Introduction
Myocardial infarction (MI), which is predominantly
caused by myocardial ischaemia, leads to large areas of myocardial
cell necrosis and apoptosis (1).
Cardiac remodelling after MI is characterized by inflammation,
fibrosis and cardiac hypertrophy in the remaining myocardium
(2). This adverse cardiac
remodelling eventually leads to heart failure (3,4). To
date, although emergency coronary artery revascularization can save
the life of patients with acute MI (AMI) and numerous anti-heart
failure treatments (such as angiotensin receptor antagonists and β
adrenergic receptor blockers) designed to suppress adverse cardiac
remodelling exist, the incidence of heart failure (with an
estimated prevalence of >37.7 million individuals globally) and
mortality caused by AMI remains high at an incidence of ~10%
worldwide (4,5). Therefore, there is a demand to
identify novel effective treatment strategies for MI and MI-induced
heart failure.
Autophagy is an important process of energy
metabolism. Under conditions of energy deprivation, autophagy is
activated, which degrades intracellular organelles and proteins
into amino acids and fatty acids to produce energy substrates for
the recycling of energy (6). Basal
levels of autophagy are essential for the maintenance of cardiac
function. Autophagy is activated to protect cardiomyocytes from
ischaemia or ischaemia/reperfusion injury under conditions of
short-term myocardial ischaemia stress (7). However, under chronic myocardial
hypoxia, excessive activation of autophagy aggravates necrosis and
apoptosis in myocardial cells, which in turn accelerates the
adverse progression of myocardial remodelling (7). Therefore, investigation into the
modulation of autophagy activation is an important topic for
slowing the progression of myocardial remodelling after MI to
prevent heart failure.
Meteorin-β (Metrnβ), which is also known as
interleukin (IL)-41, is a secretory protein that is mainly secreted
by skeletal muscle and adipose tissue (8). Previous studies have revealed that it
likely regulates energy metabolism in skeletal muscle and adipose
tissues (9,10). However, other studies have revealed
that Metrnβ is closely associated with immunity, inflammation,
obesity and diabetes (9,10). In addition, recent studies have
reported that Metrnβ serves a role in cardiovascular diseases
(11,12). Following isoproterenol stimulation,
Metrnβ-knockout mice exhibited myocardial hypertrophy, fibrosis and
heart failure. However, Metrnβ overexpression could inhibit this
type of myocardial remodelling induced by isoproterenol (11). In addition, another study reported
that myocardial cells could also secrete Metrnβ, and that the
concentration of Metrnβ in the human serum was correlated (Pearson
correlation) with the prognosis of heart failure (12). However, the role of Metrnβ in MI
remains to be fully elucidated. Therefore, the present study aimed
to explore the role and underlying mechanism of Metrnβ in MI. In
the present study, the effects and the underlying mechanism of
Metrnβ on MI-induced cardiac remodelling were explored.
Materials and methods
Animals
C57BL6J male 8-week-old mice (24-27 g) were
purchased from Beijing Huafukang Biotechnology Co., Ltd. The
animals were housed with a maximum of six other mice in
individually-ventilated cages (with a floor area of 542
cm2 and bedded with corncob). The animals were allowed
free access to food and water and were maintained on a 12 h
light/dark cycle in a controlled temperature (20-25˚C), humidity
(50±5%) and specific pathogen-free environment (13). In total, 90 mice were used, among
which 16 mice (n=6 for the sham group; n=10 for the MI group) were
used to detect alterations in the Metrnβ expression levels after
being subjected to MI. The other 74 mice were divided into four
groups: Adeno-associated virus 9 (AAV9)-negative control (NC)-sham
group (n=12); AAV9-Metrnβ-sham group (n=12); AAV9-NC-MI group
(n=25); and AAV9-Metrnβ-MI group (n=25). Mice in the MI groups were
subjected to left anterior descending (LAD) coronary artery
ligation surgery. For AAV9 injection, mice were subjected to
retro-orbital venous plexus injection of either AAV9-NC or
AAV9-Metrnβ 2 weeks before surgery. Animal health and behaviour
were monitored daily. Being incapable of maintaining normal
activities (n=3, after MI surgery) or eating on their own (n=1)
were the humane endpoints used to determine when the animals should
be sacrificed to minimize suffering. During and after the ligation
operation, 26 mice died from MI. The remaining 60 mice were
sacrificed at the end of the scheduled experiment to collect the
heart tissue. Mice were euthanized by cervical dislocation under
anaesthesia (2% isoflurane) and mortality was verified by the
cessation of breathing and heartbeat. The animal experiments were
performed (Sep. 2021 to Dec. 2022) according to the Animal
Research: Reporting of in vivo Experiments guidelines
(14) and were approved by the
Animal Care and Use Committee of the First Affiliated Hospital of
Zhengzhou University (approval no. 2021-02623).
Animal model of MI
LAD was conducted according to a previously
published protocol (15). Briefly,
anaesthesia was induced in mice by 2% isoflurane and maintained
with 1.5% isoflurane through a face mask, before the mice were
placed in a supine position. The thorax between the left third and
fourth ribs was then opened. After the pericardium was excised, 7-0
threads were ligated at the proximal end of the left coronary
artery. For the sham operation, all of the procedures were the same
except ligation. After surgery, buprenorphine (0.1 mg/kg,
subcutaneously injected) was used for postoperative analgesia in
mice. The success of the MI was evaluated by the increased cardiac
troponin T (cTnT) levels in the blood samples 12 h after LAD (three
times that of the sham group). When sampling the heart tissues, the
atrium was first removed to expose the ventricular cavities before
the thinner right ventricular tissues were removed to collect the
left ventricular tissues.
AAV9-Metrnβ and the control AAV9-NC
construction and viral delivery
AAV9-Metrnβ and the AAV9-NC [also AAV9-green
fluorescent protein (AAV9-GFP)], both with TnT promoters, were
purchased from the Vigene Bioscience Company. The plasmids
pCI-Metrnβ-his6 and an empty vector (pCI-NC) were used for
AAV9-Metrnβ and AAV9-NC, respectively. Mice were randomly assigned
to receive either 60-80 µl AAV9-Metrnβ or AAV9-NC at
5.0-6.5x1013 gene copy/ml in sterile PBS at 37˚C by
injection into the retro-orbital venous plexus 2 weeks before LAD
surgery, as described in a previous study (14). Briefly, 2% isoflurane was used to
induce and maintain anaesthesia in mice and a drop of ophthalmic
anaesthetic (0.5% proparacaine hydrochloride ophthalmic solution)
was placed on the eye that received the injection.
Echocardiography measurements
Echocardiography was conducted according to
previously published guidance (15). Mice were anesthetized with 1.5-2.0%
isoflurane to induce and maintain anaesthesia. Echocardiography
with a Mylab 30CV (Esaote) was used to measure cardiac function 2
weeks post MI. The M Doppler uses a 15-mHz probe to detect and
record cardiac function. The left ventricular (LV) ejection
fraction (LVEF) and LV fractional shortening (LVFS) were calculated
after data collection. The software MyLab™ Desk version 3 (Esaote)
was used to calculate HR and LVEF. LVFS was calculated using the
following formula:
LVFS (%)=(LVEDd-LVESd) x(100%/LVEDd); LVEF
(%)=(LVEDd3-LVESd3)
x(100%/LVEDd3). After echocardiography, mice were then
subjected to haemodynamic detection.
Pressure-volume measurements
Haemodynamic detection was conducted according to a
previous study 2 weeks post MI (16). Anaesthesia in mice was induced with
2% isoflurane and maintained with 1.5% isoflurane (17,18).
A 1.4 French (4.5 mm) Millar catheter transducer (Millar, Inc.) was
transferred from the right carotid artery in the right of the neck
to the left ventricle. A Millar pressure-volume system (MPVS-400;
Millar, Inc.) and a Powerlab/4SP A/D converter (AD instruments,
Inc.) were used to continuously record the pressure signals and
heart rate. Heart rate, end systolic volume, end diastolic volume,
maximal rate of systolic pressure increment (+dp/dt) and diastolic
pressure decrement (-dp/dt) and cardiac output were calculated and
corrected according to in vitro and in vivo volume
calibrations with PVAN 2.3 software (ADInstruments, Ltd.). The
hearts were collected after haemodynamic detection.
H&E and picrosirius red (PSR)
staining
H&E and PSR staining were conducted according to
previously published guidance (19,20).
Briefly, heart sections (4-5 µm thick) were prepared, and sections
stained with H&E were separated by 500 µm to determine infarct
size. Image-Pro Plus, version 6.0 (Media Cybernetics, Inc.), was
used to calculate MI size. Cross-sectional areas of cardiomyocytes
were observed using FITC-combined wheat germ agglutinin (WGA) and
cTnT (Invitrogen; Thermo Fisher Scientific, Inc.) fluorescence
staining. The nuclei were stained with 4',6-diamino-2-phenylindole
(DAPI). Image-Pro Plus (version 6.0) was used to measure the
myocardial cell cross-sectional area.
Several sections of each heart (4-5-mm thick) were
prepared and stained with PSR for collagen deposition analysis, and
they were then visualized with light microscopy. The LV collagen
volume fraction was calculated from the PSR-stained sections as the
area stained by PSR divided by the total area. Image-Pro Plus
(version 6.0) was used to measure the myocardial cell
cross-sectional area and LV collagen fraction.
Enzyme-linked immunosorbent assay
(ELISA) detection of inflammatory cytokines
Heart tissue samples and cell samples were lysed and
tested using ELISA. The inflammatory cytokine indicators included
tumour necrosis factor α (TNFα; LEGEND MAX™ Mouse TNF-α ELISA kit;
cat. no. #430907), IL-1β (ELISA MAX™ Deluxe Set Mouse IL-1α; cat.
no. #433404) and IL-6 (LEGEND MAX™ Mouse IL-6 ELISA kit; cat. no.
