitophagy in AP, to the best of the authors' knowledge.
The data indicate that activated MCU may promote mitophagy by
regulating the PINK1/Parkin pathway in PDECs in AP in
vitro.
Studies have demonstrated that mitochondrial
Ca2+ overload plays a crucial role in the initiation of
acinar cell death in AP (2,3,17).
As an essential channel for mitochondrial Ca2+ uptake,
the MCU may play an important role during mitophagy in AP. The
results in the present study showed that CAE increased MCU
overexpression, mitochondrial Ca2+ accumulation, and MMP
depolarization. RR is a potent inhibitor of MCU (33). In the present study, the inhibition
of the MCU by RR efficiently attenuated mitochondrial
Ca2+ accumulation and restored MMP depolarization. These
results demonstrated that reducing MCU-dependent mitochondrial
Ca2+ uptake could improve MMP depolarization in PEDCs in
AP. Notably, RR could not decrease the mitochondrial
Ca2+ level in normal cells in the present study.
Similarly, in our previous study of AP in rats, RR inhibited
mitochondrial Ca2+ overload in the AP group, but did not
reduce mitochondrial Ca2+ in the control group (21). Moreover, in Jiang et al
(34), RR inhibited mitochondrial
Ca2+ overload promoted by aconitine, but did not reduce
mitochondrial Ca2+ in mouse hippocampal neuron HT22
cells. These phenomena may be due to the fact that RR does not
interfere with the cytoplasmic Ca2+ signal propagation
to the mitochondria in normal cells (27). In addition, the gating of MCU by
the mitochondrial calcium uptake 1 may have a barrier effect on RR
in normal cells (35).
Mitochondrial dysfunction and mitophagy have been
extensively studied in acinar cells in AP (17,36,37).
However, the presence of mitophagy in PDECs in AP and its
underlying mechanisms remain unclear. LC3-II as a molecular marker
of autophagy represents the relative number of autophagosomes and
p62 as an autophagy substrate is negatively correlated with
autophagy (38-40).
A reduction in the expression of TOMM20, which is a translocase of
the outer mitochondrial membrane 20, indicates that impaired
mitochondria may be removed by mitophagy. Our previous study found
that CAE induced autophagy activation in the early stage of AP
(8). RR mainly inhibits
mitochondrial Ca2+ and is recognized as a potent
inhibitor of MCU. Therefore, RR may have a significant effect on
mitophagy, which is one of the important forms of autophagy. In the
present study, the elevated expression of LC3-Ⅱ and the decreased
expression of p62 in CAE-treated cells suggested the presence and
active process of autophagy. In addition, the increased
colocalization of a mitochondrial marker (TOMM20) and LC3 as well
as the decreased expression of TOMM20 in the present study
suggested the increased level of mitophagy in CAE-treated cells.
These results were supported by both the TEM and fluorescence
microscopy results, where mitophagosomes were observed using TEM,
and an increased colocalization of mitochondria and lysosomes was
observed using fluorescence microscopy. These results indicated the
presence of excessive mitophagy in PEDCs in this in vitro
model of AP. Notably, according to the expression of TOMM20 and
autophagy markers, changes in mitophagosomes and colocalization of
TOMM20 and LC3 in CAE+RR-treated cells, RR was able to inhibit
excessive mitophagy in PEDCs in AP. Similarly, Liu et al
(41) found that the inhibition of
the MCU attenuated excessive mitophagy and autophagic cell death by
cadmium in liver cells. Yu et al (42) found that the inhibition of the MCU
by Ru360 can inhibit hyperactivated mitophagy and protect
neurocytes from ischemia/reperfusion injury. Another study has
suggested that depolarized mitochondria are the initiating factor
for mitophagy (43). In the
present study, the MCU inhibitor RR decreased MCU expression,
reduced mitochondrial Ca2+ overload and inhibited
mitochondrial depolarization in PDECs in AP. Therefore, it can be
hypothesized that the MCU can promote mitochondrial Ca2+
uptake and mitochondrial depolarization and further lead to
mitophagy. However, whether RR exerts effects on other types of
autophagy has not been reported and needs further study.
