Introduction
Pulmonary cystic disease is a circular, thin-walled,
well-defined attenuation area in the lung parenchyma (1). The wall thickness is <2 mm and the
space is usually filled with gas or liquid (2). The cystic wall is typically comprised
of epithelial cells or fibrous tissue, which needs to be
differentiated from cystic diseases, such as emphysema, bullae,
cavitary disease and honeycomb lung (3). Diffuse cystic lung diseases (DCLDs)
occur when there are multiple cystic lesions in the lung parenchyma
caused by different diseases (4),
including Yersinia lung, Staphylococcal pneumonia,
lung Langerhans histiocytosis, lymphangioleiomyomatosis,
lymphocytic interstitial pneumonia (LIP) and Birt-Hogg-Dube (BHD)
syndrome. Different DCLDs have similar clinical manifestations, but
the treatment and intervention methods after diagnosis are somewhat
different (5). Therefore,
delineating the diagnosis and identifying the causes of different
DCLDs are essential for selecting the subsequent treatment
strategy.
Desquamative interstitial pneumonia (DIP) is a rare
form of interstitial lung disease. This disease was first defined
and named by Liebow and Billingsley (6) in 1965 and was proposed to exhibit
various features, such as alveolar infiltrates derived from
alveolar epithelial cell desquamations, relatively slight alveolar
thickening, monotonous homogeneity of the lesions and a typical but
non-specific ground glass appearance of the lung margins. However,
subsequent studies have shown that the infiltrates represent not
desquamation but instead pigmented macrophages (7,8). DIP
is commonly associated with smoking-related lung disease. Previous
studies have shown that about 90% or more of patients had a history
of smoking. However, close to 10% of the population still develops
DIP due to systemic diseases, infections,
environmental/occupational exposure to hazardous substances, and
medications (9,10). Therefore, the association between
smoking and DIP is not as robust as that with several other
smoking-related lung diseases. For example, 100% patients with
interstitial lung disease associated with respiratory bronchitis
have been documented to be smokers (11). This suggests that other factors,
such as systemic diseases, infections, environmental or
occupational exposure to harmful substances and medications, may
also be associated with DIP.
In terms of clinical diagnosis, desquamative
interstitial pneumonia (DIP) shares many features with other
diffuse cystic lung diseases (DCLDs) such as
lymphangioleiomyomatosis (LAM) and pulmonary Langerhans cell
histiocytosis (PLCH). It is necessary to review the diagnosis of
the different DCLDs (10). DIP is
characterized by the accumulation of pigmented macrophages in the
distal airways and spaces (7).
Specific manifestations of LAM include cystic changes and LAM
cytosis, while chest CT manifestations of PLCH usually include a
combination of nodules and cysts. Nodules are typically 1-10 mm in
size and are generally located in the small bronchi (4). Furthermore, alveolar filling tends to
be more diffuse and homogeneous in DIP, which is frequently
accompanied with inflammatory cell-induced septal thickening, mild
interstitial fibrosis and/or centrilobular emphysema (3). At present, the diagnosis of DIP
mainly relies on CT and biopsy. Other pathological or laboratory
tests are only sufficient to serve as auxiliary but non-specific
indicators (3). Treatment for DIP
mainly entails glucocorticoid therapy, though there is currently no
consensus on its indications, duration or dosage (12).
We present a case of a patient diagnosed with DIP
and outline his diagnostic and therapeutic journey with the aim of
expanding the case pool of patients with DIP and providing clinical
data for the diagnosis of DIP.
Case report
A 73-year-old male patient, who worked as a farmer
in Weifang, China, was admitted to the Department of Respiratory
and Critical Care Medicine of Weifang Second People's Hospital
(Weifang, China) in June 2019 due to chest tightness, shortness of
breath and intermittent fever for 6 months. At 6 months before
admission, the patient had developed chest tightness and shortness
of breath, which aggravated after physical activity. This was
accompanied with intermittent fever at 37.5˚C, without any obvious
regularity. Further medical examination results on admission are
provided in Table I; the results
showed that the patient's respiratory rate was slightly higher than
the reference value, indicating the need for further evaluation of
pulmonary ventilation. The patient had no chest pain or hemoptysis,
no nocturnal paroxysmal dyspnea, cough or pink foam-like sputum.
After oral treatment with Chai Hu oral liquid (10 ml twice a day),
the patient's symptoms of chest tightness and shortness of breath
were still not relieved, so she came to our hospital. The patient
had a history of smoking for 60 years (20 cigarettes per day) and
had quit smoking for 6 months. The patient had no family history of
hereditary diseases.
 | Table IMedical examination results on
admission. |
Table I
Medical examination results on
admission.
