Introduction
Sepsis is a systemic inflammatory response syndrome
caused by a dysregulated host response to infection, and can
progress to multi-organ dysfunction and even death, which has
become the leading cause of mortality in critically ill patients
(1,2). It is estimated that ~30 million
individuals suffer from sepsis and >6 million patients die from
sepsis worldwide annually, causing a large burden to the global
health care system (3). It has
been reported that, in 2020, the incidence of sepsis among
intensive care unit patients in 44 hospitals in China was up to
20.6%, and the mortality rate was as high as 35.5% (4). There is currently no targeted and
effective therapeutic strategy for sepsis (5). Therefore, in-depth studies to
understand the pathogenesis of sepsis and identify effective
prevention and treatment strategies are of significant
importance.
The pathogenesis of sepsis involves highly complex
and integrated responses, including the host immune response,
circulatory abnormalities, endothelial dysfunction and organ-organ
crosstalk (6). Previous studies
have demonstrated that the vascular endothelium is the main target
of pathogens, microbial toxins or endogenous danger signals, and
thus, sepsis-induced endothelial dysfunction is considered as an
important pathogenetic mechanism for the development of sepsis
(6,7).
The GRB2-associated binders (GABs) are a highly
conserved class of scaffolding proteins, including GAB1, GAB2 and
GAB3(8). GABs are involved in
signal transduction, mainly through the activation of the classical
signaling pathways SH2 domain-containing tyrosine phosphatase
2/RAS/ERK and PI3K/AKT, and through the coupling between membrane
receptors and signaling proteins, thereby regulating a series of
biological responses, such as cell proliferation, angiogenesis and
the inflammatory response (8). As
the most widely distributed and abundant member of the GAB family,
GAB1 has received widespread attention due to its biological
functions. It has been demonstrated that GAB1 is aberrantly
downregulated in patients with sepsis, and upregulation of GAB1
expression can alleviate sepsis-induced lung injury and renal
injury by inhibiting apoptosis, oxidative stress and inflammatory
responses, and thus, GAB1 is considered to be a key regulator of
sepsis (9-11).
In addition, existing evidence has also revealed the important
regulatory role of GAB1 in the expression of key transcription
factors for endothelial homeostasis and vascular cell adhesion
molecule-1, the production of proinflammatory cytokines to
endothelium-associated neovascularization and the inflammatory
response (12). However, to the
best of our knowledge, whether GAB1 is involved in endothelial
dysfunction-associated pathogenetic mechanisms for sepsis remains
unclear.
Therefore, the present study aimed to explore the
biological role of GAB1 in sepsis-mediated endothelial dysfunction,
as well as its potential mechanism of action, offering novel
insights for developing endothelium-specific therapies for the
treatment of sepsis.
Materials and methods
Cell culture and treatment
HUVECs (cat. no. iCell-h110; iCell Bioscience, Inc.)
were incubated with specific culture medium (cat. no.
iCell-h110-001b; iCell Bioscience, Inc.) supplemented with 10% FBS
(Thermo Fisher Scientific, Inc.), 1% endothelial cell growth
supplements (ECGS; iCell Bioscience, Inc.) and 1%
penicillin/streptomycin (Thermo Fisher Scientific, Inc.) in a
humified incubator with 5% CO2 at 37˚C. To simulate
endothelial cell injury under sepsis conditions, HUVECs were
exposed to 2.5, 5 and 10 µg/ml lipopolysaccharide (LPS;
Sigma-Aldrich; Merck KGaA) for 24 h at 37˚C. LPS at the
concentration of 10 µg/ml was utilized in the following functional
experiments.
Reverse transcription-quantitative
PCR
Total RNA was isolated from HUVECs using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), followed by detection of its purity and concentration using
a NanoDrop ND-1000 spectrophotometer. Total RNA (1 µg) was reverse
transcribed into cDNA using the PrimeScript RT reagent kit (Takara
Bio, Inc.) according to the manufacturer's instructions.
Subsequently, quantitative PCR was conducted with the application
of Power SYBR Master Mix (Applied Biosystems; Thermo Fisher
Scientific, Inc.) on the ABI 7500 PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The thermocycling program was as
follows: 10 min at 95˚C; 40 cycles of 2 sec at 95˚C; 20 sec at 60˚C
and 10 sec at 70˚C. The following primer sequences were used: GAB1
forward, 5'-ACCACCACGACAACATTCCA-3' and reverse,
5'-CGCTGGCTTGACTTTTCTGT-3'; suppressor of cytokine signaling 3
(SOCS3) forward, 5'-ATCCTGGTGACATGCTCCTC-3' and reverse,
5'-GGCACCAGGTAGACTTTGGA-3'; and GAPDH forward,
5'-CAGGAGGCATTGCTGATGAT-3' and reverse, 5'-GAAGGCTGGGGCTCATTT-3'.
The relative expression levels of the target gene were calculated
using the 2-ΔΔCq method (13), and GAPDH served as the internal
control.
Western blotting
Total protein was isolated from HUVECs using RIPA
lysis buffer (Thermo Fisher Scientific, Inc.), followed by
quantification of the protein concentration using a BCA Protein
Assay kit (Thermo Fisher Scientific, Inc.). Subsequently, equal
amounts of protein (40 µg/lane) were separated by 10% SDS-PAGE, and
transferred onto PVDF membranes (MilliporeSigma). Membranes were
then blocked with 5% skimmed milk at room temperature for 2 h, and
probed with primary antibodies against GAB1 (1:1,000; cat. no.
ab59362; Abcam), Bcl2 (1:1,000; cat. no. ab32124; Abcam), Bax
(1:1,000; cat. no. ab32503; Abcam), cleaved caspase-3 (1:1,000;
cat. no. 9661; Cell Signaling Technology, Inc.), caspase-3
(1:5,000; cat. no. ab32351; Abcam), endothelial nitric oxide (NO)
synthase (eNOS; 1:1,000; cat. no. ab199956; Abcam), phosphorylated
(p-)eNOS (1:1,000; cat. no. ab215717; Abcam), SOCS3 (1:1,000; cat.
no. ab16030; Abcam), p-Janus kinase 2 (JAK2) (1:1,000; cat. no.
ab32101; Abcam), JAK2 (1:5,000; cat. no. ab108596; Abcam), p-STAT3
(1:1,000; cat. no. 9131; Cell Signaling Technology, Inc.), STAT3
(1:2,000; cat. no. 4904; Cell Signaling Technology, Inc.) and GAPDH
(1:2,500; cat. no. ab9485; Abcam) at 4˚C overnight. On the
following day, after three washes with TBS with 10% Tween-20, the
membranes were incubated with HRP-conjugated goat anti-rabbit IgG
antibody (1:2,000; cat. no. ab6721; Abcam) at room temperature for
2 h. Finally, blots were visualized using an enhanced
chemiluminescence kit (Thermo Fisher Scientific, Inc.), and the
band intensity was semi-quantified using ImageJ software (version
1.8.0; National Institutes of Health).
