Introduction
Erdheim-Chester disease (ECD), also known as
non-Langerhans histiocytosis, is a rare histiocytic neoplasm in
which >50% of patients have BRAF mutations (1-3).
The disease is prevalent in middle-aged and older adults, with a
median age of onset of 55 years (4). The 5-year survival rate of the
disease is 68%, and most deaths result from involvement of vital
organs, including the heart and central nervous system (5). Symmetrical osteosclerosis of the long
bones of both lower extremities occurs in >95% of patients, only
half of whom may experience bone pain. In addition, ECD may be
accompanied by other systemic manifestations, such as oliguria and
dysuria in perirenal infiltration, periaortic inflammation in
aortic involvement, myocarditis, arrhythmia, heart failure and even
myocardial infarction in cardiac involvement, ataxia and diabetes
insipidus in neurological involvement and macular tumors in skin
involvement (3). Misdiagnosis is
common as the condition affects multiple systems and manifests
variably in different patients (6).
Typical pathologic features are that the lesion
tissue is rich in foamy histiocytes surrounded by fibrosis or
xanthogranuloma and Touton giant cells are often seen (2). Whereas immunohistochemistry (IHC)
results show positive for CD68 and CD163 and negative for CD1a and
Langerin (CD207) (2,3). Notably, a review of the literature
revealed that pathological examination of some patients with ECD
may only show fibrosis with nonspecific inflammatory cell
infiltration while lacking typically foamy cells (3,7,8).
Therefore, the diagnosis needs to be based on histopathology,
combined with a comprehensive evaluation of clinical features,
imaging and the exclusion of other diseases (2).
Pegylated interferon α (PEG-IFN-α) agents remain the
first-line treatment for the disease; however, drugs which target
mutation sites such as BRAF, MEK and mTOR inhibitors can improve
survival in patients with positive genetic tests such as BRAF-V600E
(3,9). These targeted agents often have
toxicities associated with inhibition of the MAPK pathway (e.g.,
inducing a systemic inflammatory response) and thus have to be
discontinued. However, combining PEG-IFN-α agents with interleukin
1 inhibitors (e.g., anakinra) can prolong the use of targeted
agents by controlling toxicity and effectively mitigating the
inflammatory response (10,11).
Furthermore, corticosteroids can be used as adjuvants to improve
acute symptoms associated with tissue swelling; however, they
cannot improve survival rates (3,12).
Owing to the multifocal nature and insensitivity of ECD to
radiation, extensive surgery and radiotherapy are not recommended
(13). Clinicians should increase
awareness and diagnostic sensitivity of ECD, actively biopsy all
patients with suspected ECD and all of their affected tissues, and
perform genetic testing of diseased tissues to guide treatment
(3). The options for therapy need
to be individualized based on the sites of involvement, degree of
disease progression, and genetic results (14). To aid in addressing the gaps in
knowledge, the present study discussed a case report which details
the diagnosis and treatment process of a patient with ECD.
Case report
Presentation
In early 2017, a 31-year-old man was admitted to the
cardiology unit of Liaocheng People's Hospital (Shandong, China)
and found to have a moderate asymptomatic pericardial effusion.
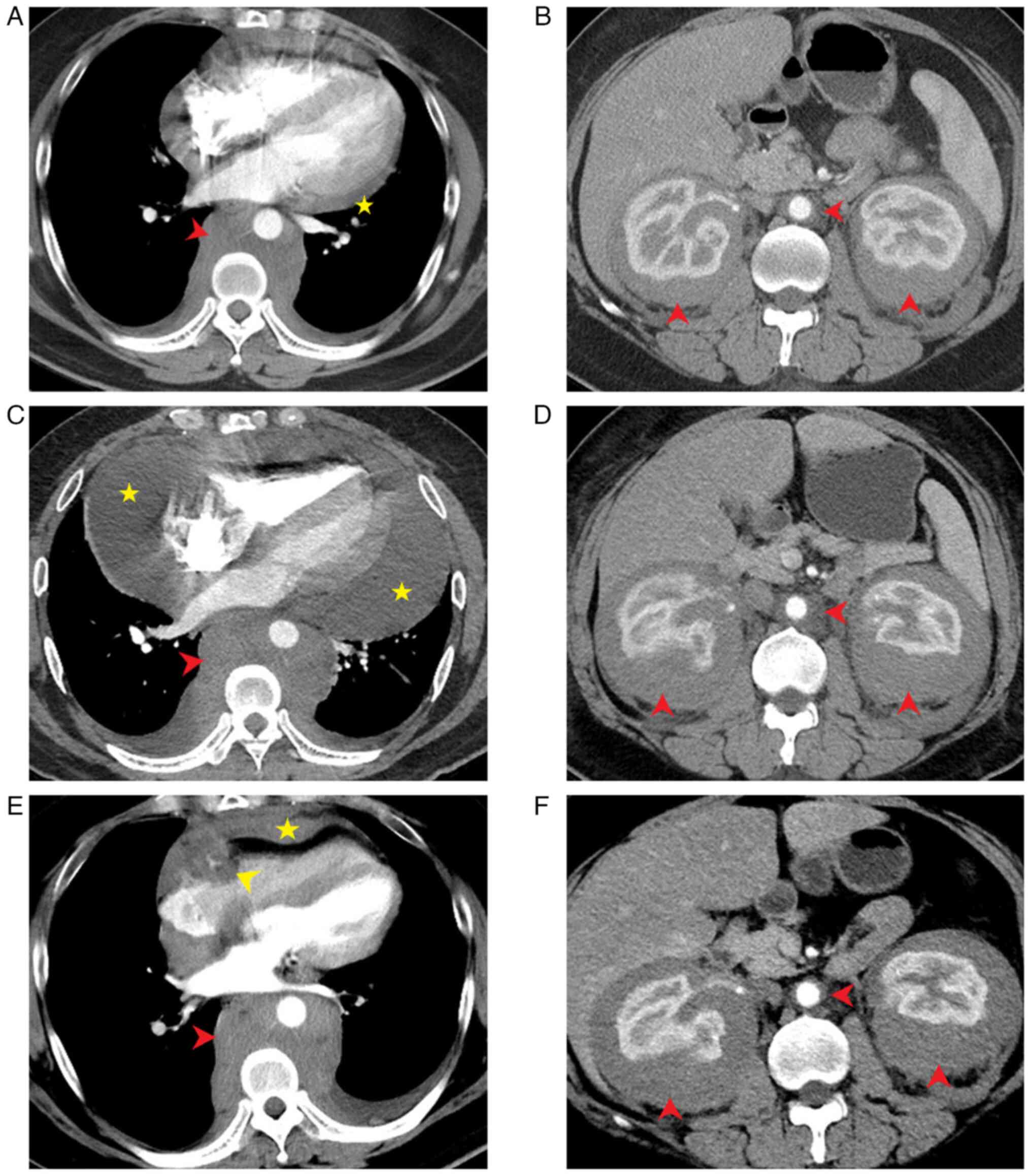
Subsequently, a CT scan of the patient's chest, abdomen and pelvis
was performed and the image showed aortic, mediastinal, and
pericardial thickening and pleural effusions. Symptomatic
treatments, including torasemide and spironolactone for diuresis,
leading to a reduction of pericardial effusion; valsartan
hydrochlorothiazide to lower blood pressure; metformin to lower
glucose; and fluvastatin to lower lipids, were performed (the
discharge records of that time did not contain detailed records of
the exact dosages of the medications, or the number of days of
their use; thus, it was not possible to present them in detail in
the present study). However, the treatments provided no
improvements regarding the pericardial effusion or retroperitoneal
mass. Following this, the patient was transferred to the First
Affiliated Hospital of Shandong First Medical University (Shandong,
China) for a mediastinal tissue biopsy which revealed fibrous
connective tissue with small vessel hyperplasia and an infiltrate
of acute-chronic inflammatory cells (images from this hospital were
unavailable). The patient was diagnosed with suspected fibrous
mediastinitis. The patient was not hospitalized for treatment and
arrived at the Rheumatology Unit of the First Affiliated Hospital
of Shandong First Medical University (Shandong, China) in April
2017 for further examination.