The PINK1/Parkin pathway plays an important role in
the removal of dysfunctional mitochondria. First, PINK1 accumulates
and is phosphorylated in the outer membrane of impaired
mitochondria. Subsequently, phosphorylated PINK1 mediates Parkin
translocation to the outer mitochondrial membrane and activates its
ubiquitin ligase activity, causing the ubiquitination of a range of
mitochondrial proteins and the formation of autophagosomes
(13,43,44).
Zhang et al (15) found
that PINK1 and Parkin were elevated in the CAE-AP mouse model as
well as the arginine-SAP mouse model at 24 h, but were decreased in
the arginine-SAP mouse model at 48 and 72 h, suggesting that
mitophagy is activated in the early stages of AP but inhibited in
the late stages. The data in the present study showed that CAE
promoted the expression of PINK1 and Parkin in HPDE6-C7 cells. In
addition, the immunofluorescence results showed an increased
translocation of Parkin to the mitochondrial outer membrane in
CAE-treated cells. These changes were all reduced when the cells
were treated with RR. Together, these results demonstrated that the
MCU may promote mitophagy by regulating the PINK1/Parkin pathway in
PDECs in AP. However, one limitation of the present study was the
lack of manipulation experiments directly involving the PINK/Parkin
pathway, such as the use of PINK1 inhibitors and knockout/knockdown
of PINK1 or Parkin.
Either impaired mitophagy or excessive mitophagy has
an important effect on cell death (45,46).
The current study supported the hypothesis that mitochondrial
Ca2+ overload by the MCU causes the accumulation of
depolarized mitochondria and activates excessive mitophagy in PDECs
in AP. Excessive mitophagy can lead to an imbalance in oxidative
phosphorylation, oxidative stress and eventual cell death (47). Therefore, the present study
provided a possible mechanism by which autophagic cell death of
PDECs may disrupt the PDMB and aggravate AP. However, an important
limitation of the present study was that cell death inhibitors,
such as 3-MA (for autophagic cell death) (48), z-VAD (for apoptosis) (49) and Nec-1 (for necrosis) (50), were not used to detect the presence
of autophagic cell death.
In addition to the inhibitor RR, the MCU activator
spermine was used in the rat model of AP in our previous study
(51). The results of the
pre-experiment showed that spermine at different concentrations
(2.5, 5.0, and 10.0 mg/kg) had no effect on the pathological score
of pancreatic tissue and the amylase levels in the rat model of AP.
In addition, RR effectively inhibits the MCU but also inhibits a
range of other channels, including the two-pore-domain
K+ channels (tandem pore domain acid-sensitive
K+ channel 3, Twik-related K+ channel 2 and
Twik-related arachidonic acid-stimulated K+ channel),
transient receptor potential receptor vanilloid subfamily, calcium
homeostasis modulator channels, ryanodine receptor, and Piezo
channels (25,52). Therefore, a superior way to
investigate the role of MCU would be to use MCU knockout mice or
transfection of MCU short interfering RNA. Future work will assess
the regulatory effect of the MCU on mitophagy in AP using the above
methods.
In conclusion, the present study suggested that
MCU-associated mitochondrial Ca2+ overload may induce
mitophagy by regulating the PINK1/Parkin pathway in PDECs in AP
in vitro. MCU-associated mitochondrial Ca2+
overload and mitophagy are attractive therapeutic targets for AP in
the future.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81970558)
and a Scientific Research Project of the Guangxi Administration of
Traditional Chinese Medicine (grant no. GXZYA20220227).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YL and HYY were the main contributors to the present
study. YL performed experimental work, analyzed data and wrote the
manuscript. HYY revised the manuscript. GDT and HYY conceived the
present study and obtained funding. NM and YYQ designed the
experimental procedures. MTX, XLX and LL performed cell culture and
collected experimental data. YL and GDT confirm the authenticity of
all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shah AP, Mourad MM and Bramhall SR: Acute
pancreatitis: Current perspectives on diagnosis and management. J
Inflamm Res. 11:77–85. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Feng S, Wei Q, Hu Q, Huang X, Zhou X, Luo
G, Deng M and Lu M: Research progress on the relationship between
acute pancreatitis and calcium overload in acinar cells. Dig Dis
Sci. 64:25–38. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Pandol SJ and Gottlieb RA: Calcium,
mitochondria and the initiation of acute pancreatitis.
