Introduction
Recent technological advances have resulted in
interest in the relationship between the microorganisms populating
the intestines and human health (1). The gut microbiota contains >100
billion microorganisms, pertaining to >100 different species of
bacteria, which have been proven to be involved in physiological
and pathophysiological host processes. In humans, one third of the
gut microbiota is common to all people, while the remaining two
thirds are unique to each individual (1).
The intestinal microbiota is comprised of numerous
and varied microorganisms that colonize the human intestinal tract.
These have evolved alongside their human hosts for thousands of
years, creating a complex relationship, with benefits for both
sides (2,3). The estimated number of intestinal
microorganisms (>1014) is ~10 times greater than the
number of human cells, and the bacterial genome is >100 times
larger than the human one (4). The
microbiome has 3.3 million non-redundant genes, whereas the entire
human genome only has 22,000 genes (5). The diversity of the microbiome is
much greater than genomic variation; two individuals are 99.9%
identical in their genomic content, but they can be 80-90%
different regarding their intestinal microbiota (5).
The microbiota offers several advantages to the host
organism, through its physiological actions. Some of these
advantages include improving and maintaining intestinal integrity,
molding the intestinal epithelium, energy production, protection
against pathogens and regulation of host immunity. When the
composition of the microbiota is altered, these functions are
modified or lost, with various pathological implications; this
state of altered microbiota is called dysbiosis (6).
Research has identified alterations of the gut
microbiota specific to chronic kidney disease (CKD) and
terminal/end-stage renal disease (ESRD), characterized by reduced
Roseburia, Akkermansia and Allobaculum, and
increased Prevotella (7).
Patients with stages 3-4 of CKD present a reduction in stool
aerobic microbial species, compared with clinically healthy
individuals. On the other hand, patients with CKD that have not yet
started dialysis treatment have been reported to exhibit an
increase in aerobic microbial species in their stool samples
(8). Furthermore, comparing
patients with ESRD and healthy individuals, significant differences
have been detected in the abundance of 190 microbial operational
taxonomic units (8,9). Some microbial species that are
normally present in the intestine of healthy individuals, such as
Lactobacillus and Prevotellaceae, have been shown to
be reduced in patients with CKD, whereas microorganisms from the
Enterobacteriaceae genus and some species of Enterococcus
are more abundant (8,10). In ESRD, increased aerobic species,
such as Enterococcus and Enterobacteriaceae, have been
observed (11). It is also known
that gut dysbiosis contributes to the increase in uremic toxin
concentrations in patients with CKD. This increase, in turn, favors
the progression of CKD (12,13).
Patients with ESRD have also been shown to exhibit reductions in
butyrate-producing microbial species, including Roseburia,
Faecalibacterium, Clostridium, Coprococcus and
Prevotella (14). This
category of patients has 19 microbial families that predominate;
out of these 19, 12 families contain urease, 5 families contain
uricase, and 3 families contain indole and p-cresyl-producing
enzymes (15).
Patients on hemodialysis also have alterations in
their gut microbiome. Proteobacteria, especially
Gammaproteobacteria; Actinobacteria; and Firmicutes, especially the
Clostridia phylum, have been reported to be present in
increased abundance, compared with in the microbiota of healthy
individuals (8).
Evidence exists of the association between
alterations in intestinal microbiota and diseases such as diabetes,
obesity, cancer, intestinal inflammatory disease, asthma,
cardiovascular disease and renal disease (16-18).
In addition, the relationship between alterations in the
microbiota, diabetes mellitus (DM) and obesity has been studied
intensively (19-21).
Chronic low-intensity inflammatory processes have been reported to
be the link between obesity and insulin resistance; however, the
causal mechanism has not yet been identified (22). In addition, inflammation in adipose
tissue can also impact the gut microbiota. Inflammatory signals may
disrupt the delicate balance of the gut microbiome, influencing the
growth and activity of specific bacterial species. This
bidirectional communication between adipose tissue and the
microbiota creates a complex relationship that can have significant
implications for metabolic health.
The microbiota can induce weight gain in patients
with diabetes or prediabetes by reducing leptin sensitivity and the
cerebral expression of obesity-suppressing neuropeptides
pro-glucagon and brain-derived neurotrophic factor (23). In addition, along with the host
genotype, the microbiota modulates insulin secretion and
diet-induced phenotypes (24) via
the effect of microbial taxa on host-secreted bile acids.
In type 2 DM (T2DM), butyrate-producing bacteria are
reduced, which affects the intestinal mucosa and increases gut
permeability. This increased gut permeability may explain the
relationship between the microbiome, T2DM and chronic inflammation
(22). Lower levels of butyrate,
and other short chain fatty acids (SCFAs), determine alterations in
glucose metabolism, body weight, energy production and homeostasis,
and in the gut barrier function (25). An altered intestinal microbiota is
a factor in the rapid progression of insulin resistance in T2DM.
The underlying mechanisms include reshaping of the intestinal
microbiome, and the modification of host metabolic and signaling
pathways (26).
The most commonly used antidiabetic medication
worldwide is metformin. This drug can affect the gut microbiome,
with elevated levels of Escherichia spp., Akkermansia
muciniphila and Subdoligranulum variabile, and lower
numbers of Intestinibacter bartlettii detected in patients
treated with metformin (27). This
alteration may result in the depletion of butyrate-producing
bacteria. Despite this, patients with T2DM treated with metformin
have been shown to exhibit increased productions of butyrate and
propionate (27).
The present study aimed to determine alterations in
the intestinal microbiota of individuals with both T2DM and ESRD,
to determine potential microbial mechanisms that affect the
development of both diseases and to identify methods of improving
the clinical state of patients within this pathological category,
through intervention on the intestinal microbiome.
Materials and methods
Patients
The present study is an observational, case-control
type study. Patient samples from ‘NC Paulescu’ National Institute
of Diabetes, Nutrition and Metabolic Diseases (Bucharest, Romania)
were collected between November 2019 and February 2020. A group of
9 patients with T2DM and ESRD (4 women and 5 men; mean age 61.9
years; age range, 44-80 years) were compared with a control group
consisting of 8 healthy individuals (4 women and 4 men; mean age
59.5 years; age range 45-74 years). All of the participants were
age matched, filled in a standardized nutritional questionnaire and
signed an informed consent form. The present study was approved by
the Ethics Commission of ‘NC Paulescu’ National Institute of
Diabetes, Nutrition and Metabolic Diseases (Bucharest, Romania;
approval no. Certif. 5911/04.10.2019). All participants gave their
written informed consent upon inclusion in the study. The research
adhered to the principles outlined in The Declaration of Helsinki
and also obtained approval from the Ethics Committee at the
University of Bucharest (Bucharest, Romania; under protocol code
CEC reg. no. 235/9.10.2019).