Subsequent CT (GE Discovery CT 750 HD; Cytiva)
images revealed a retroperitoneal soft-tissue mass growing around
the bilateral ureter, thoracic aorta, and part of the abdominal
aorta (Fig. 1A and B). Blood test results showed the presence
of an inflammatory response, as evidenced by a white blood cell
(WBC) count of 14.54x109/l (normal range:
3.5-9.5x109/l), neutrophil count of
9.99x109/l (normal range: 1.8-6.3x109/l),
erythrocyte sedimentation rate of 16 mm/h (normal range: 1-15 mm/h)
and C-reactive protein (CRP) of 9.45 mg/l (normal range: 0-3.48
mg/l). Antinuclear and antineutrophilic cytoplasmic antibodies were
negative. IgG4 levels and liver and kidney functions were within
normal limits. Physical examination revealed no palpable masses or
lymphadenopathy in the chest or abdomen. Based on these findings,
RPF was diagnosed. The comorbidities of the patient included
hypertension, type-2 diabetes and fatty liver, with no history of
cancer or autoimmune disease and no family history of cancer or
autoimmune disease. In order to reduce inflammation and shrink
fibrotic masses, the patient was administered intravenous
methylprednisolone 60 mg once daily for 11 days. Following this,
the patient was administered oral prednisone 50 mg once daily and
oral tamoxifen 10 mg three times daily for 7 days, which was
subsequently changed to 20 mg three times daily. Thereafter, the
patient presented to the First Affiliated Hospital of Shandong
First Medical University twice for follow-up examinations. CT (GE
Discovery CT 750 HD; Cytiva) showed that the retroperitoneal mass
was unchanged. After 6 months, the patient felt that there was no
improvement in his condition and discontinued oral corticosteroid
therapy without consulting the physician.
The patient remained asymptomatic until November
2021, when the patient developed decreased urine output, bilateral
eyelid edema and fever. A physical examination revealed distant
heart sounds. Blood tests were unremarkable other than a WBC count
of 13.21x109/l, neutrophil count of
10.0x109/l, CRP level of 19.4 mg/l and aspartate
aminotransferase level of 18.6 U/l (normal range: 9-50 U/l). CT (GE
Discovery CT 750 HD; Cytiva) images revealed expansion of the
periaortic soft tissue mass (Fig.
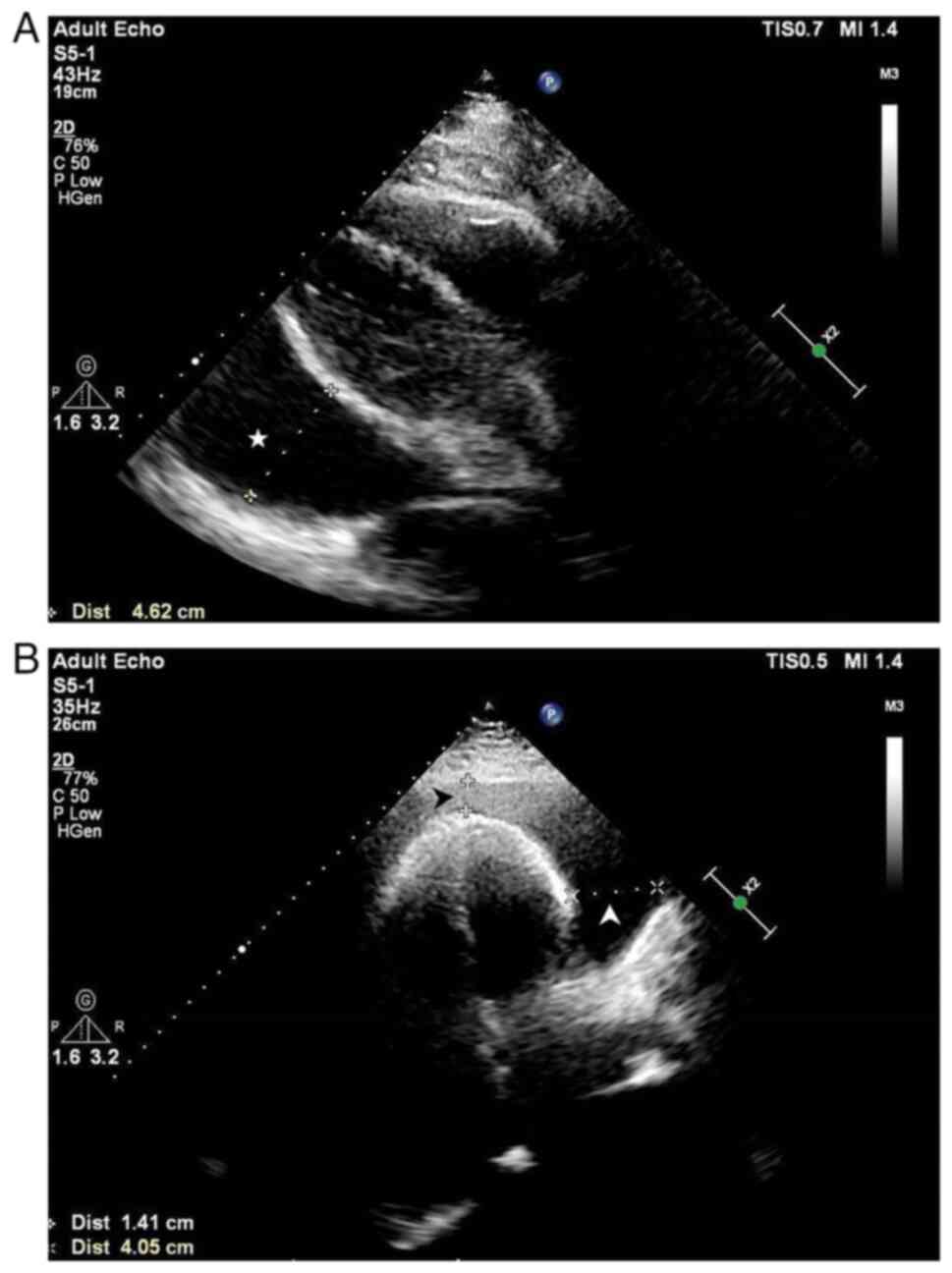
1C and D). Echocardiography
(Philips EPIQ 7C; Philips Medical Systems B.V.) revealed a large
pericardial effusion with a heart swing syndrome (Fig. 2). No findings indicated any
evidence of malignancy or tuberculosis. Subsequently, the patient
received treatment for RPF, comprising intravenous
methylprednisolone 40 mg (once daily) and oral tamoxifen 10 mg
(twice daily). Pericardiocentesis was performed, allowing the
drainage of 1,500 ml of pericardial fluid. The image obtained from
a repeat cardiac ultrasound scan suggested trace pericardial
effusion, thickening of the right ventricular lateral epicardium
layer (to ~7 mm), and reduced right ventricular systolic function.
After applying the above regimen for 6 days, the patient was
discharged with improvement, and the hormone therapies were
switched to oral prednisone 50 mg (once daily), with a reduction of
1 tablet per week, and oral tamoxifen was adjusted to 20 mg (twice
daily), with the addition of oral cyclophosphamide (once every
other day) as a second-line agent for long-term maintenance
therapy, and the patient was advised to repeat the examination
after 1 month.
