Introduction
Chronic skin infection is caused by bacterial
infection, angiogenesis disorders, excessive inflammatory responses
and other factors, which in turn cause the deterioration of the
skin tissue microenvironment (1).
Chronic infection and blood circulation disorders are the main
factors that can adversely affect wound healing and the reason that
the patient's infectious lesions remain unhealed for a long time
(2). Clinically, the treatment of
refractory chronic skin infections remains a challenge.
Simultaneously, refractory skin infections fail to heal, thereby
escalating hospitalization and treatment costs, and consequently
augmenting the medical burden on patients. Hypoxia-inducible factor
1α (HIF1A) is commonly found in human cells and is usually
expressed under normoxic conditions. However, this protein is
quickly broken down by oxygen-dependent ubiquitin proteases within
cells and only remains stable at hypoxic conditions. HIF1A can
regulate the expression of genes that allow cells to adapt to
hypoxic conditions (3-5).
RNA sequencing is commonly used to study gene expression and
identify new RNAs (6), making it a
key tool for analyzing the transcriptome (7). Research on the molecular mechanisms
underlying the differences in HIF1A expression between normal
tissues and chronically infected skin tissues is required. Based on
gene sequencing and bioinformatics analysis of R language data
model, differentially expressing genes (DEGs) in tissues from
different groups can be screened (8,9). In
the present study, skin tissues were collected from healthy human
individuals and patients with chronic infection and used for RNA
sequencing and immunohistochemical detection. A data model based on
R language was used for bioinformatics analysis to find
characteristic genes associated with chronic skin infection.
Materials and methods
Sample collection and storage
The present study received approval from the Ethics
Committee of the Affiliated Hospital of Southwest Medical
University (approval no. KY2022206; Luzhou, China). Prior to
participation, all patients (5 males and 2 females; age range,
46-61 years) and volunteers (1 male and 3 females; age range, 37-62
years) provided written informed consent either personally or
through their legal representatives. Between May 2022 and May 2023,
the Affiliated Hospital of Southwest Medical University gathered
seven samples of skin wound tissues from individuals with chronic
skin infection, along with four samples of healthy skin tissue from
volunteers, which were preserved at -80˚C for RNA sequencing.
Additionally, another set of specimens, also comprising four cases
in the healthy group and seven cases in the chronic infection
group, were washed with pre-cooled PBS and fixed in 4%
paraformaldehyde for immunohistochemical detection. Patients with
chronic skin infections included post-traumatic infections and
diabetic foot infections.
High-throughput mRNA sequencing
(RNA-Seq)
RNA sequencing was performed on tissue samples from
chronic skin infection wounds and healthy individuals using the
Illumina Novaseq 6000 sequencing platform (Illumina, Inc.). The
Illumina Truseq RNA sample prep Kit (Illumina, Inc.) was used to
generate the library and sequence all transcribed mRNA, involving
the following six steps: Total RNA extraction; mRNA enrichment with
Oligo dT; mRNA fragmentation; cDNA reverse synthesis; adapter
ligation; and Illumina sequencing. The specific sequencing process
entailed during the extraction was as follows: i) Total RNA from
tissue samples, assessment of RNA concentration and purity using
Nanodrop 2000 (NanoDrop Technologies; Thermo Fisher Scientific,
Inc.), determination of RNA integrity through agarose gel
electrophoresis and calculation of RNA integrity number using
Agilent 2100 (Agilent Technologies, Inc.); ii) mRNAs were isolated
from total RNA by A-T base pairing with polyA using oligo(dT) beads
(500 times; cat. no. YH-RCZ-05; Shanghai Majorbio Pharmaceutical
Technology Co., Ltd.) to analyze the transcriptomic information,
based on the presence of a polyA tail structure at the 3' end of
eukaryotic mRNA; iii) the mRNA was fragmented using a fragmentation
buffer [DNA Purification and Fragment Screening Kit (magnetic bead
method); cat. no. C03-050; Shanghai Meiji Zhuanghua
Biopharmaceutical Technology Co., Ltd.], resulting in the isolation
of small fragments ~300 bp in length through magnetic bead
screening for sequencing; iv) A six-base random primer (random
hexamers) is introduced during reverse transcription to generate
single-stranded cDNA from mRNA as a template, followed by
two-stranded synthesis to establish a stable double-stranded
structure; v) end repair mix was added to the double-stranded cDNA
structures to homogenize cohesive ends, followed by the addition of
an ‘A’ base at the 3' end for joining Y-shaped joints; vi) the
cDNAs obtained through PCR amplifications using Phusion DNA
polymerase (NEB) for 15 cycles according to the manufacturer's
instructions; recovered with 2% agarose gel, quantified using
TBS380 (PicoGreen; NovaSeq Reagent Kit; Illumina, Inc.) and run in
a data ratio; and vii) bridge PCR was then amplified on cBot to
generate clusters, which were subsequently sequenced on the
Illumina platform (Illumina, Inc.) using a PE library with a
reading length of 2x150 bp.
Screening for DEGs
The sequencing data, which were subjected to quality
control measures [log2(CPM+4)], were analyzed using the online
software idep.96 (http://bioinformatics.sdstate.edu/idep/) (10) and a network tool (http://bioinformatics.sdstate.edu/idep/)
based on the R language (http://www.R-project.org/). Data quality was
standardized by applying log2 (CPM)+4(11), before the DESeq2 differential
screening method [log2 fold-change (FC)≥1 and q-value
≤0.05] (12) was used to identify
DEGs between the healthy skin group and the chronically infected
skin group.
Gene ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) functional enrichment
analysis
GO analysis is a typical batch gene data analysis
method. The GO examination of genes involved the analysis of
biological process (BP), cellular component (CC) and molecular
function (MF) aspects, was performed for categorizing and
identifying groups of genes in subsequent stages (13). KEGG is a resource that
systematically examines gene signaling pathways and connects
genomic and functional data (14).
The chosen DEGs were assessed using the database to study GO
functions and KEGG pathways, with the results being visualized
through online tools utilizing R language (https://cloud.oebiotech.com/spa#/bio/detail?number=e267d1a2-4303-44d7-a6db-d2bd2ad59e3e).
Protein-protein interaction (PPI)
analysis
Core genes were identified through the construction
of a PPI network utilizing the STRING database (https://cn.string-db.org/) (15) to understand how genes interacted at
the protein level. Potential key targets were identified through
this analysis. DEGs were then filtered based on their node degree
and network, with a threshold comprehensive score of >0.4. The
present study focused on the functional interactions among target
genes and other genes associated with adapting to hypoxia,
angiogenesis, cell adhesion and cell migration.
