Introduction
Mushrooms are good food materials whose
immunomodulatory potential has been investigated. Sparassis
crispa (Wulf.) Fr. (Aphyllophoromycetideae) is an edible
mushroom used for a natural medicine that recently became
cultivatable in Korea and Japan (1). Recently, S. crispa has been
reported to have many biological effects such as antitumor,
anti-angiogenic, anti-metastatic, and wound healing (1–3).
The major component of S. crispa is β-D-glucan, a
polysaccharide of D-glucose comprising a β-(1→3)-D-glucan backbone
(3,4). According to previous research, the
β-glucan content of S. crispa was confirmed to be immense,
up to 43.6% of the dry weight (4). S. crispa derived β-glucan has
been known for several beneficial effects, however its allergic
responses have not been clarified.
Mast cells are broadly distributed throughout
mammalian tissues and play various functions as regulators of
allergic inflammation, such as asthma, atopic dermatitis, eczema,
and sinusitis. Mast cells have been considered not only in the
association of immediate type hypersensitivity but also in late
reaction, like inflammatory responses (5,6).
Immediate type hypersensitivity is mediated by histamine released
in response to the antigen cross-linking of immunoglobulin E (IgE)
bound to FcɛRI on the mast cells (7). After activation of mast cells, the
process of degranulation is triggered which results in the
releasing of mediators, such as products of arachidonic acid
metabolism, cytokines, proteases and histamine (8,9).
In mast cell-mediated inflammatory responses, histamine is one of
the most characterized and important mediators implicated in the
acute phase of immediate hypersensitivity (10,11). Mast cell activation is initiated
by phosphorylation of tyrosine kinase which leads to activation of
protein kinase C, mitogen-activated protein kinases (MAPKs),
nuclear factor (NF)-κB, and expression of proinflammatory cytokines
(8,12).
In the present study, we investigated the effect of
the water extract of S. crispa (WESC) on the systemic and
local allergic reaction and histamine release from mast cells. The
intracellular calcium content was investigated to clarify the
mechanism by which WESC inhibited histamine release from mast
cells. In addition, the effect of WESC on phorbol 12-myristate
13-acetate (PMA) and calcium ionophore A23187 (PMACI)-induced gene
expression and production of proinflammatory cytokines, and the
role of NF-κB and MAPKs in this effect were investigated using
human mast cells (HMC-1). In order to determine the amount of
active compounds of WESC, we confirmed the contents of β-glucan in
WESC.
Materials and methods
Animals
The original stock of male imprinting control region
(ICR) mice (6 weeks) and male Sprague-Dawley rats (8 weeks) were
purchased from the Dae-Han Experimental Animal Center (Daejeon,
Korea). The animals were housed 5 per cage in a laminar air flow
room maintained under a temperature of 22±2°C and relative humidity
of 55±5°C throughout the study. The care and treatment of the mice
were in accordance with the guidelines established by the Public
Health Service Policy on the Humane Care and Use of Laboratory
Animals and were approved by the Institutional Animal Care and Use
Committee.
Reagents and cell culture
Compound 48/80, anti-dinitrophenyl (DNP) IgE,
DNP-human serum albumin (HSA), phorbol 12-myristate 13-acetate
(PMA), and calcium ionophore A23187 were purchased from Sigma
Chemical Co. (St. Louis, MO, USA). The human mast cell line (HMC-1)
was grown in Iscove’s media (Life Technologies, Grand Island, NY,
USA) supplemented with 10% fetal bovine serum at 37°C in 5%
CO2. Passages 4–8 of HMC-1 cells were used throughout
the study.
Preparation of WESC
The Sparassis crispa was purchased from the
oriental drug store, Bohwa Dang (Jeonbuk, Korea) and identified by
D.K. Kim, College of Pharmacy, Woosuk University. A voucher
specimen was deposited at the Herbarium of the College of Pharmacy,
Woosuk University. The S. crispa was ground (400 x g, 30
sec) at room temperature using Micro Hammer-Cutter Mill (Culatti
Co., Zurich, Switzerland). The particle size was 0.5–2 mM after
grinding. The plant sample (100 x g) was extracted twice with
purified water (500 ml) at 70°C for 5 h in a water bath. The
extract was passed through filter paper and the filtrate was
lyophilized using a 0.45 μm syringe filter. The dried extract was
dissolved in saline or Tyrode buffer A (HEPES 10 mM, NaCl 130 mM,
KCl 5 mM, CaCl2 1.4 mM, MgCl2 1 mM, glucose
1.4 mM, 0.1% bovine serum albumin) before use.
Compound 48/80-induced systemic
anaphylaxis
Mice were given an intraperitoneal injection of 8
mg/kg body weight (BW) of the mast cell degranulator compound
48/80. WESC was administered intraperitoneally at doses of 10–1,000
mg/kg BW 1 h before the injection of compound 48/80 (n=10/group).
Mortality was monitored for 1 h after induction of anaphylactic
shock. After the mortality test, blood was obtained from the heart
of each mouse to measure serum histamine contents.
Passive cutaneous anaphylaxis (PCA)
An IgE-dependent cutaneous reaction was carried out
as described previously (13).
Briefly, mice were injected intradermally with 0.5 μg of anti-DNP
IgE. After 48 h, each mouse (n=10/group) received an injection of 1
μg of DNP-HSA containing 4% Evans blue (1:4) via the tail vein.
Thirty minutes after the challenge, the mice were sacrificed and
the dorsal skin (diameter, 1 cm) was removed for measurement of the
pigmented area.
Preparation of serum and histamine
determination
Preparation of serum and determination of histamine
contents were examined as previously described (14). Briefly, serum was withdrawn and
the histamine contents were measured by the
o-phthaldialdehyde spectrofluorometric procedure. The
fluorescent intensity was measured at emission 438 nm and
excitation 353 nm using a spectrofluorometer.