After 6 months of irregular oral treatment with the
above medications, a repeat CT scan (Fig. 1B and E) showed no significant changes in the
retroperitoneal mass in the patient. However, it was observed that
the patient's aortic involvement was circumferential, which was
inconsistent with an RPF diagnosis, as RPF only involves the
anterior and lateral parts of the aorta. Thus, it was considered
that RPF might not be the most appropriate diagnosis for the
present patient. The case was reviewed to develop new diagnostic
and treatment options. During a review of the CT images, diffusely
proliferating soft tissues involving the heart, thoracic and
abdominal aorta and both kidneys were observed (Fig. 1A-F; red arrows). Specifically, the
right atrioventricular groove pseudotumor, ‘coated aorta’ and
‘hairy kidney’ signs were observed, which are the typical imaging
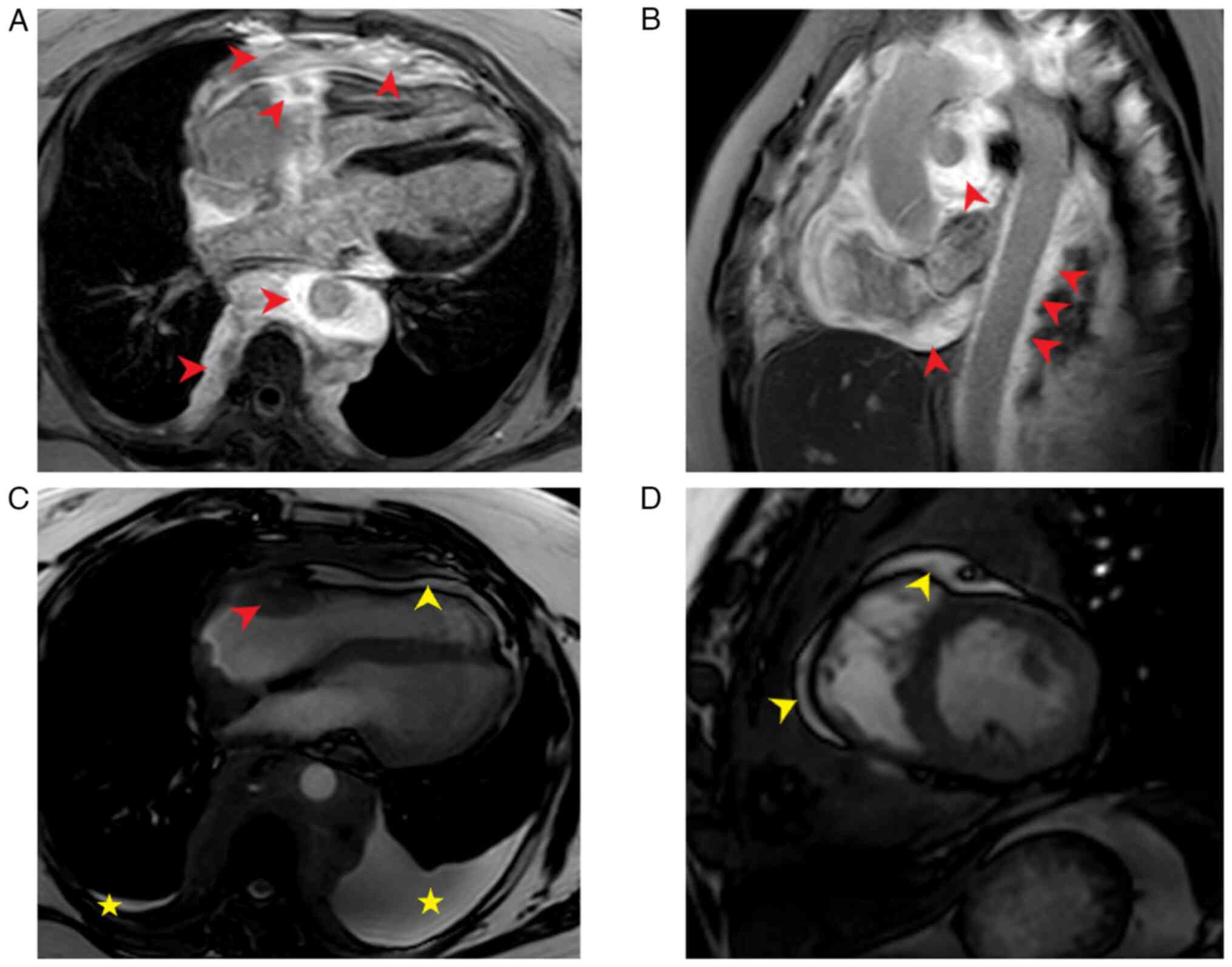
manifestations of ECD. Therefore, cardiac magnetic resonance (CMR;
Philips Ingenia CX 3.0T; Philips Medical Systems B.V.) imaging,
whole-body bone scanning (GE Infinia; Cytiva) and a biopsy of the
perirenal tissue were performed. CMR imaging revealed a diffuse
long T1 and slightly prolonged T2 signal around the mediastinum and
pericardium. Lesion tissues encircled the large mediastinal vessels
and the heart, resulting in significant cardiac compression. The
right coronary artery was involved and the left ventricular
diastolic function was visibly restricted. The findings were
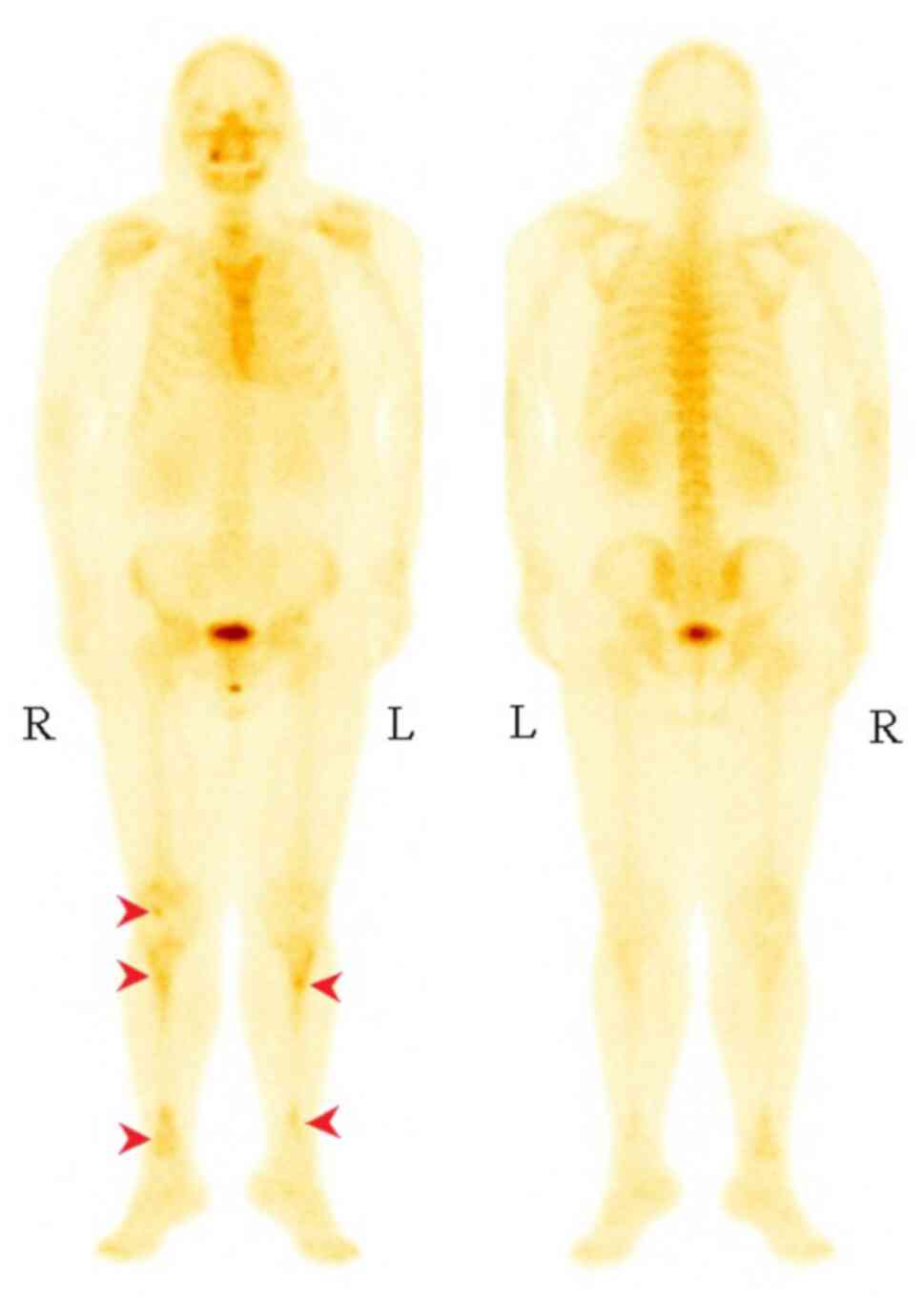
consistent with constrictive pericarditis (Fig. 3). Whole-body anteroposterior planar
imaging performed via technetium methylene diphosphonate
single-photon emission CT (99mTc-MDP-SPECT/CT) revealed increased
bone metabolism in the right lower femur and bilateral tibia and
slightly increased bone metabolism in the left sixth anterior rib,
thoracolumbar body, bilateral sacroiliac joint and bilateral hip
joint (Fig. 4). While puncturing
the perirenal tissue, it was found that the tissues were very
dense. Due to the difficult penetration of the puncture needle to
penetrate deep into the tissue, a small quantity of tissue samples
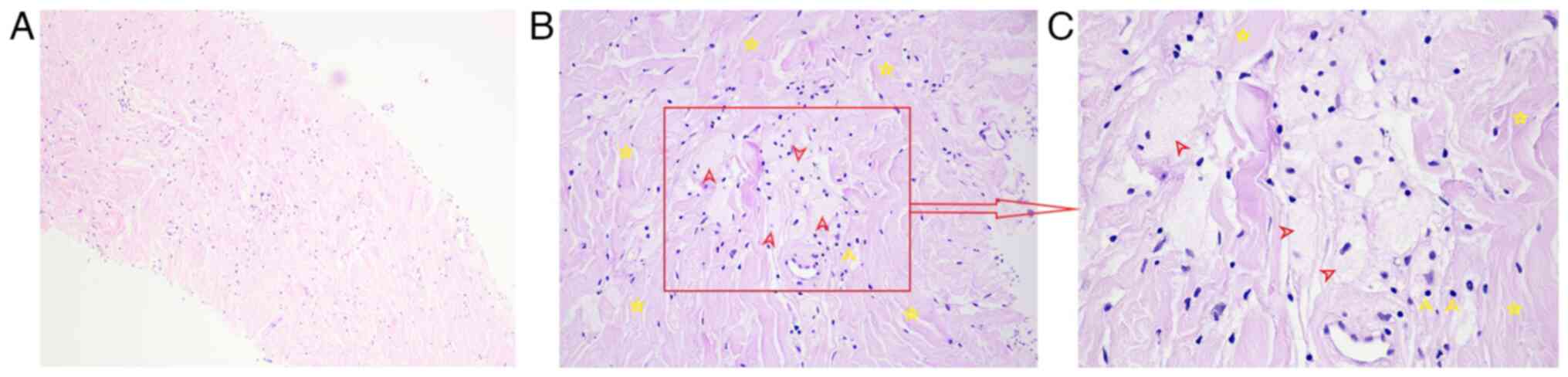
were obtained from the lesion surface. The pathological result
showed hyperplastic dense fibrous connective tissue with scattered
individual lymphocytes and phagocytes (HE staining, magnification,
x100; Fig. 5A). To avoid the risks
associated with multiple invasive surgeries, biopsies were not
performed on all diseased tissues. Since no histiocytes were found,
IHC or genetic testing were not performed on the pathologic
tissues. Thus, differential diagnosis was critical.
Differential diagnosis
The patient's physical examination and ancillary