Immunohistochemistry
The present study adhered to the typical protocols
of immunohistochemistry, including collecting materials, preparing
sections, conducting antigen-antibody reactions and developing
colors to identify the presence of HIF1A expression in healthy skin
samples from donors and in wound tissues from patients with chronic
skin infections. The specific procedures for immunohistochemistry
were as follows: i) Skin tissues were fixed in 4% paraformaldehyde,
embedded in paraffin wax and cut into 5-mm slices. The slices were
then pressed onto anti-slip plates and placed on a 60˚C microtome
for 2 h. ii) The sections were incubated twice in xylene and
dehydrated in gradient ethanol solutions. iii) After microwave
treatment in citrate buffer (pH 6.0), the slides were incubated in
3% hydrogen peroxide for 15 min to inhibit endogenous peroxidase
activity. They were then blocked with 10% normal goat serum (cat.
no. A0208; Beyotime Institute of Biotechnology) and incubated at
room temperature for 10 min. iv) Sections were treated with primary
antibodies (HIF-1α polyclonal antibody; 1:70; cat. no. 20960-1-AP;
Proteintech Group, Inc.) as per instructions and incubated
overnight at 4˚C. After rewarming for 30 min at room temperature,
they were incubated with HRP-labeled secondary antibody
[HRP-labeled goat anti-rabbit IgG (HL); 1:70; cat. no. A0208;
Beyotime Institute of Biotechnology) for 60 min. Specimens were
then washed three times in PBS for 5 min each. v) Sections were
colored with DAB for 1-10 min, then washed twice in distilled water
for 5 min each. Hematoxylin counterstaining was performed for 5
min, followed by dehydration with gradient alcohol and clearing
with xylene twice for 5 min each. A neutral gummy film was applied
for capturing images. Images were viewed using an inverted light
microscope and captured at a magnification of x100 and x200. ImageJ
(version 1.8.0_172) processing software (National Institutes of
Health) was used for the semi-quantitative analysis of
immunohistochemical staining and to determine the average optical
density (AOD) value.
Statistical analysis
Statistical analysis of experimental data was
conducted using SPSS 26.0 software (IBM Corp). The experimental
results consist of measurements that follow a normal distribution
and are represented by the mean ± standard deviation. Unpaired
t-tests were used for comparing between the two means.
Results
Quality control of genetic data
obtained from sequencing
The gene data set obtained through RNA-seq underwent