T2DM was defined according to the American Diabetes
Association criteria: A glycated hemoglobin level ≥6.5% (48
mmol/mol); a fasting plasma glucose ≥126 mg/dl (7.0 mmol/l); a 2-h
plasma glucose during 75-g oral glucose tolerance test ≥200 mg/dl
(11.1 mmol/l); and a random plasma glucose ≥200 mg/dl (11.1 mmol/l)
(28). CKD was defined in
accordance with The Kidney Disease Improving Global Outcomes
foundation guidelines as the presence of two factors: Glomerular
filtration rate (GFR) <60 ml/min and albumin >30 mg per gram
of creatinine, along with abnormalities in kidney structure or
function for >3 months. ESRD was defined as a GFR of <15
ml/min. (29). Inclusion criteria
for participants in the present study were: Diagnosed with T2DM,
>18 years of age, and diagnosed with CKD in renal replacement
therapy. Exclusion criteria for participants in the study were:
Non-renal disease-caused anemia; acute hemorrhaging or history of
hemorrhaging in the last 3 months; blood transfusion in the last 3
months; acute inflammatory or infectious diseases; acute vascular
pathology; and current immunosuppressive therapy. The patient
characteristics are listed in Table
I.
 | Table ICharacteristics of the patient group
with type 2 diabetes mellitus and end-stage renal disease included
in the present study. |
Table I
Characteristics of the patient group
with type 2 diabetes mellitus and end-stage renal disease included
in the present study.
| | Value |
|---|
| Characteristic | T2DM | Control |
|---|
| Sex, n (%) | | |
|
Female | 4(44) | 4(50) |
|
Male | 5(56) | 4(50) |
| Age,
yearsa | 61.9 (3.6) | 59.5 (2.4) |
| Diabetes duration,
yearsa | 16.7 (3.3) | - |
| Insulin,
U/daya | 21.4 (8.7) | - |
| Hemoglobin,
g/dla,b | 10.4 (0.7) | 12.2 (0.6) |
| Cholesterol,
mg/dla,c | 152.2 (22.1) | 128 (15.1) |
| Triglycerides,
mg/dla,d | 192.9 (43.2) | 145 (15.0) |
| Creatinine,
mg/dla,e | 6.0 (0.8) | 0.8 (11.0) |
| Urea,
mg/dla,f | 139.3 (17.1) | 35 (10.2) |
| Calcium,
mg/dla,g | 8.8 (0.3) | 8.9 (0.4) |
| Phosphate,
mg/dla,h | 5.0 (0.4) | 2.9 (0.2) |
| Albumin,
g/dla,i | 3.9 (0.1) | 3.6 (0.1) |
| Total protein,
g/dla,j | 6.9 (0.3) | 6.6 (0.1) |
Microbiota analysis
Microbiota analysis was performed using
culture-independent techniques, since the majority of members of
the gut microbiota are not cultivatable (3,4).
Bacterial DNA was extracted from stool samples using a commercial
kit (Qiagen Stool Mini Kit; Qiagen, Inc.). The DNA concentrations
were quantified utilizing the Qubit 4 fluorometer (Thermo Fisher
Scientific, Inc.). To investigate the gut microbiota, 16S ribosomal
RNA (rRNA) and 18S rRNA primers were used, and quantitative PCR was
performed. The sequences of the aforementioned primers are
presented in Table II. For the
quantification of the various bacterial and fungal populations, the
following was used: 9 ng DNA isolated from the stool samples, SYBR
Green 2X (Applied Biosystems; Thermo Fisher Scientific, Inc.) and
16S/18S rRNA primers (2.5 nM). Amplification was performed on a
Viia7 instrument (Applied Biosystems; Thermo Fisher Scientific,
Inc.). Each sample was analyzed in triplicate. Samples without DNA
template served as negative controls. Samples were incubated at
95˚C for 5 min, followed by 40 cycles at 95˚C for 10 sec, 60˚C for
30 sec and 72˚C for 1 sec. The microbial relative quantification
was performed using the 16S/18S rRNA threshold cycle values for
normalization (universal 16S and 18S primers were used for
normalization), and using the control group for comparison
according to the 2-ΔΔCq method
(30). Species relative abundance
was calculated as fold change compared to the healthy control.
| Table IIPrimer sequences used for the
amplification of 16S and 18S rRNA genes. |
Table II
Primer sequences used for the
amplification of 16S and 18S rRNA genes.
| A, Bacterial 16S
rRNA |
|---|
| First author,
year | Microbial
population | Primer | Primer sequence,
5'-3' | (Refs.) |
|---|