Cell transfection
The full-length coding sequence of GAB1 was cloned
into the pcDNA3.1 vector (Sangon Biotech Co., Ltd.) to construct a
GAB1-overexpressing vector (Ov-GAB1), and the empty pcDNA3.1 vector
acted as the negative control (Ov-NC). Small interfering RNA
(siRNA) targeting SOCS3, including siRNA-SOCS3-1 (sense,
5'-CCUGGUGGGACGAUAGCAACC-3'; antisense,
5'-GGACCACCCUGCUAUCGUUGG-3') and siRNA-SOCS3-2 (sense,
5'-AACAAGUUCCGUUGGAAAGUU-3'; antisense,
5'-UUGUUCAAGGCAACCUUUCAA-3'), were also obtained from Sangon
Biotech Co., Ltd., and non-targeting siRNA acted as the negative
control (siRNA-NC; sense, 5'-UUCUCCGAACGUGUCACGUTT-3'; antisense,
5'-ACGUGACACGUUCGGAGAATT-3'). Upon achieving 60-70% confluence,
HUVECs were transfected with 50 nM SOCS3 siRNA, 50 nM siRNA-NC, 10
µg Ov-GAB1 or 10 µg Ov-NC at 37˚C using Lipofectamine®
3000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according
to the manufacturer's instructions. In brief, the aforementioned
vectors and the Lipofectamine® 3000 reagent were
separately diluted in Opti-MEM. Subsequently, the two dilutions
were mixed for 20 min, and then added to each well. Cells were
incubated with the mixture for 6 h before the medium was changed.
After 48 h, the transfection efficiency was determined via reverse
transcription-quantitative PCR and western blotting as
aforementioned and were used for subsequent experiments.
Cell viability assay
Cell viability was assessed using a Cell Counting
Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.) assay according
to the manufacturer's instructions. In brief, HUVECs were
inoculated into 96-well plates (5x103 cells/well) and
cultured at 37˚C with 5% CO2. Cells were treated with
LPS (2.5, 5 and 10 µg/ml) for 24 h at 37˚C, and 10 µM CCK-8
solution was added to each well for an additional incubation for 2
h. Finally, the absorbance at 450 nm of each well was detected
using a microplate reader. Relative cell viability (%) was
calculated as follows: [Treated optical density
(OD)A450-blank ODA450]/(control
ODA450-blank ODA450) x100%.
TUNEL assay
The apoptotic cells were assessed using a One Step
TUNEL Detection kit (Beyotime Institute of Biotechnology) according
to the manufacturer's instructions. In brief, cells were fixed with
4% paraformaldehyde for 5 min at room temperature and then
incubated with 0.3% Triton X-100 for 5 min at room temperature.
After washing with PBS, cells were incubated with TUNEL reagent for
60 min at 37˚C in the dark. Finally, cells were stained with 1
mg/ml DAPI solution (Invitrogen; Thermo Fisher Scientific, Inc.)
for 10 min in the dark at room temperature, washed with PBS and
mounted in glycerol. Images were captured in three randomly
selected fields of view using an inverted fluorescence microscope
(Olympus Corporation). The cell apoptosis rate (%) was calculated
as follows: Number of apoptotic positive cells/total number of
cells.
Measurement of cytokine concentrations
and NO levels
The culture medium was harvested and centrifuged at
500 x g for 5 min at 4˚C, and the supernatant was collected. The
concentrations of the inflammatory cytokines, including TNF-α,
IL-1β and IL-6, in the supernatant were detected using their
corresponding commercial ELISA kits (TNF-α, cat. no. PT518; IL-1β,
cat. no. PI305; IL-6, cat. no. PI330; Beyotime Institute of
Biotechnology) according to the manufacturer's instructions. The
absorbance at 450 nm was detected using a microplate reader. The
levels of NO in the culture medium were detected using a commercial
kit (cat. no. BC1475; Beijing Solarbio Science & Technology
Co., Ltd.) according to the manufacturer's protocol, and the
absorbance at 550 nm was detected using a microplate reader.
Wound healing assay
HUVECs were inoculated into 6-well plates and
cultured at 37˚C with 5% CO2. Upon reaching 100%
confluence, a wound was generated using a 200-µl sterile
micropipette tip. Cells were washed with PBS to remove the
scratched cells, and incubated with serum-free medium for 24 h.
Images at 0 and 24 h were captured under an inverted light
microscope (Olympus Corporation). The relative migration rate
(%)=(wound width at 0 h-wound width at 24 h)/wound width at 0 h
x100.
Co-immunoprecipitation (Co-IP)
assay
The interaction between GAB1 and SOCS3 was confirmed
using a Co-IP assay. In brief, the total protein was extracted
using RIPA lysis buffer (Thermo Fisher Scientific, Inc.) and the
supernatant was collected after centrifugation at 13,000 x g for 10
min at 4˚C. The lysed protein samples (500 µg) were then incubated
with 2 µg anti-IgG, IP-indicated antibodies against GAB1 (1:100;
cat. no. ab133486; Abcam), SOCS3 (1:30; cat. no. ab280884; Abcam),
and untreated proteins as an input control. The mixtures were
incubated with 50 µg Protein A/G PLUS-Agarose (Santa Cruz
Biotechnology, Inc.) at 4˚C for 6 h. After the IP reaction, agarose
beads were centrifuged at 1,000 x g for 3 min at 4˚C to the bottom
of the tube. The supernatant was then carefully absorbed, and the
agarose beads were washed three times with 1 ml lysis buffer.
Subsequently, the immunoprecipitated protein complex was boiled and
denatured, and western blotting with the anti-GAB1 and anti-SOCS3
antibodies was carried out as aforementioned to detect the
precipitated protein.
Bioinformatics tools
By searching for ‘GAB1’ and selecting ‘Homo sapiens’
in Biogrid version 4.4.232 (https://thebiogrid.org/) and entering ‘GAB1’ as the
protein ID in the FpClass (http://dcv.uhnres.utoronto.ca/FPCLASS/ppis/) database
(threshold value>0.25), the interaction between GAB1 and SOCS3
was predicted.