Determination of intracellular
calcium
The intracellular calcium was measured with the use
of the fluorescence indicator Fluo-3/AM (Molecular Probes, Eugene,
OR, USA). HMC-1 cells were pre-incubated with Fluo-3/AM for 30 min
at 37°C. After washing the dye from the cell surface, the cells
were treated with WESC for 10 min before adding PMACI. It was
excited at 488 nm, and the emission was filtered with 515 nm by
flow cytometer (BD Biosciences Pharmingen, San Diego, CA, USA) and
visualized by a fluorescence microscope Olympus BX51 (Olympus,
Center Valley, PA, USA).
RNA extraction and mRNA detection
The total cellular RNA was isolated from the cells
(1x106/well in a 24-well plate) after stimulation with
PMA (20 nM) and A23187 (1 μM) with or without WESC for 2 h using
TRI reagent (Molecular Research Center, Cincinnati, OH, USA)
according to the manufacturer’s protocol. The first strand
complementary DNA (cDNA) was synthesized using the Superscript II
reverse-transcriptase (Invitrogen, Carlsbad, CA, USA). A reverse
transcriptase polymerase chain reaction (RT-PCR) was used to
analyze the expression of mRNA for TNF-α, IL-6, and β-actin
(internal control). The conditions for the reverse transcription
and PCR steps were similar to those described previously (13). The amplified products were
separated by electrophoresis on a 2% agarose gel containing
ethidium bromide, documented using a Kodak DC 290 digital camera
and digitized with UN-SCAN-IT software (Silk Scientific, Inc.,
Orem, UT, USA). The band intensity was normalized to that of
β-actin in the same sample.
Enzyme-linked immunosorbent assay
(ELISA)
The secretion of TNF-α and IL-6 was measured by the
modification of an enzyme-linked immunosorbent assay (ELISA) as
previously described (15). HMC-1
cells were cultured with media and resuspended in Tyrode buffer A.
The cells were sensitized with PMACI for 8 h in the absence or
presence of WESC. ELISA was performed by coating 96-well plates
with 6.25 ng/well of monoclonal antibody with specificity for TNF-α
and IL-6, respectively.
Western blot analysis
HMC-1 cells were washed 3 times with PBS and
resuspended in lysis buffer. Samples were electrophoresed using 12%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis, as
described elsewhere (16), and
then transferred to a nitrocellulose membrane. The nucleus and
cytosolic p65 NF-κB and IκBα was assayed using anti-NF-κB (p65) and
anti-IκBα antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA). The phosphorylation of ERK and p38 was determined using
anti-phospho-p38 and anti-phospho-ERK antibodies (Cell Signaling
Technology, Inc., Beverly, MA, USA). Immunodetection was performed
the using Supersignal West Pico chemiluminescent substrate (Thermo
Fisher Scientific, Waltham, MA, USA).
Transient transfection and luciferase
activity assay
For transient transfection, HMC-1 cells were seeded
at 2x106 in a 6-well plate 1 day before transient
transfection. The expression vectors containing the NF-κB
luciferase reporter construct (pNF-κB-LUC, plasmid containing NF-κB
binding site; Stantagen, Grand Island, NY, USA) were transfected
with serum- and antibiotics-free Iscove’s medium containing 8 μl
Lipofectamine 2000 reagent (Invitrogen). After 5 h of incubation,
the medium was replaced with Iscove’s medium containing 10% FBS and
antibiotics. Cells were allowed to recover at 37°C for 30 h and
subsequently were stimulated as indicated. Cell lysates were
prepared and assayed for luciferase activity using the Luciferase
Assay System (Promega, Madison, WI, USA), according to the
manufacturer’s instructions.
Measurement of β-glucan in WESC
β-glucan contents in WESC were determined using a
mushroom β-glucan assay kit (K-YBGL; Megazyme International,
Wicklow, Ireland) according to the manufacturer’s protocol. The
lyophilized extract of S. crispa (100 mg) and 1.5 ml of 37%
hydrochloric acid (v/v, 10 N) were mixed vigorously, and incubated
at 30°C for 45 min. The materials were mixed with 10 ml distilled
water and incubated at 100°C for 2 h. After centrifugation at 1,500
x g for 10 min, 0.1 ml aliquots were combined with 0.1 ml of a
mixture of exo-(1–3)-β-glucanase (20 U/ml) plus
β-glucosidase (4 U/ml) in 200 mM sodium acetate buffer (pH 5.0) and
incubated at 40°C for 60 min. To measure total glucan content, 3 ml
of glucose oxidase/peroxidase mixture (GOPOD) was added and
incubated at 40°C for 20 min. The absorbance of samples was
analyzed spectrophotometrically at 510 nm against the reagent blank
using a spectrophotometer (Shimadzu, UV-1201). To measure the
α-glucan content, 2 ml of 2 M KOH was added, and the phytoglycogen
and starch were dissolved by stirring for 20 min in an ice water
bath. The suspension was added to 8 ml of 1.2 M sodium acetate
buffer (pH 3.8), mixed with 0.2 ml of amyloglucosidase (16,300
U/ml) plus invertase (500 U/ml), and incubated in a water bath for
30 min at 40°C with intermittent mixing on a vortex stirrer. Tubes
were centrifuged (10 min, 1,500 x g), and 0.1 ml aliquots were
combined with 0.1 ml of sodium acetate buffer (200 mM, pH 5.0) plus
3 ml of GOPOD reagent and incubated for at 40°C for 30 min. The
absorbance of samples was analyzed at 510 nm. The β-glucan content
was determined by subtracting the α-glucan from the total glucan
content.
Statistical analysis
Statistical analyses were performed using SAS
statistical software (SAS Institute, Inc., Cary, NC, USA).
Treatment effects were analyzed using analysis of variance,
followed by Duncan’s multiple range tests. P<0.05 indicated
significance.