| Yang YW, 2015 | Actinobacteria | Act664F |
TGTAGCGGTGGAATGCGC | (56) |
| | | Act941R |
AATTAAGCCACATGCTCCGCT | |
| Yang YW, 2015 |
Deferribacteraceae | Defer1115F |
CTATTTCCAGTTGCTAACGG | (56) |
| | | Defer1265R |
GAGATGCTTCCCTCTGATTATG | |
| Yang YW, 2015 |
Verrucomicrobiota | Ver1165F |
TCAGGTCAGTATGGCCCTTAT | (56) |
| | | Ver1263R |
CAGTTTTCAGGATTTCCTCCGCC | |
| Yang YW, 2015 | Tenericutes | Ten662F |
ATGTGTAGCGGTAAAATGCGTAA | (56) |
| | | Ten862R |
CATACTTGCGTACGTACTACT | |
| Yang YW, 2015 |
Betaproteobacteria | Beta979F |
AACGCGAAAAACCTTACCTACC | (56) |
| | | Beta1130R |
TGCCCTTTCGTAGCAACTAGTG | |
| Yang YW, 2015 |
Epsilonproteobacteria | Epsilon940F |
TAGGCTTGACATTGATAGAATC | (56) |
| | | Epsilon1129R |
CTTACGAAGGCAGTCTCCTTA | |
| Yang YW, 2015 |
Gammaproteobacteria | Gamma877F |
GCTAACGCATTAAGTACCCCG | (56) |
| | | Gamma1066R |
GCCATGCAGCACCTGTCT | |
| Gomes-Neto JC,
2017 |
Mucispirillum spp. | MCSP F |
TCTCTTCGGGGATGATTAAAC | (38) |
| | | MCSP R |
AACTTTTCCTATATAAACATGCAC | |
| DeBruyn JM,
2011 |
Gemmatimonadetes | Gem F |
GAATGCGTAGAGATCC | (57) |
| | | Gem R |
CCGTCAATTCATTTGAGTTT | |
| Ferreira RB,
2011 | Eubacteria:
Universal primer for 16S rRNA | UniF340 |
ACTCCTACGGGAGGCAGCAGT | (58) |
| | | UniR514 |
ATTACCGCGGCTGCTGGC | |
| Dong Y, 2022 |
Lactobacillusspp. | LabF362 |
AGCAGTAGGGAATTCTTCCA | (59) |
| | | LabR677 |
CACCGCTACACATGGAG | |
| Rinttilä T,
2004 | BPP | F |
GGTGTCGGCTTAAGTGCCAT | (60) |
| | | R |
CGGACGTAAGGGCCGTGC | |
| Matsuki T,
2004 | Clostridium
leptum | F |
GCACAAGCAGTGGAGT | (44) |
| | | R |
CTTCCTCCGTTTTGTCAA | |
| Furet JP, 2009 | Clostridium
coccoides | F | GACGCCGCGTGAAGG
A | (61) |
| | | R | AGCCCCAGCCTTTCACAT
C | |
| Noratto GD,
2014 | Ruminococcus
spp. | F |
ACTGAGAGGTTGAACGGCCA | (62) |
| | | R |
CCTTTACACCCAGTAATTCCGGA | |
| Guo X, 2008 | Firmicutes | Firm934F |
GGAGCATGTGGTTTAATTCGAAGCA | (63) |
| | | Firm 1060R |
AGCTGACGACAACCATGCAC | |
| Guo X, 2008 | Bacteroidetes | Bact934F |
GGAACATGTGGTTTAATTCGATGAT | (63) |
| | | Bact1060R |
AGCTGACGACAACCATGCAG | |
| Ou J, 2013 |
Faecalibacterium spp. | F |
CCCTTCAGTGCCGCAGT | (64) |
| | | R |
GTCGCAGGATGTCAAGAC | |
| Pircalabioru G,
2022 |
Butyricicoccus spp. | F |
ACCTGAAGAATAAGCTCC | (65) |
| | | R |
GATAACGCTTGCTCCCTACGT | |
| Matsuki, 2004 |
Enterobacteriaceae | Uni515F | GTG CCA GCM GCC GCG
GTAA | (44) |
| | | Ent826R | GCC TCA AGG GCA CAA
CCT CCA AG | |
| B, Fungi 18 S
rRNA |
| Loeffler J,
2000 | 18S rRNA universal
primer | F |
ATTGGAGGGCAAGTCTGGTG | (66) |
| | | R |
CCGATCCCTAGTCGGCATAG | |
| Sokol H, 2017 |
Saccharomyces spp. | F |
AGGAGTGCGGTTCTTTG | (67) |
| | | R |
TACTTACCGAGGCAAGCTACA | |
| | Aspergillus
spp. | F |
GTGGAGTGATTTGTCTGCTTAATTG | (67) |
| | | R |
TCTAAGGGCATCACAGACCTGTT | |
| Frykman PK,
2015 | Candida
spp. | F |
TTTATCAACTTGTCACACCAGA | (68) |
| | | R |
ATCCCGCCTTACCACTACCG | |
Data points are presented as mean ± SEM, and graphs
were generated using GraphPad 5 software (Dotmatics). Differences
between the groups were computed using the Mann-Whitney test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
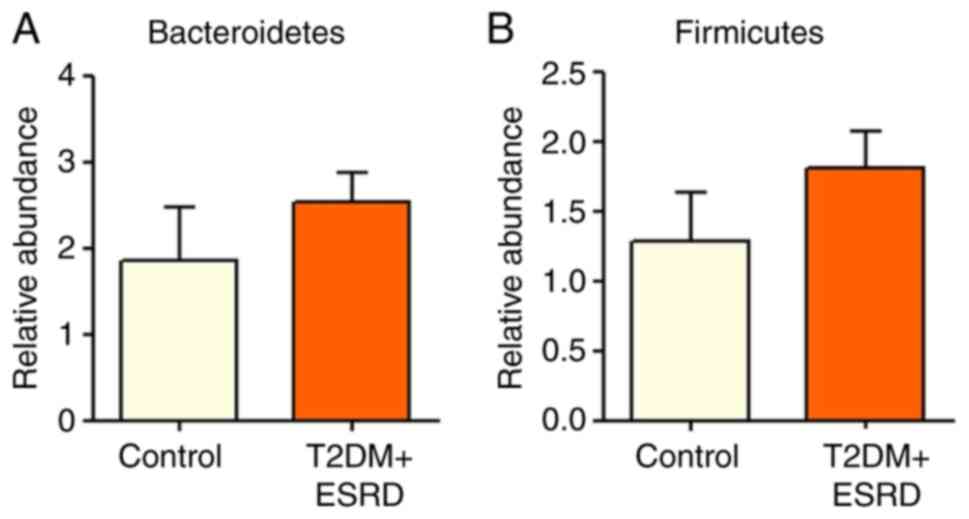
There were no significant differences in the
abundance of major bacterial phyla of the intestinal microbiota,
Bacteroidetes and Firmicutes, between the two patient groups
(Fig. 1). The intestinal
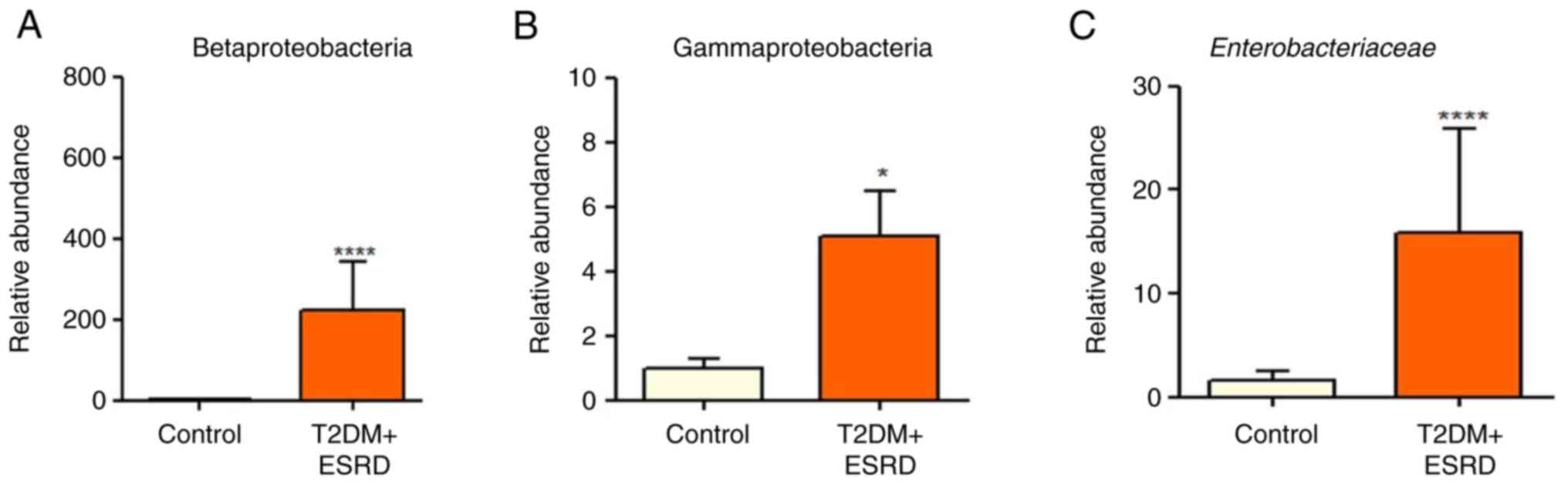
microbiota of the individuals with T2DM and ESRD was characterized
by increased levels of Gammaproteobacteria (Fig. 2B). In addition, all patients
exhibited a significantly elevated abundance of
Enterobacteriaceae (Fig.
2C). This microbial population is already associated with the
existence of an inflammatory process in the colon, and high levels
of Enterobacteriaceae are an indicator of intestinal
dysbiosis. Additionally, the Betaproteobacteria phylum was
significantly more abundant in the stool samples from the patients
with T2DM and ESRD, compared with that in the control group
(Fig. 2A).