Pancreatology. 22:838–845. 2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hegyi P, Pandol S, Venglovecz V and
Rakonczay Z Jr: The acinar-ductal tango in the pathogenesis of
acute pancreatitis. Gut. 60:544–552. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Maleth J and Hegyi P: Calcium signaling in
pancreatic ductal epithelial cells: An old friend and a nasty
enemy. Cell Calcium. 55:337–345. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Hegyi P and Rakonczay Z Jr: The role of
pancreatic ducts in the pathogenesis of acute pancreatitis.
Pancreatology. 15 (Suppl 4):S13–S17. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wen L, Javed TA, Yimlamai D, Mukherjee A,
Xiao X and Husain SZ: Transient high pressure in pancreatic ducts
promotes inflammation and alters tight junctions via calcineurin
signaling in mice. Gastroenterology. 155:1250–1263.e5.
2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yang H, Liang Z, Xie J, Wu Q, Qin Y, Zhang
S and Tang G: Gelsolin inhibits autophagy by regulating actin
depolymerization in pancreatic ductal epithelial cells in acute
pancreatitis. Braz J Med Biol Res. 56(e12279)2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yang HY, Liang ZH, Xie JL, Wu Q, Qin YY,
Zhang SY and Tang GD: Gelsolin impairs barrier function in
pancreatic ductal epithelial cells by actin filament
depolymerization in hypertriglyceridemia-induced pancreatitis in
vitro. Exp Ther Med. 23(290)2022.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Konok GP and Thompson AG: Pancreatic
ductal mucosa as a protective barrier in the pathogenesis of
pancreatitis. Am J Surg. 117:18–23. 1969.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Freedman SD, Kern HF and Scheele GA:
Pancreatic acinar cell dysfunction in CFTR(-/-) mice is associated
with impairments in luminal pH and endocytosis. Gastroenterology.
121:950–957. 2001.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Bhoomagoud M, Jung T, Atladottir J,
Kolodecik TR, Shugrue C, Chaudhuri A, Thrower EC and Gorelick FS:
Reducing extracellular pH sensitizes the acinar cell to
secretagogue-induced pancreatitis responses in rats.
Gastroenterology. 137:1083–1092. 2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Onishi M, Yamano K, Sato M, Matsuda N and
Okamoto K: Molecular mechanisms and physiological functions of
mitophagy. EMBO J. 40(e104705)2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Perrone M, Patergnani S, Di Mambro T,
Palumbo L, Wieckowski MR, Giorgi C and Pinton P: Calcium
homeostasis in the control of mitophagy. Antioxid Redox Signal.
38:581–598. 2023.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhang J, Huang W, He Q, Deng T, Wu B,
Huang F, Bi J, Jin Y, Sun H, Zhang Q and Shi K: PINK1/PARK2
dependent mitophagy effectively suppresses NLRP3 inflammasome to
alleviate acute pancreatitis. Free Radic Biol Med. 166:147–164.
2021.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Vanasco V, Ropolo A, Grasso D, Ojeda DS,
García MN, Vico TA, Orquera T, Quarleri J, Alvarez S and Vaccaro
MI: Mitochondrial Dynamics and VMP1-Related Selective mitophagy in
experimental acute pancreatitis. Front Cell Dev Biol.
9(640094)2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Mukherjee R, Mareninova OA, Odinokova IV,
Huang W, Murphy J, Chvanov M, Javed MA, Wen L, Booth DM, Cane MC,
et al: Mechanism of mitochondrial permeability transition pore
induction and damage in the pancreas: Inhibition prevents acute
pancreatitis by protecting production of ATP. Gut. 65:1333–1346.
2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Baughman JM, Perocchi F, Girgis HS,
Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L,
Goldberger O, Bogorad RL, et al: Integrative genomics identifies
MCU as an essential component of the mitochondrial calcium
uniporter. Nature. 476:341–345. 2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Guan L, Che Z, Meng X, Yu Y, Li M, Yu Z,
Shi H, Yang D and Yu M: MCU Up-regulation contributes to myocardial
ischemia-reperfusion Injury through calpain/OPA-1-mediated
mitochondrial fusion/mitophagy Inhibition. J Cell Mol Med.