logarithmic processing and was subjected to quality control using
the online analysis software idep.96 (http://bioinformatics.sdstate.edu/idep/), which
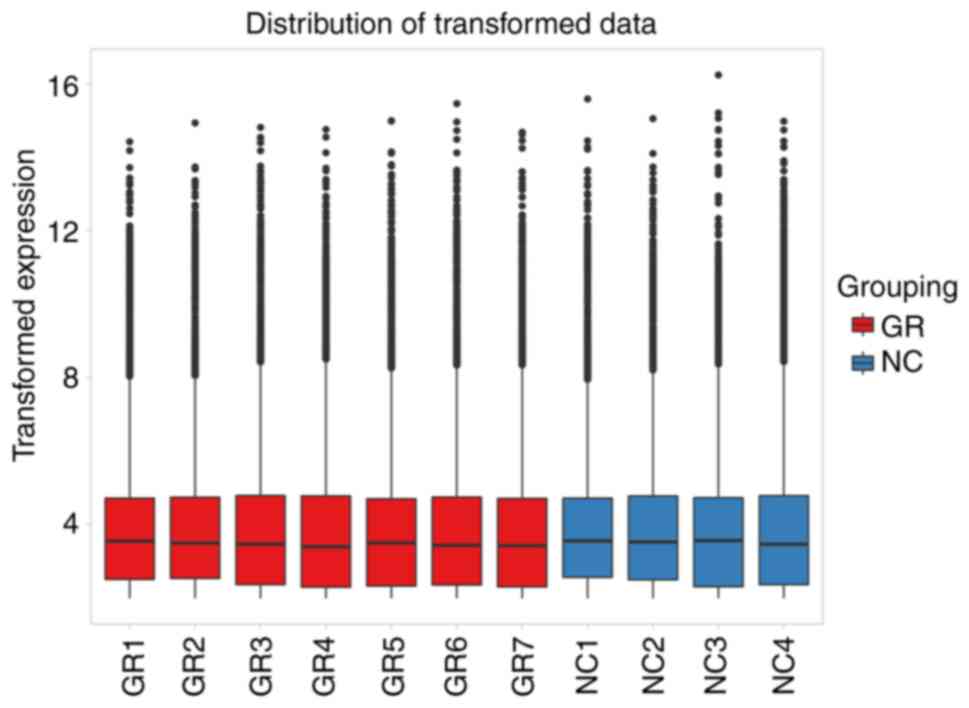
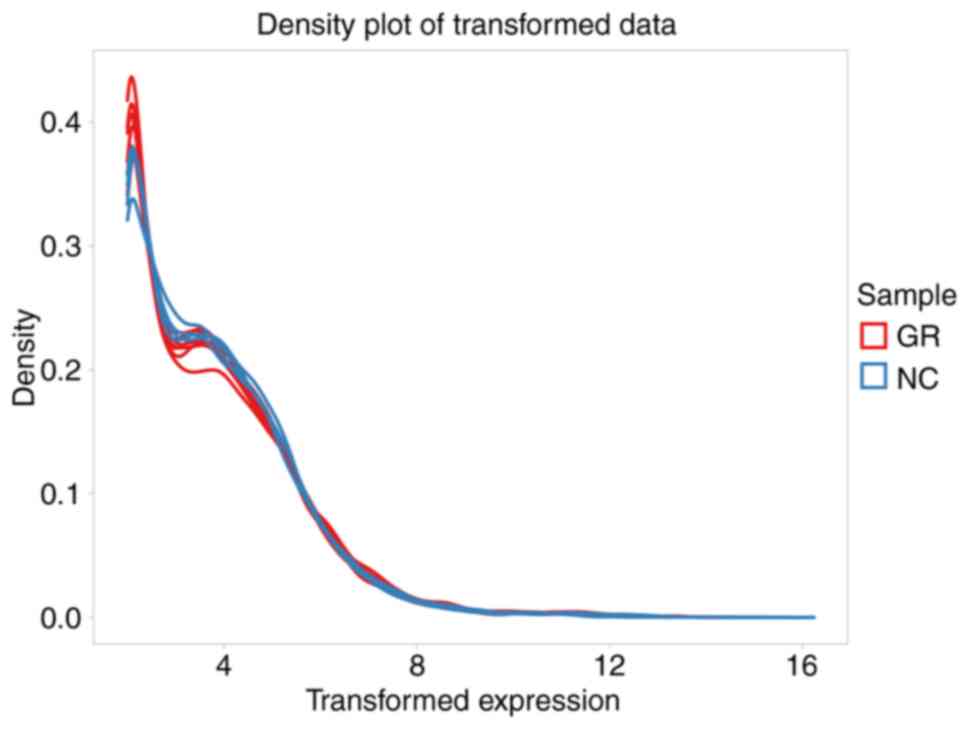
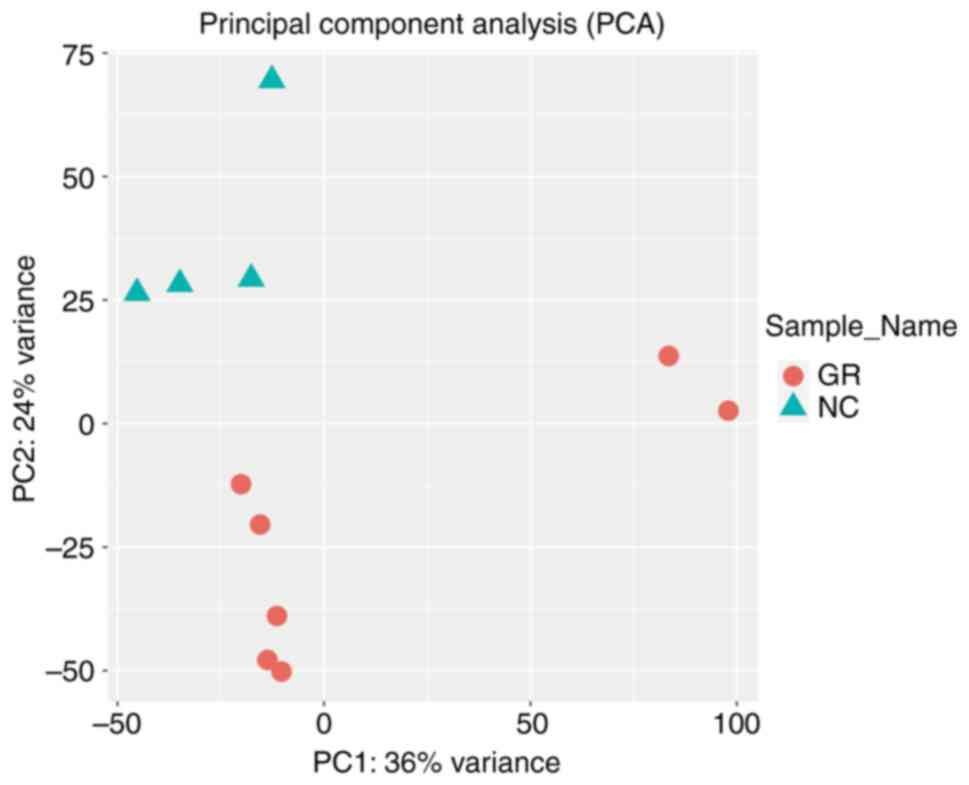
primarily involved generating box plots for two datasets (Fig. 1), data density plot (Fig. 2) and principal component analysis
plot (Fig. 3). The analysis
results indicated that the data were comparable and the quality
control of the data was satisfactory.
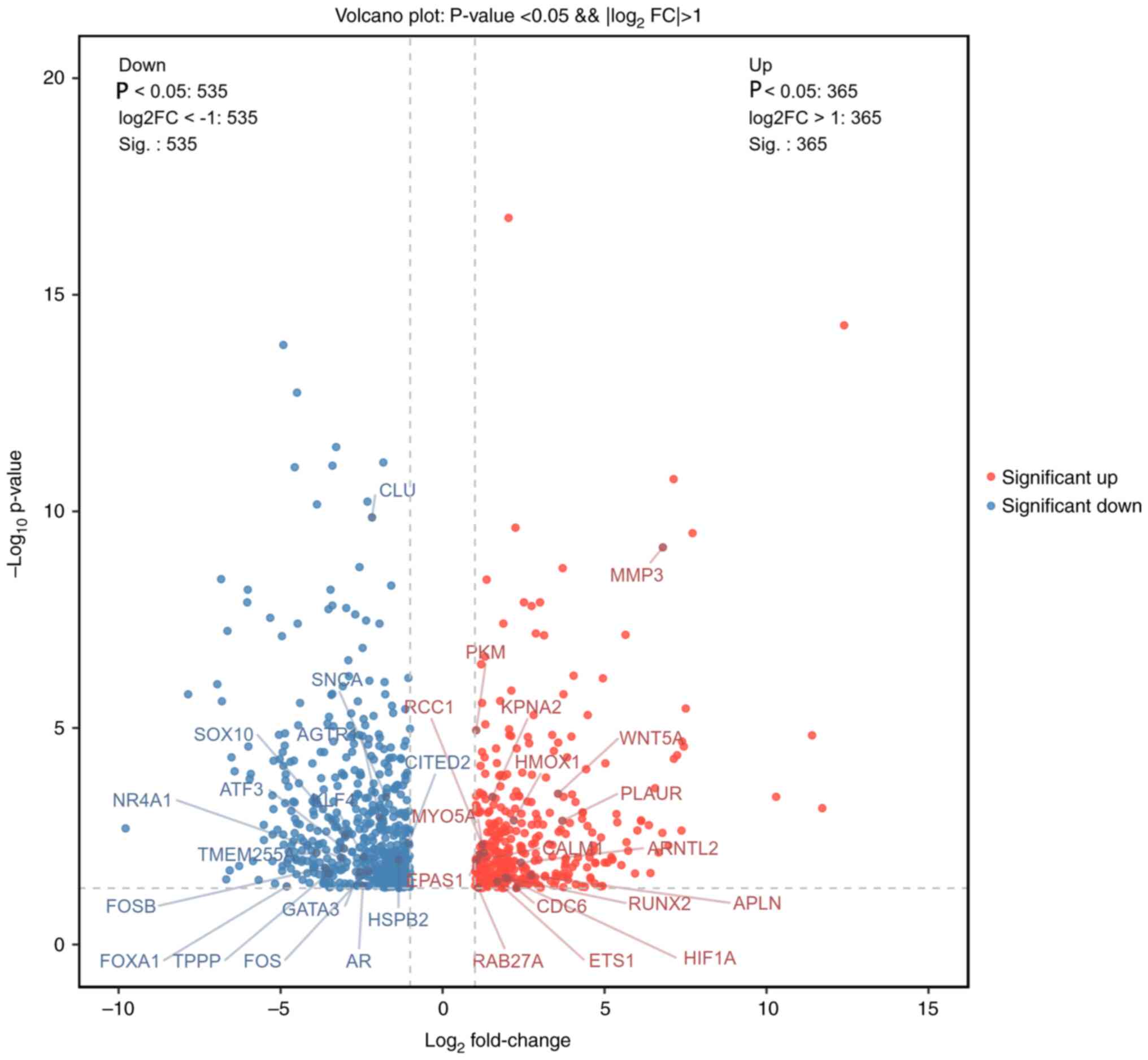
Differential gene screening
After the initial dataset was preprocessed using
logarithmic transformation, it was analyzed using the online tool
idep 0.96 (http://bioinformatics.sdstate.edu/idep/) with the
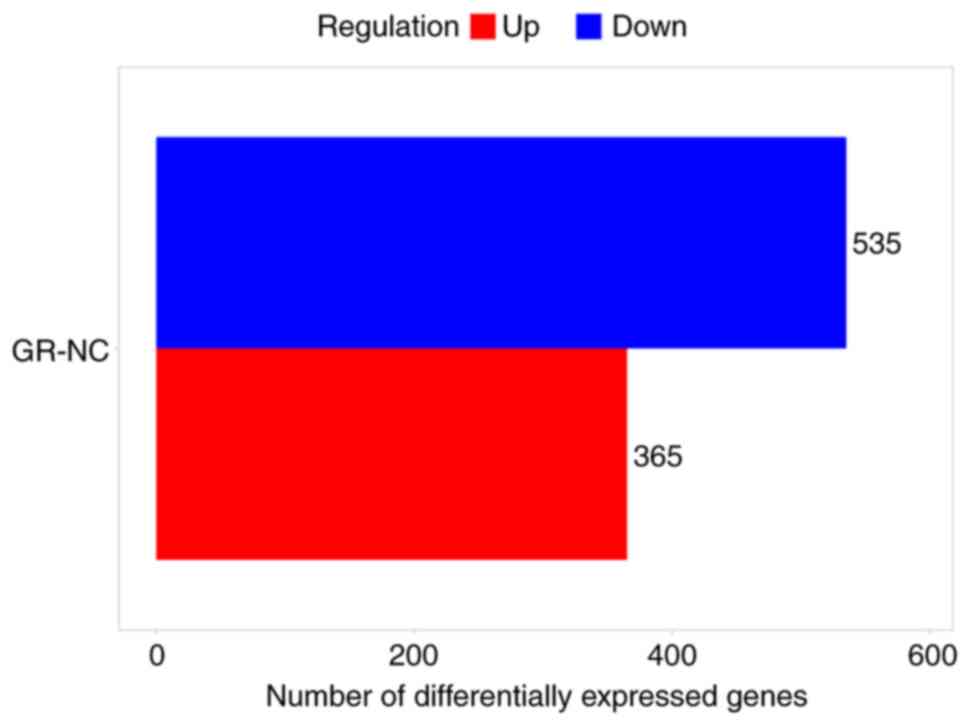
criteria of FC>4 and P<0.05. In both the chronic infection
group and healthy group, 900 DEGs were found, with 365 genes being
upregulated and 565 genes being downregulated. The DEGs were found
to be evenly distributed between the groups. The details of DEGs
are shown in the bar chart (Fig.
4) and volcano chart (Fig.
5).
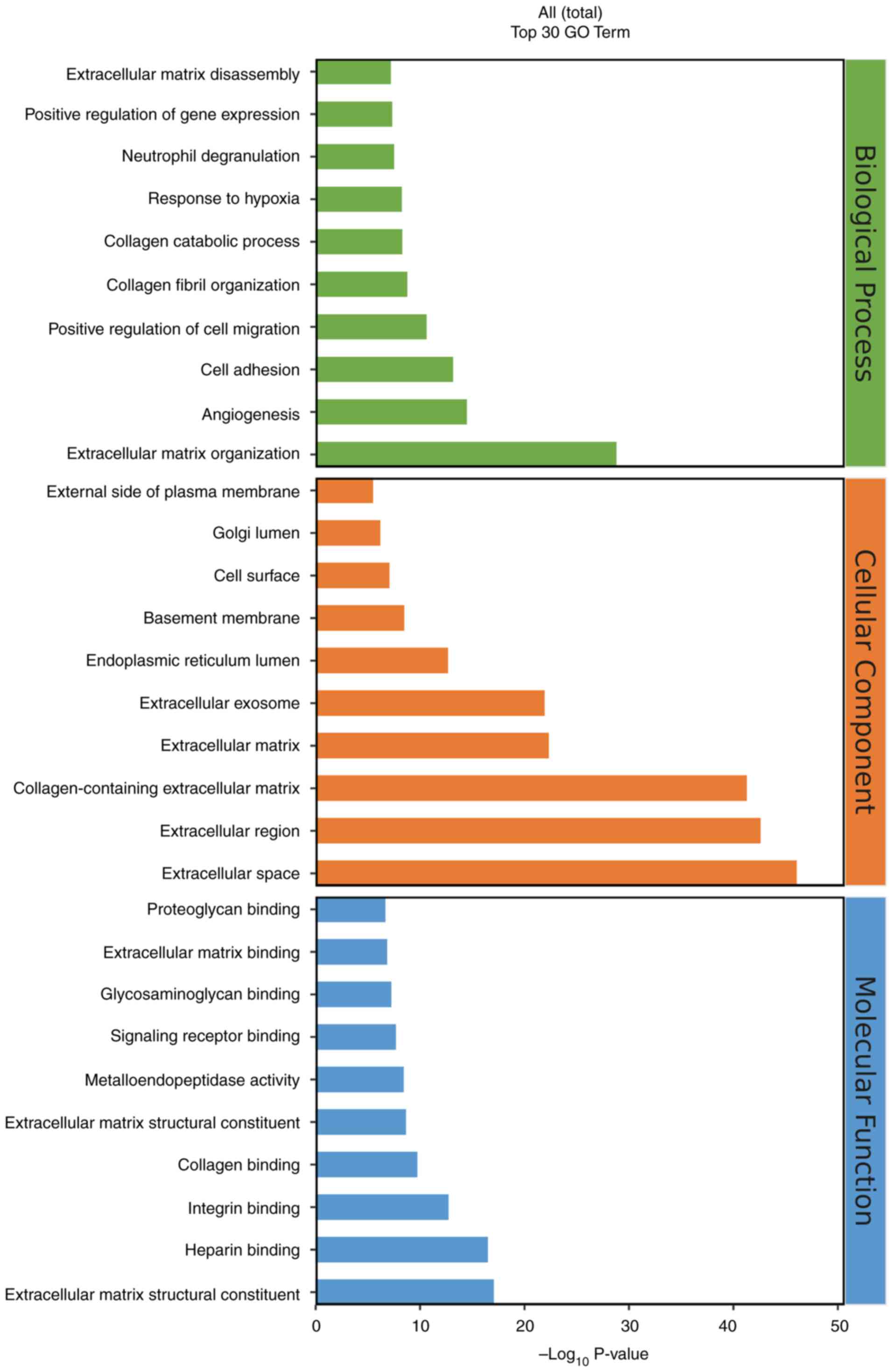
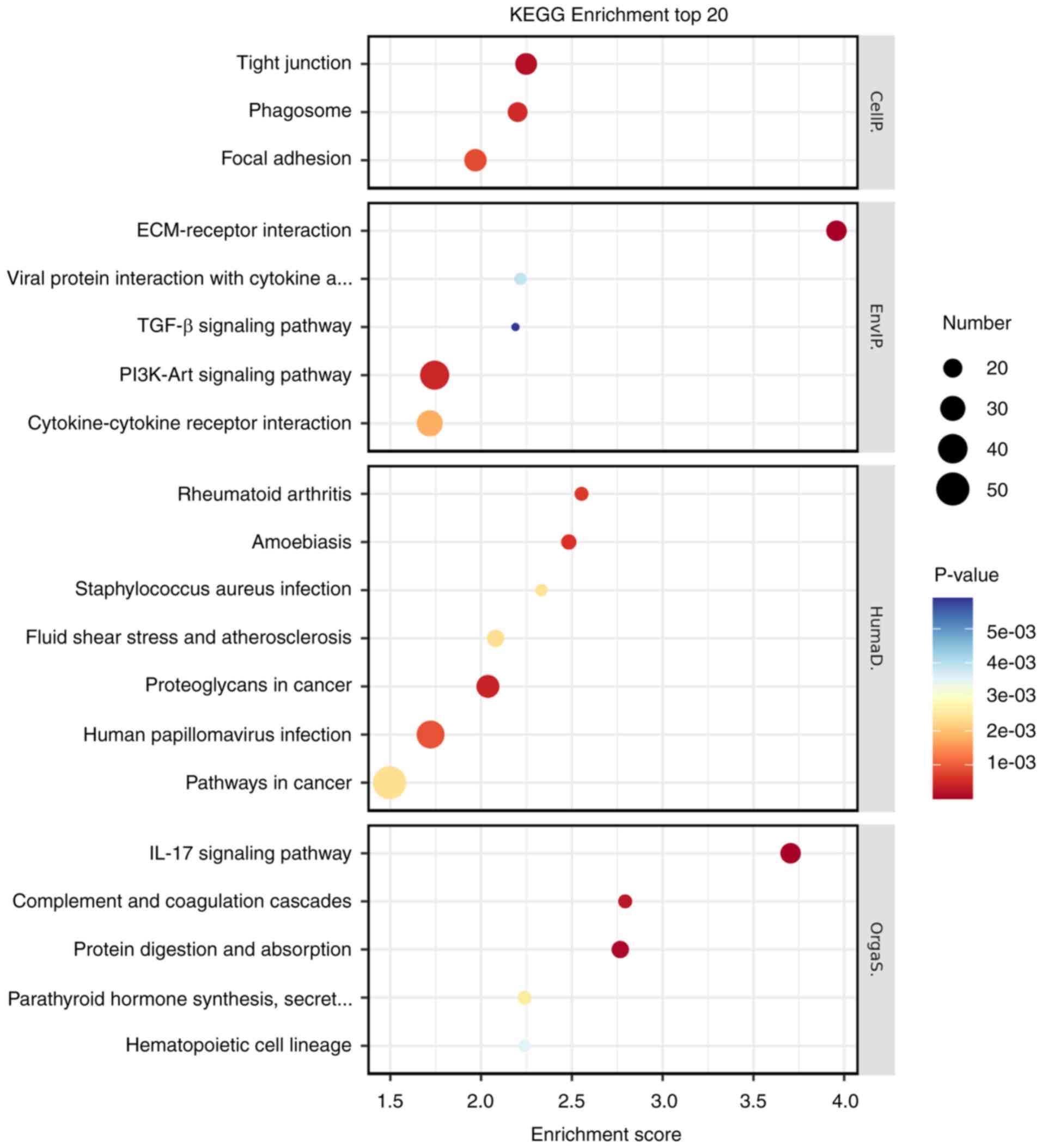
Enrichment analysis of differential
genes