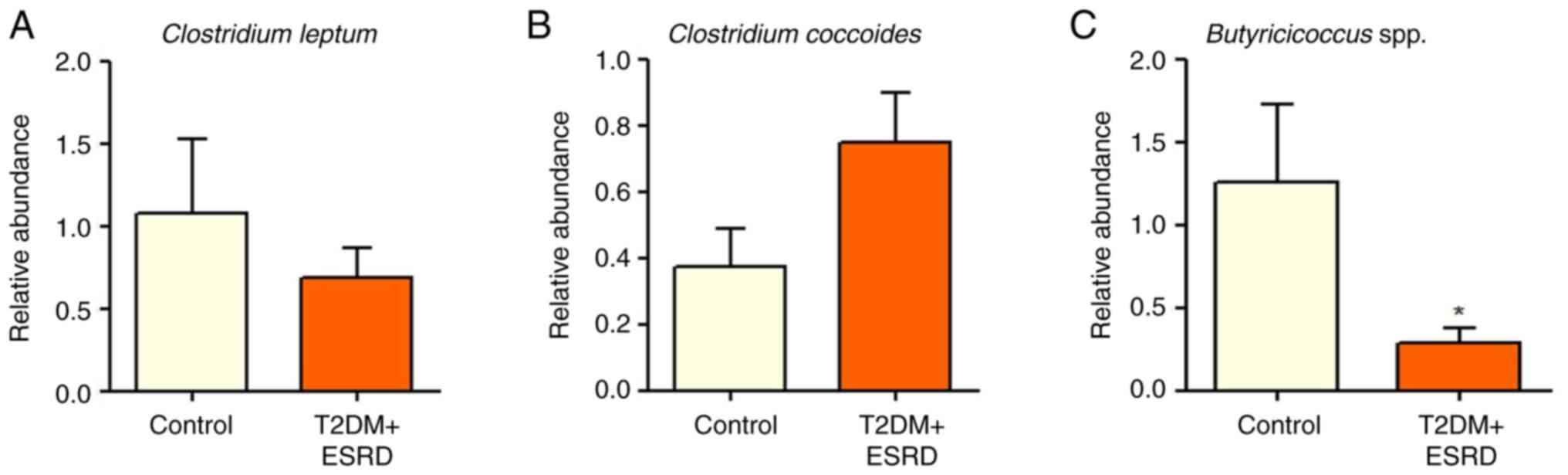
The Clostridium genus (represented by
Clostridium leptum and Clostridium coccoides) did not
show any significant differences between the two groups (Fig. 3A and B). The present study also analyzed the
abundance of Butyricicoccus spp., since this is a genus with
an important role in intestinal homeostasis, which has been
implicated in the production of SCFAs, especially butyrate
(31). Notably, the gut microbiota
of individuals with T2DM and ESRD was characterized by
significantly reduced levels of Butyricicoccus (Fig. 3C).
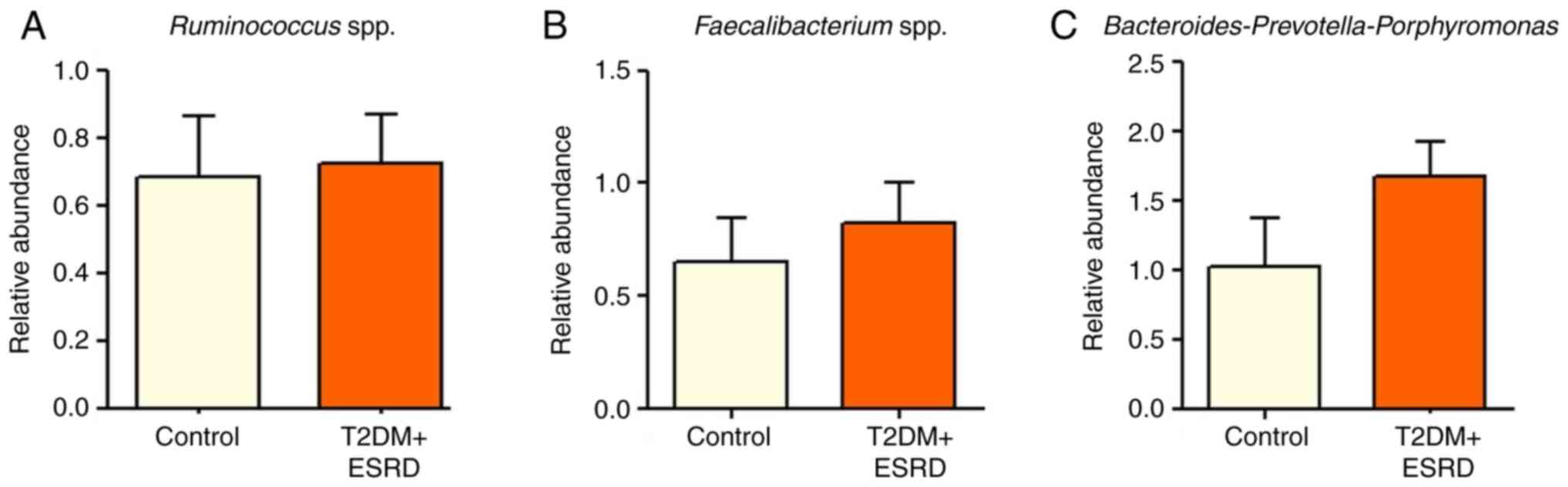
There were no statistically significant differences
in the abundance of Ruminococcus spp.,
Faecalibacterium spp. and
Bacteroides-Prevotella-Porphyromonas between patients with
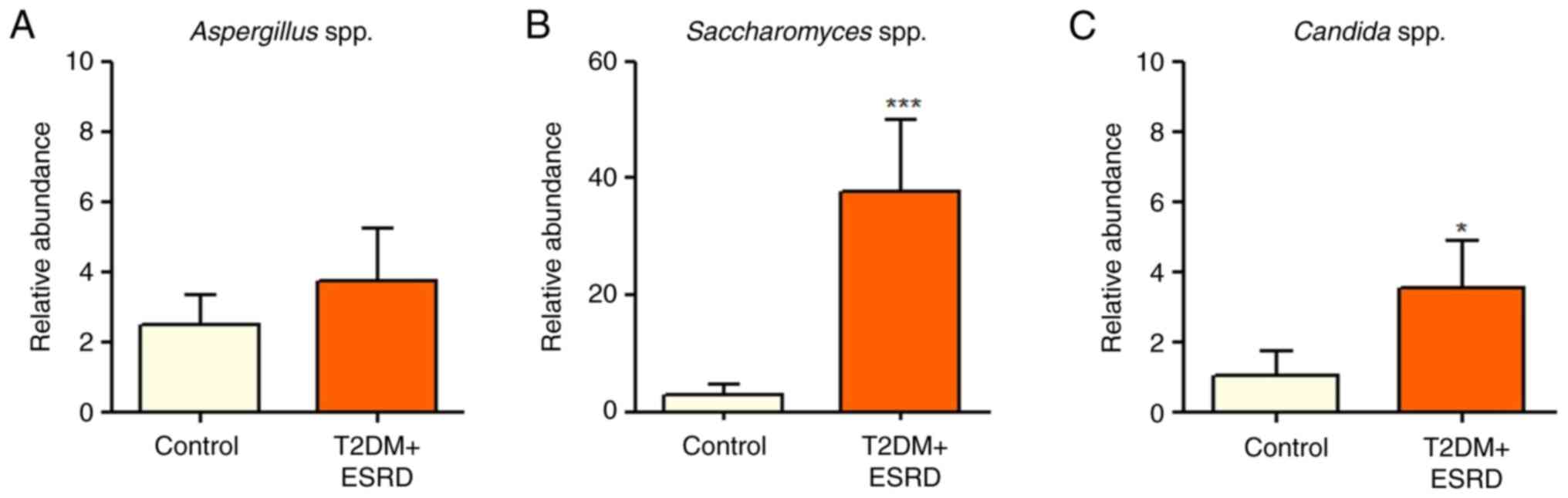
T2DM and ESRD and healthy individuals (Fig. 4). The present study also analyzed
the major fungal populations, including Candida spp.,
Aspergillus spp. and Saccharomyces spp. While there
were no statistically significant differences between the two
groups regarding Aspergillus spp., both Candida spp.