| Parameter | Value | Reference range |
|---|
| Body temperature,
˚C | 36.8 | 36.3-37.2 |
| Heart rate, beats per
min | 85 | 60-100 |
| Respiratory rate,
breaths per min | 22 | 15-20 |
| Blood pressure,
mmHg | 130/80 | 90/60-120/80 |
The results of the blood gas analysis and the
reference ranges are shown in Table
II and values were as follows: Oxygen partial pressure, 81
mmHg; carbon dioxide partial pressure, 34 mmHg; pH, 7.44; the
results were not abnormal. Furthermore, the leukocyte count was
9.82x109/l, the lymphocyte count was
2.62x109/l, the neutrophil percentage was 65.6%, the
blood sedimentation rate was 60 mm/h, hypersensitive C-reactive
protein level was 36.20 mg/l, blood glucose level was 6.91 mmol/l
and calcitonin level was 0.1 ng/ml. The results indicated that the
lymphocyte count was higher than the reference range, and it was
considered that the patient may have a lung infection. A small
number of gram-positive cocci were detected in the patient's sputum
smear test, while throat swabs tested negative for influenza A and
B viruses and cytomegalovirus. Pulmonary function examination
indicated the following: Forced expiratory volume in 1 sec (FEV1),
12.39 l; FEV1/forced vital capacity, 73.64%; and carbon monoxide
diffusing capacity (DLCO; actual value/predicted value), 68.2%,
which indicated small airway dysfunction and decreased diffusion
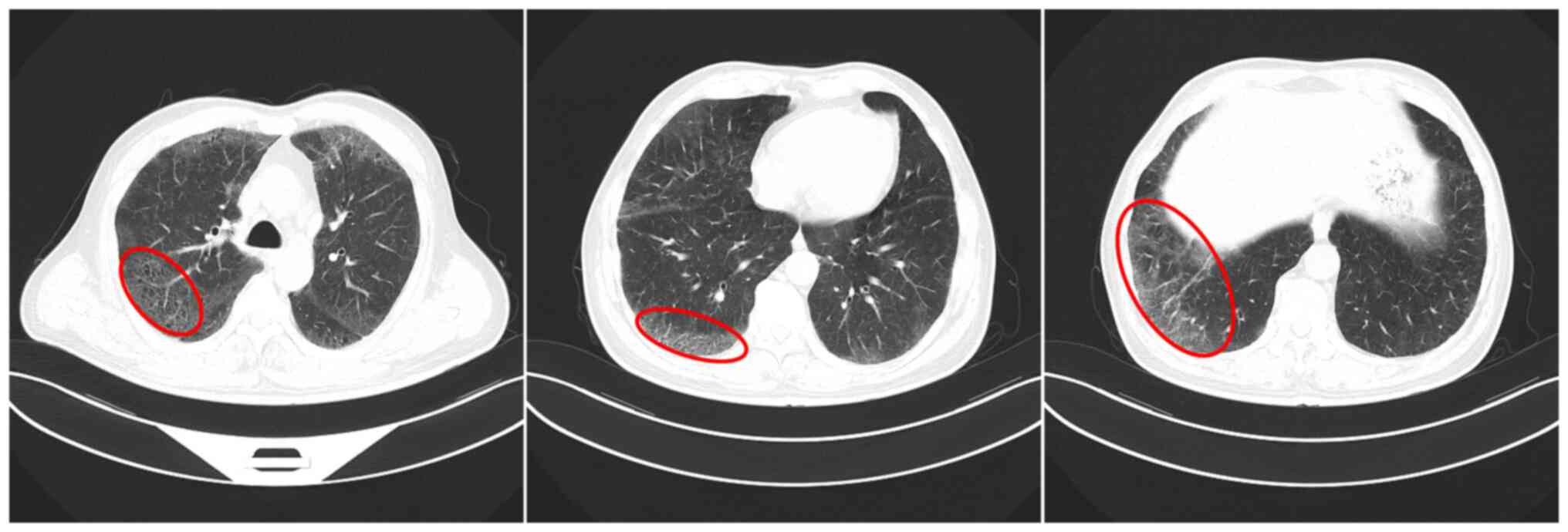
capacity. Chest CT (Optima CT660; GE HealthCare; slice interval, 5
mm; 120 kV) revealed diffuse ground glass shadows in both lungs,
with clear margins under the pleura, cystic cavity in some parts
and a solid nodule in the outer basal segment of the left lower
lobe (Fig. 1).
| Table IIResults of diagnostic tests. |
Table II
Results of diagnostic tests.
| Parameter | Value | Reference
range |
|---|
| Blood gas | | |
|
Oxygen
partial pressure, mmHg | 81 | 80-100 |
|
Carbon
dioxide partial pressure, mmHg | 34 | 35-45 |
|
pH | 7.44 | 7.35-7.45 |
| Blood biochemical
analysis | | |
|
White blood
cell count, 109/l | 9.82 | 3.5-9.5 |
|
Lymphocyte
count, 109/l | 2.62 | 0.8-4 |
|
Neutrophil
percentage, % | 65.6 | 50-75 |
|
Blood
sedimentation, mm/h | 60 | 0-20 |
|
Ultrasensitive
C-reactive protein, mg/l | 36.20 | 0-10.0 |
|
Blood
glucose, mmol/l | 6.91 | 3.9-6.1 |
|
Calcitonin,
ng/ml | 0.1 | 0-0.05 |
| Pathogenetic
testing | | |
|
Gram-positive
cocci | + | - |
|
Influenza A
virus | - | - |
|
Influenza B
virus | - | - |
|
Cytomegalovirus | - | - |
| Pulmonary
function | | |
|
FEV1, l | 12.39 | >92 |
|
FEV1/forced
vital capacity, % | 73.64 | >75 |
|
Carbon
monoxide diffusing capacity, % | 68.2 | 26.5-36.9 |
In conjunction with the patient's medical history,
laboratory tests and imaging findings, the diagnosis indicated a
noninfectious disease, such as Langerhans histiocytosis,
lymphocytic interstitial pneumonia, Birt-Hogg-Dubé syndrome, and
DIP. In addition, a panel of possible non-infectious diseases, such
as Langerhans histiocytosis, LIP, lung manifestations of BHD
syndrome and DIP, were considered. Therefore, bronchoscopy and
alveolar lavage fluid (BALF) cell pathogen culture, in addition to
transbronchial lung biopsy (TBLB) histopathology, were performed
(13).