After screening the 900 DEGs, an analysis was
conducted on their GO functions and KEGG pathways using a database,
before the results were visualized with the R language-based
oebiotech online tool. The examination revealed that the DEGs
primarily participate in biological activities such as ‘response to
hypoxia’, ‘angiogenesis’ and ‘cell adhesion’ (Fig. 6). Additionally, they served a role
in various signaling pathways, including TGF-β, PI3K-Akt and IL-17
(Fig. 7).
PPI network
A total of 1,104 DEGs were used to create the PPI
network. From this network, 10 genes were identified as being
central to the network, involved in various biological processes
such as angiogenesis, protein modification, intracellular signal
transduction, cell biosynthetic processes, catabolism regulation,
organ morphogenesis and movement regulation (Fig. 8).
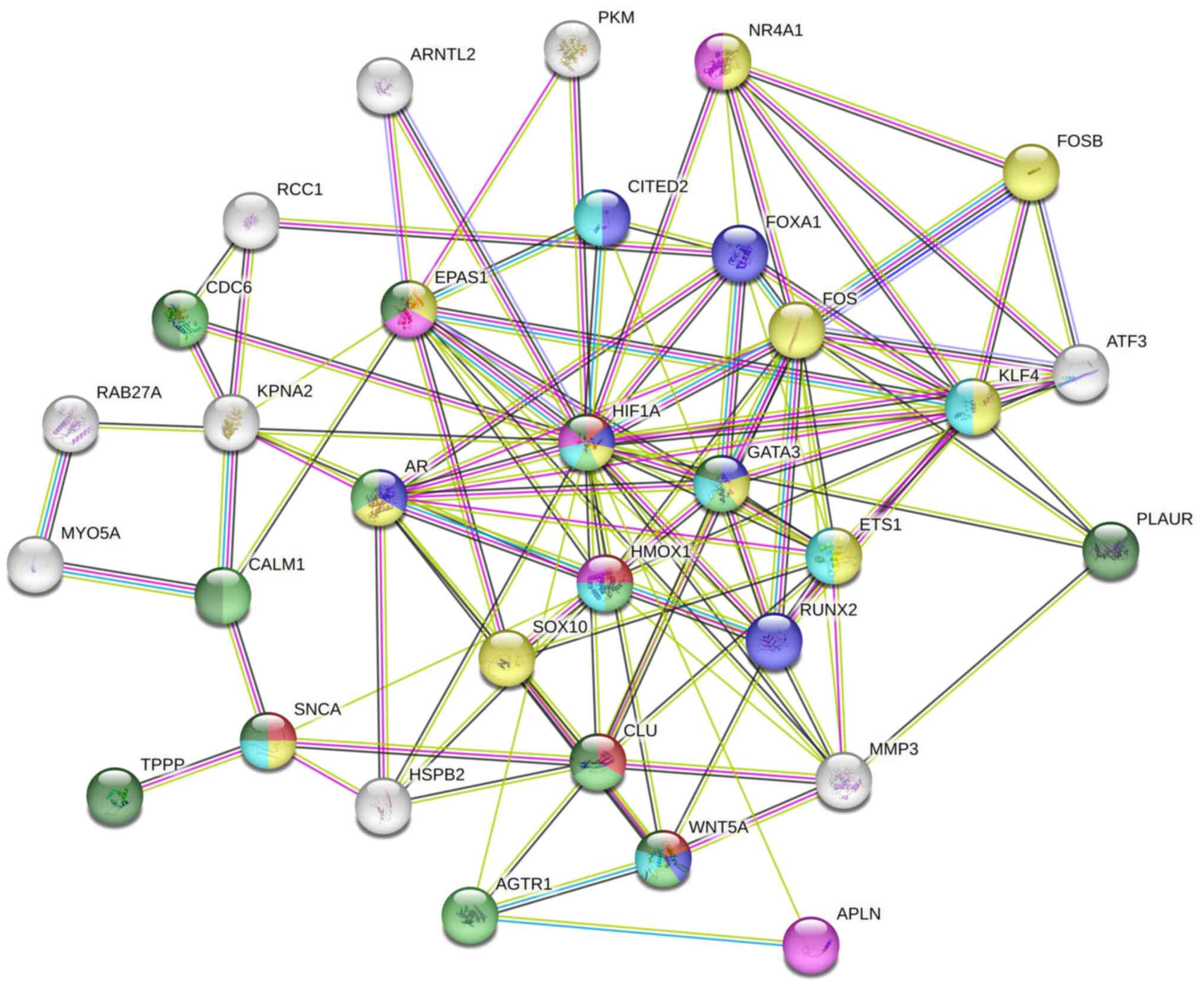 | Figure 8PPI network analysis diagram. Each dot
symbolizes a gene, including 10 core genes (HIF1A,
GATA3, HMOX1, ETS1, AR, CLU,
RUNX2, AR, KLF and CDC6) situated
centrally within the PPI network. PPI, protein-protein
interaction. |
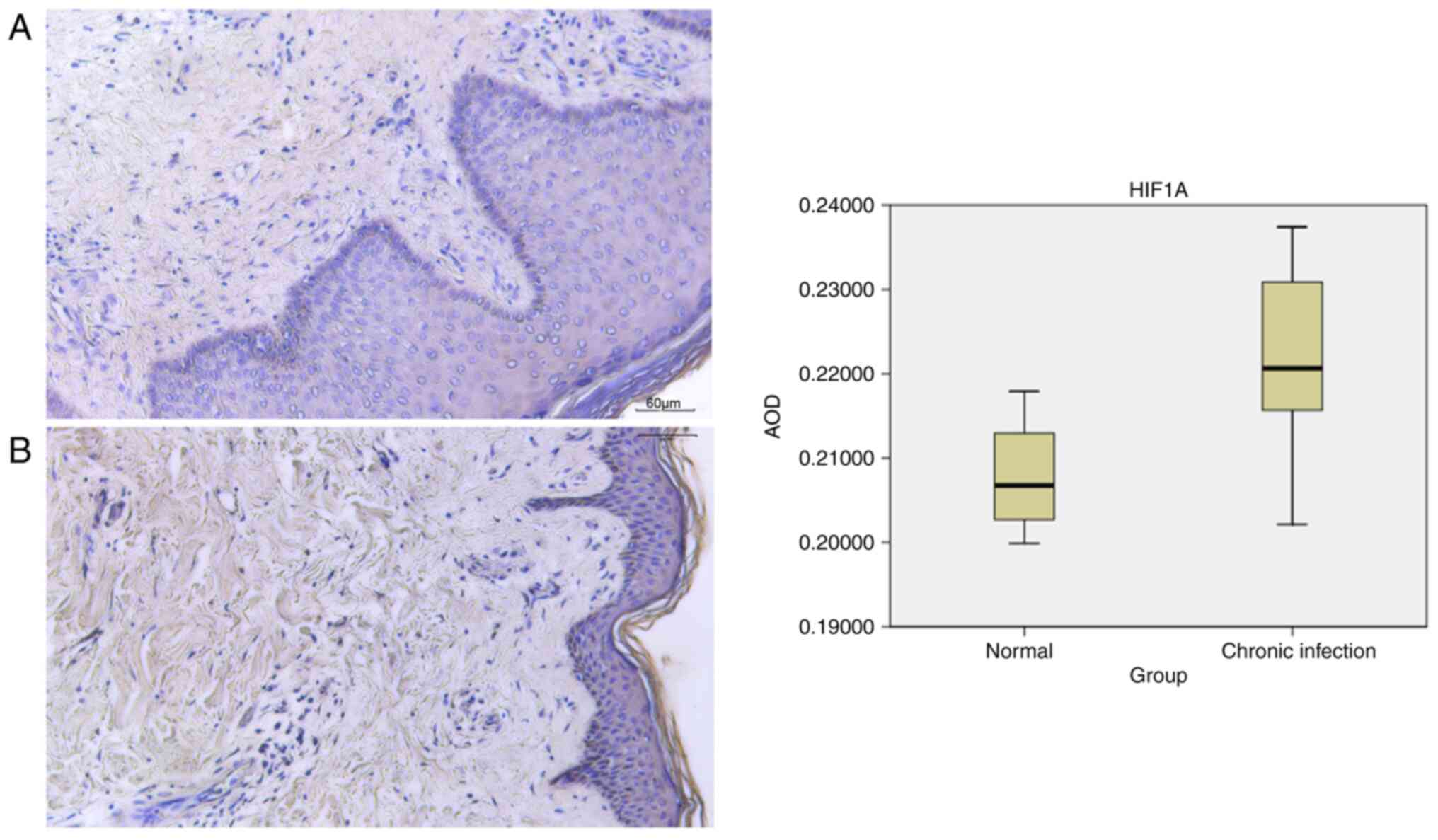
Immunohistochemistry
Healthy skin tissues exhibit distinct layers, such
as the epidermis, containing the stratum corneum, stratum lucidum,
stratum granulosum, stratum spinosum and basal cell sublayers, in
addition to the dermis with its papillary and reticular layers.
Immunohistochemistry was conducted on samples obtained from four
individuals in the healthy skin group and seven individuals in the
chronic skin infection group to assess the presence of HIF1A
expression. In the chronic infection group, observations revealed
thickening of the epidermal cuticle, destruction of the basal
layer, proliferation of fibrous tissue and disorganized arrangement
of muscle fibers. Analysis indicated the expression of HIF1A in the
dermis. Semi-quantitative analysis of HIF1A was performed using
ImageJ (version 1.8.0_172) software (National Institutes of Health)
to determine the AOD value as a measure of HIF1A expression. The
mean ± standard deviation was used to describe the experimental
data. The healthy group had an AOD value of 0.207828±0.007539,
whilst the chronic infection group had a value of
0.221970±0.009962. The data from the experiment followed a normal
distribution. A comparison between groups was conducted using an
unpaired t-test, revealing a statistical difference (P<0.05;
Fig. 9). These findings suggest
that HIF1A was highly expressed in the chronic infection group.