and Saccharomyces spp. were significantly increased in
patients with T2DM and ESRD compared with in the control group
(Fig. 5). A series of microbial
phyla and populations were not identified through quantitative PCR
(Cq value >38) in the patient group studied:
Mucispirillum, Verrucomicrobiota, Deferribacteraceae and
Tenericutes. This may be due to a very low abundance of these
species in the observed individuals. In this situation, the
aforementioned bacterial populations could be identified only
through future advanced sequencing techniques.
Discussion
To the best of our knowledge, the present study is
one of few studies that has focused on gut microbiome alterations
in T2DM and ESRD (32,33). There is ample information regarding
the characteristics of the intestinal microbiome in T2DM, and in
CKD and ESRD, respectively; however, not much data exists regarding
patients suffering from both pathologies.
Type 1 DM and T2DM are major causes of CKD, along
with hypertension and glomerulonephritis; however, all mechanisms
involved in the pathophysiology of CKD are not yet known. Even so,
it is well known that CKD contributes to alterations in the
composition and function of the gut microbiota, resulting in
dysbiosis. In addition, increasing evidence has suggested that the
various toxins produced by the microbiota, including cytokines and
non-self-components (i.e., uremic toxins, such as indoxyl sulfate,
p-cresyl sulfate and trimethylamine-N-oxide), along with
neuroendocrine molecules released in the intestines, can lead to
the appearance and development of CKD. This bidirectionality is the
subject of a number of recent research projects (34,35),
such as the present study.
The present study on microbial DNA identified
statistically significant increases in the abundance of
Gammaproteobacteria, Betaproteobacteria and
Enterobacteriaceae in the intestinal microbiota of
individuals with T2DM and ESRD, compared with those in the healthy
individuals in the control group. Proteobacteria is one of the most
abundant intestinal phyla (36),
characterized by the heterogeneity of the gram-negative bacteria
that compose it. Genetic analysis of 16S rRNA allowed the division
of the Proteobacteria phylum into six classes: Alphaproteobacteria,
Betaproteobacteria, Gammaproteobacteria, Deltaproteobacteria,
Epsilonproteobacteria and Zetaproteobacteria (37). A number of human pathogens
associated with various infectious diseases belong to this phylum:
Brucella and Rickettsia in the Alphaproteobacteria
class, Bordetella and Neisseria in the
Betaproteobacteria class, Escherichia, Shigella,
Salmonella and Yersinia in class Gammaproteobacteria,
and Helicobacter in class Epsilonproteobacteria. Besides the
intestines, these bacteria are also found on the skin, and in the
mouth, respiratory tract, stomach and vagina (38).
Studies performed on patients with metabolic
syndrome have confirmed the presence of alterations in the gut
microbiota of individuals with both T2DM and prediabetes (39). Specifically, the alterations
manifest through a significant increase in unknown species of the
Enterobacteriaceae family (39). The present study confirmed these
findings, implicating not only Enterobacteriaceae, but also
Betaproteobacteria and Gammaproteobacteria. Notably, the increases
in the abundance of these bacteria may result in the maintenance of
a low intensity inflammatory reaction in the digestive tract and
also in other parts of the body. This can be explained by the
existence of anatomical (the portal venous system) and functional
connections between the liver and the intestines; the gut-liver
axis. The liver is the first organ confronted with high levels of
lipopolysaccharide (LPS) produced by the aforementioned bacteria,
which were revealed to exhibit an increased abundance in the
present patient group. The Küpffer macrophages recognize LPS, along
with lipoteichoic acid (produced by gram-positive bacteria), as
pathogen-associated molecular pattern fragments that bind to
endocytosis receptors and Toll-like receptors (TLRs; e.g. TLR4 and
TLR2). The result of this binding is the activation of an
inflammatory process in the hepatic parenchyma, which is
accompanied by inflammatory cytokine release (IL-1β, TNF-α, IL-6
and IL-8). These cytokines act as signaling molecules, both locally
(autocrine and paracrine stimulation of Küpffer macrophages,
hepatocytes, endothelial cells, hepatic stellate cells and the
hepatic lymphocyte population) and at a distance (stimulation of
the bone marrow, hypothalamus and lymphoid structures throughout
the rest of the body). The low intensity inflammatory reaction thus
produced is not only present at the hepatic level, but also in the
pancreas and kidneys and, in the long term, can even induce
anatomical (fibrosis) and functional alterations (organ
insufficiency) or cancer (40).
On the other hand, some gut microbiome metabolites,
such as SCFAs (especially butyric and acetic acids), have an
anti-inflammatory effect, obtained through an epigenetic mechanism
involving the suppressor T cells (41). Moreover, SCFAs bind to G
protein-coupled receptors and trigger various mechanisms that
control obesity. These SCFAs can modulate an array of signaling
pathways inside the hepatic and intestinal cells, by binding
directly to transcription factors, with the purpose of maintaining
homeostasis. When the gut microbiota produces excessive amounts of
secondary biliary acids (deoxycholic acid and lithocholic acid),
intracellular signaling becomes exaggeratedly intense and a stress
response or even malignant transformation can appear (42). The present study demonstrated that
the intestinal microbiota of individuals with T2DM and ESRD was
characterized by significantly reduced levels of Butyricicoccus
spp. Thus, it may be concluded that patients with this type of
disease have a gut microbiota that produces significantly reduced
amounts of butyric acid. This, in turn, implies a diminished
anti-inflammatory effect on the intestinal and hepatic cells
(42).
Intestinal dysbiosis is associated with an increased
expression of genes that encode enzymes of the carbohydrate
metabolic pathways. This will give bacteria an increased capacity
to extract energy from the dietary products ingested by the host
and will generate an excessive accumulation of adipose tissue
(43). Although T2DM is mainly
associated with obesity, metagenomics has also identified
particularities of the fecal microbiota in patients with T2DM. Some
of the studies published on this subject have identified positive
correlations between improved glycemic control or insulin
resistance and specific gut microbiome compositions (43,44).
Research performed on Chinese patients with T2DM has shown a
moderate degree of intestinal dysbiosis, with a lower abundance of
butyrate-producing species and an increased number of several
opportunistic classes of bacteria, such as
Enterobacteriaceae (45)
The present study also indicated a significant
increase in some fungal populations: Candida spp. and
Saccharomyces spp. It is well known that candidiasis has a
higher prevalence among patients with DM (both type 1 and 2). This
increased abundance of Candida spp. in diabetes may be
explained through several mechanisms, depending on the type of
inflammatory reaction involved (local or systemic). Diabetes favors
fungal proliferation through hyperglycemia (46), an excessive secretion of some lytic
enzymes (47,48), and also through the
immunosuppressive state that it creates (48,49).
