Introduction
Multiple myeloma (MM) is one of the most common
malignant tumours in the blood system and is characterized by the
clonal proliferation of malignant plasma cells in the bone marrow
and symptoms related to anaemia, immunosuppression, bone
destruction and renal failure (1).
In recent decades, treatment strategies have greatly advanced, but
MM remains an incurable malignant disease. Considerable evidence
has indicated that epigenetic mechanisms are generally involved in
the pathogenesis of MM, and these mechanisms can regulate gene
expression at the level of DNA and chromatin structural
modifications, RNA stability, and transcriptional activity; related
modification types include RNA modifications, DNA methylations,
histone covalent modifications, and non-coding RNA regulation
(2-4).
Furthermore, epitranscriptomics is a newly emerging field that
focuses mainly on the effects of chemical modifications carried by
RNAs and their associated regulators on gene expression (5).
An increasing number of studies have revealed the
critical roles of N1-methyladenosine (m1A) modification
and its regulators in cancer development, and strategies involving
m1A modification and m1A-related regulators have received attention
(6,7). m1A, which appears on the first
nitrogen atom of adenosine in RNA, is highly enriched in the 5'UTR
and tends to be located in highly structured regions (8). Similar to DNA and protein
modifications, m1A modification is a type of dynamic reversible
process (9). The process of m1A
methylation is catalysed by methyltransferases (‘writers’)
consisting of tRNA methyltransferase 10 homolog C (TRMT10C), tRNA
methyltransferase 61B (TRMT61B), tRNA methyltransferase 6
non-catalytic subunit (TRMT6) and tRNA methyltransferase 61A
(TRMT61A), whereas the m1A removal process is mediated by
demethylases, including Alkb homolog 1, histone H2A dioxygenase
(ALKBH1) and AlkB homolog 3, α-ketoglutarate dependent dioxygenase
(ALKBH3) (‘erasers’) (10). In
addition, a group of specific RNA-binding proteins composed of YTH
domain-containing family protein (YTHD)F1/F2/F3/C1 (‘readers’) can
recognize the m1A motif, thus affecting m1A functions (11). The levels of proteins involved in
m1A modification are generally greater in gastrointestinal cancer
cells than in normal cells (12).
The reader protein YTHDF2, which is highly expressed in MM, can
promote myeloma cell proliferation through
EGR1/p21cip1/waf1/CDK2-cyclin E1 axis-mediated cell
cycle transition (13).
Methyltransferase 3, N6-adenosine-methyltransferase
complex catalytic subunit, as a writer, facilitates multiple
myeloma tumorigenesis by enhancing YY1 stability and
pri-microRNA-27 maturation (14).
TRMT6/61A-dependent base methylation of tRNA-derived fragments
regulates tRF-3 silencing activity and the unfolded protein
response to promote bladder cancer (15). In addition, cooperation among
different methylation molecules has been gradually revealed. For
example, AlkB homolog 5, RNA demethylase can regulate STAT3
activity to affect the proliferation and tumorigenicity of
osteosarcoma in a YTHDF2-dependent manner (16). FTO α-ketoglutarate dependent
dioxygenase also acts as a demethylase to promote MM cell
proliferation, migration and invasion in a YTHDF2-dependent manner
by targeting heat shock transcription factor 1/heat shock proteins
(17). However, only a small
number of studies have revealed the involvement of m1A-related
genes in the oncogenesis and progression of MM (18). Research focused on the systematic
understanding of the role of m1A in MM is warranted.
In the present study, gene expression data and
clinical information from the Gene Expression Omnibus (GEO)
database were used to explore differentially expressed and
prognosis-related m1A regulators, and preliminary verification was
carried out.
Materials and methods
MM dataset source and
preprocessing
Public gene expression data and full clinical
annotations were searched in the GEO database (https://www.ncbi.nlm.nih.gov/geo/). In total,
three eligible MM cohorts (GSE13591, GSE47552 and GSE24080) were
included (19-21).
Specifically, 133 patients with MM and 4 healthy volunteers in
GSE13591 and 41 patients with MM and 5 healthy volunteers in
GSE47552 were selected for differential gene screening and
subsequent analysis. The data were analyzed with the R (v.4.1.2;
http://www.R-project.org/) software and R
Bioconductor package (https://bioconductor.org/biocLite.R).
Analysis of m1A regulator gene
expression
The mRNA expression of m1A regulators in normal
plasma and MM bone marrow plasma cells was analyzed based on
samples from GSE13591 and GSE47552. Another dataset (GSE24080) with
prolific clinical information was subsequently used to elucidate
alterations in m1A-related regulatory genes in MM samples. The data
were analyzed with the limma R package (http://bioinf.wehi.edu.au/limma).
Unsupervised clustering for 10 m1A
regulators
Unsupervised clustering analysis was applied to
identify distinct m1A modification patterns based on the expression
of m1A regulators, and patients were classified for further
analysis. The number of clusters and their stability are determined
by the consensus clustering algorithm (22). The Consensus ClusterPlus package
(https://bioconductor.org/packages/ConsensusClusterPlus/)
was used to perform the aforementioned steps.
Single-sample gene set enrichment
analysis (ssGSEA)
The infiltration levels of the different immune cell
populations were determined via ssGSEA via the gene set variation
analysis (GSVA) R Bioconductor package ((https://github.com/rcastelo/GSVA) with default
parameters. The ssGSEA algorithm is a rank-based method that
defines a score representing the degree of absolute enrichment of a
particular gene set in each sample. The enrichment scores were used
to represent the relative abundance of each infiltrating immune
cell in each sample.
GSVA and functional annotation
GSVA is a non-parametric and unsupervised method
that can easily adapt to the analysis of RNA-seq data (23). GSVA enrichment analysis was
performed to investigate the differences in biological processes
associated with m1A modification patterns via the GSVA R package.
The gene sets of ‘c2.cp.kegg.v6.2.-symbols’ were downloaded from
the MSigDB database (https://www.gsea-msigdb.org/gsea/msigdb) for GSVA.
Adjusted P-values of <0.05 were considered statistically
significant. The clusterProfiler R package (https://yulab-smu.top/contribution-knowledge-mining/)
was used to perform functional annotation for m1A-related genes,
with cut-off values of |log fold change (FC)| >0.1 and an
adjusted P-value of <0.05.
Generation of an m1A gene
signature
An m1A gene signature was constructed to quantify
the m1A modification pattern. Information on the genes in the
signature was used to calculate the m1A score for each MM patient.
To identify signature genes, overlapping genes from differentially
expressed genes (DEGs) identified in different m1A clusters were
first extracted, and the significance criterion was set as an
adjusted P-value of <0.001. Unsupervised clustering analysis of
these overlapping genes was subsequently performed. The consensus
clustering algorithm was used to determine the number and stability
of gene clusters, and the patients were divided into three groups
for further analysis. Next, univariate regression analysis was used
to analyze the prognosis of each overlapping gene and the genes
with significant prognostic value were extracted for further
analysis. Principal component (PC)1 and PC2 were selected as the
gene signature variables, and PC analysis (PCA) was used to
construct a scoring model. The m1Ascore was then defined using a
method similar to the gene expression grade index (24,25):
m1Ascore = Σ
(PC1i+PC2i)
where i is the expression of m1A phenotype-related
genes. The feature importance was assessed by the cos2 value and
the contributions of the variables.
Validation of the gene signature
The ability of the m1A score to differentiate
between patients with MM and controls was assessed in four
validation cohorts, GSE24870, GSE27838, GSE46053 and GSE113295
(26-29),
via receiver operating characteristic (ROC) curve analysis. The
role of m1A regulatory factors in the pathogenesis of MM was also
verified via the same method. Visualization of the results was
performed via the pROC package (https://xrobin.github.io/pROC/).
Immunohistochemical (IHC)
staining
Immunohistochemistry was conducted on bone marrow
tissue sections as previously described (30). Bone marrow biopsy sections were
obtained from 25 patients with MM at the Department of Oncology and
Hematology of the Second Affiliated Hospital of Shandong University
of Traditional Chinese Medicine (Jinan, China) and the Department
of Hematology of Affiliated Hospital of Shandong University of
Traditional Chinese Medicine (Jinan, China) from July 2020 to July
2023. The sections were fixed with formalin, embedded in paraffin,
and cut into 4-µm thick sections. The sections were then incubated
with 3% H2O2 formaldehyde at room temperature
for 10 min, subjected to antigen retrieval in a microwave, cooled
at room temperature, and incubated with 10% sheep serum for 10 min.