Results
Effect of WESC on systemic and local
anaphylaxis
To determine the effect of WESC in allergic
reaction, an in vivo model of a systemic anaphylaxis was
used. Compound 48/80 (8 mg/kg) was used as a model of induction for
a systemic fatal allergic reaction. After the intraperitoneal
injection of compound 48/80, the mice were monitored for 1 h, after
which the mortality rate was determined. Injection of compound
48/80 into mice induced fatal shock in 100% of the animals. When
WESC was intraperitoneally pretreated at doses ranging from 10 to
1,000 mg/kg for 1 h, the mortality was dose-dependently reduced.
WESC completely inhibited compound 48/80-induced fatal shock at
1,000 mg/kg (Table I). In
addition, the mortality of mice administered with WESC (1,000
mg/kg) 5, 10, 15 and 20 min after compound 48/80 injection
time-dependently increased (Table
II).
 | Table IEffect of WESC on compound
48/80-induced systemic anaphylaxis. |
Table I
Effect of WESC on compound
48/80-induced systemic anaphylaxis.
| WESC treatment
(mg/kg, BW) | Compound 48/80 (8
mg/kg, BW) | Mortality (%) |
|---|
| None (saline) | + | 100 |
| 10 | + | 100 |
| 100 | + | 70 |
| 500 | + | 20 |
| 1000 | + | 0 |
| 1000 | - | 0 |
| Table IITime-dependent effects of WESC on
compound 48/80-induced systemic anaphylaxis. |
Table II
Time-dependent effects of WESC on
compound 48/80-induced systemic anaphylaxis.
| WESC treatment
(mg/kg, BW) | Compound 48/80 (8
mg/kg, BW) | Time (min) | Mortality (%) |
|---|
| None (saline) | + | | 100 |
| 1000 | + | 0 | 0 |
| 1000 | + | 5 | 0 |
| 1000 | + | 10 | 30 |
| 1000 | + | 15 | 50 |
| 1000 | + | 20 | 100 |
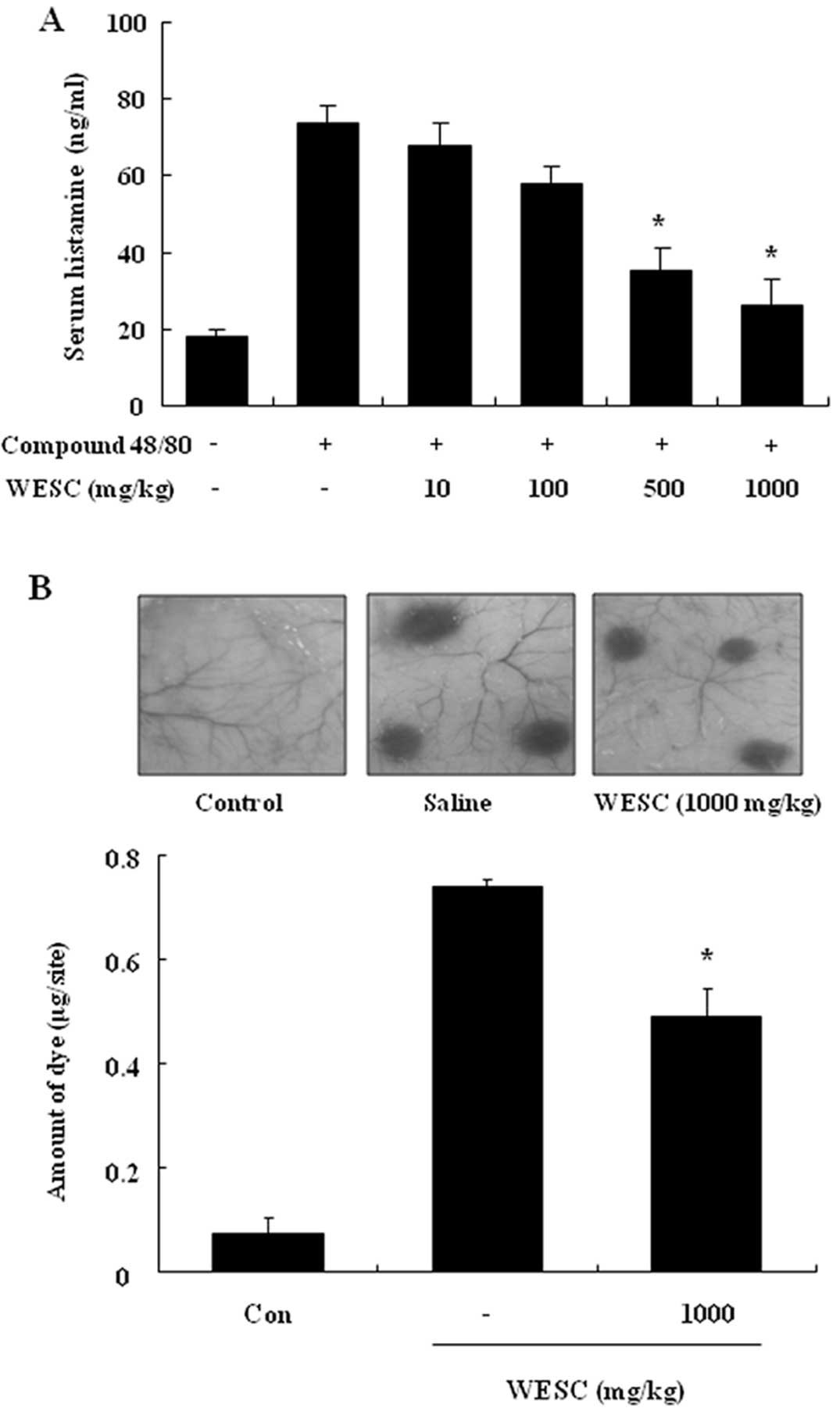
The effect of WESC on the compound 48/80-induced
serum histamine release was investigated. The histamine level
caused by compound 48/80 was decreased by WESC in a dose-dependent
manner (Fig. 1A). To confirm the
anti-allergic effects of WESC, we used a passive cutaneous
anaphylaxis (PCA) model induced by anti-DNP-IgE and DNP-HSA. To
compare to amount of dye with control, the left dorsal skin of
these mice was injected with saline alone. WESC was
intraperitoneally administered 1 h prior to the challenge with
antigen. WESC dose-dependently inhibited PCA reaction (Fig. 1B).