Statistical analysis
All data are presented as the mean ± standard
deviation. GraphPad Prism software 9.0 (Dotmatics) was used to
perform statistical analysis. All experiments were independently
repeated in triplicate. One-way ANOVA followed by Tukey's post hoc
test was performed to compare the differences among groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
GAB1 is downregulated in
LPS-challenged HUVECs
HUVECs were exposed to 2.5, 5 and 10 µg/ml LPS for
24 h to construct an in vitro model of sepsis-induced
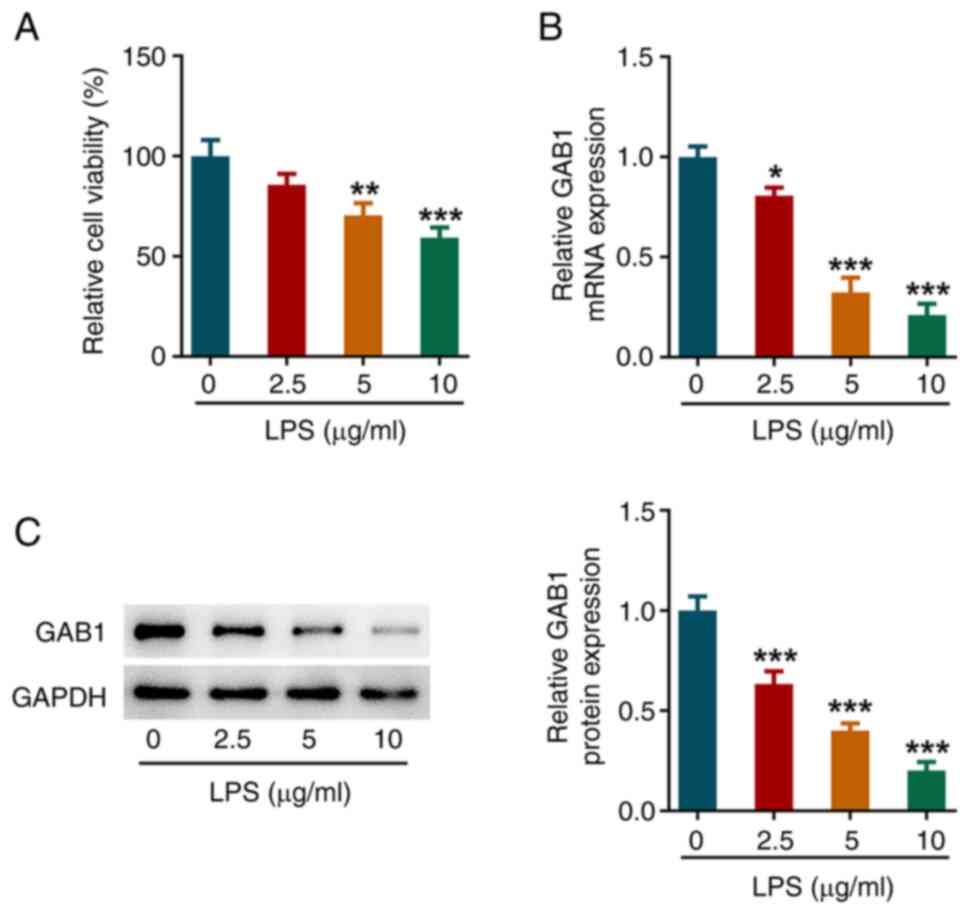
endothelial cell injury. As shown in Fig. 1A, cell viability was significantly
reduced following treatment with 5 and 10 µg/ml LPS. Furthermore,
the mRNA and protein expression levels of GAB1 were significantly
decreased following LPS exposure in a concentration-dependent
manner (Fig. 1B and C).
GAB1 mitigates LPS-induced cell
viability decrease and apoptosis in HUVECs
To explore the regulatory role of GAB1 underlying
sepsis-mediated endothelial cell injury, a gain-of-function
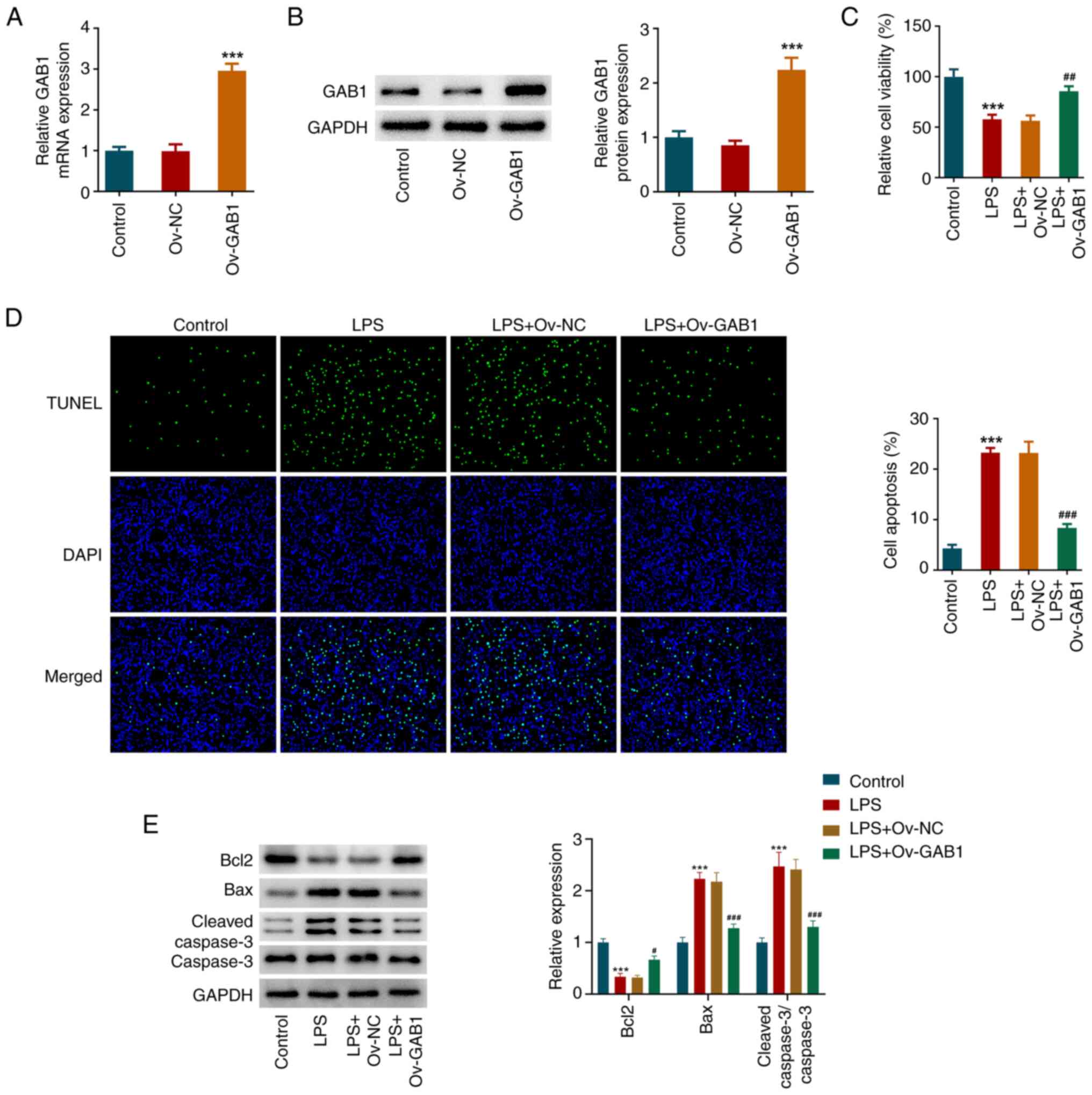
experiment was performed. As shown in Fig. 2A and B, compared with those in the Ov-NC group,
both the mRNA and protein expression levels of GAB1 were
significantly increased in the Ov-GAB1 group. Subsequently, the
normal HUVECs and GAB1-overexpressing HUVECs were exposed to LPS
for 24 h, and the CCK-8 assay showed that GAB1 overexpression
partly counteracted LPS-induced cell viability decrease in HUVECs
(Fig. 2C). In addition, it was
observable from the TUNEL assay that LPS stimulation caused an
elevation of apoptosis in HUVECs, which was partly abolished by
GAB1 overexpression (Fig. 2D). The
downregulated protein expression levels of Bcl2 and the upregulated
protein levels of Bax and cleaved caspase-3 in the LPS group
compared with the control group further confirmed the high
apoptosis rate of LPS-exposed HUVECs. However, these changes were
partly weakened by GAB1 overexpression (Fig. 2E), suggesting that GAB1 could
partly inhibit LPS-induced apoptosis in HUVECs.