Some of these conditions that are favorable for fungal growth
include: An easier adhesion to epithelial cells, an increased
salivary level of glucose, a reduced salivary flow, microvascular
degeneration and an altered neutrophil anti-Candida capacity
(48,50). All of these factors disturb the
equilibrium between the fungi and their host, and transform
Candida from a commensal species to a pathogenic species. It
has also been observed that an inadequate glycemic control
increases the risk of candidiasis (51).
On the other hand, fungi from the
Saccharomyces family are known to have beneficial effects on
patients with DM, by improving blood-sugar levels, dyslipidemia,
alveolar bone destruction, hepatic inflammation (as shown by a
reduction in the transaminase serum levels) and by modulation of
the immune response (as shown by the reduction of serum TNF-α
levels) (52-54).
A previous study performed on C57BL/6 mice with
streptozotocin-induced diabetes demonstrated that administrating
Saccharomyces boulardii through intraperitoneal injections
during an 8-week period reduced hepatic hydropic degeneration and
hepatic vascular congestion, diminished oxidative stress (by
reducing carbonylated proteins, and increasing the activity of the
antioxidant enzymes superoxide dismutase and glutathione
peroxidase) and normalized concentrations of the
renin-angiotensin-aldosterone system peptides (affecting both the
liver and the kidneys) (55).
The most interesting aspect of the present pilot
study is the research population it focused on: Individuals that
suffer from both T2DM and ESRD in renal replacement therapy.
Studies have already been performed that have focused on the
alterations of the intestinal microbiome in both diabetes and
CKD/ESRD, but there are very few, if any, studies that have focused
on patients with this specific association of pathologies (32,33).
Thus, the present results could shed further light upon the complex
interrelations that appear between the two aforementioned
pathologies and the gut microbiome. Potentially, in the future, we
may offer new methods of targeting the development and progression
of T2DM and ESRD, focusing on the intestinal microbiome.
One limitation of the present study is the small
sample size. The healthy control group consisted of only 8
individuals, since subjects were age-matched and must have
completed a nutritional questionnaire in order to avoid the impact
of variables such as age and diet on the gut microbiome. In
addition, when using cohorts containing older individuals, it is
difficult to find matched healthy controls without other
comorbidities that may also affect the gut microbiome (such as
obesity, osteoporosis, cancer and autoimmune disease). More
conclusive results may be obtained if the period of the study was
extended and if more participants were recruited. This would allow
for obtaining more significant results, and also observing the
progression of the diseases and the appearance of potential
complications, in relation to the changes in the gut
microbiota.
In conclusion, the alterations observed in the
present study suggested that local, regional (liver, pancreas and
kidneys) and systemic inflammatory processes occurred in the
present patient group. Because the present study consisted of
patients in the advanced stages of disease, it is difficult to
specify cause and effect; however, undoubtedly, gut microbiome
alterations may have a role in very complex pathophysiological
phenomena.
Acknowledgements
The authors would like to thank Professor Marieta
Costache (Department of Biochemistry and Molecular Biology,
University of Bucharest) for access to the qunatititative PCR
machine.
Funding
Funding: This research was funded by UEFISCDI [project ID
PN-III-P1-1.1-PD-2019-0499, grant no. 224/2021 and Academia
Oamenilor de Stiinta din Romania (AOSR) Teams grant no.
288/20.02.2022].
Availability of data and materials
The data generated in the present study are not
publicly available due to them containing information that could
compromise research participant privacy/consent but may be
requested from the corresponding author.
Authors' contributions
MT was involved in drafting the manuscript, data
analysis and patient recruitment. GGP was involved in study design,
funding acquisition, data analysis and manuscript editing. OS was
involved in data analysis, supervision, manuscript writing and
editing. MT, GGP and OS confirmed the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was conducted according to the guidelines
of The Declaration of Helsinki and was approved by the Ethics
Committee of University of Bucharest (protocol code CEC reg. no.
235/9.10.2019). The present study was approved by the Ethics
Commission of ‘NC Paulescu’ National Institute of Diabetes,
Nutrition and Metabolic Diseases (Bucharest, Romania; approval no.
Certif. 5911/04.10.2019). All participants gave their written
informed consent upon inclusion in the study.
Patient consent for publication
Patients provided written informed consent for data
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Luca M, Mauro MD, Mauro MD and Luca A: Gut
microbiota in Alzheimer's disease, depression, and type 2 diabetes
mellitus: The role of oxidative stress. Oxid Med Cell Longev.
2019(4730539)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Neish AS: Microbes in gastrointestinal
health and disease. Gastroenterology. 136:65–80. 2009.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bäckhed F, Ding H, Wang T, Hooper LV, Koh
GY, Nagy A, Semenkovich CF and Gordon JI: The gut microbiota as an
environmental factor that regulates fat storage. Proc Natl Acad Sci
USA. 101:15718–15723. 2004.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Gill SR, Pop M, DeBoy RT, Eckburg PB,
Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM
and Nelson KE: Metagenomic analysis of the human distal gut
microbiome. Science. 312:1355–1359. 2006.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ursell LK, Metcalf JL, Parfrey LW and
Knight R: Defining the human microbiome. Nutr Rev. 70 (Suppl
1):S38–S44. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Thursby E and Juge N: Introduction to the
human gut microbiota. Biochem J. 474:1823–1836. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yang T, Richards EM, Pepine CJ and Raizada
MK: The gut microbiota and the brain-gut-kidney axis in
hypertension and chronic kidney disease. Nat Rev Nephrol.
14:442–456. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Vaziri ND, Wong J, Pahl M, Piceno YM, Yuan
J, DeSantis TZ, Ni Z, Nguyen TH and Andersen GL: Chronic kidney
disease alters intestinal microbial flora. Kidney Int. 83:308–315.
2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ranganathan N, Friedman EA, Tam P, Rao V,
Ranganathan P and Dheer R: Probiotic dietary supplementation in
patients with stage 3 and 4 chronic kidney disease: A 6-month pilot
scale trial in Canada. Curr Med Res Opin. 25:1919–1930.
2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chen YY, Chen DQ, Chen L, Liu JR, Vaziri
ND, Guo Y and Zhao YY: Microbiome-metabolome reveals the
contribution of gut-kidney axis on kidney disease. J Transl Med.
17(5)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Hida M, Aiba Y, Sawamura S, Suzuki N,
Satoh T and Koga Y: Inhibition of the accumulation of uremic toxins
in the blood and their precursors in the feces after oral
administration of Lebenin, a lactic acid bacteria preparation, to
uremic patients undergoing hemodialysis. Nephron. 74:349–355.
1996.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Mahmoodpoor F, Rahbar Saadat Y, Barzegari
A, Ardalan M and Zununi Vahed SV: The impact of gut microbiota on
kidney function and pathogenesis. Biomed Pharmacother. 93:412–419.