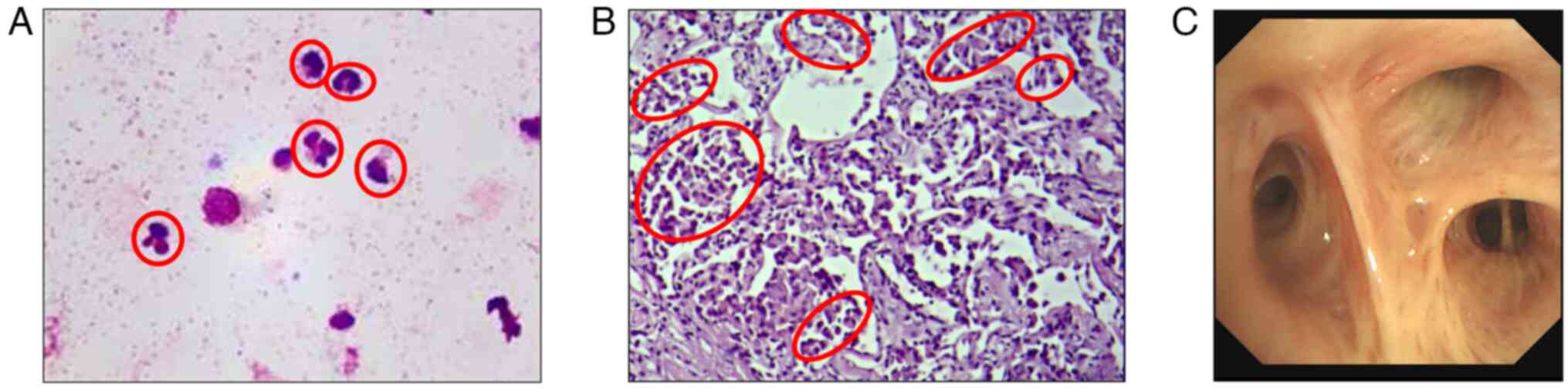
In June 2019, bronchoscopy revealed bronchial mucosa
hypertrophy and hyperemia. TBLB was performed in the upper segment
of the right upper lobe bronchus. Cytological and etiological
examination were performed using BALF. The BALF was filtered
through a 300-mesh nylon filter, acidified with glacial acetic
acid, and then subjected to microscopic hand counting of nucleated
cells (epithelial cells were excluded from the count). The filtered
BALF was centrifuged at 800 x g for 7 min. Of the supernatant, 300
µl was left to resuspend the cells, 20 µl were aspirated and a
smear was prepared from this. Staining was performed using
Wright-Giemsa Staining (Beso) for 10 min at room temperature and
400-600 non-epithelial nucleated cells were counted under an oil
microscope. The classification results were presented as a
percentage (Fig. 2). The sample
was negative for pathogens according to standard procedures.
Cytological examination showed predominantly neutrophils in the
ALF. Lung biopsy samples were examined using HE staining. The fresh
specimens were dehydrated with gradient alcohol at room
temperature, followed by sequential immersion in xylene at room
temperature and paraffin at 65˚C for embedding. Afterward, the
pathological tissues were sliced into 5-µm thin sections and then
consecutively immersed in xylene, anhydrous ethanol, 95% ethanol,
80% ethanol and running water at room temperature. They were
stained with hematoxylin for 5 min and eosin for 1 min, dehydrated
using a gradient of alcohol, and finally sealed with neutral resin.
The pathological results showed that the alveolar septum was
slightly wider, where small quantities of lymphocytes and
neutrophils had infiltrated the interstitial tissue, with
macrophages accumulating in the alveolar cavity.