Effect of WESC on histamine release and
intracellular calcium
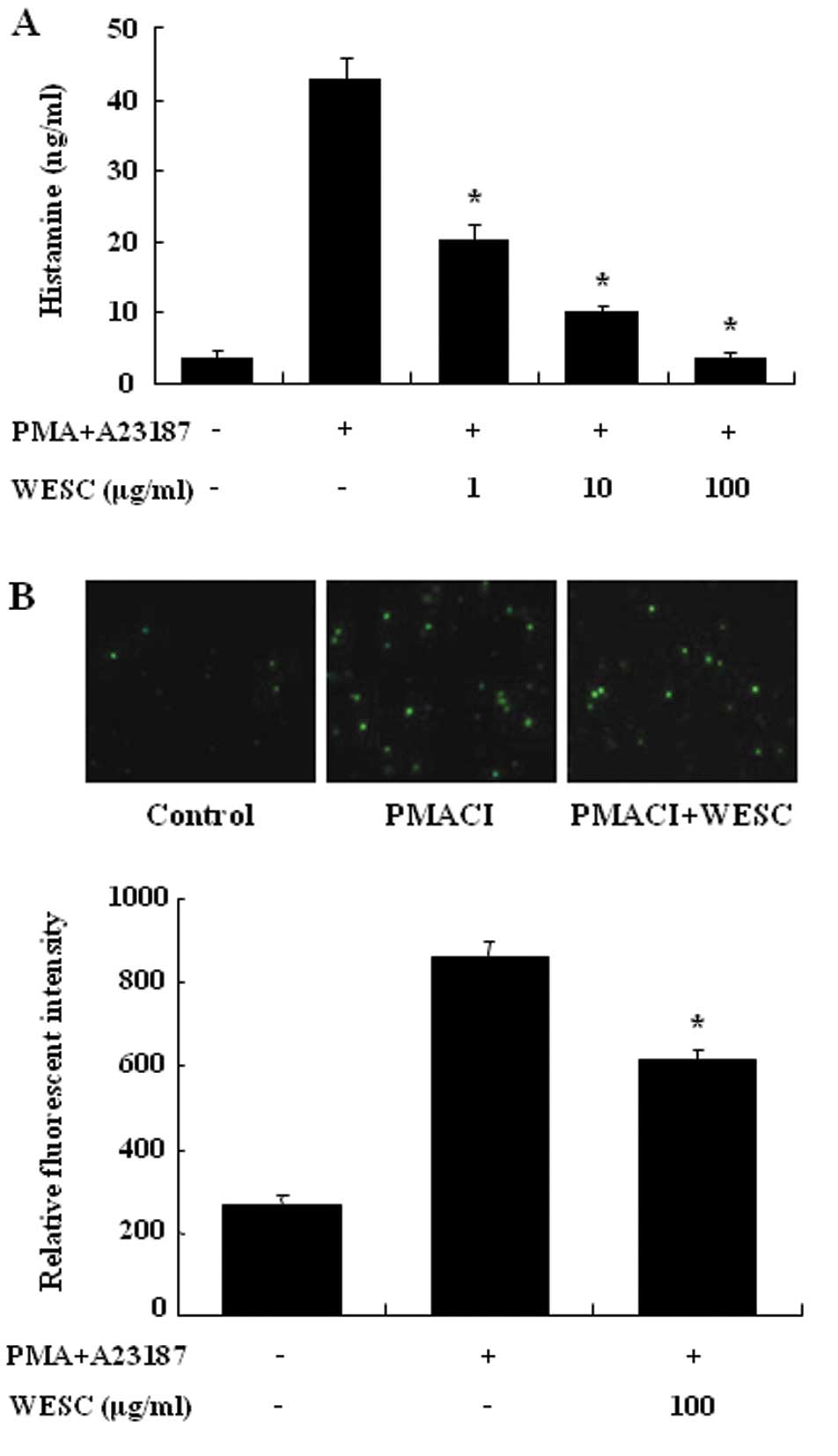
We estimated the reducing effects of WESC on the
histamine release from PMACI-induced HMC-1 cells. Mast cells
released a high level of histamine when simulated with PMACI
(Fig. 2A). When WESC was
pretreated for 30 min, histamine was dose-dependently inhibited in
PMACI-induced HMC-1. Up to 1 mg/ml of WESC did not show
cytotoxicity (data not shown). To investigate the mechanism
responsible for the reduction of histamine after WESC treatment, we
assayed the intracellular calcium levels. Calcium movements across
membranes of mast cells are critical to histamine release (17). When HMC-1 cells were stimulated
with PMACI, intracellular calcium levels were significantly
elevated (Fig. 2B). WESC (100
μg/ml) decreased the intracellular calcium level. The levels of
intracellular calcium were also assessed by the relative
fluorescent intensity.