findings did not support conditions, such as malignancy,
tuberculosis, heart failure and liver disease, and other diseases
that may result in pericardial or pleural effusions, or diseases,
such as lymphoma, sarcoma, IgG4-related disease, and multiple
myeloma, which may result in retroperitoneal masses. The
‘coated-aorta’ on CT can exclude diseases involving the aortic
wall, such as Takayasu disease, Horton disease, and infectious
aortitis. In differentiating RPF from ECD, the presentation of
bilateral lower extremity bone hypermetabolism, ‘hairy kidney’, and
circumferential changes of aorta on CT were more supportive of an
ECD diagnosis. At this point, the diagnosis was still uncertain.
Thus, the pathological images were reviewed again and some typical
foamy histiocytes of ECD surrounded by fibrosis were found when the
magnification was adjusted to x200 and x400 (Fig. 5B and C). Hematological and cranial examinations
did not reveal any involvement of the adrenal glands, pituitary
gland, central nervous system, or orbit. Finally, the patient was
diagnosed with ECD. IFN-α therapy was recommended; however, the
patient declined this option and chose to continue hormone and
cyclophosphamide treatment with regular follow-up CT scans.
Outcome and follow-up
A recent CT (GE Revolution CT; Cytiva) performed in
February 2023 revealed smaller lesions around the thoracoabdominal
aorta compared with the previous findings (Fig. 1E and F). After re-communication, the patient
consented to the administration of subcutaneous PEG-IFN-α-2b 135u
(once weekly), oral prednisone 20 mg (once daily), oral tamoxifen
10 mg (twice daily) and oral cyclophosphamide 50 mg (once every
other day) from February 2023. Until January 2024, the patient has
consistently adhered to this regimen of treatment without
experiencing any discomfort. The patient has been advised to come
to the hospital for review at the patient's convenience.
Radiographic technique
The present study selected images of the
phase-sensitive inversion recovery turbo field echo breath-hold
(PSIR-TFE-BH) sequence and the sense-balanced turbo field
echo-BH-cine sequence obtained using Philips Ingenia CX 3.0T
scanners (Philips Medical Systems B.V.). The PSIR-TFE-BH parameters
were as follows: Repetition time, 6.13 msec; echo time, 2.99 msec;
slice thickness, 2 mm; scan percentage: 74.1%; TFE factor, 20; TFE
shots, 6; field of view (FOV), 300 mm, 233.3 Hz; ACQ matrix MxP,
188x139; and scan time, 13 sec. The sBTFE-BH-cine parameters were
as follows: repetition time, 2.7 msec; echo time, 1.36 msec; slice
thickness, 3 mm; scan percentage: 94.7%; TFE factor, 18; TFE shots,
5; FOV, 300 mm, 1442.6 Hz; ACQ matrix MxP, 152x144; and scan time,
6 sec. The GE Infinia camera system (Cytiva) was used for SPECT.