23:7830–7843. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chen Z, Zhou Q, Chen J, Yang Y, Chen W,
Mao H, Ouyang X, Zhang K, Tang M, Yan J, et al: MCU-dependent
mitochondrial calcium uptake-induced mitophagy contributes to
apelin-13-stimulated VSMCs proliferation. Vascul Pharmacol.
144(106979)2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Qin Y, Yang H, Wu Q, Xie J, Meng N, Lei Y
and Tang G: Effects of inhibiting the mitochondrial calcium
uniporter on the oxidative stress in rats with acute pancreatitis.
China J Modern Med. 32:1–7. 2022.
|
|
22
|
Lerch MM and Gorelick FS: Models of acute
and chronic pancreatitis. Gastroenterology. 144:1180–1193.
2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wei B, Gong Y, Yang H, Zhou J, Su Z and
Liang Z: Role of tumor necrosis factor receptorassociated factor 6
in pyroptosis during acute pancreatitis. Mol Med Rep.
24(848)2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang J, Qin M, Wu Q, Yang H, Wei B, Xie J,
Qin Y, Liang Z and Huang J: Effects of lipolysis-stimulated
lipoprotein receptor on tight junctions of pancreatic ductal
epithelial cells in hypertriglyceridemic acute pancreatitis. Biomed
Res Int. 2022(4234186)2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Marta K, Hasan P, Rodriguez-Prados M,
Paillard M and Hajnoczky G: Pharmacological inhibition of the
mitochondrial Ca(2+) uniporter: Relevance for pathophysiology and
human therapy. J Mol Cell Cardiol. 151:135–144. 2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Colombo PM and Rascio N: Ruthenium red
staining for electron microscopy of plant material. J Ultrastruct
Res. 60:135–139. 1977.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hajnóczky G, Csordás G, Das S,
Garcia-Perez C, Saotome M, Sinha Roy S and Yi M: Mitochondrial
calcium signalling and cell death: Approaches for assessing the
role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium.
40:553–560. 2006.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y,
Markhard AL, Grabarek Z, Kong L, Liu Z, Ouyang B, et al:
Architecture of the mitochondrial calcium uniporter. Nature.
533:269–273. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wen E, Xin G, Su W, Li S, Zhang Y, Dong Y,
Yang X, Wan C, Chen Z, Yu X, et al: Activation of TLR4 induces
severe acute pancreatitis-associated spleen injury via
ROS-disrupted mitophagy pathway. Mol Immunol. 142:63–75.
2022.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Piplani H, Marek-Iannucci S, Sin J, Hou J,
Takahashi T, Sharma A, de Freitas Germano J, Waldron RT,
Saadaeijahromi H, Song Y, et al: Simvastatin induces autophagic
flux to restore cerulein-impaired phagosome-lysosome fusion in
acute pancreatitis. Biochim Biophys Acta Mol Basis Dis.
1865(165530)2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Chvanov M, Voronina S, Zhang X, Telnova S,
Chard R, Ouyang Y, Armstrong J, Tanton H, Awais M, Latawiec D, et
al: Knockout of the mitochondrial calcium uniporter strongly
suppresses stimulus-metabolism coupling in pancreatic acinar cells
but does not reduce severity of experimental acute pancreatitis.
Cells. 9(1407)2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yu X, Dai C, Zhao X, Huang Q, He X, Zhang
R, Lin Z and Shen Y: Ruthenium red attenuates acute pancreatitis by
inhibiting MCU and improving mitochondrial function. Biochem
Biophys Res Commun. 635:236–243. 2022.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Kirichok Y, Krapivinsky G and Clapham DE:
The mitochondrial calcium uniporter is a highly selective ion
channel. Nature. 427:360–364. 2004.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Jiang C, Shen J, Wang C, Huang Y, Wang L,
Yang Y, Hu W, Li P and Wu H: Mechanism of aconitine mediated
neuronal apoptosis induced by mitochondrial calcium overload caused
by MCU. Toxicol Lett. 384:86–95. 2023.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Rodríguez-Prados M, Huang KT, Márta K,
Paillard M, Csordás G, Joseph SK and Hajnóczky G: MICU1 controls
the sensitivity of the mitochondrial Ca2+ uniporter to
activators and inhibitors. Cell Chem Biol. 30:606–617.e4.