Effect of WESC on the expression and
secretion of pro-inflammatory cytokines
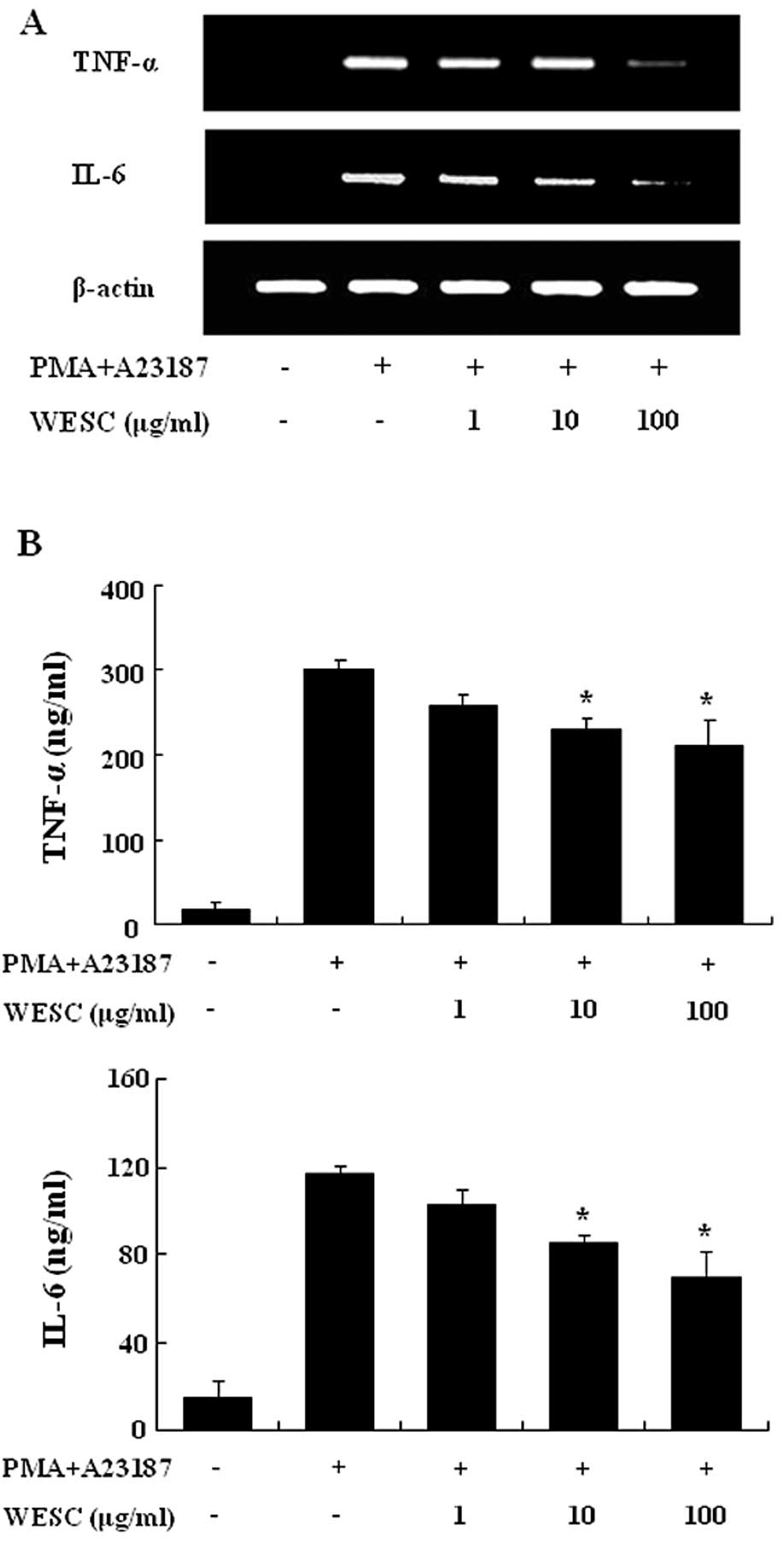
We investigated the inhibitory effect of WESC on the
expression of proinflammatory cytokines such as TNF-α and IL-6. The
HMC-1 cell line is a useful tool for researching the cytokine
activation pathway (18,19). Previously we reported that gene
expression of TNF-α and IL-6 peaked 4 h after treatment of PMACI
(20). Consequently, HMC-1 cells
were stimulated by PMACI during 4 h, and the cells were
preincubated with WESC for 30 min. Fig. 3A shows that the expression of
proinflammatory cytokines was inhibited by WESC. To confirm the
correlation of mRNA expression with protein production, we measured
the secretion of TNF-α and IL-6. When HMC-1 cells were stimulated
with PMACI for 8 h, the secretion of cytokines was remarkably
induced. WESC dose-dependently inhibited the secretion of TNF-α and
IL-6 in PMACI-stimulated HMC-1 cells (Fig. 3B).