2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Lau WL, Savoj J, Nakata MB and Vaziri ND:
Altered microbiome in chronic kidney disease: Systemic effects of
gut-derived uremic toxins. Clin Sci (Lon). 132:509–522.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jiang S, Xie S, Lv D, Wang P, He H, Zhang
T, Zhou Y, Lin Q, Zhou H, Jiang J, et al: Alteration of the gut
microbiota in Chinese population with chronic kidney disease. Sci
Rep. 7(2870)2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wong J, Piceno YM, DeSantis TZ, Pahl M,
Andersen GL and Vaziri ND: Expansion of urease- and
uricase-containing, indole- and p-cresol-forming and contraction of
short-chain fatty acid-producing intestinal microbiota in ESRD. Am
J Nephrol. 39:230–237. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ramezani A and Raj DS: The gut microbiome,
kidney disease, and targeted interventions. J Am Soc Nephrol.
25:657–670. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Dunne JL, Triplett EW, Gevers D, Xavier R,
Insel R, Danska J and Atkinson MA: The intestinal microbiome in
type 1 diabetes. Clin Exp Immunol. 177:30–37. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Al Khodor S and Shatat IF: Gut microbiome
and kidney disease: A bidirectional relationship. Pediatr Nephrol.
32:921–931. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Patterson E, Ryan PM, Cryan JF, Dinan TG,
Ross RP, Fitzgerald GF and Stanton C: Gut microbiota, obesity and
diabetes. Postgrad Med J. 92:286–300. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Salgaço MK, Oliveira LGS, Costa GN,
Bianchi F and Sivieri K: Relationship between gut microbiota,
probiotics, and type 2 diabetes mellitus. Appl Microbiol
Biotechnol. 103:9229–9238. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Xu Q, Ni JJ, Han BX, Yan SS, Wei XT, Feng
GJ, Zhang H, Zhang L, Li B and Pei YF: Causal relationship between
gut microbiota and autoimmune diseases: A two-sample mendelian
randomization study. Front Immunol. 12(746998)2022.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Estrada V and Gonzalez N: Gut microbiota
in diabetes and HIV: Inflammation is the link. EBioMedicine.
38:17–18. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Schéle E, Grahnemo L, Anesten F, Halleń A,
Backhed F and Jansson JO: The gut microbiota reduces leptin
sensitivity and the expression of the obesity-suppressing
neuropeptides proglucagon (Gcg) and brain-derived neurotrophic
factor (Bdnf) in the central nervous system. Endocrinology.
154:3643–3651. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kreznar JH, Keller MP, Traeger LL,
Rabaglia ME, Schueler KL, Stapleton DS, Zhao W, Vivas EI, Yandell
BS, Broman AT, et al: Host genotype and gut microbiome modulate
insulin secretion and diet-induced metabolic phenotypes. Cell Rep.
18:1739–1750. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wahlström A, Sayin SI, Marschall HU and
Bäckhed F: Intestinal crosstalk between bile acids and microbiota
and its impact on host metabolism. Cell Metab. 24:41–50.
2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Sharma S and Tripathi P: Gut microbiome
and type 2 diabetes: Where we are and where to go? J Nutr Biochem.
63:101–108. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Rosario D, Benfeitas R, Bidkhori G, Zhang
C, Uhlen M, Shoaie S and Mardinoglu A: Understanding the
representative gut microbiota dysbiosis in metformin-treated Type 2
diabetes patients using genome-scale metabolic modeling. Front
Physiol. 9(775)2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
American Diabetes Association. Standards
of care in diabetes-2023 abridged for primary care providers. Clin
Diabetes. 41:4–31. 2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Acosta-Ochoa I, Bustamante-Munguira J,
Mendiluce-Herrero A, Bustamante-Bustamante J and Coca-Rojo A:
Impact on outcomes across KDIGO-2012 AKI criteria according to
baseline renal function. J Clin Med. 8(1323)2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Takada T, Watanabe K, Makino H and Kushiro
A: Reclassification of Eubacterium desmolans as Butyricicoccus
desmolans comb. nov., and description of Butyricicoccus
faecihominis sp. nov., a butyrate-producing bacterium from human
faeces. Int J Syst Evol Microbiol. 66:4125–4131. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Koshida T, Gohda T, Sugimoto T, Asahara T,
Asao R, Ohsawa I, Gotoh H, Murakoshi M, Suzuki Y and Yamashiro Y:
Gut microbiome and microbiome-derived metabolites in patients with
end-stage kidney disease. Int J Mol Sci. 24(11456)2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sabatino A, Regolisti G, Cosola C,
Gesualdo L and Fiaccadori E: Intestinal microbiota in type 2
diabetes and chronic kidney disease. Curr Diab Rep.
17(16)2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Kim SM and Song IH: The clinical impact of
gut microbiota in chronic kidney disease. Korean J Intern Med.
35:1305–1316. 2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kambay M, Onal EM, Afsar B, Dagel T,
Yerlikaya A, Covic A and Vaziri ND: The crosstalk of gut microbiota
and chronic kidney disease: Role of inflammation, proteinuria,
hypertension, and diabetes mellitus. Int Urol Nephrol.
50:1453–1466. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Rizzatti G, Lopetuso LR, Gibiino G, Binda
C and Gasbarrini A: Proteobacteria: A common factor in human
diseases. Biomed Res Int. 2017(9351507)2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Dang H, Chen R, Wang L, Shao S, Dai L, Ye
Y, Guo L, Huang G and Klotz MG: Molecular characterization of
putative biocorroding microbiota with a novel niche detection of
Epsilon- and Zetaproteobacteria in Pacific Ocean coastal seawaters.
Environ Microbiol. 13:3059–3074. 2011.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Gomes-Neto JC, Mantz S, Held K, Sinha R,
Segura Munoz RR, Schmaltz R, Benson AK, Walter J and Ramer-Tait AE:
A real-time PCR assay for accurate quantification of the individual
members of the altered schaedler flora microbiota in gnotobiotic
mice. J Microbiol Methods. 135:52–62. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lambeth SM, Carson T, Lowe J, Ramaraj T,
Leff JW, Luo L, Bell CJ and Shah VO: Composition, diversity and
abundance of gut microbiome in prediabetes and type 2 diabetes. J
Diabetes Obes. 2:1–7. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Ohtani N and Kawada N: Role of the
gut-liver axis in liver inflammation, fibrosis, and cancer: A
special focus on the gut microbiota relationship. Hepatol Commun.
3:456–470. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Furusawa Y, Obata Y, Fukuda S, Endo TA,
Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et
al: Commensal microbe-derived butyrate induces the differentiation
of colonic regulatory T cells. Nature. 504:446–450. 2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Bäckhed F, Ley RE, Sonnenburg JL, Peterson
DA and Gordon JI: Host-bacterial mutualism in the human intestine.
Science. 307:1915–1920. 2005.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Gérard C and Vidal H: Impact of gut
microbiota on host glycemic control. Front Endocrinol (Lousanne).
10(29)2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Matsuki T, Watanabe K, Fujimoto J, Takada
T and Tanaka R: Use of 16S rRNA gene-targeted group-specific
primers for real-time PCR analysis of predominant bacteria in human
feces. Appl Environ Microbiol. 70:7220–7228. 2004.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F,
Liang S, Zhang W, Guan Y, Shen D, et al: A metagenome-wide
association study of gut microbiota in type 2 diabetes. Nature.