The sections were subsequently incubated overnight at 4˚C with a
primary YTHDF2 monoclonal antibody (1:2,000; cat. no. EPR23544-19;
Abcam). Biotinylated secondary antibodies (1:2,000; cat. no.
ab97080; Abcam) were labelled with streptavidin peroxidase
solution, added to the slides, and incubated for 30 min at 37˚C.
Finally, the samples were stained (room temperature, 3 min) with a
Simple DAB Stain Kit (Abcam) and counterstained with haematoxylin
(room temperature, 4 min) according to the manufacturer's
instructions. Finally, images were captured directly by optical
microscopy (x200) (Leica DM2700 M; Lecia Microsystems GmbH) and
analyzed by Image-Pro (Version 7.0; Media Cybernetics, Inc.). All
patients provided written informed consent and the protocols were
approved from the Ethics Committee of the Second Affiliated
Hospital of Shandong University of Traditional Chinese Medicine
(Jinan, China; approval no. 2023 Ethics Review-KY-001) and the
Ethics Committee of the Affiliated Hospital of Shandong University
of Traditional Chinese Medicine (Jinan, China; approval no. 2020
Ethics Review-KY-010).
Cell culture and transfection
The human MM cell line U266 (cat. no. TIB-196;
American Type Culture Collection) was cultured in RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc.) containing 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.) and 1% streptomycin-penicillin
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37˚C in a 5%
humidified CO2 atmosphere. The cells were divided into 5
groups: i) ‘NC’ refers to MM cells without any treatment; ii)
‘si-NC’ refers to MM cells transfected with RNAi that is not
homologous to the YTHDF2 sequence; iii) ‘si-YTHDF2’ refers to MM
cells transfected with YTHDF2 RNAi; iv) ‘oe-NC’ refers to MM cells
transfected with blank plasmid; and v) ‘oe-YTHDF2’ refers to MM
cells transfected with the YTHDF2 overexpression plasmid. YTHDF2
RNAi (si-YTHDF2) and YTHDF2-overexpressing vectors (oe-YTHDF2) were
designed and synthesized by Shandong Jiehelix Biotechnology Co.,
Ltd. The sequences of siRNA, including the negative controls are
presented in Table SI. U266 cells
(1x106 cells/well) were seeded into 6-well plates after
transfection with the vectors (50 nM siRNA or 2 µg plasmid DNA) via
the Lipofectamine™ 3000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). After 72 h of transfection (37˚C, 5%
CO2), reverse transcription-quantitative PCR (RT-qPCR)
was performed to verify the success of the transfection. Total RNA
was extracted using SPARKeasy Improved Cell RNAKi (Shandong Sikejie
Biotechnology Co., Ltd.), and the concentration and purity of
extracted RNA were measured with a NanoDrop 2000 spectrophotometer
(Thermo Fisher Scientific, Inc.). SPARKscript II All-in-one RT
SuperMix for qPCR Kit and VeritiPro PCR instrument (Thermo Fisher
Scientific, Inc.) were used for RT of RNA into cDNA. The RT program
was conducted according to the manufacturer's instructions at 50˚C
for 15 min and 85˚C for 5 sec. SYBR Green PCR Master Mix (Beijing
Solarbio Science & Technology Co., Ltd.) and a Step-One Plus
Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.) was used for qPCR analysis. The qPCR program with temperature
protocol was conducted according to the manufacturer's instructions
as follows: Initial denaturation at 95˚C for 5 min, followed by 40
cycles of denaturation at 95˚C for 30 sec, annealing at 61˚C for 30
sec and extension at 72˚C for 1 min. For the measurement of YTHDF2
mRNA levels, the primers used were as follows: Forward,
5'-GCAAGCAATGTTCCAAAAG-3' and reverse, 5'-GCAATATCAGCCCAAGATG-3'.
The relative mRNA levels were normalized to ACTB, the primers used
for ACTB were as follows: Forward, 5'-TGACGTGGACATCCGCAAAG-3' and
reverse, 5'-CTGGAAGGTGGACAGCGAGG-3'. The relative mRNA expression
levels were calculated using the 2-ΔΔCq method (31).
Cell viability assay
Cell viability was determined via the use of Cell
Counting Kit-8 (CCK-8) reagents (Beyotime Institute of
Biotechnology, Shanghai, China) according to the manufacturer's
instructions. Briefly, U266 cells were seeded onto 96-well plates
(1,000 cells/well) in the presence of medium containing 10% FBS and
penicillin-streptomycin (5,000 U/ml). After 24 h of culture at 37˚C
in a 5% humidified CO2 atmosphere, CCK-8 reagent (cat.
no. CT0001-B; Shandong Sikejie Biotechnology Co., Ltd.) was added,
and the mixture was incubated for 2 h. The absorption at 450 nm was
determined with a microplate reader (Tecan Group, Ltd.).
Cell apoptosis assay
The samples were analyzed with a FACSCalibur flow
cytometer (BD Biosciences) equipped with an argon laser. A total of
10,000 events were collected for each sample. U266 cells were
collected and counted, with the cell concentration adjusted to
1x106 cells/ml, before being resuspended in a cell
suspension containing annexin V-PE and 7-AAD (1:100; Dalian Meilun
Biology Technology Co., Ltd.). Apoptosis was determined according
to the number of Annexin V-PE-positive cells, whereas necrosis was
determined according to the 7-AAD staining rate via CellQuest
software (version 5.1; BD Biosciences).
Total m1A RNA methylation
quantification
Total m1A RNA methylation was quantified as
previously described (32). TRIzol
(Shandong Sikejie Biotechnology Co., Ltd.) was used to extract
total RNA from U266 cells according to the instructions. Total m1A
levels in U266 cells were then measured via the Human m1A ELISA kit
(cat. no. YJ7129203; Shanghai Enzyme-linked Biotechnology Co.,
Ltd.) for m1A RNA methylation. In brief, RNA (200 ng) was added to
each well, followed by antibody capture and detection. Following
incubation, the m1A signal was finally calculated from the
calibration curve.
Bioinformatics analysis
Kyoto Encyclopedia of Genes and Genomes enrichment
analysis was performed via the Metascape platform (https://metascape.org/). ClueGO (http://apps.cytoscape.org/apps/cluego)
was conducted with Cytoscape (version 3.8.2) to explore the
biological processes associated with the genes. Pearson correlation
analysis was performed and visualized via the corrplot (https://github.com/taiyun/corrplot; v.0.92) and
ggplot2 R packages (https://ggplot2.tidyverse.org; v.3.3.5). A correlation
coefficient absolute value of >0.7 and P<0.5 were used as the
standards to screen related genes. The RMbase database (https://rna.sysu.edu.cn/rmbase3) was used to
predict whether YTHDF2 could modify SRSF10 by m1A methylation.