#431307). The aforementioned ELISA kits were purchased from
BioLegend, Inc. The ELISA kit for Metrnβ (Mouse
Meteorin-like/METRNL DuoSet ELISA; cat. no. DY6679) was purchased
from R&D Systems China Co., Ltd. The concentration of each
sample was calculated using the standard curve method.
cTnT levels in blood samples
The venous blood from tail vein (100-200 µl) of mice
was collected 12 h after the LAD operation, and serum cTnT
concentrations were detected after 200 x g centrifugation for 15
min at 4˚C. All steps were performed according to the
manufacturer's protocols of the cardiac troponin assay kit (cat.
no. #H149-4-1; Nanjing Jiancheng Bioengineering Institute). The
ELISA plate reader (Synergy HT; Agilent Technologies, Inc.) was
used for detection.
Cell culture
H9c2 rat cardiomyocytes were purchased from The Cell
Bank of Type Culture Collection of The Chinese Academy of Sciences.
Dulbecco's minimum essential medium (DMEM) (cat. no. C11995; Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% foetal bovine
serum (FBS; Thermo Fisher Scientific, Inc.) was used for cell
culture at 37˚C (95% O2 and 5% CO2) (21,22).
A cell hypoxia model was induced for 24 h at 37˚C by culturing with
5% O2, 5% CO2 and 90% nitrogen. Control cells
were cultured in a normal atmosphere at 37˚C, 5% CO2 and
95% air (21% O2 and 78% nitrogen).
Transfection and treatments
After culture with FBS-free DMEM for 12 h at 37˚C,
cells were transfected with adenovirus overexpress with Metrnβ
(Ad-Metrnβ; MOI=50; diluted with PBS) for 24 h at 37˚C to
overexpress Metrnβ. Adenovirus carrying a control vector (Ad-NC;
MOI=50) was used as control (pAd-CMV-V5 vector; Invitrogen; Thermo
Fisher Scientific, Inc.). To silence Metrnβ expression, cells were
transfected with the small interfering RNA (siRNA) against Metrnβ
(Metrnβ siRNA; 50 nM; Guangzhou RiboBio Co., Ltd.) for 24 h at 37˚C
using the Lipo 6000™ transfection reagent (Beyotime Institute of
Biotechnology). A scramble siRNA (scRNA) was used as control. The
transfected sequences used were as follows: Metrnβ siRNA (sense
5'-CACGCTTTAGTGACTTTCAAA-3' and antisense
5'-TTTGAAAGTCACTAAAGCGTG-3'); and scRNA (sense
5'-TTCTCCGAACGTGTCACGT-3' and antisense 5'-ACGTGACACGTTCGGAGAA-3').
Cells were used for subsequent functional experiments at 24 h
post-transfection.
In addition, cells were treated with bafilomycin A1
(BAF; 100 nM; MedChemExpress) or rapamycin (10 mM; MedChemExpress)
for 12 h at 37˚C to block autophagosome degradation or activate
autophagy, respectively, before being transfected with Ad-Metrnβ
for 24 h.
Cell Counting Kit-8 (CCK-8) assay
H9c2 cell viability was detected using the CCK-8 Kit
(cat. no. HY-K0301; MedChemExpress) according to the manufacturer's
protocols. Briefly, the cell density was adjusted to
1x105/ml and 96-well plates were used for cell culture
(overnight at 37˚C with 95% O2 and 5% CO2).
In total, 10 µl CCK-8 solution was added to each well and then
cultured for another 3 h at 37˚C. An ELISA plate reader (Synergy
HT; BioTek Instruments, Inc.) was used to determine the absorbance
at 450 nm. Each group has triplicates.
Western blotting
H9c2 cells and heart tissues were lysed in
radioimmunoprecipitation (RIPA) lysis buffer [720 µl RIPA; 20 µl
PMSF (1 mM); 100 µl cOmplete (cat. no. 04693124001; Roche
Diagnostics); 100 µl PhosSTOP (cat. no. 04906837001; Roche
Diagnostics); 50 µl NaF (1 mM); 10 µl Na3VO4;
per ml]. Protein concentrations of the heart tissues and cells were
determined using the BCA Protein Detection kit (Beyotime Institute
of Biotechnology). Total protein was isolated using 4-12% gels and
sodium dodecyl sulphate polyacrylamide gel electrophoresis (50
µg/sample) and transferred to polyvinylidene difluoride membranes
(Merck KGaA). Following blocking with 5% skimmed milk powder for 2
h at room temperature, the membranes were then incubated with
primary antibodies (all from Cell Signaling Technology, Inc.)
against LC3 (cat. no. #2775; 1:1,000), p62 (cat. no. #23214;
1:1,000) or GAPDH (cat. no. #2118; 1:1,000) at 4˚C overnight. The
membranes were then incubated with the HRP-conjugated goat
anti-rabbit IgG secondary antibody (cat. no. ab6721; 1:2,000;
Abcam) for 1 h at room temperature. Protein band visualization was
performed using Clarity™ Western ECL Substrate (cat. no. #1705060;
Bio-Rad Laboratories, Inc.). ChemiDoc MP imaging system (Bio-Rad
Laboratories, Inc.) was used for imaging. Band intensities were
semi-quantified using ImageJ software (v1.8.0.112; National
Institutes of Health).
Reverse transcription-quantitative
PCR
Total mRNA from the left ventricle of the heart
tissues was extracted from frozen, pulverized mouse cardiac tissue
using TRIzol™ (cat. no. 15596-026; Thermo Fisher Scientific, Inc.).
cDNA was synthesized using a reverse transcription kit (Roche
Diagnostic) at 70˚C for 5 min. The LightCycler® 480 SYBR
Green I kit (Roche Diagnostics) was used for amplification.
Following an initial 5 min denaturation step at 95˚C, a total of 42
primer-extension cycles were carried out. Each cycle consisted of a
10 sec denaturation step at 95˚C, a 20 sec annealing step at 60˚C
and a 20 sec incubation at 72˚C for extension. Subsequently, a
final extension step was performed at 72˚C for 10 min. The relative
expression level of indicated genes was compared with that of GAPDH
expression fold changes. The 2-ΔΔCq method was used for
quantification (14). The primers
used are listed in Table I.
 | Table IPrimer sequences used for reverse
transcriptase-quantitative PCR. |
Table I
Primer sequences used for reverse
transcriptase-quantitative PCR.
| mRNA | Forward
(5'-3') | Reverse
(5'-3') |
|---|
| ANP |
ACCTGCTAGACCACCTGGAG |
CCTTGGCTGTTATCTTCGGTACCGG |
| BNP |
GTCAGTCGTTTGGGCTGTAAC |
AGACCCAGGCAGAGTCAGAA |
| Collagen I |
AGGCTTCAGTGGTTTGGATG |
CACCAACAGCACCATCGTTA |
| Collagen III |
AAGGCTGCAAGATGGATGCT |
GTGCTTACGTGGGACAGTCA |
| α-SMA |
AACACGGCATCATCACCAAC |
ACCAGTTGTACGTCCAGAGG |
| GAPDH |
ACTCCACTCACGGCAAATTC |
TCTCCATGGTGGTGAAGACA |
Metrnβ concentration detection
Serum and heart tissue samples were first collected.
Heart tissues were lysed in RIPA lysis buffer before
centrifugation. After centrifugation with 900 x g for 15 min at
4˚C, an ELISA kit for Metrnβ (Mouse Meteorin-like/METRNL DuoSet
ELISA; cat. no. DY6679) was purchased from R&D Systems China
Co., Ltd., and an ELISA plate reader were used for detection.
Lactate dehydrogenase (LDH) release
and caspase-3 activity
After hypoxia or control treatments, the cell
culture medium was collected and centrifuged (100 x g for 10 min at
4˚C) and the LDH detection kit (cat. no. A020-2-2; Nanjing
Jiancheng Bioengineering Institute) was used to detect LDH
level.
Caspase-3 activity was evaluated using the Caspase-3
Activity Assay kit (cat. no. #5723; Cell Signaling Technology,
Inc.) after washing. An ELISA plate reader was used for detecting
fluorescence at 380 nm excitation and at 450 nm emission. Caspase-3
activity was calculated as fluorometric signal
excitation/emission=380/450 nm and as the fold-change to the
control group.
TUNEL staining
TUNEL staining was performed as described in our
previous study (23). Briefly,
after the cells were fixed with RCL2® (cat. no.
RCL2-CS24L; ALPHELYS) at room temperature for 5 min, TUNEL staining
was performed using the Apo-Direct TUNEL Assay kit (cat. no.
APT110; MilliporeSigma) for 1 h at room temperature. The cells on
coverslips were mounted onto glass slides with culture media. The
nuclei were labelled with 0.29 µM DAPI at room temperature for 5
min, and the percentage of TUNEL-positive cells was calculated
using microscopy. The outline of 40 cells from each group were
visualized by fluorescence microscopy (BX51TRF; Olympus
Corporation). For each slide 10 fields of view were observed.
Immunofluorescence staining and
autophagic flux analysis
Cells were fixed with 0.2% RCL2® (cat.
no. RCL2-CS24L; ALPHELYS) at room temperature for 5 min,
immobilized, sealed with 10% goat serum (Absin Bioscience, Inc.) at
room temperature for 30 min, and incubated overnight at 37˚C with
antibody against LC3 (cat. no. 2775; 1:500; Cell Signaling
Technology, Inc.). The cells were then incubated with Alexa Fluor™
488 goat anti-rabbit IgG (cat. no. A31627; 1:10,000; Invitrogen;
Thermo Fisher Scientific, Inc.) secondary antibody for 1 h at 37˚C.
The nuclei were stained with 0.29 µM DAPI at room temperature for 5
min. Images were captured using an Olympus DX51 fluorescence
microscope (Olympus Corporation). Image Pro Plus (version 6.0) was
used to quantify fluorescence intensity.
To detect changes in autophagic flow, cells were
infected with monomeric red fluorescent protein (mRFP)-GFP-LC3
adenovirus (Ad-mRFP-GFP-LC3; MOI, 100) for 8 h at 37˚C before 24 h
of hypoxic culturing. The number of red and green puncta in the
cells was counted (10 fields of view/dish) after images were
captured with a fluorescence microscope.