GAB1 mitigates LPS-induced
inflammation and endothelial dysfunction in HUVECs
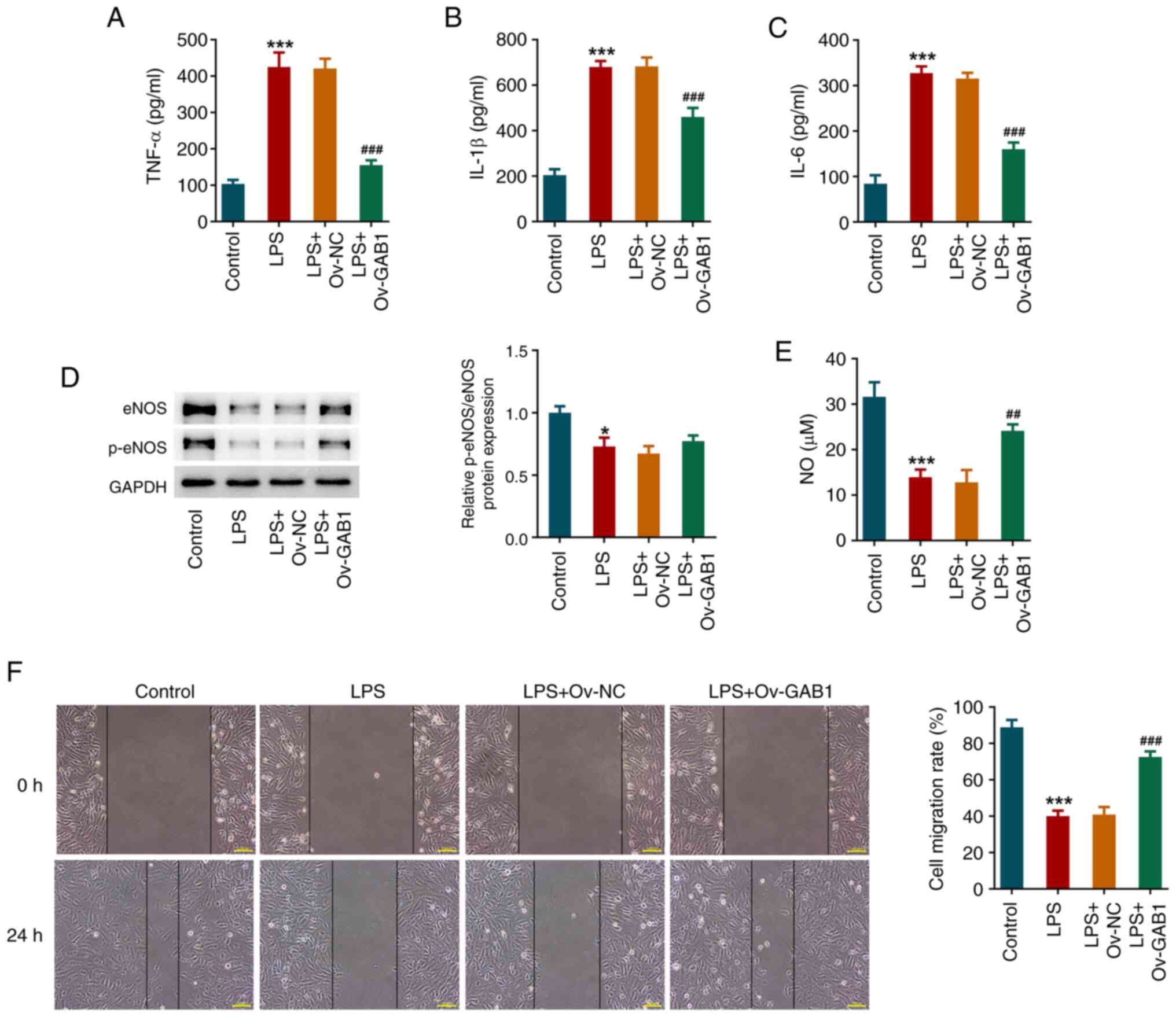
An ELISA revealed that, after LPS exposure, the
concentrations of TNF-α, IL-1β and IL-6 in the culture medium of
HUVECs were notably elevated, which were all decreased in the LPS +
Ov-GAB1 group (Fig. 3A-C). eNOS
and p-eNOS levels were significantly reduced following LPS
stimulation; however, GAB1 overexpression partly restricted this
reduction (Fig. 3D).
Overexpressing eNOS protein is an important approach to promote NO
production (14). Accordingly,
compared with that in the control group, the NO level in
LPS-induced HUVECs was significantly decreased, while GAB1
overexpression notably elevated NO levels (Fig. 3E). In addition, the results of the
wound healing assay revealed that LPS induction significantly
weakened the migration of HUVECs, which was partly counteracted by
GAB1 overexpression (Fig. 3F).
SOCS3 knockdown partly weakens the
impacts of GAB1 overexpression on cell viability, apoptosis,
inflammation and endothelial function in LPS-induced HUVECs
The present study also attempted to identify
GAB1-interacting proteins to explain its regulatory mechanisms.
Based on Biogrid and FpClass database, it was found that there may
be a protein-protein interaction between GAB1 and SOCS3, which was
then verified using a Co-IP assay (Fig. 4A). SOCS3 expression was also
downregulated in LPS-induced HUVECs, while GAB1 overexpression
increased SOCS3 expression (Fig.
4B). Therefore, to understand the role of SOCS3 in
GAB1-mediated endothelial function, HUVECs were transfected with
siRNA-SOCS3-1/2 or siRNA-NC to knock down SOCS3. As shown in
Fig. 4C and D, compared with those in the siRNA-NC
group, the mRNA and protein expression levels of SOCS3 were
significantly decreased in the siRNA-SOCS3-1 and siRNA-SOCS3-2
groups. Due to the superior transfection efficacy, siRNA-SOCS3-1
was used in the subsequent gain-of-function and loss-of-function
experiments. As shown in Fig. 4E,
HUVECs were transfected with Ov-GAB1 alone or co-transfected with
siRNA-NC or siRNA-SOCS3, followed by LPS stimulation. The elevated
cell viability caused by GAB1 overexpression in LPS-exposed HUVECs
was partly reduced following additional SOCS3 knockdown.