Statistical analysis
The survival curves for the prognostic analysis were
generated via the Kaplan-Meier method, and log-rank tests were used
to identify the significance of differences. A univariate Cox
regression model was adopted to calculate hazard ratios (HRs) for
m1A regulators and m1A phenotype-related genes. The independent
prognostic factors were ascertained through a multivariable Cox
regression model. Patients with detailed clinical data were
eligible for inclusion in the final multivariate prognostic
analysis. The data processing was performed in R (https://www.r-project.org/; v.4.1.2) software. Two-way
ordered categorical variables with different attributes were used
to evaluate the differences in the different ISS stage groups among
the three clusters using Kruskal-Wallis test and Pearson's
correlation analysis. The experimental results were statistically
analyzed using SPSS Statistics (version 27.0: IBM Corp.) and
GraphPad Prism (Version 10.0; GraphPad; Dotmatics). If each group
of data had a normal distribution and homogeneity of variance,
quantitative data were presented as the mean ± standard deviation
(x̄±s). Differences between two groups were analyzed by Student's
t-test, differences among three groups were analyzed by one-way
ANOVA with Least Significant Difference post hoc test, differences
among >3 groups were analyzed by one-way ANOVA and
Kruskal-Wallis test with Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Landscape of genetic variations in m1A
regulators in MM
First, the differential expression of m1A regulators
between normal plasma (NP; n=9) and MM (n=174) bone marrow plasma
cells was summarized. Compared with those in the NP samples, the
expression of the reader genes YTHDF1, YTHDF2 and YTHDF3 and the
writer genes TRMT61A and TRMT61B were upregulated in the MM
samples, whereas the expression of the eraser genes ALKBH1 and
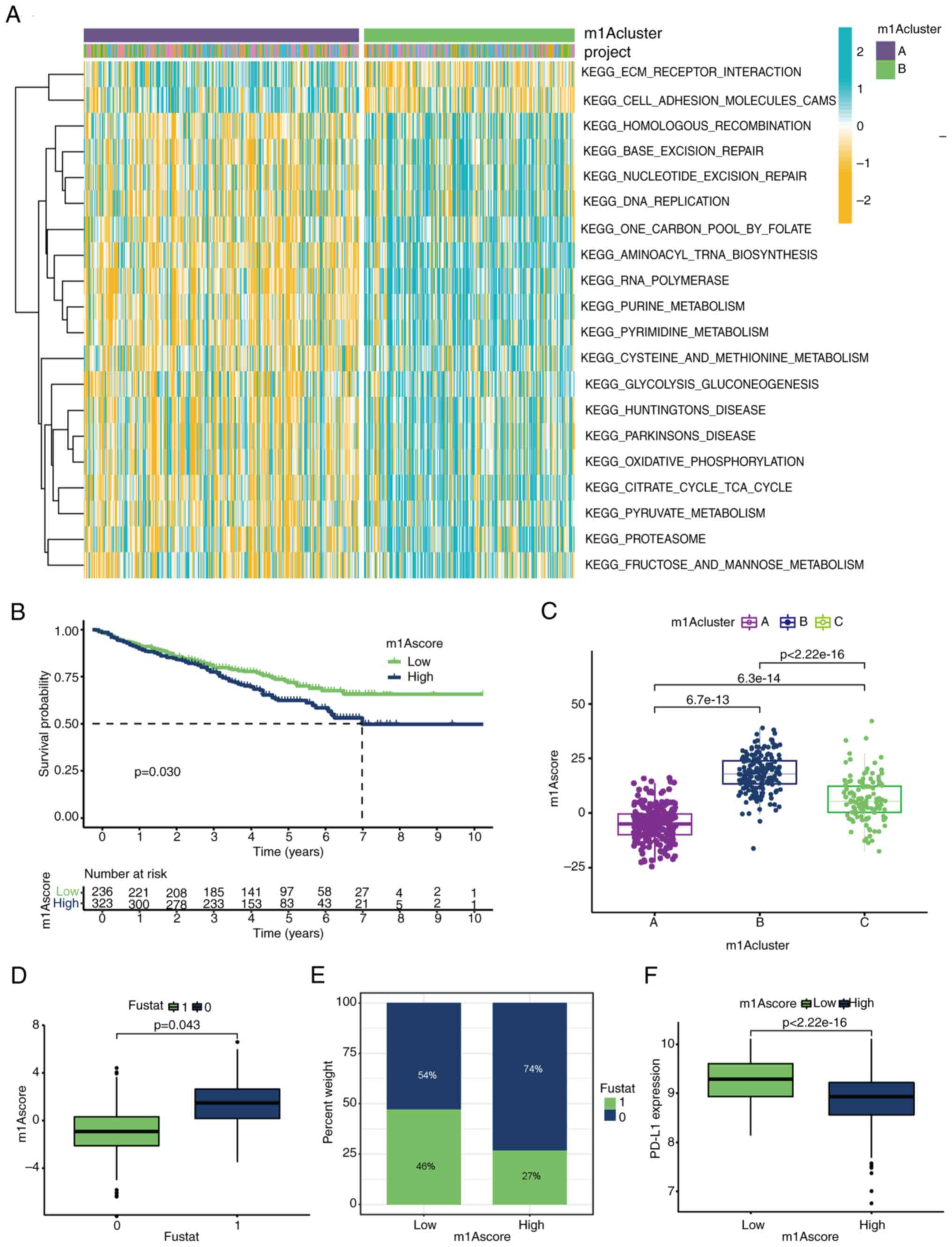
YTHDC1 were significantly downregulated (Fig. 1A). Another GEO dataset (GSE24080;
n=559) with the most comprehensive clinical annotation was
subsequently used to further investigate the prognostic value of
m1A regulators. Multivariate survival analysis was performed via
the Cox proportional hazard model. The results shown in the forest
plot revealed that among the seven differentially expressed m1A
regulators, YTHDF2 and YTHDF3 could be independent prognostic
factors (Fig. 1B). A subsequent
univariate Cox regression model revealed the prognostic value of 10
m1A regulators in patients with MM. The Kaplan-Meier survival curve
revealed six regulators that were significantly associated with
survival. High expression of five regulators (YTHDF1/2/3, TRMT6 and
TRMT61B), especially YTHDF2, was associated with shorter overall
survival, whereas YTHDC1 exhibited the opposite pattern (Fig. 1C-H). In addition, the comprehensive
landscape of m1A regulator interactions, connections and their
prognostic significance in patients with MM was depicted in the m1A
regulator network (Fig. 1I). The
network diagram revealed that YTHDF2 was significantly positively
correlated with the methyltransferase TRMT61B and negatively
correlated with the demethylase ALKBH1. These analyses indicated
that the variable expression of m1A regulators, especially the high
expression of the reader gene YTHDF2, may play a crucial role in MM
occurrence and progression.
 | Figure 1Landscape of genetic variations of
m1A regulators in MM. (A) Alterations of m1A regulatory genes in
patients with MM. (B) The forest map of m1A regulators. High
expression of TRMT10C, TRMT61B, YTHDF2 and YTHDF3 were associated
with poor prognosis in patients with MM. (C-H) High expression of
(C) TRMT6, (D) TRMT61B, (F) YTHDF1, (G) YTHDF2 and (H) YTHDF3 is
associated with poor prognosis in patients with MM. (E) Low
expression of YTHDC1 is associated with a worse prognosis. (I) The
interaction among m1A regulators in MM. The circle size represents
the effect of each regulator on the prognosis, and the range of
values was calculated by the log-rank test. Purple dots in the
circle indicate risk factors for prognosis; green dots in the
circle indicate protective factors for prognosis. The lines linking
regulators showed their interactions, and the thickness shows the
correlation strength between regulators. Negative correlations are
marked with blue, and positive correlations are marked with red.