According to previous reports on DIP, patients with
this disease tended to present with restrictive ventilatory
dysfunction, where imaging typically shows bilateral diffuse
ground-glass shadows with large numbers of macrophages detectable
in the BALF (14). In the present
report, the patient exhibited significant pulmonary restriction and
reduced diffusion capacity upon admission. Imaging tests showed
diffuse ground glass shadows in both lungs with subpleural foci and
clear margins. Biopsy of the lung tissue revealed slightly widened
alveolar septa, interstitial infiltration of a few lymphocytes and
neutrophils and multiple macrophage aggregates in the alveolar
lumen, all of which were shown according to the results of BALF and
TBLB pathology. These histological features are characteristic of
DIP. The patient was instructed to quit smoking and inhale
budesonide and formoterol fumarate powder for inhalation (15,16)
(160 µg/bid; AstraZeneca) for 38 days, during which time the
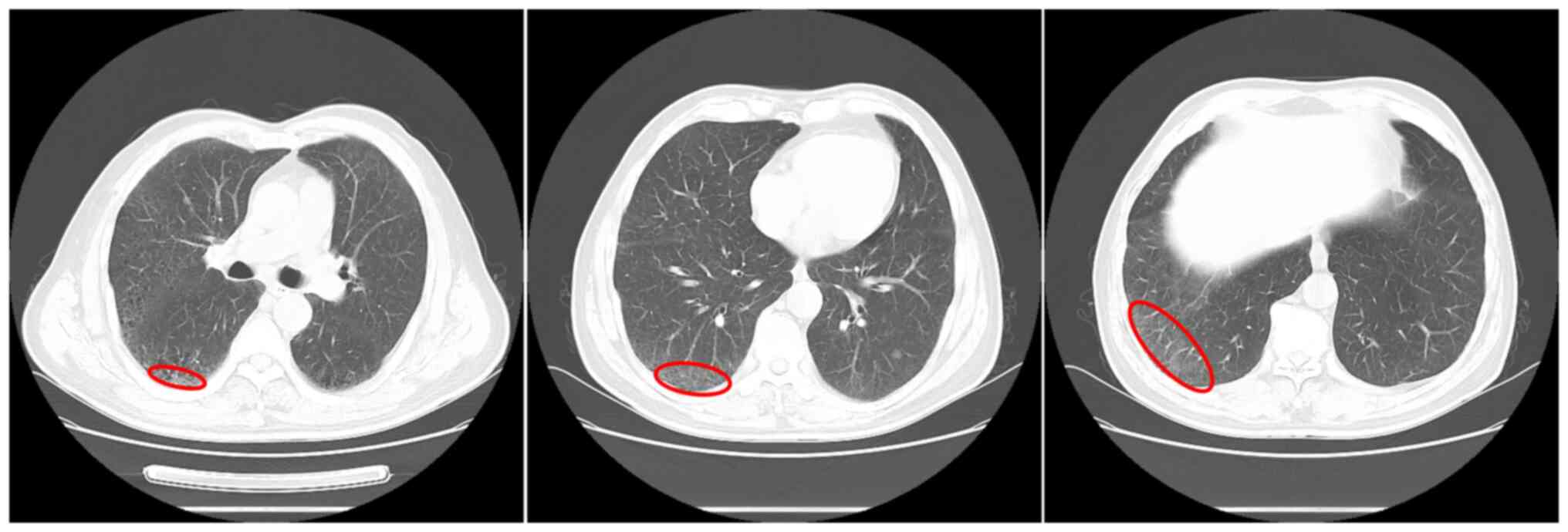
patient remained abstinent from smoking. Chest CT scan was
reperformed in July 2019, where the ground glass shadow was further
reduced (Fig. 3). Lung function
examination suggested that the FEV1 was 12.64 l, the DLCO (actual
value/predicted value) was 76.2%, the FEV1/FVC was 71.74% and lung
function had improved. In August 2019, the patient had no fever,
and the cough and shortness of breath had improved significantly.
The patient was able to breathe normally and no longer felt any
chest tightness. The patient was also instructed to gradually
reduce the use of the drug to prevent a rebound phenomenon. The
patient was followed up by telephone at 1, 3 and 6 months after
discharge. The patient discontinued medication and successfully
maintained smoking cessation within 1 month after discharge. There
was no recurrence of the disease.
Discussion
DIP is a rare group of lung diseases of unknown
etiology and belongs to the category of interstitial pneumonia that
was first reported by Liebow and Billingsley (6) in 1965. A large number of cells, which
was considered to be epithelial cells at the time (they were later
shown to be macrophages), were initially observed in the alveolar
lumen of patients with DIP. This feature was at first proposed to
be due to pulmonary epithelial cell desquamation, giving rise to
its name (6). In non-smoking
patients, possible causes of DIP include hepatitis C virus or
cytomegalovirus infection, chronic mold exposure and inorganic
particle accumulation, such as welding, diesel fumes and
metalworking (17,18). However, long-term exposure to
mycotoxins or dust, in addition to the long-term use of various
drugs, such as sirolimus, furantoin and/or illicit drugs, such as
marijuana, have also been reported to contribute to the development
of DIP. In pediatric patients, the cause is more likely to be
familial, especially if there was a history of hereditary
interstitial pneumonia (19).
Although DIP is part of the DCLD family, treatment
options for individual cases of DCLDs can drastically vary.