Furthermore, the inhibitory effects of GAB1 overexpression on
TUNEL-positive cells and the protein levels of Bcl2, Bax and
cleaved caspase-3 in LPS-exposed HUVECs were significantly
abolished by additional SOCS3 knockdown (Fig. 4F and G).
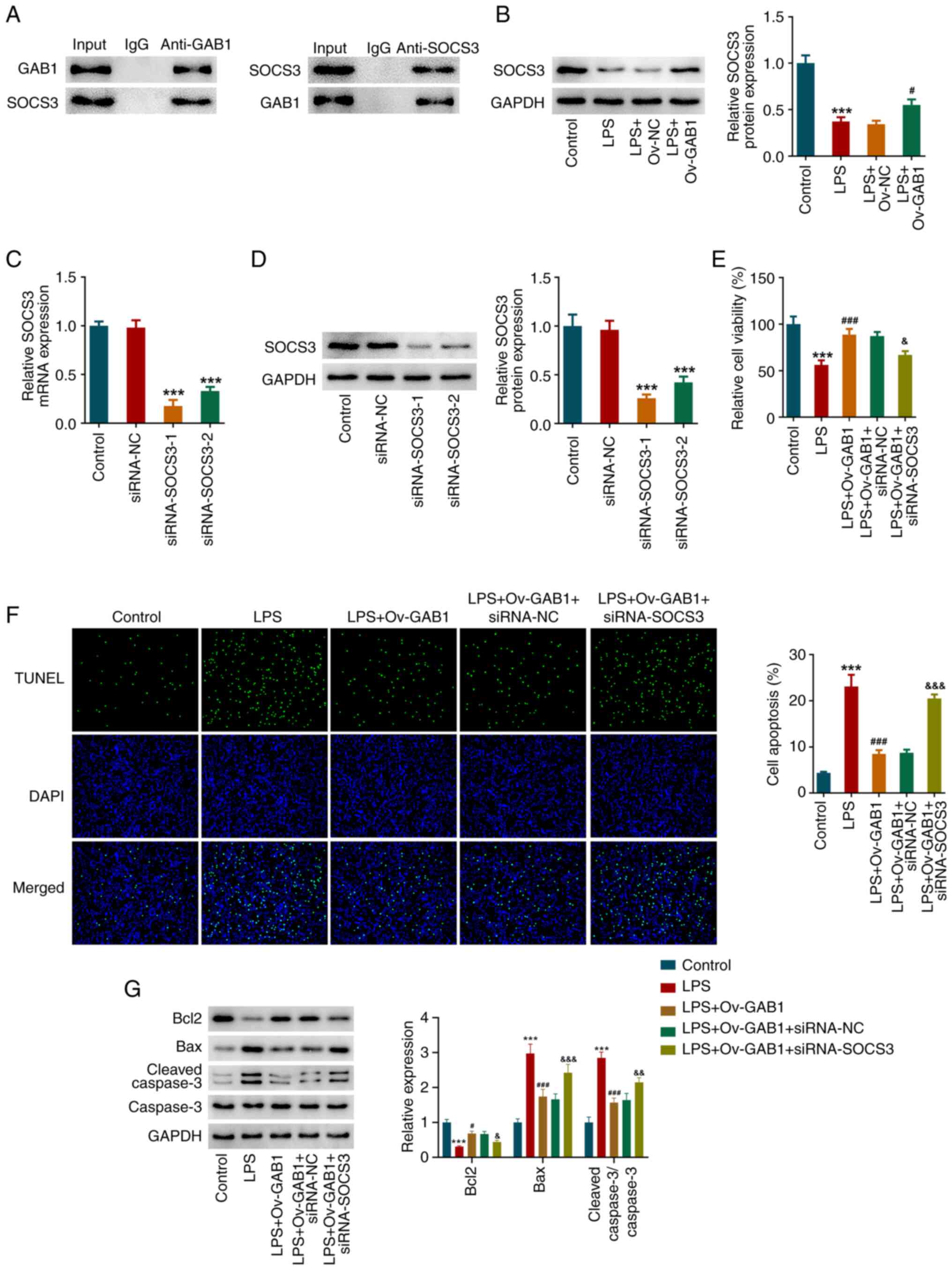 | Figure 4SOCS3 knockdown partially weakens the
impacts of GAB1 overexpression on cell viability and apoptosis in
LPS-induced HUVECs. (A) In HUVECs, a co-immunoprecipitation assay
was performed to verify the protein-protein interaction between
GAB1 and SOCS3. (B) HUVECs were transfected with Ov-NC or Ov-GAB1,
followed by LPS stimulation. HUVECs without any treatment served as
the control group. The protein expression levels of SOCS3 were
detected using western blotting. ***P<0.001 vs.
control; #P<0.05 vs. LPS + Ov-NC. (C) HUVECs were
transfected with siRNA-SOCS3-1/2 or siRNA-NC to knock down SOCS3,
and the mRNA levels of SOCS3 were detected using reverse
transcription-quantitative PCR. (D) Protein expression levels of
SOCS3 were detected using western blotting.
***P<0.001 vs. siRNA-NC. (E) HUVECs were transfected
with Ov-GAB1 alone or co-transfected with siRNA-NC/siRNA-SOCS3,
followed by LPS stimulation, and a Cell Counting Kit-8 assay was
performed to assess cell viability. (F) A TUNEL assay was conducted
to examine cell apoptosis; magnification, x200. (G) Expression
levels of apoptosis-related proteins were detected using western
blotting. ***P<0.001 vs. control;
#P<0.05 and ###P<0.001 vs. LPS;
&P<0.05, &&P<0.01 and
&&&P<0.001 vs. LPS + Ov-GAB1 + siRNA-NC.
GAB1, GRB2-associated binder 1; LPS, lipopolysaccharide; Ov-GAB1,
GAB1-overexpressing vector; Ov-NC, scramble pcDNA3.1 vector;
siRNA-NC, scramble small interfering RNA; siRNA-SOCS3, small
interfering RNA targeting SOCS3; SOCS3, suppressor of cytokine
signaling 3. |
In addition, SOCS3 knockdown also weakened the
anti-inflammatory activity of GAB1 overexpression in LPS-exposed
HUVECs, as demonstrated by the upregulated levels of TNF-α, IL-1β
and IL-6 in the LPS + Ov-GAB1 + siRNA-SOCS3 group compared with the
LPS + Ov-GAB1 + siRNA-NC group (Fig.
5A-C). Furthermore, the increase in the protein levels of
p-eNOS and eNOS, NO levels, and cell migration in LPS-exposed
HUVECs following GAB1 overexpression was partly counteracted by
SOCS3 knockdown (Fig. 5D-F).