Adult mouse cardiomyocyte/cardiac
macrophage isolation
Adult mouse cardiomyocytes were obtained from mice 2
weeks post-MI using the Langendorff method according to a previous
study (24). The mouse hearts were
removed and attached to the Langendorff perfusion system (Radnoti;
ADInstruments, Ltd.) in 1x basal solution with 10 mM d-(+)-Glucose,
10 mM 2,3-butanedione monoxime and 5 mM taurine. The pH was
adjusted to 7.4 at 37˚C and filtered with a bottle top filtration
unit. The heart was then digested using a circulating enzyme
digestion solution (collagenase type 2; CAS no. 9001-12-1; AbMole
Bioscience Inc.; the pH was adjusted to 7.4 at 37˚C with a final
concentration of 525 U/ml) at 37˚C for 15-20 min. The physiological
morphology of cardiomyocytes was then observed though microscope,
with a ‘brick-like’ appearance considered to be ‘healthy’. The
isolated cardiomyocytes were then filtered through a 250 µm filter
and seeded onto culture dishes coated with 20 mM HEPEs, 4 mM
NaHCO3, 0.1 mg/ml bovine serum albumin (AbMole
Bioscience Inc.) and 10 µg/ml laminin (Abcam) at 37˚C for 12 h.
Adult mouse hearts were removed from mice post-MI
and digested with 100 µg/ml collagenase II at 37˚C for 30 min a
total of five times (25). The
cells were then filtered through a 250 µm filter and re-suspended
in culture medium. Non-specific binding was blocked with TruStain
FcX antibody (cat. no. 101320; 1:1,000; BioLegend, Inc.) at 37˚C
for 15 min. The macrophages (106) were then washed with
10 ml FACS buffer and labelled with the following antibodies (all
from BioLegend, Inc.): CD45-PerCPCy5.5 (2 µg/ml) and F4/80-PE (6
µg/ml) The macrophages were sorted using flow cytometry (Fig. S1) (BD FACSCanto II flow cytometer;
BD Biosciences).
Statistical analysis
All data are expressed as the mean ± standard
deviation. All data were normally distributed, which was confirmed
by the Shapiro-Wilk test. The differences between two groups were
analysed using an unpaired Student's t-test. One-way ANOVA analysis
followed by Tukey's post hoc test was used to compare the four
groups, whereas two-way ANOVA analysis followed by Tukey's post hoc
test was used to compare the four/six groups with two variables.
P<0.05 was considered to indicate a statistically significant
difference. Each experiment was repeated three times for the in
vitro H9c2 experiments.
Results
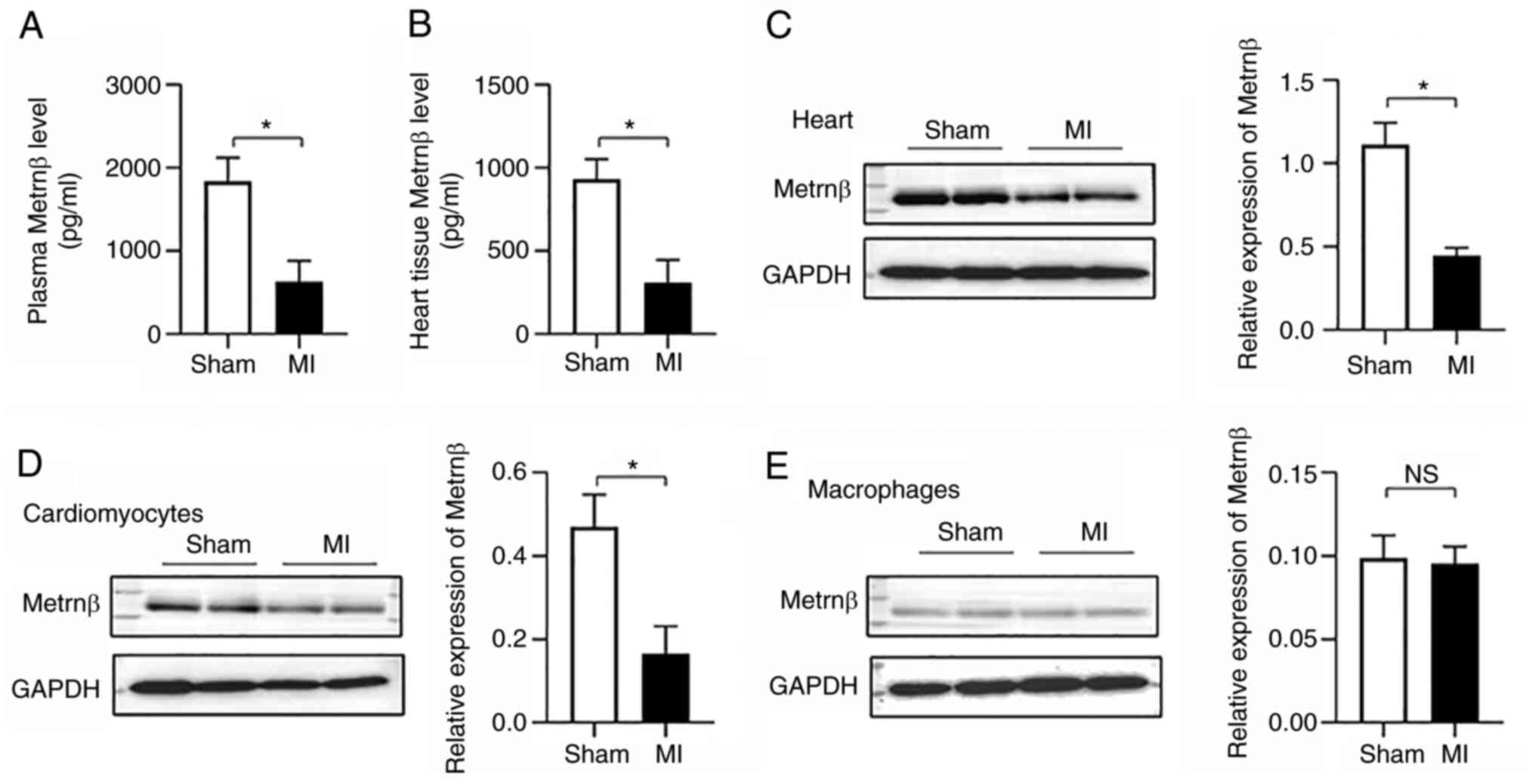
Metrnβ is downregulated upon MI in
mice
The levels of Metrnβ were detected in mice that were
subjected to MI. There was no difference in the HRs between the
sham and MI groups within the experimental series (Table II). The results indicated that
Metrnβ expression levels were significantly reduced in the plasma
(1887±284 vs. 630±252 pg/ml; P<0.01, sham vs. MI, respectively)
(Fig. 1A) and in the heart tissue
(931±120 vs. 309±137 pg/ml; P<0.01, sham vs. MI, respectively)
(Fig. 1B and C) of mice in response to MI stress.
Metrnβ is reported as mainly secreted by macrophages (10). The expression levels of Metrnβ on
cardiomyocytes isolated from MI heart tissue and the expression
levels of Metrnβ on macrophages isolated from MI heart tissue were
therefore detected in the present study. The Metrnβ levels in
cardiomyocytes isolated from MI heart tissue were significantly
decreased (Fig. 1D). The Metrnβ
levels in macrophages demonstrated no significant difference when
the MI samples were compared with the control samples (Figs. 1E and S1). Thus, these data suggested that the
decreased Metrnβ levels were mainly due to reduced expression in
the cardiomyocytes.
| Table IIHeart rate of mice used for detection
of Meteorin-β protein expression levels. |
Table II
Heart rate of mice used for detection
of Meteorin-β protein expression levels.
| Experimental
series | Sham (bpm) | MI (bpm) |
|---|
| Mice for collecting
heart tissue | 482±7 | 480±10 |
| Mice for collecting
cardiomyocytes | 476±12 | 468±14 |
| Mice for collecting
macrophages | 465±9 | 478±12 |
Metrnβ overexpression improves cardiac
parameters post MI
To further investigate the role of Metrnβ in MI,
Metrnβ was overexpressed using AAV9, which has been reported to
target the heart, specifically cardiomyocytes (26). Mice were also subjected to LAD
surgery 2 weeks after injection with AAV9 (Fig. 2A). Successful overexpression was
confirmed by Metrnβ protein level measurements, and AAV9-Metrnβ
increased the plasma Metrnβ levels by greater than two times
compared with the AAV9-NC in the sham treatment group as well as in
the MI treatment group (Fig. 2B).
Additionally, the Metrnβ protein levels in the heart tissues were
also increased in the AAV9-Metrnβ group compared with the AAV9-NC
group in both the sham and MI treatment groups (Fig. 2C). Furthermore, 2 weeks after MI
surgery, the level of Metrnβ in cardiomyocytes isolated from mouse
hearts was significantly increased in the two AAV9-Metrnβ groups
compared with the two respective AAV9-NC groups (Fig. 2D). The cTnT levels were increased
at 12 h post-MI in the two MI treatment groups, and there was no
difference between AAV9-Metrnβ and AAV9-NC, which suggested MI
occurred to the same extent in both of these MI groups (Fig. 2E). Metrnβ overexpression markedly
increased the post-MI survival rate (Fig. 2F). The cell death parameters were
examined, and Metrnβ overexpression significantly lowered the
infarct size (Fig. 2G).
Furthermore, hypotrophy parameters were investigated and revealed
that Metrnβ overexpression inhibited the MI-induced increase in the
cross-sectional area, as well as atrial natriuretic peptide (ANP)
and B-type natriuretic peptide (BNP) gene transcription (Fig. 2H and I).