Effect of WESC on the activation of NF-κB
and MAPKs
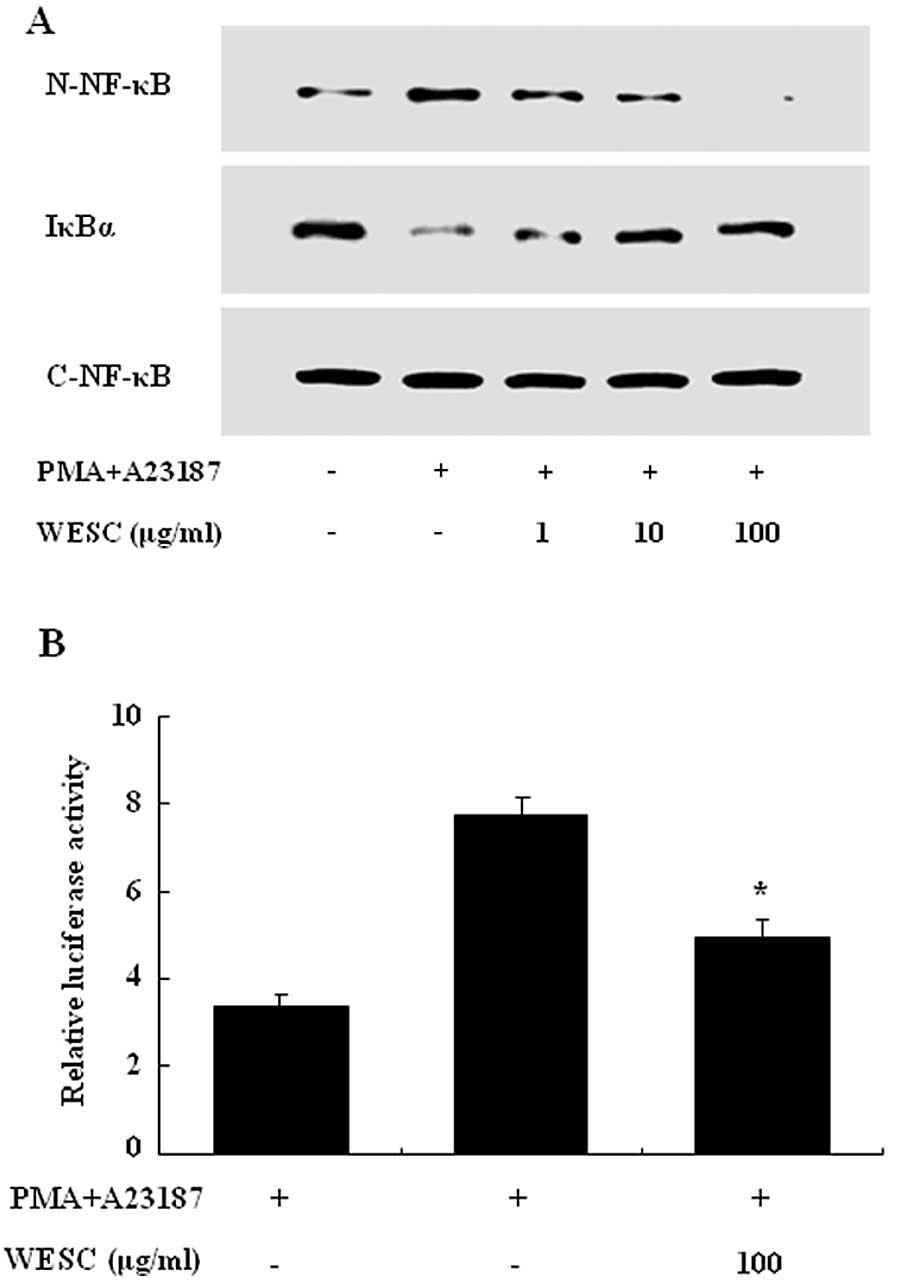
To investigate the intracellular mechanism
responsible for the inhibitory effect of WESC on the expression of
proinflammatory cytokines, we examined the effect of WESC on the
activation of transcription factors, NF-κB and MAPKs. NF-κB is an
important transcriptional regulator of inflammatory cytokines and
plays a crucial role in immune and inflammatory responses.
Stimulation of HMC-1 with PMACI induced the nuclear translocation
of p65 NF-κB and degradation of IκBα after 2 h of incubation. WESC
inhibited the PMACI-induced nuclear translocation of NF-κB and
degradation of IκBα (Fig. 4A). To
confirm the inhibitory effect of WESC on the NF-κB activation, we
examined the effect of WESC on the NF-κB-dependent gene reporter
assay. HMC-1 cells were transiently transfected with a
NF-κB-luciferase reporter construct or an empty vector. Exposure of
cells to PMACI increased the luciferase activity in the cells
transfected with the NF-κB-luciferase reporter construct (Fig. 4B). WESC significantly reduced
PMACI-induced luciferase activity.
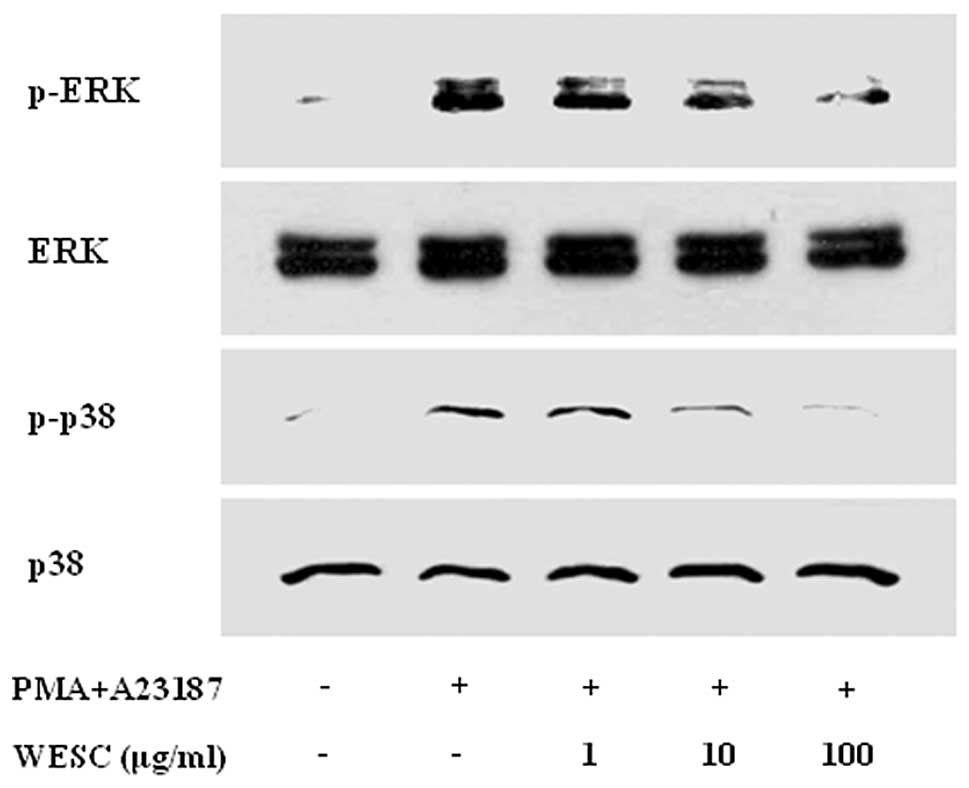
The MAPK signaling cascades also regulate important
cellular processes including gene expression, cell proliferation,
and cell survival and death (21). Previously we documented that PMACI
activates all three types of MAPKs such as p38, JNK, and ERK at
15–30 min in HMC-1 (22). In the
present results, stimulation of cells with PMACI induced
phosphorylation of p38, JNK, and ERK, and WESC markedly attenuated
PMACI-induced phosphorylation of ERK and p38 MAPK (Fig. 5). However WESC did not affect the
phosphorylation of JNK (data not shown).