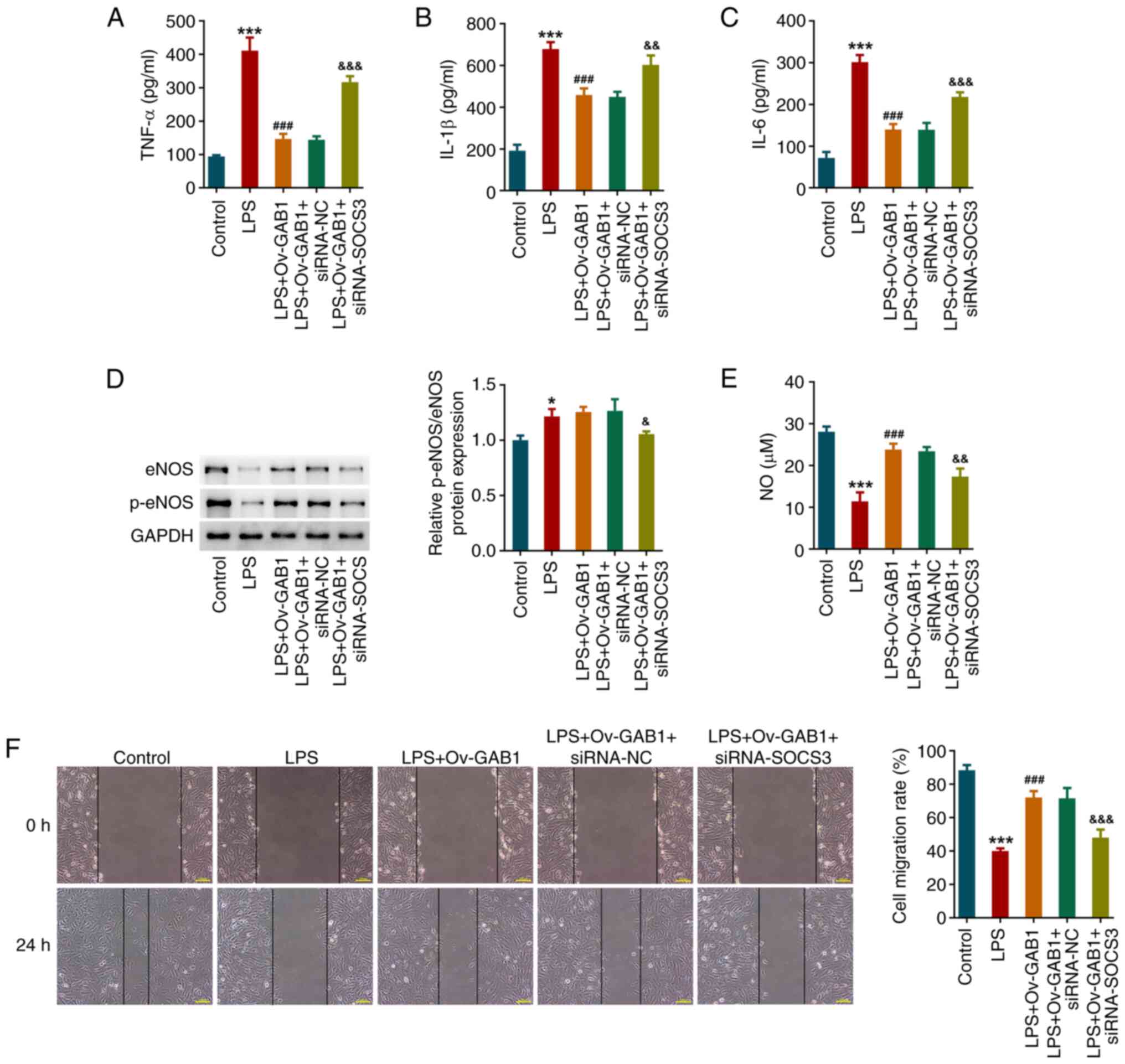 | Figure 5SOCS3 knockdown partially weakens the
impacts of GAB1 overexpression on inflammation and endothelial
function in LPS-induced HUVECs. Concentrations of (A) TNF-α, (B)
IL-1β and (C) IL-6 in the culture medium of HUVECs were detected
using ELISA kits. (D) Protein levels of eNOS and p-eNOS were
examined using western blotting. (E) NO levels in the culture
medium of HUVECs were examined. (F) Cell migration was assessed
using a wound healing assay. Scale bar, 100 µm.
*P<0.05 and ***P<0.001 vs. control;
###P<0.001 vs. LPS; &P<0.05,
&&P<0.01 and
&&&P<0.001 vs. LPS + Ov-GAB1 + siRNA-NC.
eNOS, endothelial NO synthase; GAB1, GRB2-associated binder 1; LPS,
lipopolysaccharide; NO, nitric oxide; Ov-GAB1, GAB1-overexpressing
vector; p-, phosphorylated; siRNA-NC, scramble small interfering
RNA; siRNA-SOCS3, small interfering RNA targeting SOCS3; SOCS3,
suppressor of cytokine signaling 3. |
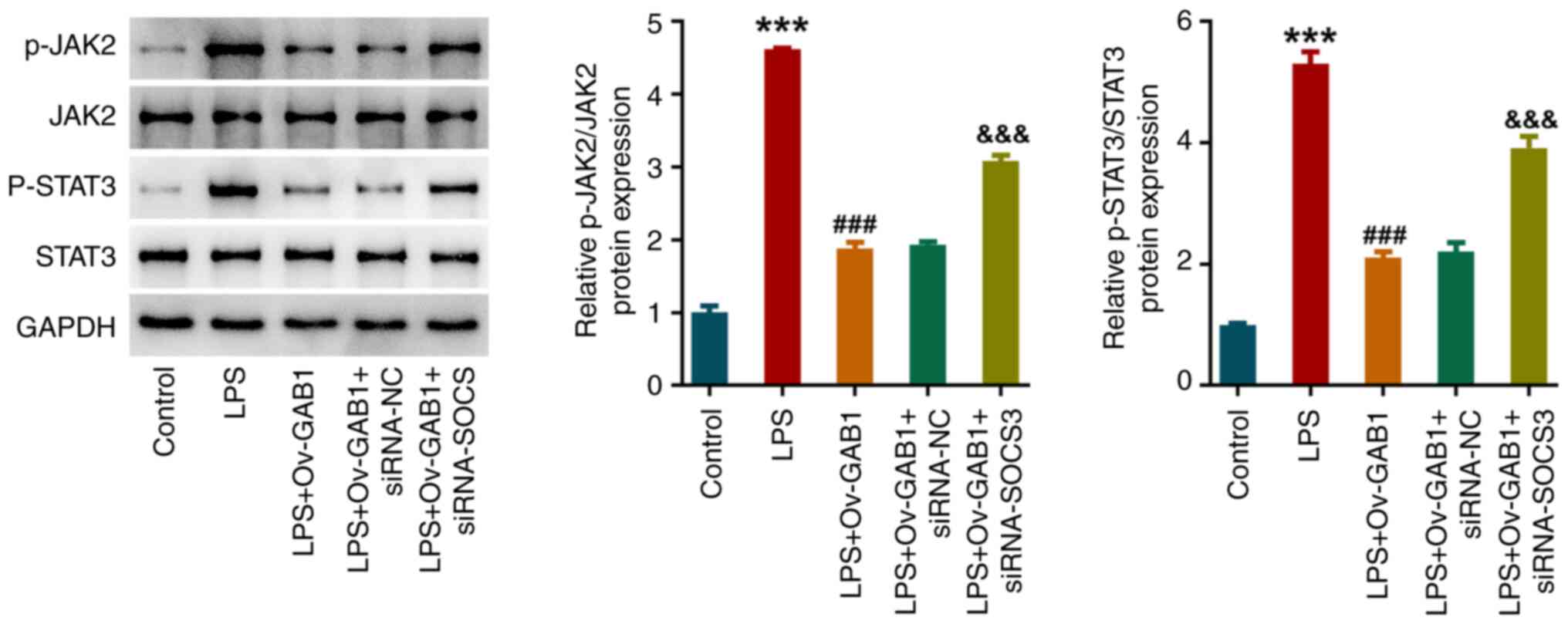
GAB1 and SOCS3 regulate JAK2/STAT3
signaling in LPS-induced HUVECs
The changes in JAK2/STAT3 signaling underlying the
regulation of the GAB1/SOCS3 axis in LPS-exposed HUVECs were also
explored. As shown in Fig. 6,
compared with those in the control group, the protein levels of
p-JAK2 and p-STAT3 were significantly increased in the LPS group,
indicating that LPS triggered the activation of JAK2/STAT3
signaling in HUVECs. However, the activation of JAK2/STAT3
signaling in LPS-exposed HUVECs was inhibited by GAB1
overexpression, which was then partly abolished by additional SOCS3
knockdown, suggesting that JAK2/STAT3 signaling might be involved
in the regulation of the GAB1/SOCS3 axis in LPS-exposed HUVECs.