Therefore, accurate diagnosis of DIP is critical for the treatment
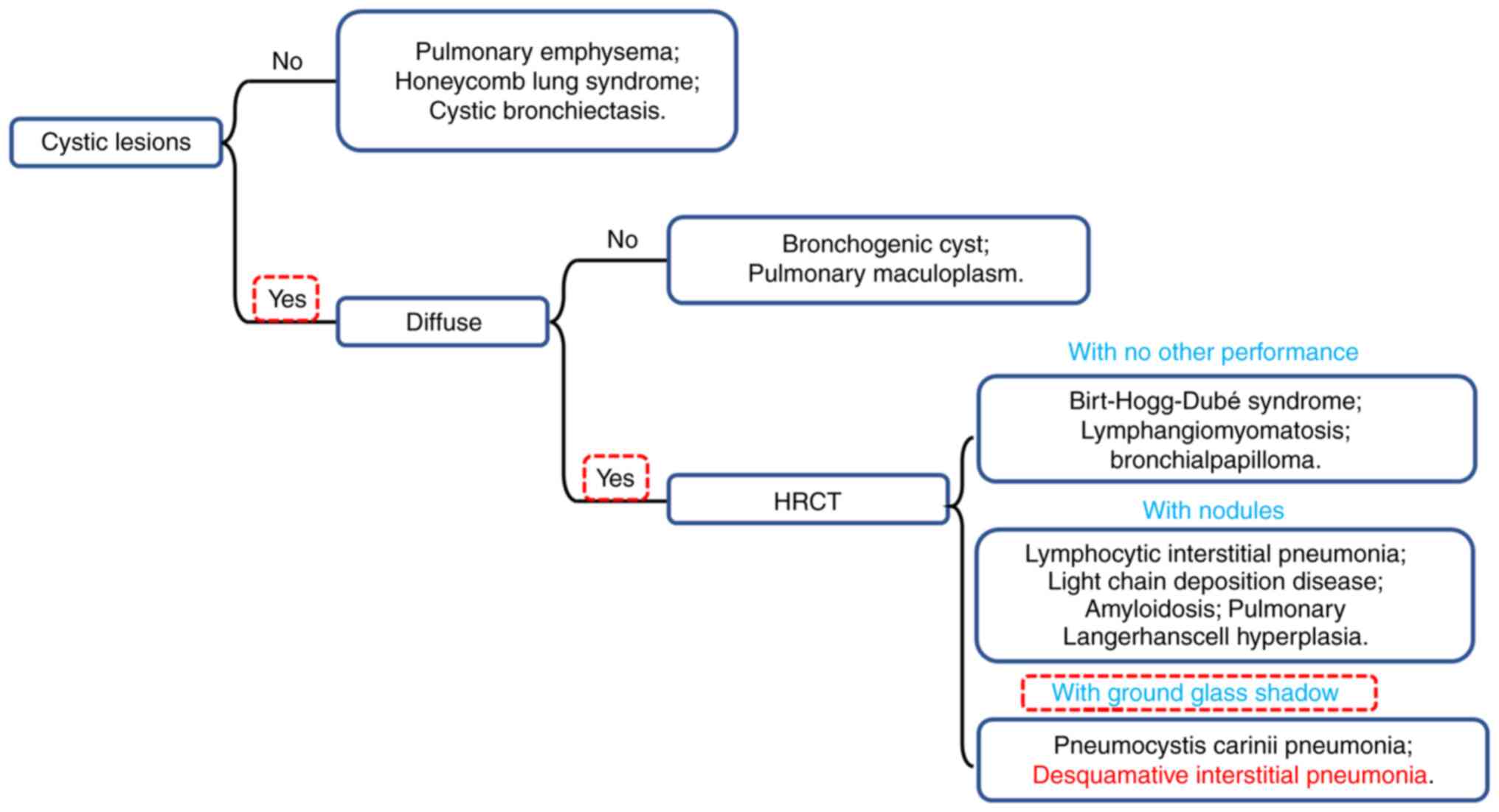
of DIP. This case summarizes the features of various pulmonary
diseases that are similar to the manifestations of DIP and a flow
chart for confirming the diagnosis of DIP was generated, which
would facilitate the accurate diagnosis of suspected DIP in future
cases (Fig. 4 and Table III). The gold standard for
diagnosing DIP is open chest biopsy, but this procedure can pose
greater harm to the patient. Therefore, methods such as
high-resolution CT and bronchoscopy have become the primary
criteria for its differential diagnosis. DIP is characterized by
bilateral areas of grossly glassy opacity in the lungs on imaging
and pathologically by macrophage infiltration of the alveoli.
Currently, the main treatment approach involves counseling patients
to stop smoking and using glucocorticoids (20).
| Table IIIDiagnostic features and treatment
options for related diseases. |
Table III
Diagnostic features and treatment
options for related diseases.
| Disease | Pathogenesis | Main
symptoms/features | Accompanying
symptoms | Treatment
options | (Refs.) |
|---|
| Yersinia
pneumonia | Yersinia
enterocolitica infection | Swollen and painful
lymph nodes, highly contagious | Sepsis without
pustules, hyperthermia, bleeding tendency | Third generation
cephalosporins | (34) |
|
Staphylococcal pneumonia |
Staphylococcus infection | Massive hemoptysis,
rapid accumulation of pleural effusion, acute respiratory distress
and leukopenia | Peripheral
circulatory collapse and corresponding organ damage | Cefazolin, Zocillin
or Ceftazolin | (35) |
| Pulmonary
Langerhans cell histiocytosis | Closely related to
tobacco exposure | Eosinophilic
granuloma with Langerhans cell infiltration and destruction of the
distal airway | Extrapulmonary
lysis lesions, skin lesions or central nervous system involvement
and skeletal system | Smoking cessation
and treatment based on clarabine or cytarabine | (36,37) |
|
Lymphangioleiomyomatosis | Cause of disease
not known | Dyspnea, cough,
chest pain, and hemoptysis; spontaneous pneumothorax is
common. | Axial lymph node
enlargement in the pelvis, abdomen and chest, smooth muscle tumors
in the abdominal lymphatics | Macrolide
immunosuppressants | (38,39) |
| Lymphocytic
interstitial pneumonia | Autoimmune and
environmental factors are involved | The main
respiratory manifestation is dry cough | Bronchospasm and
cough | Glucocorticoids,
steroids and γ-globulin therapy | (1,40) |
| Birt-Hogg-Dubé
syndrome | Germline mutations
in the folliculin gene on chromosome 17p11.2 | Intrapulmonary
cysts in both lungs and secondary spontaneous pneumothorax | Multiple, yellowish
or white, prominent flat, round, smooth papules 1-5 mm in diameter
on the face, neck and upper trunk | There is no known
effective treatment | (41) |
The characteristic lesion in patients with DIP, as
observed by CT, is a bilateral ground glass shadow in the lungs,
occurring mostly in the peripheral and lower lung regions.