Discussion
Anaphylaxis is a life-threatening syndrome induced
by a sudden systemic release of inflammatory mediators, such as
histamine, various cytokines and lipid-derived mediators (23). Using in vitro and in
vivo models, we showed that WESC has anti-allergic properties.
WESC inhibited compound 48/80-induced systemic allergic reaction
(anaphylaxis) and serum histamine release in mice. These results
indicate that mast cell-mediated immediate-type allergic reactions
are inhibited by WESC. In addition, WESC-administered mice were
protected from IgE-mediated PCA, which is one of the most important
in vivo models of anaphylaxis in a local allergic reaction.
This finding suggests that WESC might be useful in the treatment of
allergic disease, particularly skin reactions.
Histamine was originally considered as a mediator of
acute inflammatory and immediate hypersensitivity responses.
Recently, it has been reported that histamine affects chronic
inflammation and regulates several essential events of the immune
response, such as immune cell maturation, polarization, and
lymphocyte responsiveness (24).
Many reports have established that stimulation of mast cells with
compound 48/80 or IgE initiates the activation of signal
transduction pathways, which lead to histamine release. Several
studies have shown that compound 48/80 and other polybasic compound
are able, apparently directly, to activate G-proteins (25). Compound 48/80 increases the
permeability of the lipid bilayer membrane by causing a
perturbation in the membrane. These reports indicate that the
increase in membrane permeability may be an essential trigger for
the release of the mediator from mast cells. In this sense,
anti-allergic agents having a membrane-stabilizing action may be
desirable (26). WESC might
stabilize the lipid bilayer membrane, thus preventing the compound
48/80-induced membrane perturbation.
Intracellular calcium plays an important role in the
release of histamine and the expression of cytokines. Calcium
movements across the membranes of mast cells represent a major
target for efficient anti-allergic drugs, as these are essential
events linking stimulation to secretion. In our results, WESC
decreased the intracellular calcium level in mast cells. We suggest
that the decreased intracellular calcium levels may be involved in
the inhibitory effect of WESC on histamine release.
The HMC-1 cell line is one of the useful cells for
studying cytokine activation pathways (8). The various types of cytokines
produced by HMC-1 with PMACI stimulation supports the
well-recognized role of mast cells in immediate-type
hypersensitivity. TNF-α and IL-6, the known proinflammatory
cytokines, play an important role in triggering and sustaining the
allergic inflammatory response in mast cells (27,28). Mast cells are a principal source
of TNF-α in human dermis. TNF-α has major amplifying effect in
asthmatic inflammation and potently stimulates airway epithelial
cells to produce cytokines (29).
It promote inflammation, leukocyte infiltration, chemotaxis of
neutrophils and T cells (30).
IL-6 is also produced from mast cells, and its local accumulation
is associated with PCA reaction (31). These reports indicate that the
reduction of proinflammatory cytokines from mast cells is one of
the key indicators of reduced allergic symptoms. In the present
study, WESC inhibited the expression of TNF-α and IL-6 in
PMACI-stimulated mast cells. This result suggests that the
anti-allergic effect of WESC results from its inhibition of TNF-α
and IL-6 generation from mast cells.
Expression of TNF-α and IL-6 is regulated by the
activation of the transcription factor NF-κB (32). NF-κB regulates the expression of
multiple inflammatory and immune genes and plays a critical role in
chronic inflammatory diseases. Activation of NF-κB required
phosphorylation and proteolytic degradation of the inhibitory
protein IκBα, an endogenous inhibitor that binds to NF-κB in the
cytoplasm. In PMACI-stimulated mast cells, WESC decreased the
degradation of IκBα and nuclear translocation of NF-κB. The data
demonstrated that WESC attenuates activation of NF-κB and
downstream cytokine expression such as TNF-α and IL-6. To further
identify the mechanism of WESC, we evaluated the inhibitory effect
of WESC on activation of MAPKs, such as p38, JNK and ERK in
PMACI-stimulated mast cells. The MAPK cascade is one of the
important signaling pathways in immune responses. The MAPK
signaling cascades regulate important cellular processes including
gene expression, cell proliferation, cell survival and death, and
cell mobility (21). Precise
signaling pathways in allergic diseases among three types of MAPKs
such as ERK, JNK, and p38 are still unclear. However, the induction
of inflammatory cytokine genes requires activation of the p38 MAPK
and ERK (33). In our results,
WESC decreased phosphorylation of ERK and p38 MAPKs in
PMACI-stimulated mast cells. This data suggest that WESC may
decrease cytokine production and activation of NF-κB via inhibition
of ERK and p38 MAPK.
In summary, WESC significantly reduced mast
cell-mediated allergic inflammation in in vivo and in
vitro models. In the present study, we used the whole water
extract of S. crispa, not a purified single compound.
However β-glucan is already known to be the major compound of WESC.
We examined the β-glucan content in WESC using a mushroom β-glucan
assay kit. The β-glucan content in WESC was 39.3%. Therefore, we
assume that β-glucan is responsible for the anti-allergic
inflammatory effects of WESC. In conclusion, S. crispa could
contribute to prevention or treatment of mast cell-mediated
allergic inflammatory diseases.