Discussion
The present study was undertaken to explore the
protective role and mechanism of action of GAB1 in attenuating the
endothelial dysfunction induced by LPS. The major findings of the
present study were that GAB1 could mitigate cell viability
decrease, apoptosis, inflammation and endothelial dysfunction in
LPS-induced HUVECs, an in vitro cellular model stimulating
sepsis-induced endothelial dysfunction, partly through upregulating
SOCS3, accompanied by the involvement of JAK2/STAT3 signaling.
Endothelial cells, which cover the inner wall of
blood vessels, are non-conventional immune cells of blood vessels
and constitute the basic barrier between tissues and blood, serving
a crucial role in the maintenance of the homeostasis of the
internal environment of the body (15,16).
During sepsis, endothelial cells are stimulated by a large number
of pathogens and endotoxins, leading to endothelial cell activation
and a gradual phenotypic shift toward pro-apoptosis,
pro-inflammatory, pro-adhesion and pro-coagulant phenotypes,
accompanied by the excessive release of pro-inflammatory factors
and upregulation of endothelial adhesion molecules. This leads to
impaired endothelial barrier function and an uncontrolled systemic
inflammatory response, and ultimately results in multisystem organ
dysfunction and failure (17,18).
Therefore, protection of endothelial cells is one of the mechanisms
in the current treatment of sepsis. In the present study,
LPS-exposed HUVECs were used to mimic endothelial dysfunction in
sepsis in vitro as previously proposed (19,20).
It was demonstrated that LPS exposure resulted in the excessive
production of pro-inflammatory cytokines and apoptosis of HUVECs,
while GAB1 overexpression significantly hindered these changes. In
addition, there was a reduction in p-eNOS and eNOS levels, and NO
production in HUVECs in response to LPS exposure. Upregulation of
eNOS protein is an important approach to promote NO production
(14). NO was originally
identified as a vasodilator, and the reduction of NO was regarded
as one of the critical causes of endothelial dysfunction (21). Therefore, a moderate increase in NO
production is of great importance for maintaining endothelial
function. Accordingly, elevated p-eNOS and eNOS expression and NO
production were observed following GAB1 overexpression in
LPS-exposed HUVECs, indicating that GAB1 could attenuate
LPS-mediated endothelial dysfunction, and might have a protective
effect against sepsis.
SOCS3 is a novel intracellular regulator that
negatively regulates the sustained activation of multiple
cytokine-associated signaling pathways, which in turn participates
in biological processes such as inflammation, oxidative stress,
cell proliferation and apoptosis (22,23).
SOCS3 has been shown to limit TNF-α and endotoxin-induced
endothelial dysfunction by blocking essential autocrine IL-6
signaling in human endothelial cells (24). For example, upregulation of SOCS3
protein levels was able to inhibit IL-6 signaling and repair
impairment of endothelial barrier function (25). This confirms that SOCS3 is a key
component contributing to the inhibition of endothelial lesions
during sepsis, and stabilizing endothelial SOCS3 could potentially
be an effective measure against sepsis-induced multi-organ failure
(25). Therefore, SOCS3 serves a
protective role in sepsis-induced endothelial cell injury. Notably,
in the present study, the protein-protein interaction between GAB1
and SOCS3 was verified, and GAB1 positively regulated SOCS3
expression. SOCS3 knockdown significantly weakened the inhibitory
effects of GAB1 overexpression on LPS-mediated endothelial damage,
further suggesting that the protective role of GAB1 against
LPS-induced endothelial dysfunction was partly achieved via
upregulation of SOCS3.
The JAK2/STAT3 signaling pathway is a common
signaling pathway that regulates a variety of important biological
behaviors such as cell proliferation, apoptosis, differentiation
and inflammation (26). A previous
study has shown that the development of sepsis is closely related
to the persistent activation of the JAK2/STAT3 signaling pathway,
and that modulation of the JAK2/STAT3 pathway can affect the course
of sepsis and organ dysfunction (27). For example, melatonin alleviates
sepsis-induced myocardial injury by regulating the JAK2/STAT3
signaling pathway (28), and
eupatilin effectively reduces inflammation and coagulation
dysfunction by inhibiting the JAK2/STAT3 signaling pathway, thereby
reducing the progression of sepsis-induced lung injury (29). Consistently, in the present study,
GAB1 overexpression significantly inhibited the JAK2/STAT3
signaling pathway in LPS-treated HUVECs, which may partly account
for the protective effect of GAB1 against endothelial dysfunction.
Further studies have revealed that SOCS3 is a critical negative
regulator of the JAK/STAT3 signaling pathway (30,31).
Therefore, the protective effect of GAB1 against LPS-induced
endothelial dysfunction might be achieved via regulation of the
SOCS3/JAK/STAT3 signaling pathway.