The regulator erasers, readers and writers are marked with red,
orange and gray, respectively. Differences between the MM group and
Normal group were analyzed by Student's t-test. A univariate Cox
regression model to calculate hazard ratios for m1A regulators and
the independent prognostic factors were ascertained through a
multivariable Cox regression model. The survival curves for the
prognostic analysis were generated via the Kaplan-Meier method, and
log-rank tests were used to identify the significance of
differences. *P<0.1 and **P<0.01. MM,
multiple myeloma; m1A, N1-methyladenosine; TRMT10C, tRNA
methyltransferase 10 homolog C; TRMT61B, tRNA methyltransferase
61B; YTHD, YTH domain-containing family protein; TRMT6, tRNA
methyltransferase 6 non-catalytic subunit; TRMT61A, tRNA
methyltransferase 61A; ALKBH1, Alkb homolog 1, histone H2A
dioxygenase; ALKBH3, AlkB homolog 3, α-ketoglutarate dependent
dioxygenase. |
m1A methylation patterns mediated by
10 regulators
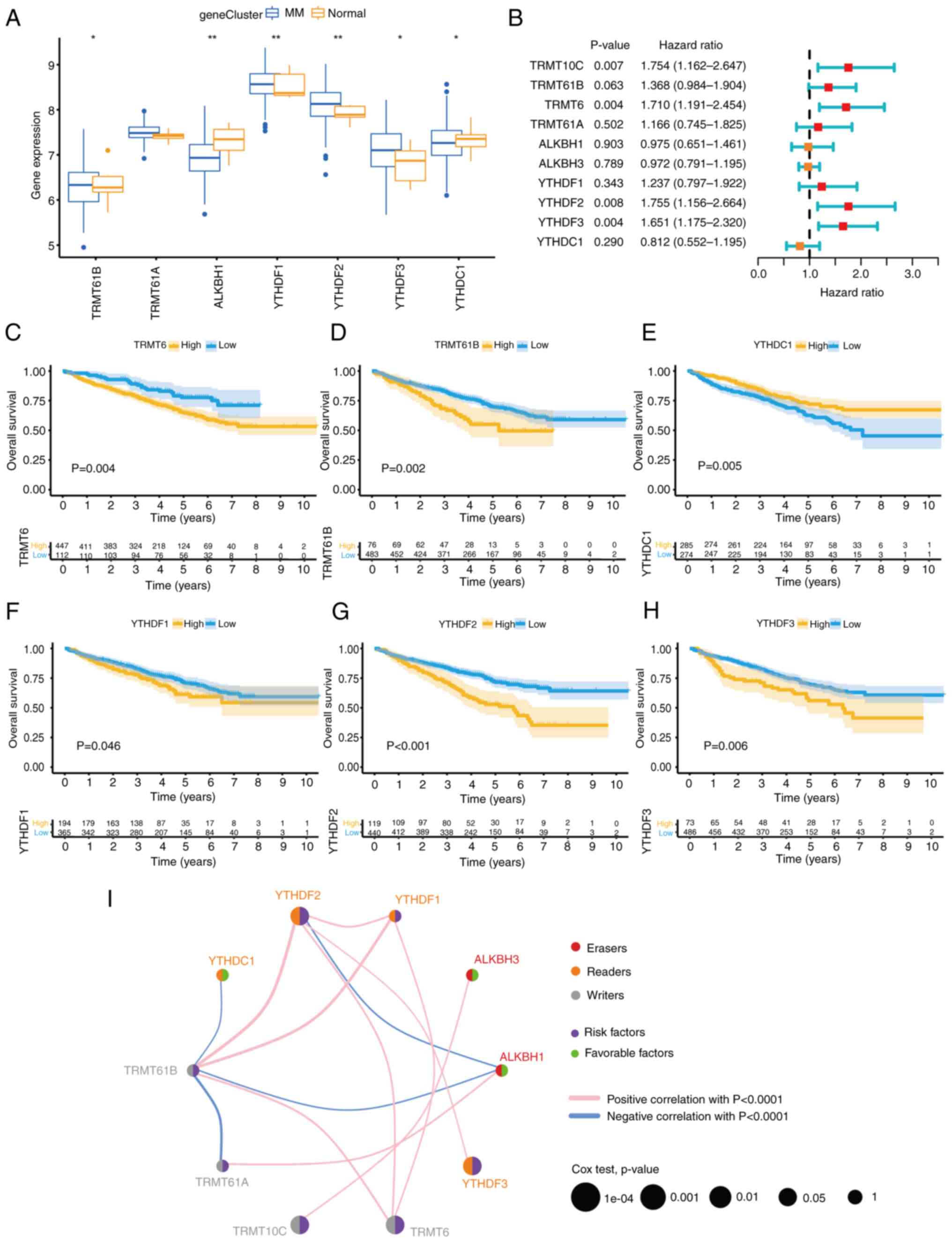
Based on the expression of 10 m1A regulators, three
distinct modification patterns were eventually identified via
unsupervised clustering: A total of 248 patients exhibited pattern
A, 190 patients exhibited pattern B, and 121 patients exhibited
pattern C; these groups were termed Clusters A-C, respectively
(Fig. 2A-D). There was a
significant distinction in the m1A transcriptional profile between
Cluster B and the other two clusters (Fig. 2C). The results of PCA feature
importance selection revealed that YTHDF2, ALKBH3, TRMT10C and
TRMT61B play important roles in clustering differentiation. As
revealed in Fig. 2D, the cos2
value in Fig. 2E was used to
measure the usefulness of a variable. The higher the value is, the
more important the variable is in the PCA. In Fig. 2E, the red dotted line represents
the average contribution, and a value higher than the average value
can be considered an important variable. As demonstrated in
Fig. 2F, Cluster B was
characterized by increased expression of YTHDF2, TRMT10C, TRMT61B,
TRMT6 and ALKBH3. Prognostic analysis of the three m1A modification
subtypes revealed a particular survival disadvantage in Cluster B
compared with Clusters A and C (Fig.
2G). Notably, Cluster C, which had better survival than Cluster
B, presented a significant decrease in the expression of YTHDF2,
indicating that YTHDF2 is likely strongly associated with poor
prognosis in patients with MM. The value of m1A clusters in risk
stratification prediction for MM was also evaluated, but there was
no statistical difference (P=0.057) in the International Staging
System risk score among different clusters (Table SII). As expected, subsequent
analyses of immune cell infiltration indicated that Cluster B had
markedly low levels of infiltration of immune cells, including
natural killer cells, macrophages, mast cells, myeloid-derived
suppressor cells and immature dendritic cells (Fig. 2H).
 | Figure 2m1A methylation modification patterns
mediated by 10 regulators. (A and B) Unsupervised clustering
analysis. The difference between different clusters was the biggest
when k=3. (C) PCA of the transcriptome profiles of the three m1A
modification patterns. (D) Variable representative analysis. The
length from the center point to the variable represents the
proportion of the variable in this dimension. (E) The contribution
of variables to PCs. The red dotted line represents the average
contribution, and higher than the average value can be considered
as an important variable. (F) Unsupervised clustering of 10 m1A
regulators. The m1Acluster, ISS staging system, sex, immunoglobulin
type and age were used as patient annotations. Blue represents high
expression of regulators, and yellow represents low expression. (G)
Survival analyses for the three m1A modification patterns. (H) The
abundance of immune cells in three m1A modification patterns.
Differences among multiple groups were analyzed by one-way ANOVA
with LSD post hoc test. The survival curves for the prognostic
analysis were generated via the Kaplan-Meier method, and log-rank
tests were used to identify the significance of differences.
*P<0.1, **P<0.01 and
***P<0.001. m1A, N1-methyladenosine; PCA,
principal component analysis; PC, principal component; ISS,
International Staging System; ns, no significance; TRMT10C, tRNA
methyltransferase 10 homolog C; TRMT61B, tRNA methyltransferase
61B; YTHD, YTH domain-containing family protein; TRMT6, tRNA
methyltransferase 6 non-catalytic subunit; TRMT61A, tRNA
methyltransferase 61A; ALKBH1, Alkb homolog 1, histone H2A
dioxygenase; ALKBH3, AlkB homolog 3, α-ketoglutarate dependent
dioxygenase. |
To further investigate the potential biological
behaviour of each m1A modification pattern, GSVA enrichment
analysis was performed between Clusters A and B, which presented
prominently different outcomes. As shown in Fig. 3A, Cluster B was markedly enriched
in various metabolic pathways, such as ‘purine metabolism’,
‘pyrimidine metabolism’, ‘citrate cycle’, ‘pyruvate metabolism’,
‘cysteine and methionine metabolism’, and ‘fructose and mannose
metabolism’. Cluster A was enriched in stromal pathways (Fig. 3A).
Prognostic value and association of
the m1A score with immunotherapy response
In the three m1A gene clusters, prominent
differences in the expression of m1A regulators were observed.
However, these analyses were based on the patient population and
could not accurately predict the pattern of m1A methylation in
individual patients. Considering the individual heterogeneity and
complexity of m1A modification, a scoring system was constructed to
quantify the m1A modification patterns of individual patients with
MM based on DEGs identified from different m1A clusters. First, the
patients were divided into low- and high-m1A score groups with a
cut-off value of -2.3971 via the survminer package (https://rpkgs.datanovia.com/survminer/index.html).
The Kruskal-Wallis test revealed that patients with a low m1A score
had better survival (Fig. 3B). A
significantly increased m1A score was detected in Cluster B, and
Cluster A presented the lowest median score (Fig. 3C). Patients with high m1A scores
had a greater survival rate than those with low m1A scores (74 vs.
54%), which again demonstrated that a low m1A score may be closely
associated with a poor prognosis (Fig.
3D and E). Immunotherapies
involving PD-L1 and PD-1 blockade have undoubtedly emerged as major
breakthroughs in cancer therapy (33). As illustrated in Fig. 3F, patients with a low m1A score
exhibited significant increased expression levels of PD-L1, which
indicated a potential response to anti-PD-L1 immunotherapy.
Validation of the m1A score in MM
The gene signature-based model was validated via
four external datasets containing MM and control samples.
Satisfactory model performance was achieved, as determined by the
ROC curves, with area under the curve (AUC) values of 0.917, 0.777,
0.667 and 0.625 for the GSE24870, GSE27838, GSE46053 and GSE113295
cohorts, respectively (Fig. 4A-D).
Subsequently, two datasets, GSE24870 and GSE27838, which exhibited
excellent predictive efficacy, were selected to further demonstrate
the important role of m1A regulators in MM and the significance of
YTHDF2, YTHDF3, TRMT10C and TRMT6 in the pathogenesis of MM was
verified (Fig. 4E and F).