2023.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Biczo G, Vegh ET, Shalbueva N, Mareninova
OA, Elperin J, Lotshaw E, Gretler S, Lugea A, Malla SR, Dawson D,
et al: Mitochondrial dysfunction, through impaired autophagy, leads
to endoplasmic reticulum stress, deregulated lipid metabolism, and
pancreatitis in animal models. Gastroenterology. 154:689–703.
2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Shalbueva N, Mareninova OA, Gerloff A,
Yuan J, Waldron RT, Pandol SJ and Gukovskaya AS: Effects of
oxidative alcohol metabolism on the mitochondrial permeability
transition pore and necrosis in a mouse model of alcoholic
pancreatitis. Gastroenterology. 144:437–446 e6. 2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Barth S, Glick D and Macleod KF:
Autophagy: Assays and artifacts. J Pathol. 221:117–124.
2010.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu
HL, Yang C and Liu HF: p62 links the autophagy pathway and the
ubiqutin-proteasome system upon ubiquitinated protein degradation.
Cell Mol Biol Lett. 21(29)2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Liu C, Li HJ, Duan WX, Duan Y, Yu Q, Zhang
T, Sun YP, Li YY, Liu YS and Xu SC: MCU upregulation overactivates
mitophagy by promoting VDAC1 dimerization and ubiquitination in the
hepatotoxicity of cadmium. Adv Sci (Weinh).
10(e2203869)2023.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Yu S, Zheng S, Leng J, Wang S, Zhao T and
Liu J: Inhibition of mitochondrial calcium uniporter protects
neurocytes from ischemia/reperfusion injury via the inhibition of
excessive mitophagy. Neurosci Lett. 628:24–29. 2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Shirihai OS, Song M and Dorn GW II: How
mitochondrial dynamism orchestrates mitophagy. Circ Res.
116:1835–1849. 2015.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Nguyen TN, Padman BS and Lazarou M:
Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends
Cell Biol. 26:733–744. 2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Sidarala V, Pearson GL, Parekh VS,
Thompson B, Christen L, Gingerich MA, Zhu J, Stromer T, Ren J, Reck
EC, et al: Mitophagy protects β cells from inflammatory damage in
diabetes. JCI Insight. 5(e141138)2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Rademaker G, Boumahd Y, Peiffer R, Anania
S, Wissocq T, Liegeois M, Luis G, Sounni NE, Agirman F,
Maloujahmoum N, et al: Myoferlin targeting triggers mitophagy and
primes ferroptosis in pancreatic cancer cells. Redox Biol.
53(102324)2022.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Kang SU, Kim DH, Lee YS, Huang M, Byeon
HK, Lee SH, Baek SJ and Kim CH: DIM-C-pPhtBu induces lysosomal
dysfunction and unfolded protein response-mediated cell death via
excessive mitophagy. Cancer Lett. 504:23–36. 2021.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Pasquier B: Autophagy inhibitors. Cell Mol
Life Sci. 73:985–1001. 2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Green DR: Caspase activation and
inhibition. Cold Spring Harb Perspect Biol.
14(a041020)2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Cao L and Mu W: Necrostatin-1 and
necroptosis inhibition: Pathophysiology and therapeutic
implications. Pharmacol Res. 163(105297)2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Qin Y YH and Tang G: Effects of activated
mitochondrial calcium uniporter on acute pancreatitis induced by
caerulein in rats. Electronic Journal of Clinical Medical
Literature. 8:8–11. 2021.
|
|
52
|
Ma J: Block by ruthenium red of the
ryanodine-activated calcium release channel of skeletal muscle. J
Gen Physiol. 102:1031–1056. 1993.PubMed/NCBI View Article : Google Scholar
|