In conclusion, to the best of our knowledge, the
present study was the first to reveal that GAB1 exerted significant
ameliorative effects on LPS-induced endothelial cell apoptosis,
inflammation and dysfunction by modulating the SOCS3/JAK2/STAT3
signaling pathway. The findings may provide preclinical data to
support the use of GAB1 as a candidate gene in targeted therapy and
drug development for the treatment of sepsis.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
GR designed the study. GR, RL, HM, GY, FD, CW, SC
and XL conducted the experiments to collect data. RL, HM, GY and FD
analyzed and interpreted the data. GR and RL wrote the manuscript.
GR and RL confirm the authenticity of all the raw data. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (sepsis-3). JAMA.
315:801–810. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Evans L, Rhodes A, Alhazzani W, Antonelli
M, Coopersmith CM, French C, Machado FR, Mcintyre L, Ostermann M,
Prescott HC, et al: Surviving sepsis campaign: International
guidelines for management of sepsis and septic shock 2021. Crit
Care Med. 49:e1063–e1143. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Seymour CW, Kennedy JN, Wang S, Chang CCH,
Elliott CF, Xu Z, Berry S, Clermont G, Cooper G, Gomez H, et al:
Derivation, validation, and potential treatment implications of
novel clinical phenotypes for sepsis. JAMA. 321:2003–2017.
2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Xie J, Wang H, Kang Y, Zhou L, Liu Z, Qin
B, Ma X, Cao X, Chen D, Lu W, et al: The epidemiology of sepsis in
Chinese ICUs: A national cross-sectional survey. Crit Care Med.
48:e209–e218. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
van der Poll T, van de Veerdonk FL,
Scicluna BP and Netea MG: The immunopathology of sepsis and
potential therapeutic targets. Nat Rev Immunol. 17:407–420.
2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lelubre C and Vincent JL: Mechanisms and
treatment of organ failure in sepsis. Nat Rev Nephrol. 14:417–427.
2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Vincent JL, Ince C and Pickkers P:
Endothelial dysfunction: A therapeutic target in bacterial sepsis?
Expert Opin Ther Targets. 25:733–748. 2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Gu H and Neel BG: The ‘Gab’ in signal
transduction. Trends Cell Biol. 13:122–130. 2003.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Sun L, Zhu H and Zhang K: GAB1 alleviates
septic lung injury by inhibiting the TLR4/NF-κB pathway. Clin Exp
Pharmacol Physiol. 49:94–103. 2022.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Qiu N, Xu X and He Y: LncRNA TUG1
alleviates sepsis-induced acute lung injury by targeting
miR-34b-5p/GAB1. BMC Pulm Med. 20(49)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Xiong Y, Wang Y, Tian H, Li Y, Xu Q and He
Z: Circ-PRKCI alleviates lipopolysaccharide-induced human kidney 2
cell injury by regulating miR-106b-5p/GAB1 axis. J Cardiovasc
Pharmacol. 78:523–533. 2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Higuchi K, Nakaoka Y, Shioyama W, Arita Y,
Hashimoto T, Yasui T, Ikeoka K, Kuroda T, Minami T, Nishida K, et
al: Endothelial Gab1 deletion accelerates angiotensin II-dependent
vascular inflammation and atherosclerosis in apolipoprotein E
knockout mice. Circ J. 76:2031–2040. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 402-408:2001.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kolluru GK, Siamwala JH and Chatterjee S:
eNOS phosphorylation in health and disease. Biochimie.
92:1186–1198. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Aird WC: The role of the endothelium in
severe sepsis and multiple organ dysfunction syndrome. Blood.
101:3765–3777. 2003.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Page AV and Liles WC: Biomarkers of
endothelial activation/dysfunction in infectious diseases.
Virulence. 4:507–516. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Joffre J, Hellman J, Ince C and
Ait-Oufella H: Endothelial responses in sepsis. Am J Respir Crit
Care Med. 202:361–370. 2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Li Z, Yin M, Zhang H, Ni W, Pierce RW,
Zhou HJ and Min W: BMX represses thrombin-PAR1-mediated endothelial
permeability and vascular leakage during early sepsis. Circ Res.
126:471–485. 2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Huang L, Li Y, Cheng Z, Lv Z, Luo S and
Xia Y: PCSK9 promotes endothelial dysfunction during sepsis via the
TLR4/MyD88/NF-κB and NLRP3 pathways. Inflammation. 46:115–128.
2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhao L, Hu J, Zheng P, Mi B, Chen Z, Zhao
X, Wu J and Wang Y: PAR1 regulates sepsis-induced vascular
endothelial barrier dysfunction by mediating ERM phosphorylation
via the RhoA/ROCK signaling pathway. Int Immunopharmacol.
124(110992)2023.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Janaszak-Jasiecka A, Siekierzycka A,
Płoska A, Dobrucki IT and Kalinowski L: Endothelial dysfunction
driven by hypoxia-the influence of oxygen deficiency on no
bioavailability. Biomolecules. 11(982)2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Carow B and Rottenberg ME: SOCS3, a major
regulator of infection and inflammation. Front Immunol.
5(58)2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Jin D, Liu Y, Sun F, Wang X, Liu X and He
Z: Restoration of skilled locomotion by sprouting corticospinal
axons induced by co-deletion of PTEN and SOCS3. Nat Commun.
6(8074)2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Martino N, Ramos RB, Lu S, Leyden K,
Tomaszek L, Sadhu S, Fredman G, Jaitovich A, Vincent PA and Adam
AP: Endothelial SOCS3 maintains homeostasis and promotes survival
in endotoxemic mice. JCI Insight. 6(e147280)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Martino N, Bossardi Ramos R, Chuy D,
Tomaszek L and Adam AP: SOCS3 limits TNF and endotoxin-induced
endothelial dysfunction by blocking a required autocrine
interleukin-6 signal in human endothelial cells. Am J Physiol Cell
Physiol. 323:C556–C569. 2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Jaśkiewicz A, Domoradzki T and Pająk B:
Targeting the JAK2/STAT3 pathway-can we compare it to the two faces
of the God Janus? Int J Mol Sci. 21(8261)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yu Q, Wang H, Li X and Leng B: Effects of
JAK2/STAT3 signaling pathway activation on sepsis-induced kidney
injury. Minerva Med. 113:350–352. 2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhen G, Liang W, Jia H and Zheng X:
Melatonin relieves sepsis-induced myocardial injury via regulating
JAK2/STAT3 signaling pathway. Minerva Med. 113:983–989.
2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lu Y, Li D, Huang Y, Sun Y, Zhou H, Ye F,
Yang H, Xu T, Quan S and Pan J: Pretreatment with eupatilin
attenuates inflammation and coagulation in sepsis by suppressing
JAK2/STAT3 signaling pathway. J Inflamm Res. 16:1027–1042.
2023.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Durham GA, Williams JJL, Nasim MT and
Palmer TM: Targeting SOCS proteins to control JAK-STAT signalling
in disease. Trends Pharmacol Sci. 40:298–308. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gao Y, Zhao H, Wang P, Wang J and Zou L:
The roles of SOCS3 and STAT3 in bacterial infection and
inflammatory diseases. Scand J Immunol. 88(e12727)2018.PubMed/NCBI View Article : Google Scholar
|