Discussion
Chronic infections in the skin typically exhibit
persistent inflammation, posing challenges for effective healing
(16). Hypoxia has been previously
identified as a contributing factor to the aggravation of
chronically infected skin tissues (17). The body response to hypoxia
involves the stimulation of angiogenesis, increasing blood flow and
enhanced oxygenation (18). A
prior study revealed that in low-oxygen conditions, endothelial
cells are key in starting angiogenesis by accumulating HIF1A to
address hypoxia (19). Hypoxia
serves as the primary stimulus for the upregulation of HIF1A
expression, which in turn serves a crucial role in orchestrating
the cellular response to hypoxia (20,21).
The expression of HIF1A is typically elevated under hypoxic
conditions, where it is involved in various processes, such as
angiogenesis and cell metabolism (18).
For the present study, specimens were collected from
individuals with diabetic skin infection, traumatic skin infection
and other chronic skin infections for analysis using
immunohistochemistry, RNA-seq and bioinformatics techniques.
Immunohistochemical analysis revealed the increased expression of
HIF1A in chronically infected skin tissues. Furthermore, RNA-seq
identified upregulated expression of the HIF1A gene in blood
samples from individuals with chronic skin infections.
Bioinformatics analysis revealed that the DEGs primarily implicated
in the PI3K-Akt, TGF-β and IL-17 signaling pathways.
Skin infections in patients with diabetes present a
challenge in wound healing due to the effects of high blood sugar
on the microvascular basement membrane and endothelial cells,
resulting in chronic ischemia and hypoxia of the tissue (22). Additionally, the formation of
chronic infection wounds in the skin tissue can lead to
vascularization disorders and hindered epithelialization (23). Fridoni et al (24) previously reported an increase in
HIF1A expression in the infected wound tissue of diabetic rats.
This phenomenon may be attributed to the significant enhancement of
HIF1A expression by human bone marrow cell MSC-EV, leading to the
transformation of macrophages from the M1 to the M2 phenotype. Guo
et al (25) and Jiang et
al (26) previously
demonstrated that increased HIF1A expression can facilitate skin
wound vascularization and accelerate the healing process of
diabetic foot ulcers by upregulating VEGF expression, suppressing
inflammation, promoting angiogenesis, and facilitating skin ulcer
healing. In a study by Liu et al (27), it was observed that HIF1A exhibited
decreased expression levels in skin tissues affected by diabetic
wounds. Similarly, Liu et al (27) reported a significant reduction in
HIF1A expression in biopsy samples from individuals with diabetic
foot infections. These findings indicate a potential association
between diminished HIF1A levels and inadequate angiogenic factors,
suggesting that reduced HIF1A expression can serve a pivotal role
in the delayed healing of wounds (28).
Angiogenesis serves an important role in the repair
of chronically infected skin wounds, by facilitating the formation
of new blood vessels that supply oxygen and nutrients necessary for
tissue regeneration (29). The
PI3K/AKT cell signaling pathway, known for its involvement in
various cellular functions, such as proliferation, autophagy,
senescence and apoptosis, may be activated by the hypoxia-induced
upregulation of HIF1A during this process (30-32).
It has been previously demonstrated that activation of the
PI3K/AKT/mTOR pathway can enhance VEGF expression by upregulating
HIF1A to promote angiogenesis (33). It is becoming accepted that VEGF is
a key angiogenic factor among the various well-known pro-angiogenic
factors (34). Activation of the
PI3K/AKT signaling pathway has been shown to result in the
upregulation of HIF1A and increased VEGF expression, subsequently
facilitating the development of new blood vessels. Inhibiting the
VEGF signaling pathway is hypothesized to suppress angiogenesis
(35). The present study used
RNA-seq and bioinformatics techniques, followed by enrichment
analysis of the identified DEGs, to reveal the enrichment of genes
in the PI3K/AKT signaling pathway.
Previous studies have demonstrated that the
HIF1A/VEGF signaling pathway can serve a crucial role in mediating
the skin wound healing process by regulating a number of key
biological processes, including inflammation, angiogenesis,
fibroblast proliferation and re-epithelialization (36). The present study also found that
HIF1A expression was different between the chronic infected skin
and healthy skin. Gene detection and immunohistochemistry analysis
in the chronic infection group revealed the high expression of
HIF1A, suggesting its potential role as a pivotal gene in the
repair process of chronically infected skin tissue. Furthermore,
functional enrichment analysis of DEGs in the chronic infection
group compared with the healthy group suggests that HIF1A may be
associated with the PI3K/AKT signaling pathway, which can
potentially contribute to the healing process of chronic infected
skin wounds through a yet unidentified mechanism.
Nevertheless, it is important to acknowledge the
limitations of the present study. The small sample size may have
restricted the ability to fully explore the phenomenon and uncover
potential mechanisms. Additionally, the present study did not
definitively establish HIF1A as the target gene for chronic
infected skin wound repair, where its underlying mechanism remains
unclear. Future studies may benefit from expanding the sample size.
It is also imperative to investigate the expression levels of HIF1A
and pathways associated with angiogenesis and downstream cytokines
in tissues of chronic skin infection wounds. The present study
research suggested that HIF1A may serve a role in the healing of
chronic infected skin wounds by modulating various processes, such
as inflammation and angiogenesis. Additionally, the present study
identified a potential association between HIF1A and the PI3K/AKT
signaling pathway. Future studies may involve the detection of
inflammatory factors, vascular markers and other relevant
molecules.
To conclude, the present study identified a
significant upregulation of HIF1A in chronically infected skin
tissue, suggesting its potential role as a pivotal gene in the
reparative mechanisms associated with chronic skin infections.
Furthermore, HIF1A may be involved in the PI3K/AKT signaling
pathway. These findings offer a foundation for future
investigations into potential biological targets for the treatment
of chronic skin infections.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Southwest
Medical University (grant no. 0903-00031431).
Availability of data and materials
The data generated in the present study may be found
in the China National GeneBank DataBase under accession number
(CNP0004833) or at the following URL: http://db.cngb.org/cnsa/project/CNP0004833_09297f74/reviewlink/.
Other data generated in the present study may be requested from the
corresponding author.
Authors' contributions
YCH and WZ designed the study. HYC, WX, MZ, LH and
YX performed the experiments. HYC and WX acquired and analyzed the
data. MZ applied for clinical ethics approval and obtained the
clinical samples. LH and YX obtained the clinical samples. HYC
wrote and revised the manuscript. YCH and WZ confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
All methods were conducted in compliance with all
applicable rules and regulations. The peripheral blood and skin
tissue samples were acquired from patients who were hospitalized in
the Affiliated Hospital of Southwest Medical University (Luzhou,
China). All experiments were authorized by the hospital's ethics
committee (approval no. KY2022206) and conformed to the Declaration
Helsinki guidelines. Written informed consent was obtained from all
subjects.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jull AB, Cullum N, Dumville JC, Westby MJ,
Deshpande S and Walker N: Honey as a topical treatment for wounds.