Examination of the BALF should reveal increased numbers of
neutrophils, eosinophils and lymphocytes. However, not all patients
with DIP exhibit an increase in all of the aforementioned mature
leukocytes. Patients may only exhibit an increase in one or more of
these cell types (10,21,22).
Previously, an open-chest biopsy was considered the most powerful
means of identifying DIP (20,23).
On chest radiography, the benign tumor typically appears nodular
and is located mostly in the lower lobes, with peripheral lesions
of variable sizes. On CT examination, such tumors frequently
contain punctate calcifications. A number of retrospective studies
have shown that positron emission tomography cannot sufficiently
differentiate between inflammation and malignancy, rendering
resection to be the only definitive diagnostic method for this type
of pathology (24,25). With the advancement of
histopathologic and transbronchial biopsy techniques, it is now
possible to diagnose DIP effectively while minimizing patient harm
compared to open biopsy, open-chest biopsy has been gradually
replaced (26). In addition, the
TBLB technique used in the present case has markedly reduced the
postoperative recovery time of patients, whilst accurately
diagnosing their disease (3). The
TBLB technique enhances the accuracy of diagnosing DIP, reduces the
likelihood of missed or incorrect diagnoses, enables more
aggressive and effective treatment and ultimately improves
prognosis, the prognosis for diffuse interstitial pneumonia (DIP)
is generally favorable, with most patients showing improvement
after quitting smoking and receiving corticosteroid therapy. The
10-year survival rate is ~70% and the mortality rate ranges from 6
to 28% (16,27). Similar to the previous cases of
DIP, hormonal therapy was chosen as the treatment regimen for the
present case. Hormonal therapy is currently the most commonly used
treatment option for DIP and has demonstrated superior clinical
benefits both in terms of short-term treatments lasting a few weeks
and long-term treatments lasting more than 6 months (3).
Although it has been 58 years since the first case
of DIP was reported, further exploration is required regarding the
causes of this disease. In a previous review by Hellemons et
al (28), case reports of DIP
between 1965 and 2019 were summarized, which proposed that the most
common clinical symptoms of this disease are dyspnea and coughing.
In addition, patients will typically exhibit pulmonary function
limitation (70%) with reduced diffusion capacity, and majority of
patients achieve the alleviation of symptoms after treatment with
glucocorticoids (13). In another
study by Craig et al (29),
who followed up 49 patients at Royal Brompton Hospital (London,
UK), with DIP, the average survival was found to be 8.8 years for
non-smokers and 7 years for smokers. Although the presence of
hazardous substance-related occupational exposures in the patient
population does not allow for a definitive conclusion that smoking
significantly reduces patient survival, smoking cessation is
generally included in treatment recommendations to prevent further
deterioration in patient condition (30). Oral glucocorticoids, such as
prednisone, have been the most commonly used treatment option for
DIP in several previous case reports (31,32).
Typically, hormone administration provides immediate relief of the
patient's dyspnea and shows an improvement effect on lung function
in the long-term control of DIP (33). In addition, depending on the
patient's condition, immunosuppressants and antimicrobials can be
supplemented into the treatment regimen to target the different
symptoms. For example, allergies or bacterial infections may be
present.
In the treatment regimen reported in the present
report, the patient was initially advised to abstain from smoking.
This step has been demonstrated to be crucial for the improvement
of DIP, as evidenced previously (3,13).
Considering that the patient was admitted with an elevated
respiratory rate and chest tightness and there was a possibility of
a gram-positive Coccobacillus infection (follow-up tests
proved that the patient was not infected with the bacteria), the
patient received continuous low-flow oxygen, anti-infective therapy
and asthma control therapy during his hospitalization. The patient
did not exhibit severe dyspnea or decreased oxygen saturation.
After 8 days of treatment, the patient was discharged after his
symptoms improved significantly. Considering that the patient was
unable to maintain oxygenation or receive nursing care after being
discharged, the patient was prescribed treatment with the
glucocorticoid aerosol Sinebicort (AstraZeneca).
Certain limitations of the present report remain.
The present report only included one case, which prevented some of
the characteristics and treatment protocols in the present report
from being applied for the wider diagnosis and treatment of all
patients with DIP. Furthermore, a follow-up telephone call was made
and the patient indicated that there had been no relapse of the
disease after discontinuing the medication, but since the patient
did not return for a follow-up visit, there was no imaging
information available to accurately verify his recovery.