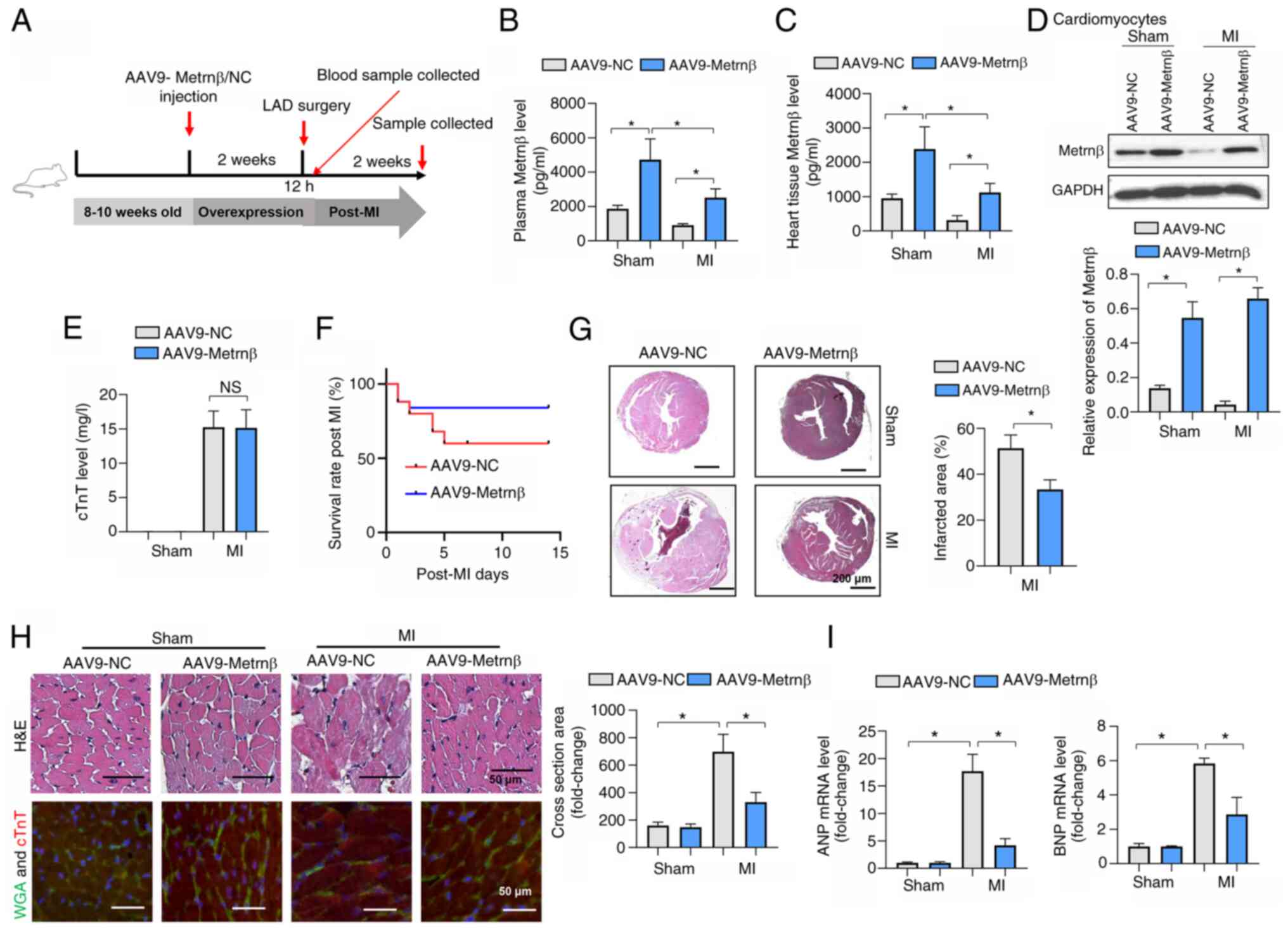 | Figure 2Metrnβ overexpression improves
cardiac parameters post MI. (A) Animal experiment timeline. (B)
Plasma levels of Metrnβ in mice after AAV9-Metrnβ or AAV9-NC
injection (n=6). (C) Levels of Metrnβ in mouse hearts after
AAV9-Metrnβ or AAV9-NC injection (n=6). (D) Levels of Metrnβ in
cardiomyocytes isolated from adult mouse hearts (n=4). (E) Serum
cTnT levels in MI mice 12 h post-LAD (n=6). (F) Survival rates of
mice 2 weeks post-MI. (G) H&E staining and quantification of
infarction size in mice (n=6). (H) H&E staining images, WGA and
cTnT co-staining images, and quantified cross-sectional area in
mice (n=6). (I) mRNA levels of ANP and BNP in mouse hearts (n=6).
*P<0.05. Student's t-test was used for comparison
between two groups. Two-way ANOVA was used for comparison among
four groups with two variables: Treatment (AAV9-NC or AAV9-Metrnβ)
and group (sham or MI). Metrnβ, meteorin-β; MI, myocardial
infarction; NS, no significant difference; NC, negative control;
LAD, left coronary artery ligation surgery; AAV9, adeno-associated
virus 9; cTnT, cardiac troponin T; WGA, wheat germ agglutinin; ANP,
atrial natriuretic peptide; BNP, B-type natriuretic peptide. |
Metrnβ overexpression protects the
heart from MI-induced fibrosis and inflammation
Cardiac fibrosis is a characteristic of adverse
cardiac remodelling post MI that leads to heart failure (27). The LV collagen volume was detected
using PSR staining. Mice in the MI group exhibited marked LV
collagen deposits 2 weeks after MI, while Metrnβ overexpression
reversed this effect (Fig. 3A and
B). The levels of fibrotic markers
were also reduced by Metrnβ overexpression compared with the levels
in the mice that received the AAV9-NC (Fig. 3B). Inflammation is the initiating
factor of cardiac fibrosis after MI (28). Proinflammatory factors were
increased in the two MI groups 2 weeks after MI. Metrnβ
overexpression reduced the increase in the levels of the
proinflammatory factors, as shown in the comparison between the
AAV9-Metrnβ group and the AAV9-NC group (Fig. 3C).
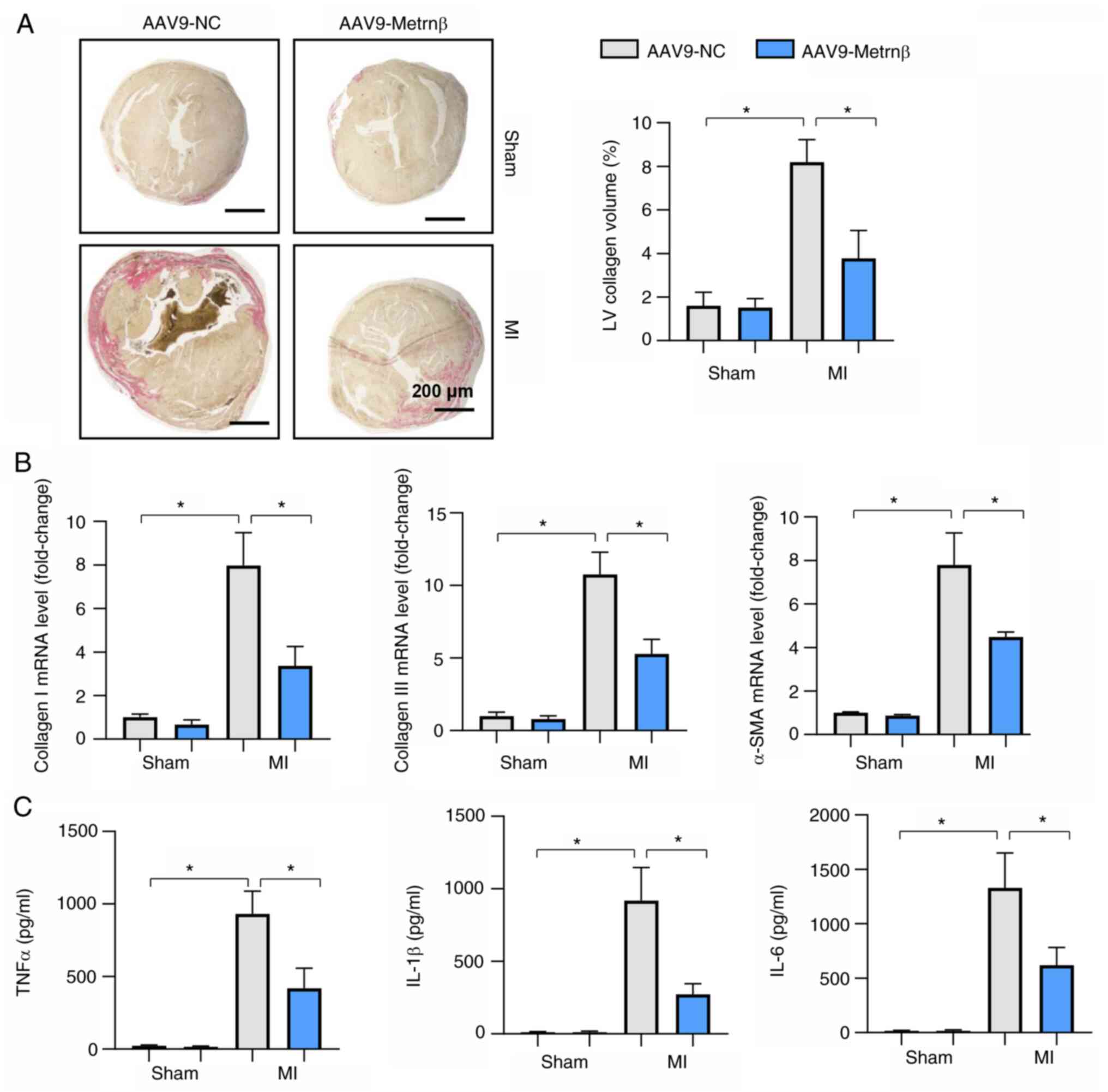 | Figure 3Metrnβ overexpression protects the
heart from MI-induced fibrosis and inflammation. (A) Picrosirius
red staining results and quantified results in mouse hearts (n=6).