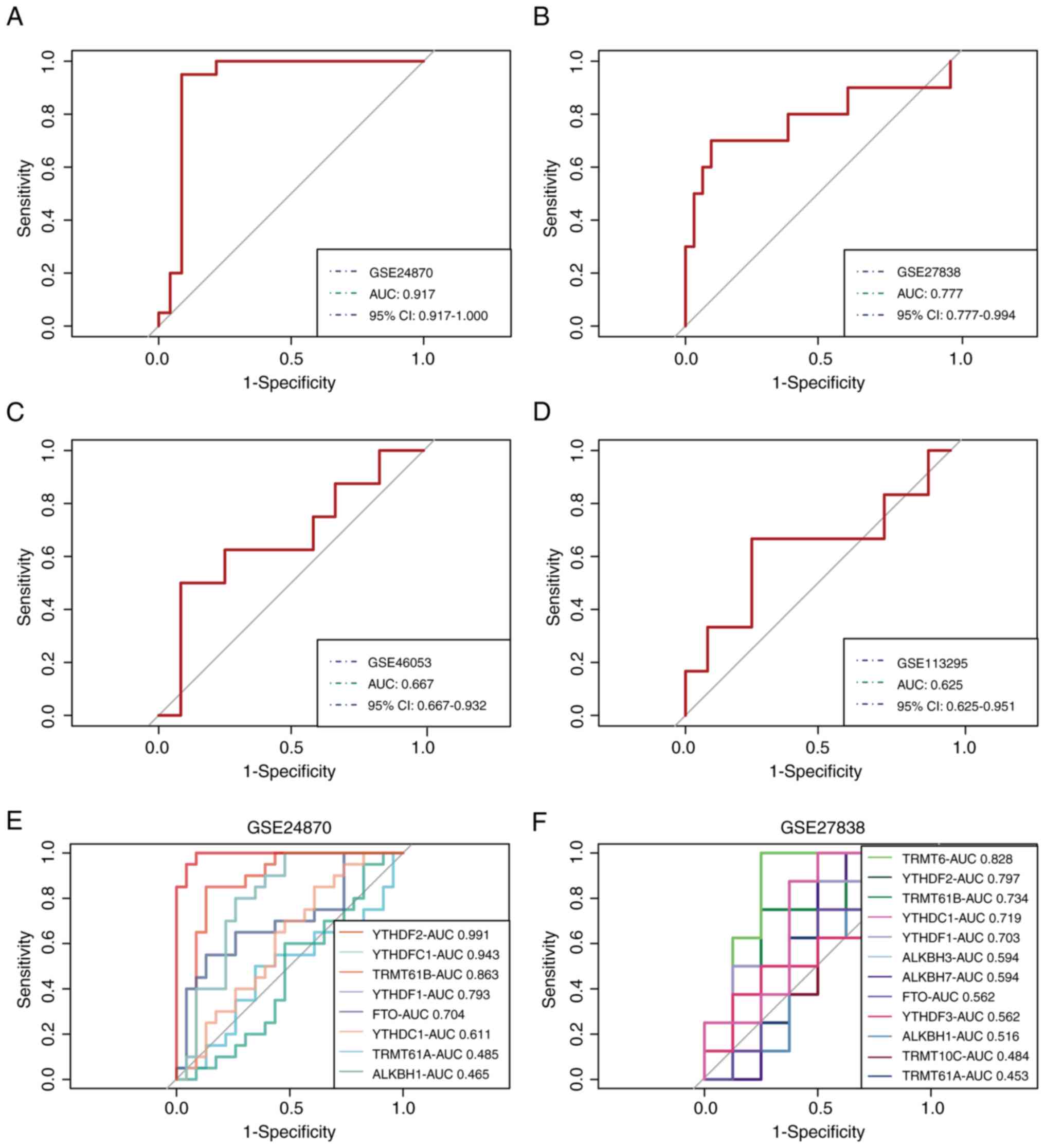 | Figure 4Validation of the m1A score in MM.
Receiver operating characteristic curves of the gene signature in
the (A) GSE24870, (B) GSE27838, (C) GSE46053 and (D) GSE113295
datasets, respectively. The role of m1A regulators in MM was
further confirmed in (E) GSE24870 and (F) GSE27838. m1A,
N1-methyladenosine; MM, multiple myeloma; AUC, area
under the curve; CI, confidence interval; TRMT10C, tRNA
methyltransferase 10 homolog C; TRMT61B, tRNA methyltransferase
61B; YTHD, YTH domain-containing family protein; TRMT6, tRNA
methyltransferase 6 non-catalytic subunit; TRMT61A, tRNA
methyltransferase 61A; ALKBH1, Alkb homolog 1, histone H2A
dioxygenase; ALKBH3, AlkB homolog 3, α-ketoglutarate dependent
dioxygenase; FTO, FTO α-ketoglutarate dependent dioxygenase. |
YTHDF2 promotes cellular proliferation
and m1A methylation in MM
Through bioinformatics analysis, it was revealed
that YTHDF2 expression is associated with poor survival in patients
with MM. A series of experiments were subsequently designed to
explore the important role of aberrantly expressed YTHDF2 in MM
pathogenesis. The IHC results revealed a significant increase in
YTHDF2 levels in the high-risk MM patient group compared with those
in the low-risk group (Fig. 5A).
To further investigate the potential mechanism of action of YTHDF2
in MM, YTHDF2 overexpression and knockdown experiments were
performed in U266 cells. CCK-8 assays verified that the si-YTHDF2
group exhibited a significantly lower growth rate than the NC and
si-NC groups (Fig. 5B). Flow
cytometric analysis revealed that the percentage of apoptotic cells
was significantly greater in si-YTHDF2 cells than in control cells
(Fig. 5C and D). As revealed in Fig. 5E, a YTHDF2-overexpressing cell
model was successfully constructed. Since YTHDF2 serves as a reader
of m1A modifications involved in multiple biological processes,
including cell differentiation and cancer progression (34), an m1A dot blot assay was performed
to assess the global mRNA m1A levels in oe-YTHDF2 cells and
si-YTHDF2 cells. An evidently increased m1A level was observed in
the oe-YTHDF2 group, whereas YTHDF2 knockdown decreased the overall
m1A level of mRNAs (Fig. 5F),
indicating that YTHDF2-mediated changes in the m1A level may be
involved in MM pathogenesis. Several databases were subsequently
used to explore the potential downstream regulatory mechanisms of
YTHDF2. First, the aim was to identify the DEGs related to YTHDF2
expression in MM and then to perform functional enrichment analysis
on the top 150 related genes associated with the peroxisome
proliferator-activated receptor ‘(PPAR) signaling pathway’,
‘protein export’ and ‘various types of N-glycan biosynthesis’
(Fig. 5G). Moreover, ClueGO
biological function analysis revealed that these YTHDF2-related
genes were enriched in ‘positive regulation of protein targeting to
the membrane’, ‘gamma-delta intraepithelial T-cell
differentiation’, and ‘positive regulation of anion channel
activity’ (Fig. 5H). Next, genes
closely related to YTHDF2 expression were screened and it was
observed that serine/arginine splicing factor 10 (SRSF10) was
significantly positively correlated with YTHDF2 expression
(Fig. 5I). SRSF10 is an
rRNA-cleaving enzyme that is significantly correlated with a
variety of tumours (Fig. 5J).
Subsequently, through the RMbase database, it was found that YTHDF2
can install m1A modifications on SRSF10, which is consistent with
the findings of a previous study (35). Survival analysis revealed that high
expression of SRSF10 indicated a poor prognosis (Fig. 5K), suggesting that YTHDF2 may play
a role in MM by methylating SRSF10; however, the underlying
mechanism needs to be explored in the future.