This case demonstrates that combining low-flow
oxygen supply with glucocorticoid therapy for treating a patient
with DIP, and using asthma-relieving medications for the patient's
apparent chest tightness, may have a more positive impact on the
patient's treatment. Although glucocorticoid therapy has become the
primary treatment for DIP, there is no consensus on the
indications, duration and dosage of hormone use. Since the number
of patients with this disease is small and there are not sufficient
clinical samples to summarize the diagnostic indicators. At Weifang
Second People's Hospital, a number of DIP cases have already been
collected, where the treatment regimens and prognosis of the
patients are planned to be documented in more detail. The present
report intends to raise awareness of the disease to reduce the rate
of missed diagnoses and misdiagnoses, promote effective treatment,
improve patient compliance, and enhance the prognosis.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by funds from the
Science and Technology Development Project of Weifang (grant nos.
2021YX070 and 2022ZJ1059), Scientific Project of Weifang Health
Commission (grant no. WFWSJK-2022-220) and the Chinese Federation
of Public Health Foundation (grant no. GWLM202043).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MQ, HZ and YZ were responsible for the conception of
this work. HZ was a major contributor in writing the manuscript.
GY, BY, SM, YW, HZ and XZ assisted with the data analysis and
drafted the discussion part of the manuscript. MQ and YZ confirm
the authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Examination images of the patient, including chest
CT, bronchoscopy and cytology, were used in this case, and patient
information had been withheld, requiring ethical approval. The
present report was approved by the ethics committee of Weifang
Second People's Hospital (Weifang, China; approval no.
RY2022-025-01).
Patient consent for publication
The patient provided written informed consent for
the publication of the manuscript including any identifying images
or data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Panchabhai TS, Farver C and Highland KB:
Lymphocytic interstitial pneumonia. Clin Chest Med. 37:463–474.
2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Radzikowska E: Update on pulmonary
Langerhans cell histiocytosis. Front Med (Lausanne).
7(582581)2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Diken ÖE, Şengül A, Beyan AC, Ayten Ö,
Mutlu LC and Okutan O: Desquamative interstitial pneumonia: Risk
factors, laboratory and bronchoalveolar lavage findings,
radiological and histopathological examination, clinical features,
treatment and prognosis. Exp Ther Med. 17:587–595. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Obaidat B, Yazdani D, Wikenheiser-Brokamp
KA and Gupta N: Diffuse cystic lung diseases. Respir Care.
65:111–126. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ryu JH, Tian X, Baqir M and Xu K: Diffuse
cystic lung diseases. Front Med. 7:316–327. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Liebow AA, Steer A and Billingsley JG:
Desquamative interstitial pneumonia. Am J Med. 39:369–404.
1965.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Godbert B, Wissler MP and Vignaud JM:
Desquamative interstitial pneumonia: An analytic review with an
emphasis on aetiology. Eur Respir Rev. 22:117–123. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hino T, Lee KS, Yoo H, Han J, Franks TJ
and Hatabu H: Interstitial lung abnormality (ILA) and nonspecific
interstitial pneumonia (NSIP). Eur J Radiol Open.
8(100336)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wells AU, Nicholson AG and Hansell DM:
Challenges in pulmonary fibrosis. 4: Smoking-induced diffuse
interstitial lung diseases. Thorax. 62:904–910. 2007.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Blin T, De Muret A, Teulier M, Ferreira M,
Vincent M, Catinon M, Legras A, Diot P and Marchand-Adam S:
Desquamative interstitial pneumonia induced by metal exposure. A
case report and literature review. Sarcoidosis Vasc Diffuse Lung
Dis. 37:79–84. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Serrano Gotarredona MP, Navarro Herrero S,
Gómez Izquierdo L and Rodríguez Portal JA: Smoking-related
interstitial lung disease. Radiologia (Engl Ed). 64 (Suppl
3):S277–S289. 2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Moreno García MJ, López-Herce Cid J,
Rodríguez Sánchez C, Alvarado Ortega F, Carrasco S and Ruza Tarrió
F: Desquamative interstitial pneumonia. An Esp Pediatr. 23:431–437.
1985.PubMed/NCBI
|
|
13
|
Medenica M and Medenica M: Desquamative
interstitial pneumonia with clinical, radiological and histologic
correlation. Radiol Case Rep. 14:505–509. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yamakawa H, Suido Y, Sadoyama S, Yamanaka
Y, Ikeda S, Kitamura H, Baba T, Okudela K, Takemura T and Ogura T:
Desquamative interstitial pneumonia complicated with IgG4-related
lung disease. Intern Med. 56:1553–1556. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Margaritopoulos GA, Harari S, Caminati A
and Antoniou KM: Smoking-related idiopathic interstitial pneumonia:
A review. Respirology. 21:57–64. 2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hagmeyer L and Randerath W:
Smoking-related interstitial lung disease. Dtsch Arztebl Int.
112:43–50. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lougheed MD, Roos JO, Waddell WR and Munt
PW: Desquamative interstitial pneumonitis and diffuse alveolar
damage in textile workers. Potential role of mycotoxins. Chest.
108:1196–1200. 1995.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Nemery B: Metal toxicity and the
respiratory tract. Eur Respir J. 3:202–219. 1990.PubMed/NCBI
|
|
19
|
Cottin V: Desquamative interstitial
pneumonia: Still orphan and not always benign. Eur Respir Rev.