The reagent used was 99mTc-MDP; route, intravenous injection; dose,
20 mCi; time elapsed, 4 h; scan area, anterior and posterior
position; collimator, low-energy high-resolution; collimator mode,
H mode; enter machine mode, enter head-first; energy peak, 140 kev;
time, 10 min. The GE Discovery CT 750 HD scanner (Cytiva) was used
for CT; scan type, helical; voltage, 120 kV; routine time, 0.7 sec;
scan duration, 9.62 sec; detector coverage. 40 mm; helical
thickness. 5 mm; coverage speed. 56.25 mm/sec; pitch, 0.984:1. The
GE Revolution CT scanner (Cytiva) was also be used; scan type,
helical; voltage, 120 kV; routine time, 0.5 sec; scan duration,
12.30 sec; detector coverage, 80 mm; helical thickness, 5 mm;
coverage speed, 158.75 mm/sec; pitch, 0.992:1. The Philips EPIQ 7C
system (Philips Medical Systems B.V.) was used with the following
specifications employed for echocardiography: model, Adult Echo;
ultrasound probe, S5-1; ultrasonic frequency, 43 Hz; penetration
distance, 19 cm; thermal index of soft tissue, 0.7; mechanical
index, 1.4.
Literature search
A search of PubMed (https://pubmed.ncbi.nlm.nih.gov) was performed using
the keywords ‘Erdheim-Chester’ or ‘non-Langerhans,’
‘retroperitoneal fibrosis’ or ‘idiopathic retroperitoneal fibrosis’
or ‘chronic peri-aortitis,’ ‘hairy kidney,’ ‘coated-aorta,’
‘pericardial effusion,’ ‘histiocytosis,’ ‘Langerhans,’ and ‘Rosai
Dorfman’, for English publications up to October 20, 2023, without
other restrictions on publication date. The reference sections of
the included articles were also searched and the titles and
abstracts from all sources screened.
Discussion
ECD is a rare form of non-Langerhans histiocytosis
characterized by mutant clonal histiocytes that provoke nonspecific
fibrosis and inflammation followed by infiltration of long bone
marrow and/or tissues of a number of other organs (3). BRAF is a common mutation site,
occurring in >50% of patients with ECD, with the primary
affected pathway being MAPK-ERK. The identification of gene
mutations provides evidence for their clonal properties (2). As systemic histiocytosis, ECD, along
with Langerhans cell histiocytosis (LCH) and Rosai-Dorfman disease
(RDD), have been classified as low-grade histiocytic/dendritic
neoplasms in the fifth edition of the World Health Organization
Classification of Hematolymphoid Tumors (1). By 2024, >1,500 cases of ECD are
estimated to have been reported in the medical literature. Early
diagnosis is challenging due to its broad and nonspecific clinical
presentation, low prevalence, inconsistent pathological findings,
and lack of clinician awareness of the disease (7,15).
The typical pathology of ECD is characterized by a
diffuse infiltrate of foamy or lipid-laden histiocytes admixed or
surrounded by fibrosis or xanthogranuloma and Touton giant cells
are often present (3,8). The histiocytes are positive for CD68
and CD163 (the macrophage or dendritic cell markers, which are also
positive in LCH and RDD), negative for CD1a and Langerin (CD207;
which are typically positive in LCH), and negative or weakly
positive for the S-100 protein (which is positive in RDD) (2). This tissue pathology can affect a
number of organs due to its susceptibility to connective, adipose,
and perivascular tissues (6). At
the molecular level, in addition to the most common BRAF600E
mutation, other MAPK pathway-related genes, such as PIK3CA,
MAP2K1, N/KRAS and MAP3K1, are also involved
(14). The presence of BRAF
mutations, which has a similar probability in ECD and LCH, but
rarely in other histiocytosis, can be useful for differentiating
ECD from other non-LCH (8,15). In a study by De Abreu et al,
histological analysis revealed a much greater extent of ECD than
that revealed by magnetic resonance imaging (MRI) or radiology
(16). Variable components include
histiocytic infiltrates and the surrounding stroma (3). It has also been suggested that the
BRAF mutation rate could be 100% if every patient with ECD
completed genetic testing (8).
Therefore, biopsies of lesion tissues and genetic testing should be
performed in all cases of ECD to confirm the diagnosis and
establish the mutational status to potentially guide therapy
(3). Moreover, the BRAF
mutations are expressed in foamy tissue and Touton giant cells, but
not in fibroblasts, lymphocytes, or endothelial cells, suggesting a
need for multiple core biopsies and to expand the scope of lesion
biopsies to optimize tissue yield for histopathological review and
molecular testing (3,17).
In the present case, tissue biopsies were performed
at different times on two different lesion sites that showed only
fibrosis with inflammatory cell infiltration, lacking the typical
histiocytes of ECD, which poses a diagnostic challenge. The
literature was reviewed (3,7,8)
and it was found that the ECD histopathology may lack the typical
foamy histiocytes and show only nonspecific inflammation mixed with
fibrosis or even only fibrosis lamellae mixed with a few
histiocytes, which is consistent with both of the biopsies in the
present study. Therefore, the ECD diagnosis cannot be made solely
based on pathologic findings; instead, it necessitates
clinicoradiological manifestations and the exclusion of other
diseases (2).
ECD primarily occurs in middle-aged to older adults,
with a male:female ratio of 3:1, and can affect almost all organ
systems (4,5). Symmetric long bone involvement is the
most common symptom and can occur in 95% of cases, showing cortical
thickening, metaphyseal medullary sclerosis of the tubular bones
and coarse trabeculations on radiographs (8). It might be overlooked because only
50% patients present with bone pain (3). Although rare, the involvement of the
cranial and facial bones, clavicle, sternum, ribs, scapula, pelvis
and spine has also been reported (18). The patient in the present study did
not present with bone pain during the course of the disease;
nevertheless, a typical symmetric metabolic increase in long bones
was found after performing the 99mTc-MDP-SPECT/CT. This suggested
that clinicians should supplement the assessments with a whole-body
bone or PET-CT scan to evaluate long bone involvement as early as
possible when patients are suspected of having ECD due to other
clinical manifestations. In addition, an MRI of long bones may be
informative in revealing epiphyseal involvement and periostitis,
which are often overlooked on plain films (8).
Extraskeletal manifestations, such as perirenal
infiltrates, cardiac pseudotumors, periaortic sheaths, protruding
eyeballs, yellow tumors of the eyelids and involvement of the
lungs, nervous system and skin are present in ~50% of the patients
(19). The involvement of cardiac
or neurologic systems in ECD usually has a poor prognosis (14).
Tissue infiltration of thoracic involvement in ECD
can involve all anatomic areas from the pleura to the periaortic
region and may appear as a thickening of the visceral pleura and
pseudotumoral mass (20). The
incidence of this infiltration occurring in the cardiovascular
system is 40-70% (3,14). Vascular involvement typically shows
tissue infiltration from the ascending aorta to the iliac artery
junction, creating a ‘coated aorta’ on CT (19). Unlike Takayasu disease, Horton
disease, and infectious aortitis, which may involve the entire
aortic wall, ECD tissue infiltration involves only the space around
the outer aortic membrane. This infiltration is circumferential
rather than anterior or lateral to the aorta, sparing the posterior
aspect, as in RPF (21). It is
difficult to differentiate ECD from mediastinitis (chronic
granulomatous, fibrosing, or carcinomatous) or lymphoma when the
superior vena cava and/or pulmonary arteries are compressed by the
superior vena cava and/or pulmonary arteries (3). It needs to be differentiated in
relation to pathology and the involvement of other sites. When the
lesions involve the pericardium, ECD may be characterized by
pericardial thickening and chronic progressive pericardial effusion
with possible cardiac tamponade. Pseudotumor of the lateral wall of
the right atrium and the right atrioventricular groove may occur in
myocardial involvement, the latter of which may encircle the right
coronary artery and lead to myocardial infarction (rare but with
high mortality). Other cardiac manifestations include arrhythmias
and heart failure (14,22,23).