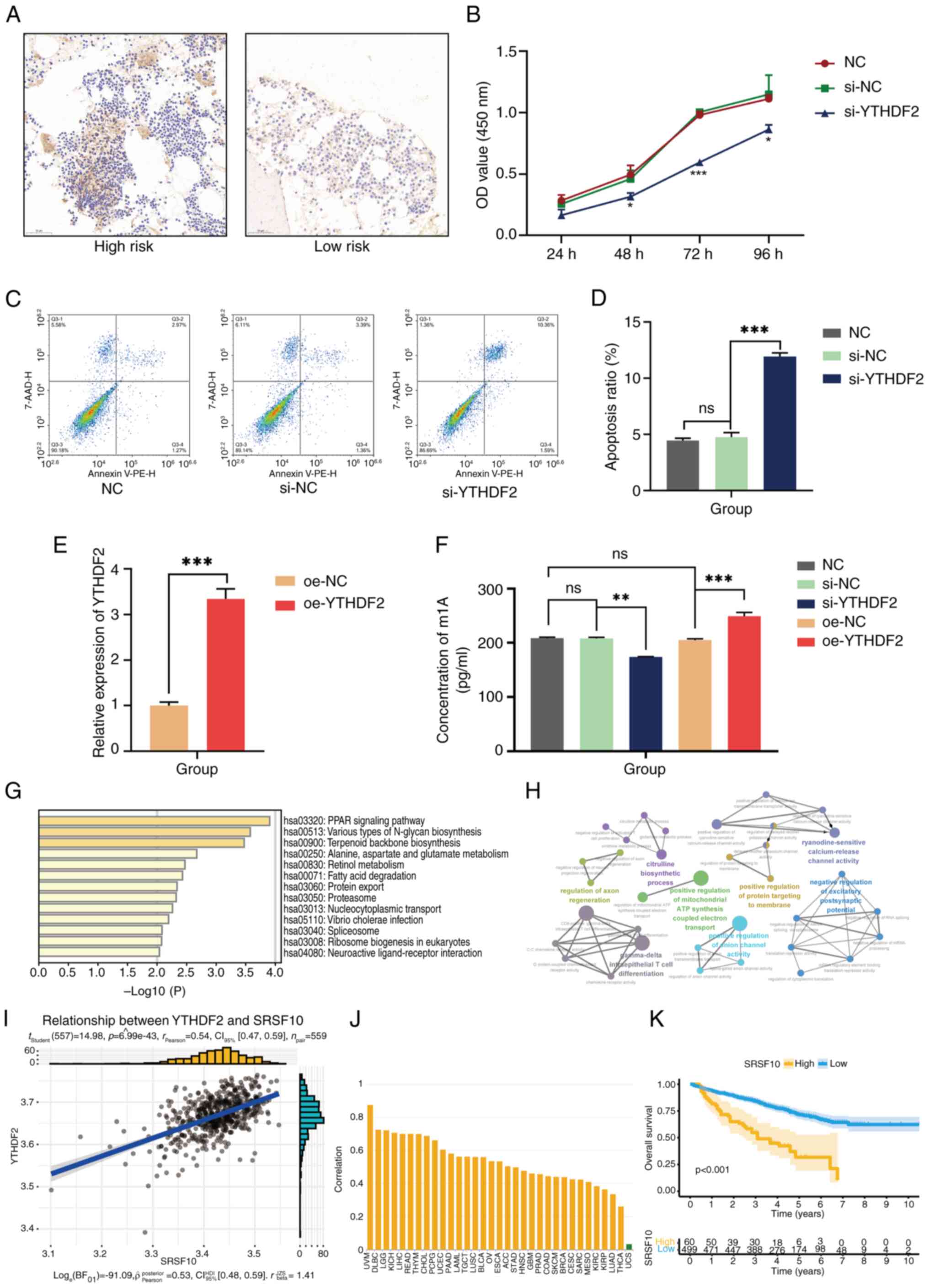 | Figure 5Preliminary experimental verification
and prediction of the downstream mechanism of YTHDF2. (A) The
expression level of YTHDF2 in pathological sections detected by
immunohistochemistry. (B) YTHDF2 silencing led to lower viability
of U266 cells. (C and D) YTHDF2 silencing was associated with a
higher apoptosis level in U266 cells. (E) Transfection efficiency
of oe-YTHDF2 was confirmed by reverse transcription-quantitative
PCR methods. (F) Detection of m1A methylation levels in si-YTHDF2
and si-YTHDF2 cells. (G) Kyoto Encyclopedia of Genes and Genomes
analysis of YTHDF2-associated genes. (H) ClueGO analysis of
YTHDF2-associated genes. (I) SRSF10 was positively correlated with
YTHDF2 expression. (J) The correlation of SRSF10 with different
tumors. (K) The high expression of SRSF10 was related to the worse
outcome in multiple myeloma. Differences between two groups were
analyzed by Student's t-test, and differences among multiple groups
were analyzed by oneway ANOVA and Kruskal–Wallis test with LSD post
hoc test or Tukey's post hoc test. *P<0.1,
**P<0.01, and ***P<0.001. ns, no
significance; YTHDF2, YTH domain-containing family protein 2; oe,
overexpressing; m1A, N1-methyladenosine; si-, small
interfering; SRSF10, serine/arginine splicing factor 10; NC,
negative control; CI, confidence interval. |
Discussion
M1A regulators govern m1A RNA methylation functions.
Some research groups have reported that m1A regulators play
important roles in tumorigenesis. In the present study, seven
differentially expressed m1A regulators from the GEO database were
identified, including TRMT61A/B, the reader YTHDF1/2/3 and the
eraser ALKBH1 (Fig. 1A). The
forest plot and Kaplan-Meier curve revealed that the expression of
m1A-related regulatory genes was a reliable prognostic indicator
for patients with MM; for example, high expression of YTHDF2 was
closely associated with shorter overall survival (Fig. 1B and G). The m1A regulator network diagram
revealed a strong positive correlation between YTHDF2 and TRMT61B
and a significant negative correlation between YTHDF1 and ALKBH1,
suggesting a comprehensive network of different m1A regulators
(Fig. 1I). Then, based on the
expression of 10 m1A-related genes, three distinct m1A modification
patterns were defined via unsupervised cluster analysis (Fig. 2A-C). PCA also revealed that YTHDF2,
TRMT10C, TRMT61B and TRMT61A played important roles in the
differentiation of the three clusters (Fig. 2D and E). Survival analysis revealed a shorter
survival time for patients with MM in Cluster B, which was
characterized by a high expression of YTHDF2, TRMT61B and TRMT10C
(Fig. 2F and G). Cluster B also exhibited lower immune
infiltration, as expected (Fig.
3F). Clusters A and B were subsequently selected for GSVA due
to the obvious difference in survival time between the two
clusters, and the main differentially enriched pathway was DNA and
RNA metabolism progression (Fig.
3A). In addition, m1A score values were established and
compared among different m1A clusters. The survival results
indicated that patients with MM with low m1A scores had longer
survival times, and the m1A score was significantly greater in
Cluster B, which was consistent with the survival rate trend in the
m1A cluster groups (Fig. 3B-E).
Moreover, it was also observed that patients with a high m1A score
had lower PD-L1 expression, suggesting the value of the m1A score
in predicting the response to anti-PD-L1 immunotherapy (Fig. 3F). Additionally, the m1A score was
validated using four external datasets, confirming its robustness
with satisfactory AUC values. The identified m1A regulators also
exhibited high accuracy in predicting MM diagnosis, suggesting that
the developed model may serve as a powerful diagnostic tool for
MM.
Considering the aforementioned bioinformatics
analysis results, the reader protein YTHDF2 attracted the attention
of the authors, due to its high prognostic value in MM. YTHDF2 can
directly interact with m1A-modified RNA within the YTH domain,
which can disrupt the stability of RNA transcripts (36,37).
A previous study revealed that YTHDF2 is overexpressed in acute
myeloid leukaemia (AML) and is considered a unique target for the
treatment of AML (38). It has
also been reported that YTHDF2 can promote MM cell proliferation
via the STAT5A/MAP2K2/p-ERK axis and that decreased YTHDF2
expression retards MM tumour growth (39). There is also a YTHDF2-dependent
mechanism by which other methylases promote cell proliferation,
migration and invasion in MM (17). First, it was verified that YTHDF2
was overexpressed in the high-risk group of patients with MM via
immunohistochemistry (Fig. 5A).
The CCK-8 and flow cytometric results demonstrated that YTHDF2
plays a proliferation-promoting and apoptosis-inhibiting role in MM
(Fig. 5B-D). Gain- and
loss-of-function studies revealed that YTHDF2 could increase the
level of m1A in U266 cells, which was consistent with previous
findings (Fig. 5F). Next, the
potential downstream mechanism of YTHDF2 was explored and it was
found that its co-expressed genes were enriched in the ‘PPAR
signaling pathway’, ‘nucleocytoplasmic transport’, and ‘positive
regulation of protein targeting to the membrane’ (Fig. 5G and H). Previous studies have confirmed the
crucial role of the PPAR signaling pathway in MM (40). PPARs are nuclear receptor proteins,
and there are three main isoforms: PPARα, PPARβ and PPARγ. PPARγ
can induce apoptosis in MM cells, and its agonist pioglitazone can
enhance the cytotoxic effect of a histone deacetylase inhibitor on
MM cells (41). However, YTHDF2
has been reported to bind to PPARs to mediate their degradation,
which may be one of the potential mechanisms by which YTHDF2
regulates MM through the PPAR pathway (42). SRSF10 was subsequently selected due
to its significant positive correlation with YTHDF2 (Fig. 5I). SRSF10 is a member of the family
of mammalian splicing regulators known as SR proteins (43,44).