Cochrane Database Syst Rev. 2015(CD005083)2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wen Q, Liu D, Wang X, Zhang Y, Fang S, Qiu
X and Chen Q: A systematic review of ozone therapy for treating
chronically refractory wounds and ulcers. Int Wound J. 19:853–870.
2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wenger RH, Stiehl DP and Camenisch G:
Integration of oxygen signaling at the consensus HRE. Sci STKE.
2005(re12)2005.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Semenza GL: Oxygen-dependent regulation of
mitochondrial respiration by hypoxia-inducible factor 1. Biochem J.
405:1–9. 2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Iyer NV, Kotch LE, Agani F, Leung SW,
Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY
and Semenza GL: Cellular and developmental control of O2
homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev.
12:149–162. 1998.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Hrdlickova R, Toloue M and Tian B: RNA-Seq
methods for transcriptome analysis. Wiley Interdiscip Rev RNA.
8(10.1002/wrna.1364)2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Shi H, Zhou Y, Jia E, Pan M, Bai Y and Ge
Q: Bias in RNA-seq library preparation: Current challenges and
solutions. Biomed Res Int. 2021(6647597)2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Andrews TS, Kiselev VY, McCarthy D and
Hemberg M: Tutorial: Guidelines for the computational analysis of
single-cell RNA sequencing data. Nat Protoc. 16:1–9.
2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Lafzi A, Moutinho C, Picelli S and Heyn H:
Tutorial: Guidelines for the experimental design of single-cell RNA
sequencing studies. Nat Protoc. 13:2742–2757. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ge SX, Son EW and Yao R: iDEP: An
integrated web application for differential expression and pathway
analysis of RNA-Seq data. BMC Bioinformatics.
19(534)2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140.
2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15(550)2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ge SX, Jung D and Yao R: ShinyGO: A
graphical gene-set enrichment tool for animals and plants.
Bioinformatics. 36:2628–2629. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ge X: iDEP Web application for RNA-Seq
data analysis. Methods Mol Biol. 2284:417–443. 2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Szklarczyk D, Gable AL, Nastou KC, Lyon D,
Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, et al:
Correction to ‘The STRING database in 2021: Customizable
protein-protein networks, and functional characterization of
user-uploaded gene/measurement sets’. Nucleic Acids Res.
49(10800)2021.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Martin P: Wound healing-aiming for perfect
skin regeneration. Science. 276:75–81. 1997.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tandara AA and Mustoe TA: Oxygen in wound
healing-more than a nutrient. World J Surg. 28:294–300.
2004.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Fong GH: Mechanisms of adaptive
angiogenesis to tissue hypoxia. Angiogenesis. 11:121–140.
2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Cash TP, Pan Y and Simon MC: Reactive
oxygen species and cellular oxygen sensing. Free Radic Biol Med.
43:1219–1225. 2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Pawar KB, Desai S, Bhonde RR, Bhole RP and
Deshmukh AA: Wound with diabetes: Present scenario and future. Curr
Diabetes Rev. 17:136–142. 2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Salazar JJ, Ennis WJ and Koh TJ: Diabetes
medications: Impact on inflammation and wound healing. J Diabetes
Complications. 30:746–752. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Thangarajah H, Yao D, Chang EI, Shi Y,
Jazayeri L, Vial IN, Galiano RD, Du XL, Grogan R, Galvez MG, et al:
The molecular basis for impaired hypoxia-induced VEGF expression in
diabetic tissues. Proc Natl Acad Sci USA. 106:13505–13510.
2009.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang X, Li R and Zhao H: Enhancing
angiogenesis: Innovative drug delivery systems to facilitate
diabetic wound healing. Biomed Pharmacother.
170(116035)2024.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Fridoni M, Kouhkheil R, Abdollhifar MA,
Amini A, Ghatrehsamani M, Ghoreishi SK, Chien S, Bayat S and Bayat
M: Improvement in infected wound healing in type 1 diabetic rat by
the synergistic effect of photobiomodulation therapy and
conditioned medium. J Cell Biochem. 120:9906–9916. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Guo J, Hu Z, Yan F, Lei S, Li T, Li X, Xu
C, Sun B, Pan C and Chen L: Angelica dahurica promoted angiogenesis
and accelerated wound healing in db/db mice via the HIF-1α/PDGF-β
signaling pathway. Free Radic Biol Med. 160:447–457.
2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Jiang W, Zhang J, Zhang X, Fan C and Huang
J: VAP-PLGA microspheres (VAP-PLGA) promote adipose-derived stem
cells (ADSCs)-induced wound healing in chronic skin ulcers in mice
via PI3K/Akt/HIF-1α pathway. Bioengineered. 12:10264–10284.
2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Liu W, Yuan Y and Liu D: Extracellular
vesicles from adipose-derived stem cells promote diabetic wound
healing via the PI3K-AKT-mTOR-HIF-1α signaling pathway. Tissue Eng
Regen Med. 18:1035–1044. 2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Veith AP, Henderson K, Spencer A, Sligar
AD and Baker AB: Therapeutic strategies for enhancing angiogenesis
in wound healing. Adv Drug Deliv Rev. 146:97–125. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Aoki M and Fujishita T: Oncogenic roles of
the PI3K/AKT/mTOR axis. Curr Top Microbiol Immunol. 407:153–189.
2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Han J, Huang C, Jiang J and Jiang D:
Activation of autophagy during farnesyl pyrophosphate synthase
inhibition is mediated through PI3K/AKT/mTOR signaling. J Int Med
Res. 48(300060519875371)2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Rai SN, Dilnashin H, Birla H, Singh SS,
Zahra W, Rathore AS, Singh BK and Singh SP: The role of PI3K/Akt
and ERK in neurodegenerative disorders. Neurotox Res. 35:775–795.
2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zubair M and Ahmad J: Role of growth
factors and cytokines in diabetic foot ulcer healing: A detailed
review. Rev Endocr Metab Disord. 20:207–217. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhu Y, Wang Y, Jia Y, Xu J and Chai Y:
Roxadustat promotes angiogenesis through HIF-1α/VEGF/VEGFR2
signaling and accelerates cutaneous wound healing in diabetic rats.
Wound Repair Regen. 27:324–334. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Hart PH and Norval M: Ultraviolet
radiation-induced immunosuppression and its relevance for skin
carcinogenesis. Photochem Photobiol Sci. 17:1872–1884.
2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Amin KN, Umapathy D, Anandharaj A,
Ravichandran J, Sasikumar CS, Chandra SKR, Kesavan R and Mohanram
RK: miR-23c regulates wound healing by targeting stromal
cell-derived factor-1α (SDF-1α/CXCL12) among patients with diabetic
foot ulcer. Microvas Res. 127(103924)2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wang Y, Zhu J, Chen J, Xu R, Groth T, Wan
H and Zhou G: The signaling pathways induced by exosomes in
promoting diabetic wound healing: A mini-review. Curr Issues Mol
Biol. 44:4960–4976. 2022.PubMed/NCBI View Article : Google Scholar
|