29(200183)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Lovrenski A, Eri Ž, Tegeltija D,
Kašiković-Lečić S and Panjković M: Desquamative interstitial
pneumonia: A case report. Srp Arh Celok Lek. 142:602–606.
2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ishimoto H, Sakamoto N, Ozasa M, Tsutsui
S, Hara A, Kido T, Yamaguchi H, Yamamoto K, Obase Y, Ishimatsu Y
and Mukae H: Idiopathic desquamative interstitial pneumonia
diagnosed using transbronchial lung cryobiopsy: A case report.
Respir Med Case Rep. 34(101523)2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sousa V and Carvalho L: DIP (desquamative
interstitial pneumonia): As a tobacco-associated disease-case
report. Rev Port Pneumol. 10:431–435. 2004.PubMed/NCBI View Article : Google Scholar : (In Portuguese).
|
|
23
|
Miyaoka M, Hatanaka K, Iwazaki M and
Nakamura N: Pulmonary adenocarcinoma mimicking desquamative
interstitial pneumonia: Report of 2 cases with genetic analysis.
Int J Surg Pathol. 26:655–659. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Neacşu F, Vârban AŞ, Simion G, Şurghie R,
Pătraşcu OM, Sajin M, Dumitru M and Vrînceanu D: Lung cancer
mimickers-a case series of seven patients and review of the
literature. Rom J Morphol Embryol. 62:697–704. 2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Furuya K, Yasumori K, Takeo S, Sakino I,
Uesugi N, Momosaki S and Muranaka T: Lung CT: Part 1, mimickers of
lung cancer-spectrum of CT findings with pathologic correlation.
AJR Am J Roentgenol. 199:W454–W463. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Batra K and Adams TN: Imaging features of
idiopathic interstitial lung diseases. J Thorac Imaging. 38 (Suppl
1):S19–S29. 2023.PubMed/NCBI View Article : Google Scholar
|
|
27
|
American Thoracic Society; European
Respiratory Society. American thoracic society/European respiratory
society international multidisciplinary consensus classification of
the idiopathic interstitial pneumonias. This joint statement of the
American thoracic society (ATS), and the European respiratory
society (ERS) was adopted by the ATS board of directors, June 2001
and by the ERS executive committee, June 2001. Am J Respir Crit
Care Med. 165:277–304. 2002.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hellemons ME, Moor CC, von der Thüsen J,
Rossius M, Odink A, Thorgersen LH, Verschakelen J, Wuyts W,
Wijsenbeek MS and Bendstrup E: Desquamative interstitial pneumonia:
A systematic review of its features and outcomes. Eur Respir Rev.
29(190181)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Craig PJ, Wells AU, Doffman S, Rassl D,
Colby TV, Hansell DM, Du Bois RM and Nicholson AG: Desquamative
interstitial pneumonia, respiratory bronchiolitis and their
relationship to smoking. Histopathology. 45:275–282.
2004.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Katzenstein AL, Mukhopadhyay S, Zanardi C
and Dexter E: Clinically occult interstitial fibrosis in smokers:
Classification and significance of a surprisingly common finding in
lobectomy specimens. Hum Pathol. 41:316–325. 2010.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ryu JH, Colby TV, Hartman TE and Vassallo
R: Smoking-related interstitial lung diseases: A concise review.
Eur Respir J. 17:122–132. 2001.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Vassallo R: Diffuse lung diseases in
cigarette smokers. Semin Respir Crit Care Med. 33:533–542.
2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Deutsch GH, Young LR, Deterding RR, Fan
LL, Dell SD, Bean JA, Brody AS, Nogee LM, Trapnell BC, Langston C,
et al: Diffuse lung disease in young children: Application of a
novel classification scheme. Am J Respir Crit Care Med.
176:1120–1128. 2007.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Jarrot PA, Mahmoudi R, Novella JL and
Manckoundia P: Pneumonia due to Yersinia enterocolitica. Med Mal
Infect. 43:38–39. 2013.PubMed/NCBI View Article : Google Scholar : (In French).
|
|
35
|
He H and Wunderink RG: Staphylococcus
aureus pneumonia in the community. Semin Respir Crit Care Med.
41:470–479. 2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Radzikowska E: Pulmonary Langerhans' cell
histiocytosis in adults. Adv Respir Med. 85:277–289.
2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Shaw B, Borchers M, Zander D and Gupta N:
Pulmonary Langerhans cell histiocytosis. Semin Respir Crit Care
Med. 41:269–279. 2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Xu KF, Xu W, Liu S, Yu J, Tian X, Yang Y,
Wang ST, Zhang W, Feng R and Zhang T: Lymphangioleiomyomatosis.
Semin Respir Crit Care Med. 41:256–268. 2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
McCarthy C, Gupta N, Johnson SR, Yu JJ and
McCormack FX: Lymphangioleiomyomatosis: Pathogenesis, clinical
features, diagnosis, and management. Lancet Respir Med.
9:1313–1327. 2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Pitt J: Lymphocytic interstitial
pneumonia. Pediatr Clin North Am. 38:89–95. 1991.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Daccord C, Good JM, Morren MA, Bonny O,
Hohl D and Lazor R: Birt-Hogg-Dubé syndrome. Eur Respir Rev.
29(200042)2020.PubMed/NCBI View Article : Google Scholar
|