(B) mRNA levels of collagen I, collagen III and α-SMA in mouse
hearts (n=6). (C) ELISA results of TNFα, IL-1β and IL-6 in mouse
hearts (n=6). *P<0.05. Two-way ANOVA was used for
comparison among four groups with two variables: Treatment (AAV9-NC
or AAV9-Metrnβ) and group (sham or MI). Metrnβ, meteorin-β; MI,
myocardial infarction; LV, left ventricular; α-SMA, α-smooth muscle
actin; AAV9, adeno-associated virus 9; NC, negative control, TNFα,
tumour necrosis factor α; IL, interleukin; ELISA, enzyme-linked
immunosorbent assay. |
Metrnβ overexpression preserved
cardiac function post MI
The progression of heart failure was revealed by
heart weight (HW) and lung weight (LW). The HW/body weight (BW)
ratio and the LW/BW ratio were increased in the two MI groups
compared with those of the two sham groups at 2 weeks post-MI,
which indicated adverse cardiac hypertrophy and lung oedema. Metrnβ
overexpression reversed the cardiac hypertrophy and lung oedema
observed in the AAV9-NC group (Fig.
4A). Cardiac function was also evaluated using echocardiography
and pressure-volume measurements. All the groups demonstrated
similar HR levels (Fig. 4B). The
LV end diastolic diameter (LVEDD) and LV end systolic diameter
(LVESD) were increased at 2 weeks after MI compared with those of
the sham group (Fig. 4B-D). The
LVEF and LVFS were decreased in the MI group compared with that of
the sham group. Metrnβ overexpression improved cardiac dilation and
function, as demonstrated by the reduced LVESD and increased LVEF
and LVFS compared with the respective AAV9-NC groups. The
pressure-volume measurement results also demonstrated that the LV
pressure increase/delay at the end systolic phase (dp/dt max, dp/dt
min) was reduced in the MI group compared with that in the sham
group, but increased in the Metrnβ-overexpressing group compared
with the AAV9-NC group (Fig. 4E;
Table III).
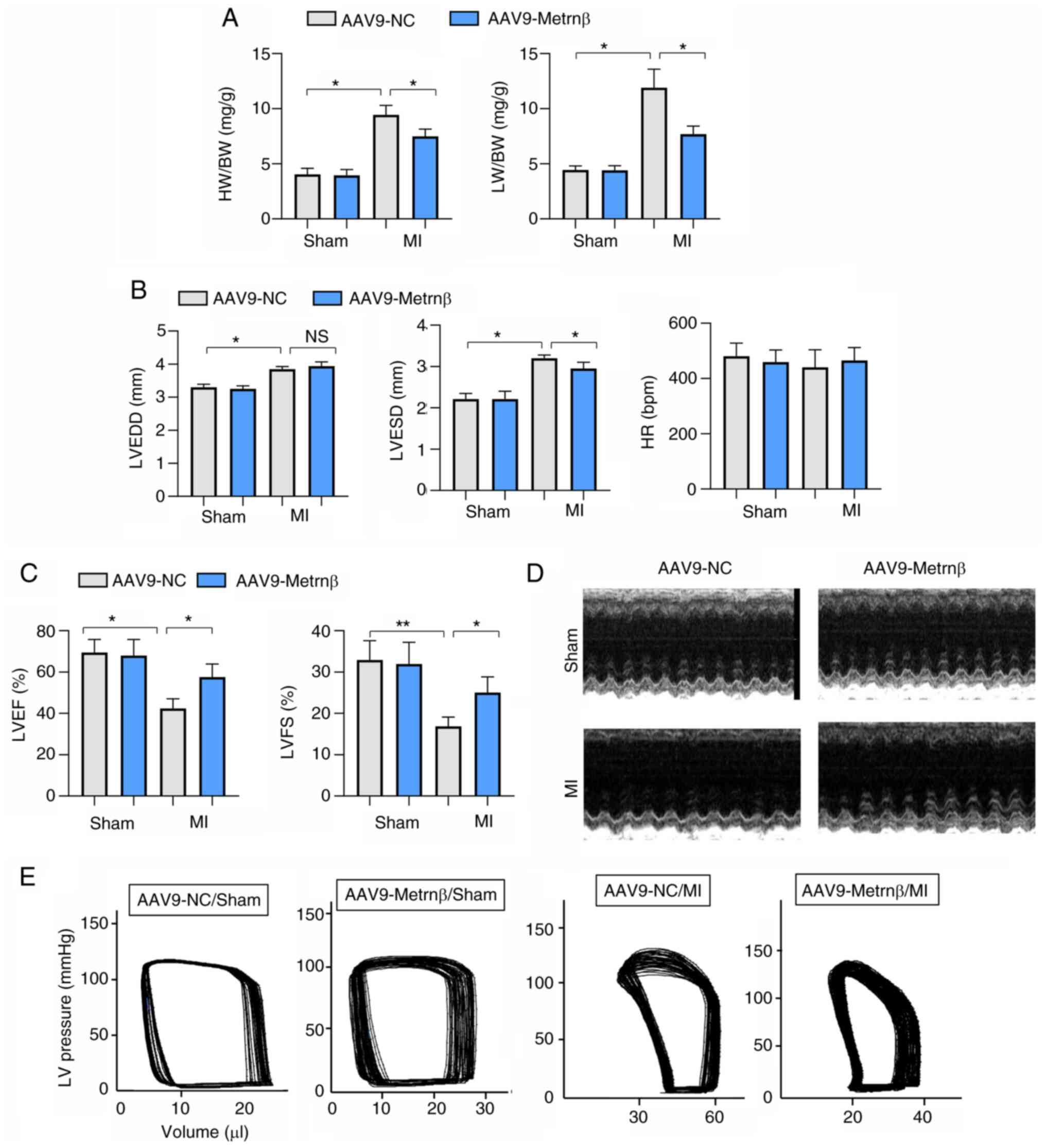 | Figure 4Metrnβ overexpression preserves
cardiac function post MI. (A) HW/BW and LW/BW in mouse hearts
(n=8). (B) Echocardiography results of LVEDD, LVESD and HR in mouse
hearts (n=6). (C) Echocardiography results of LVEF and LVFS in
mouse hearts (n=6). (D) Echocardiography results in mouse hearts
(n=6). (E) Representative images of pressure-volume measurements in
mouse hearts (n=6). *P<0.05 and
**P<0.01. Two-way ANOVA was used for comparison among
four groups with two variables: Treatment (AAV9-NC or AAV9-Metrnβ)
and group (sham or MI). Metrnβ, meteorin-β; MI, myocardial
infarction; HW, heart weight; BW, body weight; LW, lung weight; LV,
left ventricular; LVEDD, LV end diastolic diameter; LVESD, LV end
systolic diameter; HR, heart rate; LVEF, LV ejection fraction;
LVFS, LV fractional shortening; AAV9, adeno-associated virus 9; NC,
negative control; NS, no significant difference. |
| Table IIIHemodynamic parameters in mice. |
Table III
Hemodynamic parameters in mice.
| Parameter | AAV9-NC/Sham |
AAV9-Metrnβ/Sham | AAV9-NC/MI | AAV9-Metrnβ/MI |
|---|
| HR (beats per
min) | 480.45±47.52 | 459.44±43.85 | 440.34±63.82 | 465.43±47.02 |
| ESV (µl) | 11.55±2.12 | 10.63±2.24 |
46.17±2.00a |
24.03±2.87b |
| EDV (µl) | 26.87±2.38 | 26.15±1.59 |
56.65±3.96a |
36.60±2.53b |
| dp/dt max
(mmHg/sec) |
9,998.22±805.23 |
10,316.43±1,295.23 |
5,465.33±756.12a |
7,988.67±1,127.33a,b |
| dp/dt min
(mmHg/sec) |
-10,366.44±1,212.34 |
-10,244.45±1,452.45 |
-5,270.22±894.34a |
-7,251.34±659.34b |
| CO (µl/min) |
7,389.33±419.45 |
7,148.54±125.56 |
4,536.22±312.09a |
5,874.05±269.21a,b |
Metrnβ overexpression protects against
cardiomyocyte hypoxia injury
H9c2 cardiomyocytes were transfected with Ad-Metrnβ
to overexpress Metrnβ. Ad-Metrnβ did not affect the cell viability
under control or hypoxic conditions, as revealed using the CCK-8
assay (Fig. 5A). After 72 h of
transfection, the levels of Metrnβ were increased in H9c2 cells in
the Ad-Metrnβ group (Fig. 5A).
Apoptosis was induced by 24 h of hypoxia, which was revealed by
increased TUNEL-positive cells and caspase 3 activity. Metrnβ
overexpression inhibited the apoptosis induced by hypoxia (Fig. 5B and C). Furthermore, the cell injury marker,
LDH, was increased after 24 h of hypoxia in the hypoxia group.
Cells treated with the Ad-Metrnβ exhibited reduced LDH levels
compared with cells treated with the Ad-NC (Fig. 5D). Hypoxia also induced the release
of proinflammatory cytokines (increased TNFα, IL-1β and IL-6 levels
in the hypoxic group compared with the control group). The levels
of these proinflammatory cytokines were reduced in the Ad-Metrnβ
group compared with the Ad-NC group after exposure to hypoxic
conditions (Fig. 5E).