The overexpression of SRSF10 has been verified in a growing list of
cancers, as revealed in Fig. 5J,
and its contribution to the splicing of transcripts implicated in
clinically relevant pathways suggests that SRSF10 could become a
treatment target (45). Notably,
the activation of SRSF10 was found to dysregulate the
polyadenylation of PPAR, and studies have confirmed that YTHDF2
could affect alternative splicing patterns of genes to regulate
their expression in a methylation-dependent manner (46-49).
Therefore, it was hypothesized that YTHDF2 may regulate SRSF10
through m1A modification to intervene in MM by affecting the PPAR
signaling pathway. Moreover, as shown in Fig. 5K, high expression of SRSF10
predicted a shorter survival time, indicating its predictive value
for MM prognosis.
However, some limitations should be noted. First,
the differentially expressed m1A regulators in MM via the GEO
database were identified and their functions were preliminarily
predicted via bioinformatics analysis. However, the important role
of m1A regulators and the m1A score in MM still needs to be
verified in clinical trials and more experiments, such as
verification of the expression level of prognosis-related m1A
regulators in patients with MM and determination of their
predictive ability as clinical prognostic factors via Cox analysis
and ROC curves. Second, the role of YTHDF2 in the development, risk
stratification and prognosis of MM remains to be further confirmed,
and its downstream molecular regulatory mechanism remains to be
further explored. Additionally, the regulatory relationship between
YTHDF2 and SRSF10 needs to be further verified.
In conclusion, seven differentially expressed m1A
regulators and three distinct m1A modification patterns that were
associated with differences in overall survival and immune
infiltration in patients with MM were identified. The m1A score
model was subsequently constructed to quantitatively evaluate the
m1A modification patterns of individual patients, which may serve
as effective biomarkers for predicting the prognosis of patients
with MM. Moreover, the reader protein of m1A, YTHDF2, could
increase the m1A level, promote proliferation and inhibit apoptosis
in U266 cells, indicating that it may be a potentially crucial
target for MM treatment.
Supplementary Material
YTHDF2 siRNA sequences.
Two-way ordered categorical variables
with different attributes between different m1A clusters and
different risk stratifications.
Acknowledgements
The authors are grateful to Mr. Jingwei Cui (Jiaxuan
School of Jinan, Jinan, China) for providing help with data
analysis in this paper, especially in the application of R to
perform the survival analysis and functional prediction in the
bioinformatics section.
Funding
Funding: The present study was supported by the National Natural
Science Foundation of China (grant nos. 82074348 and 82274491).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author. The data generated in the
present study may be found in the Gene Expression Omnibus (GEO)
database under accession numbers GSE13591, GSE47552, GSE24080,
GSE24870, GSE27838, GSE46053 and GSE113295 at the following URL:
https://www.ncbi.nlm.nih.gov/geo/.
Authors' contributions
XC conceived the project, designed the experiments
and revised the paper. JF performed the experiments and wrote the
original paper. XH, WG and MY assisted with the experiments, and
data analysis and interpretation. All authors read and approved the
final version of the manuscript. XC, JF, XH, WG and MY confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Affiliated Hospital of Shandong University of
Traditional Chinese Medicine, Jinan, China (approval no. 2020
Ethics Review-KY-010) and the Ethics Committee of the Second
Affiliated Hospital of Shandong University of Traditional Chinese
Medicine, Jinan, China (approval no. 2023 Ethics Review-KY-001).
Written informed consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhan F, Huang Y, Colla S, Stewart JP,
Hanamura I, Gupta S, Epstein J, Yaccoby S, Sawyer J, Burington B,
et al: The molecular classification of multiple myeloma. Blood.
108:2020–2028. 2006.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Allegra A, Casciaro M, Barone P, Musolino
C and Gangemi S: Epigenetic crosstalk between malignant plasma
cells and the tumour microenvironment in multiple myeloma. Cancers
(Basel). 14(2597)2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
He L, Yu C, Qin S, Zheng E, Liu X, Liu Y,
Yu S, Liu Y, Dou X, Shang Z, et al: The proteasome component PSMD14
drives myelomagenesis through a histone deubiquitinase activity.
Mol Cell. 83:4000–4016.e6. 2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Muylaert C, Van Hemelrijck LA, Maes A, De
Veirman K, Menu E, Vanderkerken K and De Bruyne E: Aberrant DNA
methylation in multiple myeloma: A major obstacle or an
opportunity? Front Oncol. 12(979569)2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Liu R, Shen Y, Hu J, Wang X, Wu D, Zhai M,
Bai J and He A: Comprehensive Analysis of m6A RNA methylation
regulators in the prognosis and immune microenvironment of multiple
myeloma. Front Oncol. 11(731957)2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Jin Z, MacPherson K, Liu Z and Vu LP: RNA
modifications in hematological malignancies. Int J Hematol.
117:807–820. 2023.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhao BS, Roundtree IA and He C:
Post-transcriptional gene regulation by mRNA modifications. Nat Rev
Mol Cell Biol. 18:31–42. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Dominissini D, Nachtergaele S,
Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, Dai Q, Di Segni
A, Salmon-Divon M, Clark WC, et al: The dynamic
N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature.
530:441–446. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yang Y, Hsu PJ, Chen YS and Yang YG:
Dynamic transcriptomic m(6)A decoration: Writers, erasers, readers
and functions in RNA metabolism. Cell Res. 28:616–624.
2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Liu Y, Zhang S, Gao X, Ru Y, Gu X and Hu
X: Research progress of N1-methyladenosine RNA modification in
cancer. Cell Commun Signal. 22(79)2024.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zou Z, Sepich-Poore C, Zhou X, Wei J and
He C: The mechanism underlying redundant functions of the YTHDF
proteins. Genome Biol. 24(17)2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Chen Z, Qi M, Shen B, Luo G, Wu Y, Li J,
Lu Z, Zheng Z, Dai Q and Wang H: Transfer RNA demethylase ALKBH3
promotes cancer progression via induction of tRNA-derived small
RNAs. Nucleic Acids Res. 47:2533–2545. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Liu R, Miao J, Jia Y, Kong G, Hong F, Li
F, Zhai M, Zhang R, Liu J, Xu X, et al: N6-methyladenosine reader
YTHDF2 promotes multiple myeloma cell proliferation through
EGR1/p21cip1/waf1/CDK2-Cyclin E1 axis-mediated cell
cycle transition. Oncogene. 42:1607–1619. 2023.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Che F, Ye X, Wang Y, Wang X, Ma S, Tan Y,
Mao Y and Luo Z: METTL3 facilitates multiple myeloma tumorigenesis
by enhancing YY1 stability and pri-microRNA-27 maturation in
m6A-dependent manner. Cell Biol Toxicol. 39:2033–2050.
2023.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Su Z, Monshaugen I, Wilson B, Wang F,
Klungland A, Ougland R and Dutta A: TRMT6/61A-dependent base
methylation of tRNA-derived fragments regulates gene-silencing
activity and the unfolded protein response in bladder cancer. Nat
Commun. 13(2165)2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Yang Z, Cai Z, Yang C, Luo Z and Bao X:
ALKBH5 regulates STAT3 activity to affect the proliferation and
tumorigenicity of osteosarcoma via an m6A-YTHDF2-dependent manner.
EBioMedicine. 80(104019)2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Xu A, Zhang J, Zuo L, Yan H, Chen L, Zhao
F, Fan F, Xu J, Zhang B, Zhang Y, et al: FTO promotes multiple
myeloma progression by posttranscriptional activation of HSF1 in an
m6A-YTHDF2-dependent manner. Mol Ther. 30:1104–1118.
2022.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Coira IF, Rincón R and Cuendet M: The
Multiple Myeloma Landscape: Epigenetics and Non-Coding RNAs.