Since the clinical manifestations of cardiac involvement are
usually not evident, as in the present patient and the involvement
is often associated with a poor prognosis, systematic cardiac
evaluation should be performed using MRI and CT (5,8,20).
Integrated echo and cardiac MRI can recognize constriction and
characterize pericardial tissue with high sensitivity and
specificity (14). The presence of
‘coated aorta’, pericardial thickening, pericardial effusion, right
atrioventricular groove pseudotumor, encapsulated right coronary
artery, and left ventricular diastolic insufficiency on imaging in
the present case are all consistent with cardiovascular involvement
in ECD. Pleural effusion was also present, possibly related to ECD
lung involvement (5).
Of patients with ECD, >30% may present with
pseudo ‘RPF,’ which presents as a perirenal mass. This may lead to
proximal ureteral obstruction and hydronephrosis, resulting in
dysuria and urinary urgency (24).
Retroperitoneal involvement in ECD needs to be differentiated from
idiopathic retroperitoneal fibrosis (IRF) and IgG4-related RPF
because of the perinephric soft tissue rind manifestations. The
main components of IRF are fibrotic and inflammatory infiltrating
cells, which usually involve the anterolateral aspect of the
abdominal aorta and iliac arteries, wrapping around the distal
ureter and leading to manifestations, such as ureteral obstruction
and oliguria, but rarely affect the perirenal space. Lesion tissues
of IRF can also involve the inferior vena cava, usually causing
stenosis and occlusion (22,25).
Treatment consists mainly of surgical interventions to relieve
ureteral obstruction and pharmacologic therapy to induce remission
of fibroinflammation. Early application of glucocorticoids rapidly
relieves inflammation and promotes shrinkage of the mass (26). The long-term use of glucocorticoids
is limited by adverse effects; therefore, immunosuppressive agents,
such as cyclophosphamide, azathioprine, methotrexate, cyclosporine
and tamoxifen can be selected as combinations for recurrent or
maintenance therapy for IRF (25,27).
However, there is no consensus regarding the choice of
immunosuppressive agents. Wang et al (26). found that the application of a
combination of hormones and cyclophosphamide and the addition of
tamoxifen according to the patient's condition, resulted in a
reduction of the mass in most patients (27). At the time of diagnosis of RPF in
the present patient, tamoxifen, which has anti-inflammatory and
antifibrotic effects and is well tolerated, was chosen as a
co-administration and cyclophosphamide added when CT suggested that
the mass was not shrinking. IgG4-related disease is a
fibroinflammatory disease with typical pathologic features of
massive IgG4-positive plasma cell infiltration and storiform
fibrosis in the diseased tissue, as well as serologic IgG4
positivity in some patients. Among them, IgG4-related RPF commonly
involves the abdominal aorta and anterior lateral iliac artery and
rarely involves the perinephric and proximal ureters as in ECD
(28,29). Additionally, IgG4-related diseases
do not usually involve bones (4).
Contrastingly, the perinephric infiltration in the present patient
is a highly prevalent (68% of cases) as is the characteristic
radiographic manifestation of ECD, which appears as a ‘hairy
kidney’ on CT (8,24). Its lesion tissues may directly
infiltrate the renal pelvis and proximal ureter, leading to
manifestations, such as dysuria and oliguria, while the distal
ureter and pelvic cavity are usually unaffected and rarely involve
the inferior vena cava (4,22). When RPF with atypical clinical
presentation and imaging is encountered, the possibility of ECD
needs to be considered, and further pathological examination is
recommended to exclude other diseases. Palliative percutaneous
nephrostomies can be selected for patients with renal involvement
to preserve renal function (30).
Additionally, retro-orbital infiltration may
manifest as bilateral exophalmos, and endocrine involvement most
frequently manifests as diabetes insipidus. Neurological
involvement may manifest as cerebellar and pyramidal syndromes and
skin involvement may present as macular tumors in the orbital or
periorbital space. Since these manifestations were not found in the
present patients, they will not be discussed in detail.
As shown in Table
I, 20% of ECD characteristics can overlap with those of LCH;
however, the two diseases are very different in terms of pathology,
clinical features and radiologic manifestations (9). The Langerhans histiocytes of LCH tend
to migrate or differentiate from bone marrow-derived monocyte or
dendritic cells. Contrastingly, the histiocytes of ECD are mostly
associated with bone marrow-derived monocytes. Histiocyte staining
for CD1a-positive and CD207-positive can provide evidence of LCH
(2,4,9). LCH
most commonly occurs in children and can involve bones throughout
the body in 80% of cases, with radiographic manifestations of
osteolytic changes (2,8,9).
Furthermore, skin involvement manifests as red papules in the
groin, chest, abdomen and back, which can be differentiated from
cutaneous xanthomas in ECD (8).
RDD is also a non-Langerhans histiocytosis, which is more common in
children and young adults (2,6,31).
The pathological hallmark is the accumulation of characteristic
S100-positive large histiocytes showing frequent emperipolesis
(i.e., the cells are phagocytosed by the RDD histiocyte and still
survive and can exit the histiocytes) and IHC showing positivity
for CD68, CD163 and S100, and negativity for CD1a and CD207
(2,8,9,31).
The main clinical manifestations are bilateral painless cervical
lymphadenopathy (57%) and palpable masses in other lymphatic sites
(9,31). Extra-lymph node involvement,
including osteolytic or sclerotic changes in the bone,
slow-progressing and painless skin nodules or rashes and pulmonary
nodular solid lesions, can occur in 43% of cases. However, such
symptoms lack typical radiologic features. Cardiovascular and
retroperitoneal area involvement is very rare in both LCH and RDD
(8,9,31).
Considering this, the present case did not fit the typical
presentation of LCH and RDD, and these conditions were excluded as
possible diagnoses.
 | Table IClinical features of ECD, LCH and RDD
(2,4,8,10,23). |
Table I
Clinical features of ECD, LCH and RDD
(2,4,8,10,23).
| Clinical
feature | ECD | LCH | RDD |
|---|
| Bone | 95%. Bilateral,
symmetric cortical osteosclerosis of the diaphyseal and metaphyseal
regions. Only 50% of patients describe bone pain | 80%. Osteolytic
lesions of calvarium, facial bones, proximal limbs, pelvis and
scapula. Lytic skull lesions in children are the most common, which
may be asymptomatic | 5-10%. Osteolytic
or sclerotic or mixed lysogenic changes of the long bones,
vertebrae,or sacrum, often accompanied by LN changes. Bone pain is
common |
|
Retroperitoneum | 1/3. Perinephric
infiltration leading to hydronephrosis and ureteral narrowing.
‘Hairy kidney’ on CT | Reported, but
featureless | 4%. Reported, but
featureless |
| Cardiovascular | 40-70%.