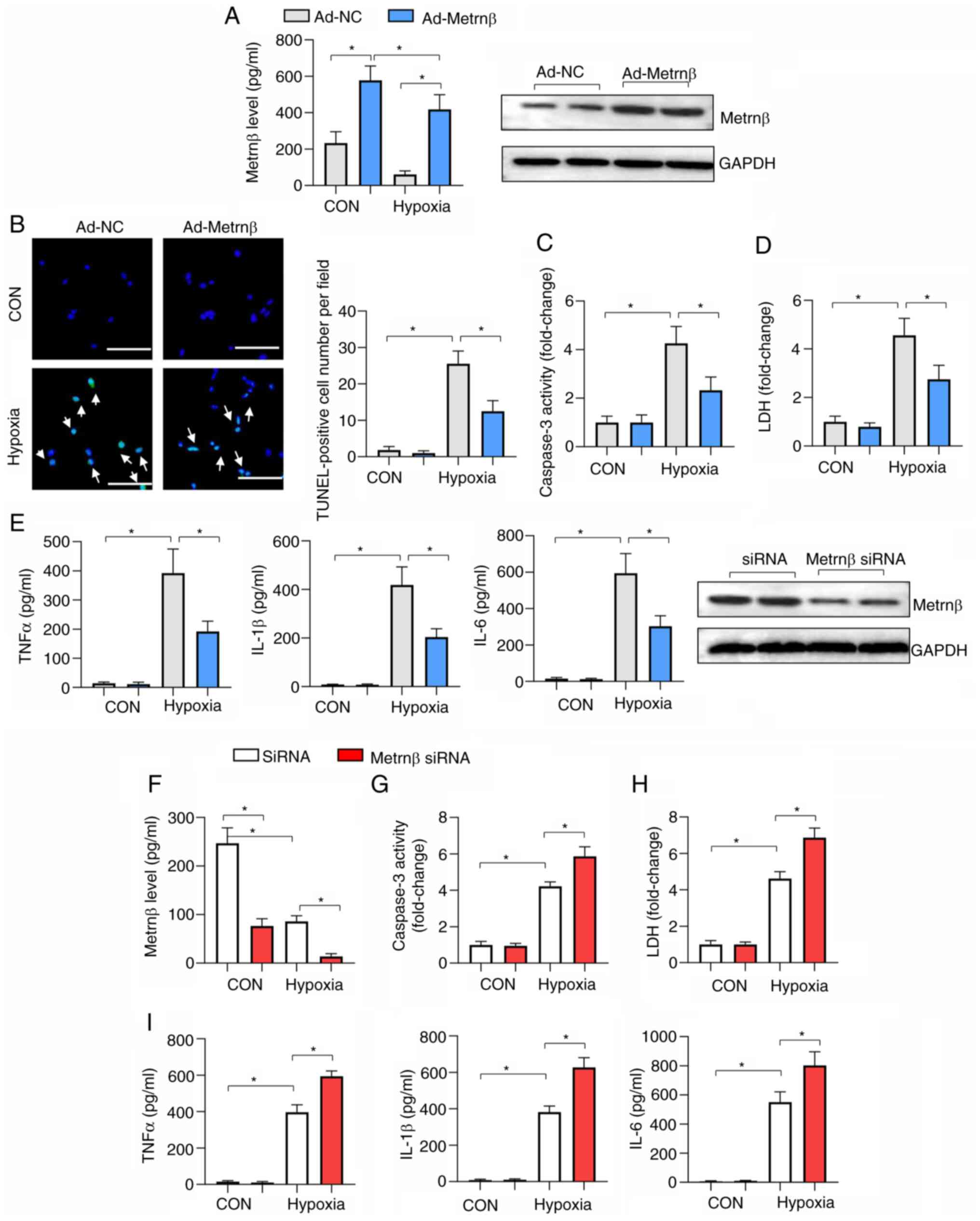 | Figure 5Metrnβ overexpression protects
against cardiomyocyte hypoxia injury. (A-E) Cells were transfected
with Ad-Metrnβ. (A) Protein levels of Metrnβ and cell viability in
cardiomyocytes (n=6). (B) TUNEL staining and quantified results in
cardiomyocytes (n=6; scale bar, 100 µm). (C) caspase 3 activity and
(D) LDH levels in cardiomyocytes (n=6). (E) ELISA results of TNFα,
IL-1β and IL-6 in cardiomyocytes (n=6). (F-I) Cells were
transfected with Metrnβ siRNA. (F) Protein levels of Metrnβ in
cardiomyocytes transfected with Metrnβ siRNA or scRNA (n=6). (G)
caspase 3 activity and (H) LDH level in cardiomyocytes (n=6). (I)
ELISA results of TNFα, IL-1β and IL-6 in cardiomyocytes (n=6).
*P<0.05. Two-way ANOVA was used for comparison among
four groups with two variables: Treatment (Ad-NC or Ad-Metrnβ) and
group (CON or hypoxia). Metrnβ, meteorin-β; Ad, adenovirus; LDH,
lactate dehydrogenase; TNFα, tumour necrosis factor α; IL,
interleukin; siRNA, small interfering RNA; NC, negative control;
CON, control; scRNA, scrambled RNA. |
To confirm the effect of Metrnβ on cardiomyocyte
hypoxic injury, cells were transfected with Metrnβ siRNA to
knockdown Metrnβ, and the decrease in the Metrnβ protein expression
levels was confirmed after transfection (Fig. 5F). Metrnβ knockdown caused
deteriorated hypoxia-induced cell injury and inflammation, as
revealed by increased caspase 3 activity, LDH levels and
pro-inflammatory cytokine levels compared with the siRNA NC
(Fig. 5G-I).
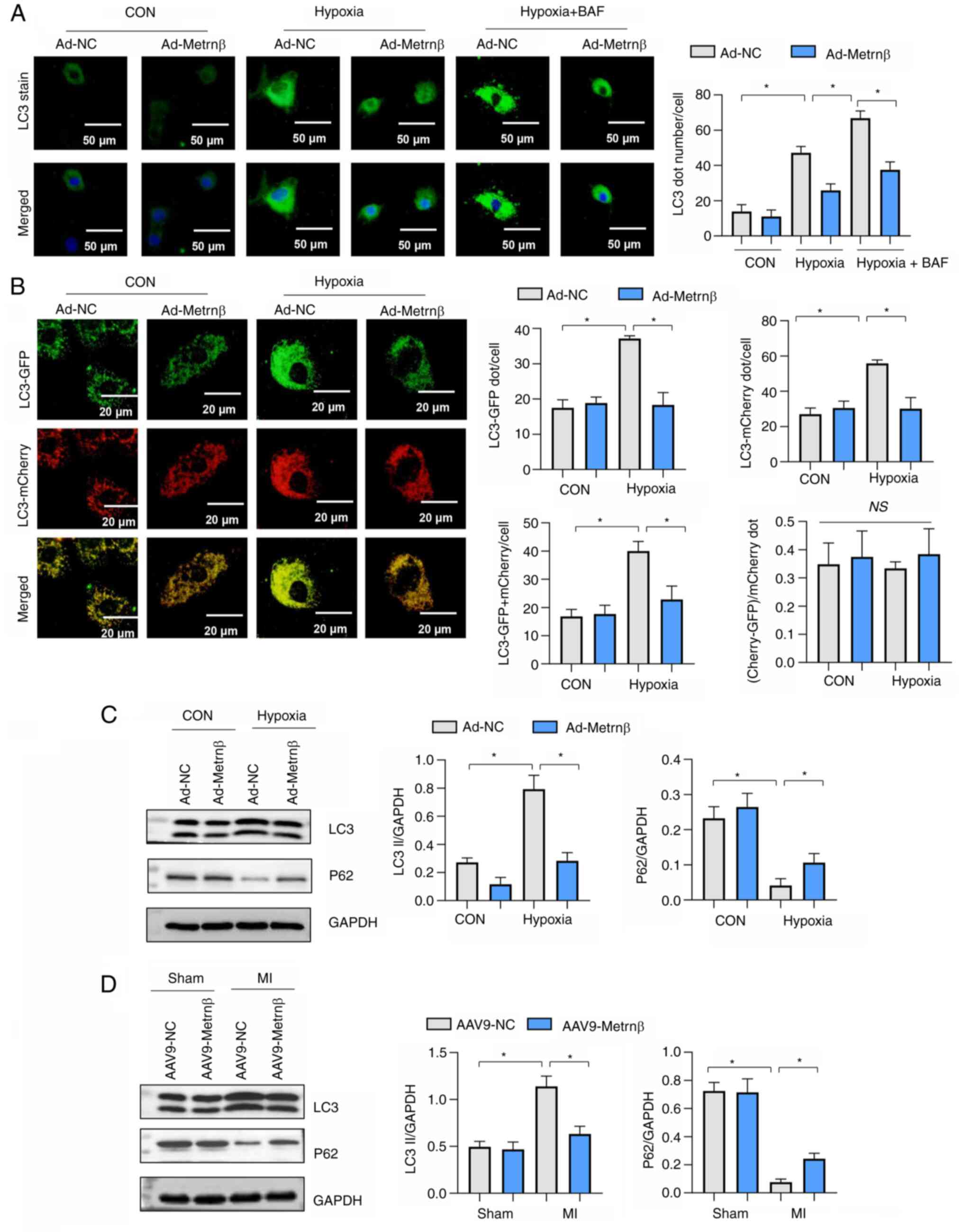
Metrnβ inhibits autophagy in
cardiomyocytes exposed to hypoxia
LC3 levels were increased in cells exposed to
hypoxia (Fig. 6A). p62 levels were
reduced in cells exposed to hypoxic conditions (Fig. 6C and D). Metrnβ overexpression reduced LC3
levels compared with the Ad-NC (Fig.
6A and B) and increased p62
protein levels under hypoxic conditions (Fig. 6C and D). However, the effects of Metrnβ
overexpression on LC3 and p62 were not significantly inhibited by
the classic autophagy inhibitor BAF, which works by blocking the
autophagosome lysosomal fusion (Fig.
6A-D). Cells were transfected with Ad-mRFP-GFP-LC3, which
served as a dual-fluorescence pH sensor for autophagic vacuoles and
revealed autolysosome formation. mRFP-stained LC3 (indicating the
formation of autophagosomes) and GFP-LC3 (representing the
remaining autophagosomes after degradation) were both revealed to
increase with hypoxic stimuli but were reduced by Metrnβ
overexpression (Fig. 6B). However,
the degradation rate (red-green puncta/red puncta) was unaltered
when Metrnβ was overexpressed under both physiological and hypoxic
conditions (Fig. 6B). Furthermore,
LC3II levels were also increased, and p62 levels were reduced in MI
heart tissue. Metrnβ overexpression reduced LC3 levels and
increased p62 protein levels under hypoxic conditions when compared
with the Ad-NC (Fig. 6C).