Cancers (Basel). 14(2348)2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Agnelli L, Mosca L, Fabris S, Lionetti M,
Andronache A, Kwee I, Todoerti K, Verdelli D, Battaglia C, Bertoni
F, et al: A SNP microarray and FISH-based procedure to detect
allelic imbalances in multiple myeloma: An integrated genomics
approach reveals a wide gene dosage effect. Genes Chromosomes
Cancer. 48:603–614. 2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
López-Corral L, Corchete LA, Sarasquete
ME, Mateos MV, García-Sanz R, Fermiñán E, Lahuerta JJ, Bladé J,
Oriol A, Teruel AI, et al: Transcriptome analysis reveals molecular
profiles associated with evolving steps of monoclonal gammopathies.
Haematologica. 99:1365–1372. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Popovici V, Chen W, Gallas BG, Hatzis C,
Shi W, Samuelson FW, Nikolsky Y, Tsyganova M, Ishkin A, Nikolskaya
T, et al: Effect of training-sample size and classification
difficulty on the accuracy of genomic predictors. Breast Cancer
Res. 12(R5)2010.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Li J, Xie L, Xie Y and Wang F: Bregmannian
consensus clustering for cancer subtypes analysis. Comput Methods
Programs Biomed. 189(105337)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hänzelmann S, Castelo R and Guinney J:
GSVA: Gene set variation analysis for microarray and RNA-seq data.
BMC Bioinformatics. 14(7)2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sotiriou C, Wirapati P, Loi S, Harris A,
Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B, et al:
Gene expression profiling in breast cancer: Understanding the
molecular basis of histologic grade to improve prognosis. J Natl
Cancer Inst. 98:262–272. 2006.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhang B, Wu Q, Li B, Wang D, Wang L and
Zhou YL: m6A regulator-mediated methylation modification patterns
and tumor microenvironment infiltration characterization in gastric
cancer. Mol Cancer. 19(53)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Bruns I, Cadeddu RP, Brueckmann I, Fröbel
J, Geyh S, Büst S, Fischer JC, Roels F, Wilk CM, Schildberg FA, et
al: Multiple myeloma-related deregulation of bone marrow-derived
CD34(+) hematopoietic stem and progenitor cells. Blood.
120:2620–2630. 2012.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Garg TK, Szmania SM, Khan JA, Hoering A,
Malbrough PA, Moreno-Bost A, Greenway AD, Lingo JD, Li X, Yaccoby
S, et al: Highly activated and expanded natural killer cells for
multiple myeloma immunotherapy. Haematologica. 97:1348–1356.
2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Garcia-Gomez A, De Las Rivas J, Ocio EM,
Díaz-Rodríguez E, Montero JC, Martín M, Blanco JF, Sanchez-Guijo
FM, Pandiella A, San Miguel JF and Garayoa M: Transcriptomic
profile induced in bone marrow mesenchymal stromal cells after
interaction with multiple myeloma cells: Implications in myeloma
progression and myeloma bone disease. Oncotarget. 5:8284–8305.
2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Liu H, He J, Koh SP, Zhong Y, Liu Z, Wang
Z, Zhang Y, Li Z, Tam BT, Lin P, et al: Reprogrammed marrow
adipocytes contribute to myeloma-induced bone disease. Sci Transl
Med. 11(eaau9087)2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Liu B, Li X, Liu F, Li F, Wei S, Liu J and
Lv Y: Expression and Significance of TRIM 28 in Squamous Carcinoma
of Esophagus. Pathol Oncol Res. 25:1645–1652. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Frapin M, Guignard S, Meistermann D, Grit
I, Moullé VS, Paillé V, Parnet P and Amarger V: Maternal protein
restriction in rats alters the expression of genes involved in
mitochondrial metabolism and epitranscriptomics in fetal
hypothalamus. Nutrients. 12(1464)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Costa F, Vescovini R, Marchica V, Storti
P, Notarfranchi L, Dalla Palma B, Toscani D, Burroughs-Garcia J,
Catarozzo MT, Sammarelli G and Giuliani N: PD-L1/PD-1 pattern of
expression within the bone marrow immune microenvironment in
smoldering myeloma and active multiple myeloma patients. Front
Immunol. 11(613007)2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Huang T, Liu Z, Zheng Y, Feng T, Gao Q and
Zeng W: YTHDF2 promotes spermagonial adhesion through modulating
MMPs decay via m(6)A/mRNA pathway. Cell Death Dis.
11(37)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xiao W, Adhikari S, Dahal U, Chen YS, Hao
YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, et al: Nuclear m(6)A
Reader YTHDC1 Regulates mRNA Splicing. Mol Cell. 61:507–519.
2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Dai X, Wang T, Gonzalez G and Wang Y:
Identification of YTH Domain-Containing Proteins as the Readers for
N1-Methyladenosine in RNA. Anal Chem. 90:6380–6384. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Seo KW and Kleiner RE: YTHDF2 Recognition
of N1-Methyladenosine (m1A)-Modified RNA Is Associated with
Transcript Destabilization. ACS Chem Biol. 15:132–139.
2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Paris J, Morgan M, Campos J, Spencer GJ,
Shmakova A, Ivanova I, Mapperley C, Lawson H, Wotherspoon DA,
Sepulveda C, et al: Targeting the RNA m6A Reader YTHDF2 selectively
compromises cancer stem cells in acute myeloid leukemia. Cell Stem
Cell. 25:137–148. e6. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Hua Z, Wei R, Guo M, Lin Z, Yu X, Li X, Gu
C and Yang Y: YTHDF2 promotes multiple myeloma cell proliferation
via STAT5A/MAP2K2/p-ERK axis. Oncogene. 41:1482–1491.
2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Sha Y, Wu J, Paul B, Zhao Y, Mathews P, Li
Z, Norris J, Wang E, McDonnell DP and Kang Y: PPAR agonists
attenuate lenalidomide's anti-myeloma activity in vitro and in
vivo. Cancer Lett. 545(215832)2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Aouali N, Broukou A, Bosseler M, Keunen O,
Schlesser V, Janji B, Palissot V, Stordeur P and Berchem G:
Epigenetic activity of peroxisome proliferator-activated receptor
gamma agonists increases the anticancer effect of histone
deacetylase inhibitors on multiple myeloma cells. PLoS One.
10(e0130339)2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Yu JT, Hu XW, Chen HY, Yang Q, Li HD, Dong
YH, Zhang Y, Wang JN, Jin J, Wu YG, et al: DNA methylation of FTO
promotes renal inflammation by enhancing m6A of PPAR-α in
alcohol-induced kidney injury. Pharmacol Res.
163(105286)2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Long JC and Caceres JF: The SR protein
family of splicing factors: Master regulators of gene expression.
Biochem J. 417:15–27. 2009.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Longman D, Johnstone IL and Cáceres JF:
Functional characterization of SR and SR-related genes in
caenorhabditis elegans. EMBO J. 19:1625–1637. 2000.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Shkreta L, Delannoy A, Salvetti A and
Chabot B: SRSF10: An atypical splicing regulator with critical
roles in stress response, organ development, and viral replication.
RNA. 27:1302–1317. 2021.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Jobbins AM, Haberman N, Artigas N, Amourda
C, Paterson HAB, Yu S, Blackford SJI, Montoya A, Dore M, Wang YF,
et al: Dysregulated RNA polyadenylation contributes to metabolic
impairment in non-alcoholic fatty liver disease. Nucleic Acids Res.
50:3379–3393. 2022.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Maimaiti A, Tuersunniyazi A, Meng X, Pei
Y, Ji W, Feng Z, Jiang L, Wang Z, Kasimu M, Wang Y and Shi X:
N6-methyladenosine RNA methylation regulator-related alternative
splicing gene signature as prognostic predictor and in immune
microenvironment characterization of patients with low-grade
glioma. Front Genet. 13(872186)2022.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Lai S, Wang Y, Li T, Dong Y, Lin Y, Wang
L, Weng S, Zhang X and Lin C: N6-methyladenosine-mediated CELF2
regulates CD44 alternative splicing affecting tumorigenesis via
ERAD pathway in pancreatic cancer. Cell Biosci.
12(125)2022.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Yuan J, Lv T, Yang J, Wu Z, Yan L, Yang J
and Shi Y: HDLBP-stabilized lncFAL inhibits ferroptosis
vulnerability by diminishing Trim69-dependent FSP1 degradation in
hepatocellular carcinoma. Redox Biol. 58(102546)2022.PubMed/NCBI View Article : Google Scholar
|