Pericarditis, pericardial effusion, cardiac tamponade, pseudotumor
of the right atrium or right atrioventricular groove. ‘Coated
aorta’ on CT | Reported, but
featureless | 0.1-0.2%. Rare and
uncharacteristic |
| Skin | Xanthelasma
(usually involving the eyelids or periorbital spaces) | 33%. Scaly
erythematous patches, Birbeck granules on electron microscopy | 10%.
slow-progressing, painless, nonpruritic reddish-brown nodules,
plaques, or papules, which may appear throughout the body |
| LN | Reported, but
featureless | 5-10%. Uncommon but
constitutes high-risk disease | 57%. Bilateral
painless cervical LN enlargement or palpable masses in other LN
sites |
| CNS | 25-50%. Tumor or
degenerative lesions; acute headache, elevated intracranial
pressure, cone system response, cognitive or behavioral
disorders | Pituitary lesions
(25%). Others (2-4%); cerebellar or brain stem lesions, dural-based
lesions, brain parenchymal lesions, and non-infiltrative
neurodegeneration | <5%. Dural-based
mass |
| Orbit | Retro orbital
xanthelasma, exophthalmos | Rare | 11%. Orbital
mass |
| Lungs | 50%. Asymptomatic
and normal on plain films. HRCT shows thickening of interlobular
septal, ground-glass opacities, or centrilobular opacities, pleural
thickening, or pleural effusion | 15%. Nodular and
cystic changes in upper and middle lobes on CT | 2%. Pulmonary
nodular consolidation in lobes of the lungs on CT. Involvement of
lower respiratory tract has 45% mortality rate |
| Liver and
spleen | Rare | 15%. Uncommon but
constitutes high-risk disease | Reported, but
featureless |
| Endocrine | 25%. DI | 20%. DI, growth
hormone deficiency, or panhypopituitarism with hypo thalamic
syndrome | Rare |
Imaging examination is crucial for the diagnosis of
ECD. As retroperitoneal and vascular involvement are often
asymptomatic, all patients with clinical suspicion of ECD should
undergo thoracoabdominal CT (21).
If the economic status of the patient permits,
18F-fluorodeoxyglucose positron emission tomography/CT (18F-FDG
PET/CT) should be performed 3-6 months after the initiation of
therapy to evaluate the metabolic response of ECD (3,32,33).
Identifying bone and bone marrow involvement and confirming the
area for CT-guided percutaneous biopsy may be important in cases of
extraskeletal involvement (32).
For patients who have already undergone FDG PET/CT before biopsy,
Goyal et al (3) recommend a
biopsy of most FDG-avid sites that are accessible and safe,
especially in cases of bony lesions.
The 5-year survival rate for ECD is 68%, and the
primary causes of death include respiratory distress, pulmonary
fibrosis and heart failure (5).
The choice of therapy should be individualized according to the
characteristics of the involved organs, disease progression and
type of gene mutation. Currently, IFN-α and PEG-IFN-α are the
first-line drugs for the treatment of ECD (3). There are no significant differences
in the potential side effects of these two drugs, which include
fever, flu-like symptoms, muscle pain and arthralgia, neurological
symptoms, gastrointestinal symptoms and myelosuppression. Notably,
IFN-α has emerged as an independent prognostic indicator of
improved survival in a multivariate analysis (34). It may exert beneficial effects
within patients with ECD by inducing the maturation and activation
of dendritic cells, immune-mediated histiocyte destruction, or
direct resistance to tissue cell proliferation (9,35).
Nevertheless, PEG-IFN-α is generally considered to have improved
toleration (9) while being easily
applicable for once-weekly administration. Therefore, it is
recommended that the patient receive PEG-IFN-α treatment. Due to
the short duration of the treatment, further follow-up is required
to fully investigate the efficacy and prognosis.
The present study presented a case of ECD involving
multiple organs. Due to the inadequate knowledge of ECD, it failed
to find the few foam cells on the second pathology result of the
patient in time, which delayed the diagnosis. The following
possibilities were considered as reasons for the low number of
typical pathologic cells of this patient: i) The patient may have
had a type of ECD that lacks typical histiocytes; ii) due to the
delayed diagnosis, the lesion tissues have developed into dense
fibrosis and the puncture needle was unable to reach deeper
tissues; therefore, the tissue obtained was atypical; iii) the
pathological tissues obtained were too small to detect sufficient
lesion cells; iv) long-term hormone and immunosuppressant therapy
may have masked the real condition of the patient. The present case
showed that clinicians must increase their knowledge of ECD to
improve diagnostic sensitivity and accuracy. In clinical practice,
the possibility of ECD should be considered if manifestations, such
as osteosclerosis (especially symmetrical long bone changes),
‘hairy kidney,’ ‘coated aorta’ and pericardial effusion of unknown
etiology, are encountered. Such patients should be subjected to
systemic screening as early as possible, aggressive biopsy of
tissues from all lesions, and refinement of genetic tests to
clarify the diagnosis and guide clinical treatment at an early
stage. Even if the pathological presentation is not sufficiently
typical, ECD cannot be completely excluded, and a comprehensive
judgment should be made in conjunction with clinical and imaging
studies, and other diseases should be ruled out. This study has
several limitations. First, due to the rarity of ECD, most
available data are obtained from case reports, and our knowledge of
ECD is lacking. Second, although a final diagnosis was made, the
lack of genetic evidence limited the treatment options. In the
future, longer follow-ups of the patient will be performed to
observe the local and systemic status and provide timely
therapeutic interventions.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Special Fund for
Flow Cytometry Lymphocyte Subgroups of Shandong Provincial Medical
Association (grant no. YXH2022ZX03223), the Open Project of
Shandong Key Laboratory of Rheumatic Disease and Translational
medicine (grant no. QYKFKT2023-7) and China Zhongguancun Precision
Medicine Science and Technology Foundation (grant no.
RCTAIIRSLE021).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
XS designed the study, drafted and revised the
manuscript, created and revised illustrations. GS provided
substantial contributions to critically revising the manuscript for
important intellectual content. MX contributed substantially by
obtaining the patients' medical records and revising the
manuscript. TL and YL contributed substantially to obtaining and
revising medical images. YH and ZW collected clinical data of the
patient, and confirm the authenticity of all the raw data. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Khoury JD, Solary E, Abla O, Akkari Y,
Alaggio R, Apperley JF, Bejar R, Berti E, Busque L, Chan JKC, et
al: The 5th edition of the World Health Organization Classification
of Haematolymphoid Tumours: Myeloid and histiocytic/dendritic
neoplasms. Leukemia. 36:1703–1719. 2022.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Emile JF, Cohen-Aubart F, Collin M,
Fraitag S, Idbaih A, Abdel-Wahab O, Rollins BJ, Donadieu J and
Haroche J: Histiocytosis. Lancet. 398:157–170. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Goyal G, Heaney ML, Collin M, Cohen-Aubart
F, Vaglio A, Durham BH, Hershkovitz-Rokah O, Girschikofsky M,
Jacobsen ED, Toyama K, et al: Erdheim-Chester disease: Consensus
recommendations for evaluation, diagnosis, and treatment in the
molecular era. Blood. 135:1929–1945. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Haroche J, Cohen-Aubart F, Rollins BJ,
Donadieu J, Charlotte F, Idbaih A, Vaglio A, Abdel-Wahab O, Emile
JF and Amoura Z: Histiocytoses: Emerging neoplasia behind
inflammation. Lancet Oncol. 18:e113–e125. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Campochiaro C, Tomelleri A, Cavalli G,
Berti A and Dagna L: Erdheim-Chester disease. Eur J Intern Med.
26:223–229. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
McClain KL, Bigenwald C, Collin M, Haroche
J, Marsh RA, Merad M, Picarsic J, Ribeiro KB and Allen CE:
Histiocytic disorders. Nat Rev Dis Primers. 7(73)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Cao XX, Sun J, Li J, Zhong DR, Niu N, Duan
MH, Liang ZY and Zhou DB: Evaluation of clinicopathologic
characteristics and the BRAF V600E mutation in Erdheim-Chester
disease among Chinese adults. Ann Hematol. 95:745–750.
2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Diamond EL, Dagna L, Hyman DM, Cavalli G,
Janku F, Estrada-Veras J, Ferrarini M, Abdel-Wahab O, Heaney ML,
Scheel PJ, et al: Consensus guidelines for the diagnosis and
clinical management of Erdheim-Chester disease. Blood. 124:483–492.