Furthermore, Metrnβ overexpression reduced LC3 levels and increased
p62 protein levels in MI heart tissue when compared with the Ad-NC
(Fig. 6D). Taken together, the
data demonstrated that the protection induced by Metrnβ was not due
to the regulation of autophagosome degradation, but through the
effect on autophagy induction.
Autophagy activation counteracts the
protective effects of Metrnβ
To confirm the effect of Metrnβ on autophagy, an
autophagy activator, rapamycin, was used. Rapamycin increased
hypoxia-induced apoptosis as revealed by the increased number of
TUNEL-positive cells, increased caspase 3 activity and increased
LDH levels (Fig. 7A-C). Rapamycin
also exacerbated hypoxia-induced cell inflammation, as revealed by
increased levels of proinflammatory factors (Fig. 7D). Metrnβ overexpression could not
reverse the rapamycin-induced effects on the cardiomyocytes
(Fig. 7A-D). These data indicate
that autophagy activation could abolish the protective effects of
Metrnβ on cardiomyocytes.
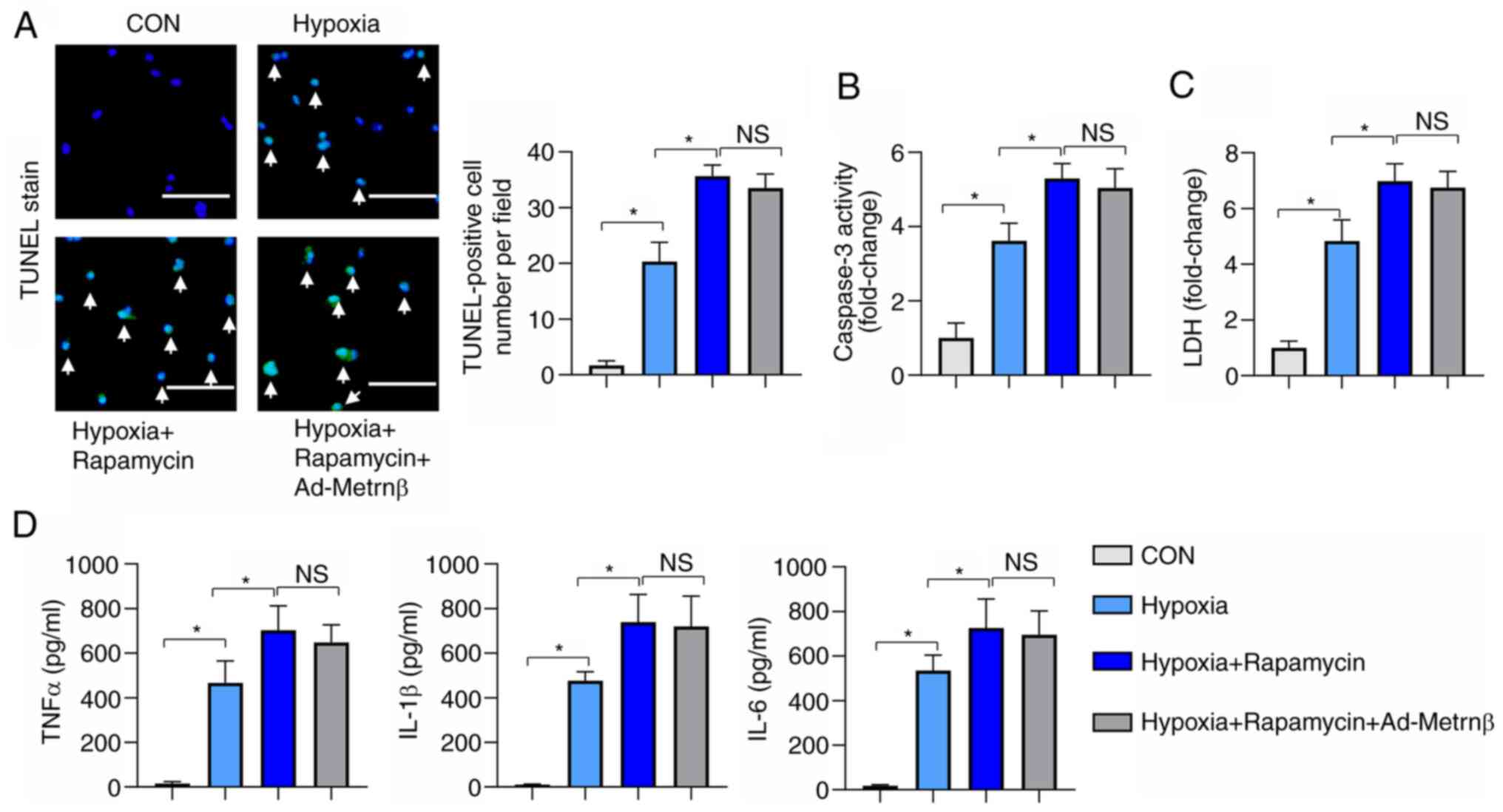 | Figure 7Autophagy activation counteracts the
protective effects of Metrnβ. H9c2 cells were treated with
rapamycin and transfected with Ad-Metrnβ. (A) TUNEL staining and
quantified results in cardiomyocytes exposed to hypoxia (n=6; scale
bar, 100 µm). (B) caspase 3 activity and (C) LDH levels in
cardiomyocytes exposed to hypoxia (n=6). (D) ELISA results of TNFα,
IL-1β and IL-6 in cardiomyocytes exposed to hypoxia (n=6).
*P<0.05. One-way ANOVA analysis followed by Tukey's
post hoc test was used for comparison among the four groups.
Metrnβ, meteorin-β; Ad, adenovirus; CON, control; TNFα, tumour
necrosis factor α; IL, interleukin; LDH, lactate dehydrogenase; NS,
no significant difference; ELISA, enzyme-linked immunosorbent
assay. |
Discussion
In the present study, the expression levels of
Metrnβ were first revealed to be reduced after MI both in plasma as
well as in heart tissues, which was localized to the
cardiomyocytes. The effect of Metrnβ on cardiac remodelling after
MI was then evaluated and it was revealed that Metrnβ could reduce
the infarct size and improve survival rates and cardiac function at
2 weeks post-MI. The present study also revealed that Metrnβ
suppressed cardiac hypertrophy, fibrosis and inflammation by
inhibiting autophagy induction but not the autophagosome
degradation after MI. Autophagy activation abolished the protective
effects of Metrnβ on cardiomyocytes under hypoxic conditions.
Therefore, Metrnβ may become a new therapeutic target for
inhibiting the progression of heart failure after MI.
MI places a significant burden on global health,
affecting >7 million individuals worldwide each year (3). In the past 10 years, early
revascularization has had far-reaching significance for the
treatment of MI (1). The 30-day
survival rate of acute ST segment elevation MI has increased to 95%
(4). However, despite
revascularization, patients with AMI have a 75% probability of
developing heart failure within 5 years (2,3). An
increasing number of patients that survive AMI develop heart
failure. Therefore, it is important to prevent or even reverse
heart failure after MI and to develop novel treatments focusing on
the prevention of heart failure. Metrnβ is a secretory protein
involved in glucose metabolism (8). A previous study revealed that a
decrease in plasma Metrnβ was associated with insulin resistance in
patients with type 2 diabetes (10). Metrnβ was also reported to inhibit
airway inflammation in house dust mite-induced allergic asthma
(24). Ushach et al
(10) reported that Metrnβ
regulated inflammatory responses in macrophages. It was also
revealed that Metrnβ knockout mice exhibited increased cytokine
production. Recently, Rupérez et al (11) revealed that Metrnβ ameliorated
cardiac dysfunction and cardiac hypertrophy in response to
isoproterenol and ageing. Hu et al (25) recently reported that by modulating
the cAMP/protein kinase A/Sirtuin 1 pathway, Metrnβ protein
inhibited doxorubicin-induced cardiotoxicity. In the present study,
the data demonstrated that Metrnβ expression levels were reduced
after MI. Furthermore, the present study revealed that Metrnβ
overexpression using AAV9-Metrnβ delivery could offer cardiac
protection to hearts subjected to MI, which was indicated by the
reduced cardiac infarct size, improved cardiac function, and
reduced cardiac hypertrophy, fibrosis and inflammatory response 2
weeks after MI. Since it was revealed that the decrease in the
Metrnβ protein levels was localized in cardiomyocytes and not in
the macrophage cells in mice heart tissues, the ex vivo
cardiomyocyte model was used to further explore whether Metrnβ
could protect the cardiomyocytes from hypoxic damage. The present
study revealed that Metrnβ overexpression in cardiomyocytes reduced
hypoxia-induced apoptosis and inflammation. Thus, the results
suggest that Metrnβ could directly affect cardiomyocytes to exert
protective effects.
Autophagy is another type of cell death.
Intracellular components (including proteins and organelles) are
digested and recovered through lysosomal degradation to maintain
energy production and protein synthesis to promote cell survival
(26). Autophagy under stress,
especially during ischaemia, starvation and β-adrenaline
stimulation, allows myocardial cells to remove damaged or misfolded
proteins, organelles and aggregates (27). Normally, autophagy can remove
damaged cells and organelles, whereas abnormal autophagy leads to
abnormal protein accumulation, which can lead to heart failure due
to a variety of causes (such as hypertension and myocardial
infarction) (7,28). Autophagy was previously revealed to
be upregulated in cardiomyocytes in heart failure (6). In addition, impaired autophagy serves
a pathological role in the progression of heart failure (7,28).