490:55–60. 2012.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Duggan S, Essig F, Hünniger K, Mokhtari Z,
Bauer L, Lehnert T, Brandes S, Häder A, Jacobsen ID, Martin R, et
al: Neutrophil activation by Candida glabrata but not Candida
albicans promotes fungal uptake by monocytes. Cell Microbiol.
17:1259–1276. 2015.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Motta-Silva AC, Aleva NA, Chavasco JK,
Armond MC, França JP and Pereira LJ: Erythematous oral candidiasis
in patients with controlled type II diabetes mellitus and complete
dentures. Mycopathologia. 169:215–223. 2010.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Rodrigues CF, Rodrigues ME and Henriques
M: Candida sp. Infections in patients with diabetes mellitus. J
Clin Med. 8(76)2019.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Balan P, B Gogineni S, Kumari NS, Shetty
V, Lakshman Rangare A, L Castelino R and Areekat KF: Candida
carriage rate and growth characteristics of saliva in diabetes
mellitus patients: A case-control study. J Dent Res Dent Clin Dent
Prospects. 9:274–279. 2015.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Kadir T, Pisiriciler R, Akyüz S, Yarat A,
Emekli N and Ipbüker A: Mycological and cytological examination of
oral candidal carriage in diabetic patients and non-diabetic
control subjects: Thorough analysis of local aetiologic and
systemic factors. J Oral Rehab. 29:452–457. 2002.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Gosiewski T, Salamon D, Szopa M, Sroka A,
Malecki MT and Bulanda M: Quantitative evaluation of fungi of the
genus Candida in the feces of adult patients with type 1 and 2
diabetes-a pilot study. Gut Pathogens. 6(43)2014.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Albuquerque RCMF, Brandão ABP, de Abreu
ICME, Ferreira FG, Santos LB, Moreira LN, Taddei CR, Aimbire F and
Cunha TS: Saccharomyces boulardii Tht 500101 changes gut microbiota
and ameliorates hyperglycaemia, dyslipidaemia, and liver
inflammation in streptozotocin-diabetic mice. Beneficial Microbes.
10:901–912. 2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Silva Vde O, Lobato RV, Andrade EF, de
Macedo CG, Napimoga JT, Napimoga MH, Messora MR, Murata RM and
Pereira LJ: B-glucans (Saccharomyces cereviseae) reduce glucose
levels and attenuate alveolar bone loss in diabetic rats with
periodontal disease. PLoS One. 10(e0134742)2015.PubMed/NCBI View Article : Google Scholar
|
|
54
|
De Sales Guilarducci J, Marcelino BAR,
Konig IFM, Orlando TM, Varaschin MS and Pereira LJ: Therapeutic
effects of different doses of prebiotic (isolated from
Saccharomyces cerevisiae) in comparison to n-3 supplement on
glycemic control, lipid profiles and immunological response in
diabetic rats. Diabetol Metab Syndr. 12(69)2020.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Barssotti L, Abreu ICME, Brandão ABP,
Albuquerque RCMF, Ferreira FG, Salgado MAC, Dias DDS, De Angelis K,
Yokota R, Casarini DE, et al: Saccharomyces boulardii modulates
oxidative stress and renin angiotensin system attenuating
diabetes-induced liver injury in mice. Sci Rep.
11(9189)2021.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Yang YW, Chen MK, Yang BY, Huang XJ, Zhang
XR, He LQ, Zhang J and Hua ZC: Use of 16S rRNA gene-targeted
group-specific primers for real-time PCR analysis of predominant
bacteria in mouse feces. Appl Environ Microbiol. 81:6749–6756.
2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
DeBruyn JM, Nixon LT, Fawaz MN, Johnson AM
and Radosevich M: Global biogeography and quantitative seasonal
dynamics of Gemmatimonadetes in soil. Appl Environ Microbiol.
77:6295–6300. 2011.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Ferreira RB, Gill N, Willing BP, Antunes
LC, Russell SL, Croxen MA and Finlay BB: The intestinal microbiota
plays a role in Salmonella-induced colitis independent of pathogen
colonization. PLoS One. 6(e20338)2011.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Dong Y, Fan H, Zhang Z, Jiang F, Li M,
Zhou H, Guo W, Zhang Z, Kang Z, Gui Y, et al: Berberine ameliorates
DSS-induced intestinal mucosal barrier dysfunction through
microbiota-dependence and Wnt/β-catenin pathway. Int JBiol Sci.
18:1381–1397. 2022.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Rinttilä T, Kassinen A, Malinen E, Krogius
L and Palva A: Development of an extensive set of 16S rDNA-targeted
primers for quantification of pathogenic and indigenous bacteria in
faecal samples by real-time PCR. J Appl Microbiol. 97:1166–1177.
2004.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Furet JP, Firmesse O, Gourmelon M,
Bridonneau C, Tap J, Mondot S, Doré J and Corthier G: Comparative
assessment of human and farm animal faecal microbiota using
real-time quantitative PCR. FEMS Microbiol Ecol. 68:351–362.
2009.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Noratto GD, Garcia-Mazcorro JF, Markel M,
Martino HS, Minamoto Y, Steiner JM, Byrne D, Suchodolski JS and
Mertens-Talcott SU: Carbohydrate-free peach (Prunus persica) and
plum (Prunus salicina) [corrected] juice affects fecal microbial
ecology in an obese animal model. PLoS One.
9(e101723)2014.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Guo X, Xia X, Tang R, Zhou J, Zhao H and
Wang K: Development of a real-time PCR method for Firmicutes and
Bacteroidetes in faeces and its application to quantify intestinal
population of obese and lean pigs. Lett Appl Microbiol. 47:367–373.
2008.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Ou J, Carbonero F, Zoetendal EG, DeLany
JP, Wang M, Newton K, Gaskins HR and O'Keefe SJ: Diet, microbiota,
and microbial metabolites in colon cancer risk in rural Africans
and African Americans. Am J Clin Nutr. 98:111–120. 2013.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Gradisteanu Pircalabioru G, Chifiriuc MC,
Picu A, Petcu LM, Trandafir M and Savu O: Snapshot into the
type-2-diabetes-associated microbiome of a Romanian cohort. Int J
Mol Sci. 23(15023)2022.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Loeffler J, Hebart H, Magga S, Schmidt D,
Klingspor L, Tollemar J, Schumacher U and Einsele H: Identification
of rare Candida species and other yeasts by polymerase chain
reaction and slot blot hybridization. Diagn Microb Infect Dis.
38:207–212. 2000.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Sokol H, Leducq V, Aschard H, Pham HP,
Jegou S, Landman C, Cohen D, Liguori G, Bourrier A and
Nion-Larmurier I: Fungal microbiota dysbiosis in IBD. Gut.
66:1039–1048. 2017.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Frykman PK, Nordenskjöld A, Kawaguchi A,
Hui TT, Granström AL, Cheng Z, Tang J, Underhill DM, Iliev I,
Funari VA, et al: Characterization of bacterial and fungal
microbiome in children with hirschsprung disease with and without a
history of enterocolitis: A multicenter study. PLoS One.
10(e0124172)2015.PubMed/NCBI View Article : Google Scholar
|