2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Papo M, Cohen-Aubart F, Trefond L, Bauvois
A, Amoura Z, Emile JF and Haroche J: Systemic Histiocytosis
(langerhans cell histiocytosis, erdheim-chester disease,
destombes-rosai-dorfman disease): From oncogenic mutations to
inflammatory disorders. Curr Oncol Rep. 21(62)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Cohen Aubart F, Emile JF, Carrat F,
Charlotte F, Benameur N, Donadieu J, Maksud P, Idbaih A, Barete S,
Hoang-Xuan K, et al: Targeted therapies in 54 patients with
Erdheim-Chester disease, including follow-up after interruption
(the LOVE study). Blood. 130:1377–1380. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Campochiaro C, Cavalli G, Farina N,
Tomelleri A, De Luca G and Dagna L: Efficacy and improved
tolerability of combination therapy with interleukin-1 blockade and
MAPK pathway inhibitors for the treatment of Erdheim-Chester
disease. Ann Rheum Dis. 81(e11)2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Arnaud L, Hervier B, Néel A, Hamidou MA,
Kahn JE, Wechsler B, Pérez-Pastor G, Blomberg B, Fuzibet JG,
Dubourguet F, et al: CNS involvement and treatment with
interferon-α are independent prognostic factors in Erdheim-Chester
disease: A multicenter survival analysis of 53 patients. Blood.
117:2778–2782. 2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Miller RC, Villà S, Kamer S, Pasquier D,
Poortmans P, Micke O and Call TG: Palliative treatment of
Erdheim-Chester disease with radiotherapy: A rare cancer network
study. Radiother Oncol. 80:323–326. 2006.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Palmisano A, Campochiaro C, Vignale D,
Tomelleri A, De Luca G, Bruno E, Monti CB, Cavalli G, Dagna L and
Esposito A: Cardiovascular involvement in Erdheim-Chester diseases
is associated with myocardial fibrosis and atrial dysfunction.
Radiol Med. 128:456–466. 2023.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Bartoli L, Angeli F, Stefanizzi A,
Fabrizio M, Paolisso P, Bergamaschi L, Broccoli A, Zinzani PL,
Galiè N, Rucci P, et al: Genetics and clinical phenotype of
Erdheim-Chester disease: A case report of constrictive pericarditis
and a systematic review of the literature. Front Cardiovasc Med.
9(876294)2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
De Abreu MR, Chung CB, Biswal S, Haghighi
P, Hesselink J and Resnick D: Erdheim-Chester disease: MR imaging,
anatomic, and histopathologic correlation of orbital involvement.
AJNR Am J Neuroradiol. 25:627–630. 2004.PubMed/NCBI
|
|
17
|
Hervier B, Haroche J, Arnaud L, Charlotte
F, Donadieu J, Néel A, Lifermann F, Villabona C, Graffin B, Hermine
O, et al: Association of both Langerhans cell histiocytosis and
Erdheim-Chester disease linked to the BRAFV600E mutation. Blood.
124:1119–1126. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wang F, Cao X, Niu N, Zhang Y, Wang Y,
Feng F and Jin Z: Multisystemic imaging findings in chinese
patients with erdheim-chester disease. AJR Am J Roentgenol.
213:1179–1186. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Haroche J, Arnaud L, Cohen-Aubart F,
Hervier B, Charlotte F, Emile JF and Amoura Z: Erdheim-Chester
disease. Curr Rheumatol Rep. 16(412)2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Brun AL, Touitou-Gottenberg D, Haroche J,
Toledano D, Cluzel P, Beigelman-Aubry C, Piette JC, Amoura Z and
Grenier PA: Erdheim-Chester disease: CT findings of thoracic
involvement. Eur Radiol. 20:2579–2587. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Nikpanah M, Kim L, Mirmomen SM, Symons R,
Papageorgiou I, Gahl WA, O'Brien K, Estrada-Veras JI and Malayeri
AA: Abdominal involvement in Erdheim-Chester disease (ECD): MRI and
CT imaging findings and their association with BRAFV600E
mutation. Eur Radiol. 28:3751–3759. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Dion E, Graef C, Haroche J, Renard-Penna
R, Cluzel P, Wechsler B, Piette JC and Grenier PA: Imaging of
thoracoabdominal involvement in Erdheim-Chester disease. AJR Am J
Roentgenol. 183:1253–1260. 2004.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Gianfreda D, Palumbo AA, Rossi E,
Buttarelli L, Manari G, Martini C, De Filippo M and Vaglio A:
Cardiac involvement in Erdheim-Chester disease: An MRI study.
Blood. 128:2468–2471. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chazal T, Pegoraro F, Manari G, Bettiol A,
Maniscalco V, Gelain E, Charlotte F, Mazor RD, Renard-Penna R,
Amoura Z, et al: Clinical phenotypes and long-term outcome of
kidney involvement in Erdheim-Chester histiocytosis. Kidney Int.
103:177–186. 2023.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Vaglio A, Palmisano A, Alberici F,
Maggiore U, Ferretti S, Cobelli R, Ferrozzi F, Corradi D, Salvarani
C and Buzio C: Prednisone versus tamoxifen in patients with
idiopathic retroperitoneal fibrosis: An open-label randomised
controlled trial. Lancet. 378:338–346. 2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang DL, Liu SB, Feng YC, Li J, Zhang T,
Wan L and Gao H: Corticosteroid combinated with cyclophosphamide in
the patients with retroperiteneal fibrosis: A clinical features and
prognostic analysis. J Clin Med. 17:18–22. 2019.
|
|
27
|
van Bommel EF, Pelkmans LG, van Damme H
and Hendriksz TR: Long-term safety and efficacy of a
tamoxifen-based treatment strategy for idiopathic retroperitoneal
fibrosis. Eur J Intern Med. 24:444–450. 2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Marando A, D'Ambrosio G, Catanzaro F, La
Rosa S and Sessa F: IgG4-related disease of the ureter: Report of
two cases and review of the literature. Virchows Arch. 462:673–678.
2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Deshpande V, Zen Y, Chan JK, Yi EE, Sato
Y, Yoshino T, Klöppel G, Heathcote JG, Khosroshahi A, Ferry JA, et
al: Consensus statement on the pathology of IgG4-related disease.
Mod Pathol. 25:1181–1192. 2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Verdalles U, Goicoechea M, García de
Vinuesa S, Mosse A and Luño J: Erdheim-Chester disease: A rare
cause of renal failure. Nephrol Dial Transplant. 22:1776–1777.
2007.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Elbaz Younes I, Sokol L and Zhang L:
Rosai-Dorfman disease between proliferation and neoplasia. Cancers
(Basel). 14(5271)2022.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Balink H, Hemmelder MH, de Graaf W and
Grond J: Scintigraphic diagnosis of Erdheim-Chester disease. J Clin
Oncol. 29:e470–e472. 2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lin E: FDG PET/CT for biopsy guidance in
Erdheim-Chester disease. Clin Nucl Med. 32:860–861. 2007.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Hervier B, Arnaud L, Charlotte F, Wechsler
B, Piette JC, Amoura Z and Haroche J: Treatment of Erdheim-Chester
disease with long-term high-dose interferon-α. Semin Arthritis
Rheum. 41:907–913. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Braiteh F, Boxrud C, Esmaeli B and
Kurzrock R: Successful treatment of Erdheim-Chester disease, a
non-Langerhans-cell histiocytosis, with interferon-alpha. Blood.
106:2992–2994. 2005.PubMed/NCBI View Article : Google Scholar
|