In the present study, augmented autophagy activation in hypoxic
cardiomyocytes and MI heart tissues was also revealed. It has been
reported that Metrnβ ameliorates diabetic cardiomyopathy via the
inactivation of cGAS/STING signalling, which is dependent on
LKB1/AMPK/ULK1-mediated autophagy (27). Therefore, the present study
hypothesized that autophagy regulation may serve a role in the
protection of Metrnβ against MI injury. It was revealed in the
present study that Metrnβ inhibited the increase in the autophagy
of cardiomyocytes in hypoxic conditions. With the administration of
BAF, an autophagosome degradation inhibitor, it was confirmed that
Metrnβ inhibited autophagy induction without affecting
autophagosome degradation. mTOR kinases are associated with protein
synthesis and autophagy, and mTORC1 inhibits catabolic processes,
including autophagy. Rapamycin is an allosteric inhibitor of
mTORC1, which activates autophagy (27). To test this relationship, rapamycin
in cardiomyocytes under hypoxic conditions was used to activate
autophagy, and aggravated cell injury was revealed upon hypoxia.
Furthermore, the protective effects of Metrnβ were abolished, which
confirmed that the cardiac protection of Metrnβ was dependent on
autophagy inhibition in MI.
In summary, Metrnβ suppresses cardiac hypertrophy,
fibrosis and inflammation after MI together with reduced autophagy.
Autophagy activation abolished the protective effects of Metrnβ on
cardiomyocytes under hypoxic conditions. Metrnβ may be a novel
therapeutic target for inhibiting the progression of heart failure
after MI by inhibiting autophagy.
Supplementary Material
Cardiac macrophage isolation. Adult
mouse hearts were removed from mice 2 weeks post-MI and digested
with 100 μg/ml collagenase IV at 37˚C for 30 min a total of
5 times. The cells were then filtered and re-suspended in culture
medium. The macrophages (106) were then washed with 10
ml FACS buffer and labelled with the following antibodies (all from
BioLegend, Inc.): CD45-PerCPCy5.5 (2 μg/ml) and F4/80-PE (6
μg/ml).
Acknowledgements
Not applicable.
Funding
Funding: This research was supported via the National Natural
Science Foundation of China (grant nos. 81600191, 81600189 and
81400323), the Medical Science and Technology Research Project of
Henan province (grant no. 201702063) and the Scientific and
Technological Project of Henan province (grant no.
172102310531).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JS and LL contributed to the conception of the study
and the design of the experiments. JS, GL and LX performed the
experiments. WZ and XZ analysed the experimental results and
revised the manuscript. JS and LL wrote and revised the manuscript.
JS and LL confirm the authenticity of all the raw data. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The animal experiments were performed according to
the Animal Research: Reporting of in vivo Experiments
guidelines and were approved by the Animal Care and Use Committee
of the First Affiliated Hospital of Zhengzhou University (approval
no. 2021-02623).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bahit MC, Kochar A and Granger CB:
Post-myocardial infarction heart failure. JACC Heart Fail.
6:179–186. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Berezin AE and Berezin AA: Adverse cardiac
remodelling after acute myocardial infarction: Old and new
biomarkers. Dis Markers. 2020(1215802)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jenča D, Melenovský V, Stehlik J, Staněk
V, Kettner J, Kautzner J, Adámková V and Wohlfahrt P: Heart failure
after myocardial infarction: Incidence and predictors. ESC Heart
Fail. 8:222–237. 2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Mouton AJ, Rivera OJ and Lindsey ML:
Myocardial infarction remodeling that progresses to heart failure:
A signaling misunderstanding. Am J Physiol Heart Circ Physiol.
315:H71–H79. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Du J, Liu Y and Fu J: Autophagy and heart
failure. Adv Exp Med Biol. 1207:223–227. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Du J, Li Y and Zhao W: Autophagy and
myocardial ischemia. Adv Exp Med Biol. 1207:217–222.
2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Liu CY, Zhang YH, Li RB, Zhou LY, An T,
Zhang RC, Zhai M, Huang Y, Yan KW, Dong YH, et al: LncRNA CAIF
inhibits autophagy and attenuates myocardial infarction by blocking
p53-mediated myocardin transcription. Nat Commun.
9(29)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Baht GS, Bareja A, Lee DE, Rao RR, Huang
R, Huebner JL, Bartlett DB, Hart CR, Gibson JR, Lanza IR, et al:
Meteorin-like facilitates skeletal muscle repair through a
Stat3/IGF-1 mechanism. Nat Metab. 2:278–289. 2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wu Q, Dan YL, He YS, Xiang K, Hu YQ, Zhao
CN, Zhong X, Wang DG and Pan HF: Circulating meteorin-like levels
in patients with type 2 diabetes mellitus: A meta-analysis. Curr
Pharm Des. 26:5732–5738. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ushach I, Arrevillaga-Boni G, Heller GN,
Pone E, Hernandez-Ruiz M, Catalan-Dibene J, Hevezi P and Zlotnik A:
Meteorin-like/Meteorin-β is a novel immunoregulatory cytokine
associated with inflammation. J Immunol. 201:3669–3676.
2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Rupérez C, Ferrer-Curriu G, Cervera-Barea
A, Florit L, Guitart-Mampel M, Garrabou G, Zamora M, Crispi F,
Fernandez-Solà J, Lupón J, et al: Meteorin-like/Meteorin-β protects
heart against cardiac dysfunction. J Exp Med.
218(e20201206)2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cai J, Wang QM, Li JW, Xu F, Bu YL, Wang
M, Lu X and Gao W: Serum meteorin-like is associated with weight
loss in the elderly patients with chronic heart failure. J Cachexia
Sarcopenia Muscle. 13:409–417. 2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Li F, Zong J, Zhang H, Zhang P, Xu L,
Liang K, Yang L, Yong H and Qian W: Orientin reduces myocardial
infarction size via eNOS/NO signaling and thus mitigates adverse
cardiac remodeling. Front Pharmacol. 8(926)2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wu QQ, Xiao Y, Duan MX, Yuan Y, Jiang XH,
Yang Z, Liao HH, Deng W and Tang QZ: Aucubin protects against
pressure overload-induced cardiac remodelling via the
beta3 -adrenoceptor-neuronal NOS cascades. Br J
Pharmacol. 175:1548–1566. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Xiao Y, Yang Z, Wu QQ, Jiang XH, Yuan Y,
Chang W, Bian ZY, Zhu JX and Tang QZ: Cucurbitacin B protects
against pressure overload induced cardiac hypertrophy. J Cell
Biochem. 118:3899–3910. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Franke WW, Dörflinger Y, Kuhn C,
Zimbelmann R, Winter-Simanowski S, Frey N and Heid H: Protein LUMA
is a cytoplasmic plaque constituent of various epithelial adherens
junctions and composite junctions of myocardial intercalated disks:
A unifying finding for cell biology and cardiology. Cell Tissue
Res. 357:159–172. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Guo Z, Tuo H, Tang N, Liu FY, Ma SQ, An P,
Yang D, Wang MY, Fan D, Yang Z and Tang QZ: Neuraminidase 1
deficiency attenuates cardiac dysfunction, oxidative stress,
fibrosis, inflammatory via AMPK-SIRT3 pathway in diabetic
cardiomyopathy mice. Int J Biol Sci. 18:826–840. 2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wan N, Liu X, Zhang XJ, Zhao Y, Hu G, Wan
F, Zhang R, Zhu X, Xia H and Li H: Toll-interacting protein
contributes to mortality following myocardial infarction through
promoting inflammation and apoptosis. Br J Pharmacol.
172:3383–3396. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wu QQ, Xu M, Yuan Y, Li FF, Yang Z, Liu Y,
Zhou MQ, Bian ZY, Deng W, Gao L, et al: Cathepsin B deficiency
attenuates cardiac remodeling in response to pressure overload via
TNF-α/ASK1/JNK pathway. Am J Physiol Heart Circ Physiol.
308:H1143–H1154. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Fan D, Yang Z, Yuan Y, Wu QQ, Xu M, Jin YG
and Tang QZ: Sesamin prevents apoptosis and inflammation after
experimental myocardial infarction by JNK and NF-κB pathways. Food
Funct. 8:2875–2885. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lomas O, Brescia M, Carnicer R, Monterisi
S, Surdo NC and Zaccolo M: Adenoviral transduction of FRET-based
biosensors for cAMP in primary adult mouse cardiomyocytes. Methods
Mol Biol. 1294:103–115. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Alonso-Herranz L, Porcuna J and Ricote M:
Isolation and purification of tissue resident macrophages for the
analysis of nuclear receptor activity. Methods Mol Biol.
1951:59–73. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Gao X, Leung TF, Wong GW, Ko WH, Cai M, He
EJ, Chu IM, Tsang MS, Chan BC, Ling J, et al: Meteorin-β/Meteorin
like/IL-41 attenuates airway inflammation in house dust
mite-induced allergic asthma. Cell Mol Immunol. 19:245–259.
2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hu C, Zhang X, Song P, Yuan YP, Kong CY,
Wu HM, Xu SC, Ma ZG and Tang QZ: Meteorin-like protein attenuates
doxorubicin-induced cardiotoxicity via activating cAMP/PKA/SIRT1
pathway. Redox Biol. 37(101747)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang D, Lv L, Xu Y, Jiang K, Chen F, Qian
J, Chen M, Liu G and Xiang Y: Cardioprotection of panax notoginseng
saponins against acute myocardial infarction and heart failure
through inducing autophagy. Biomed Pharmacother.
136(111287)2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Gao G, Chen W, Yan M, Liu J, Luo H, Wang C
and Yang P: Rapamycin regulates the balance between cardiomyocyte
apoptosis and autophagy in chronic heart failure by inhibiting mTOR
signaling. Int J Mol Med. 45:195–209. 2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Fei Q, Ma H, Zou J, Wang W, Zhu L, Deng H,
Meng M, Tan S, Zhang H, Xiao X, et al: Metformin protects against
ischaemic myocardial injury by alleviating
autophagy-ROS-NLRP3-mediated inflammatory response in macrophages.
J Mol Cell Cardiol. 145:1–13. 2020.PubMed/NCBI View Article : Google Scholar
|