Acknowledgements
This study was supported by the grant
of the Korea Healthcare Technology R&D Project, Ministry for
Health, Welfare and Family Affairs, Republic of Korea
(A090015).
References
|
1.
|
K YoshikawaN KokudoT HashimotoK YamamotoT
InoseT KimuraNovel phthalide compounds from Sparassis crispa
(Hanabiratake), Hanabiratakelide A-C, exhibiting anti-cancer
related activityBiol Pharm Bull3313551359201020686231
|
|
2.
|
AH KwonZ QiuM HashimotoK YamamotoT
KimuraEffects of medicinal mushroom (Sparassis crispa) on
wound healing in streptozotocin-induced diabetic ratsAm J
Surg197503509200918585672
|
|
3.
|
K YamamotoT KimuraA SugitachiN
MatsuuraAnti-angiogenic and anti-metastatic effects of
beta-1,3-D-glucan purified from Hanabiratake, Sparassis
crispaBiol Pharm Bull32259263200910.1248/bpb.32.25919182386
|
|
4.
|
R TadaT HaradaN Nagi-MiuraNMR
characterization of the structure of a beta-(1→3)-D-glucan isolate
from cultured fruit bodies of Sparassis crispaCarbohydr
Res342261126182007
|
|
5.
|
GH CaugheyMast cell proteases as
protective and inflammatory mediatorsAdv Exp Med
Biol716212234201110.1007/978-1-4419-9533-9_1221713659
|
|
6.
|
SJ GalliM TsaiAM PiliponskyThe development
of allergic
inflammationNature454445454200810.1038/nature0720418650915
|
|
7.
|
SJ GalliJ KalesnikoffMA GrimbaldestonAM
PiliponskyCM WilliamsM TsaiMast cells as ‘tunable’ effector and
immunoregulatory cells: recent advancesAnnu Rev
Immunol237497862005
|
|
8.
|
SH KimCD JunK SukGallic acid inhibits
histamine release and pro-inflammatory cytokine production in mast
cellsToxicol Sci91123131200610.1093/toxsci/kfj06316322071
|
|
9.
|
K AminThe role of mast cells in allergic
inflammationRespir
Med106914201210.1016/j.rmed.2011.09.00722112783
|
|
10.
|
DH LeeSH KimJS EunTY ShinMosla
dianthera inhibits mast cell-mediated allergic reactions
through the inhibition of histamine release and inflammatory
cytokine productionToxicol Appl
Pharmacol216479484200610.1016/j.taap.2006.06.007
|
|
11.
|
M TagenA ElorzaD KempurajMitochondrial
uncoupling protein 2 inhibits mast cell activation and reduces
histamine contentJ
Immunol18363136319200910.4049/jimmunol.080342219846869
|
|
12.
|
Y GwackS FeskeS SrikanthPG HoganA
RaoSignalling to transcription: store-operated Ca2+
entry and NFAT activation in lymphocytesCell
Calcium42145156200710.1016/j.ceca.2007.03.00717572487
|
|
13.
|
Y BaeS LeeSH KimChrysin suppresses mast
cell-mediated allergic inflammation: involvement of calcium,
caspase-1 and nuclear factor-kappaBToxicol Appl
Pharmacol2545664201110.1016/j.taap.2011.04.00821515303
|
|
14.
|
SH KimS LeeIK KimSuppression of mast
cell-mediated allergic reaction by Amomum xanthiodesFood
Chem Toxicol4521382144200710.1016/j.fct.2007.05.01117602813
|
|
15.
|
S LeeHS YunSH KimThe comparative effects
of meso-porous silica nanoparticles and colloidal silica on
inflammation and
apoptosisBiomaterials3294349443201110.1016/j.biomaterials.2011.08.04221889200
|
|
16.
|
S LeeK SukIK KimSignaling pathways of
bisphenol A-induced apoptosis in hippocampal neuronal cells: role
of calcium-induced reactive oxygen species, mitogen-activated
protein kinases, and nuclear factor-kappaBJ Neurosci
Res8629322942200810.1002/jnr.21739
|
|
17.
|
M EisenhutH WallaceIon channels in
inflammationPflugers
Arch461401421201110.1007/s00424-010-0917-y21279380
|
|
18.
|
TY ShinSH KimK SukAnti-allergic effects of
Lycopus lucidus on mast cell-mediated allergy modelToxicol
Appl Pharmacol209255262200510.1016/j.taap.2005.04.01115936049
|
|
19.
|
GY KimJW LeeHC RyuJD WeiCM SeongJH
KimProinflammatory cytokine IL-1beta stimulates IL-8 synthesis in
mast cells via a leukotriene B4 receptor 2-linked pathway,
contributing to angiogenesisJ
Immunol18439463954201010.4049/jimmunol.090173520194723
|
|
20.
|
HH ParkS LeeJM OhAnti-inflammatory
activity of fisetin in human mast cells (HMC-1)Pharmacol
Res553137200710.1016/j.phrs.2006.10.00217079162
|
|
21.
|
S ArbabiRV MaierMitogen-activated protein
kinasesCrit Care Med30Suppl
1S74S79200210.1097/00003246-200201001-00010
|
|
22.
|
SH KimCH ChoiSY KimJS EunTY
ShinAnti-allergic effects of Artemisia iwayomogi on mast
cell-mediated allergy modelExp Biol Med
(Maywood)2308288200515618130
|
|
23.
|
SR BodenA Wesley BurksAnaphylaxis: a
history with emphasis on food allergyImmunol
Rev242247257201110.1111/j.1600-065X.2011.01028.x21682750
|
|
24.
|
M JutelK BlaserCA AkdisHistamine in
allergic inflammation and immune modulationInt Arch Allergy
Immunol1378292200510.1159/00008510815832054
|
|
25.
|
VA PalomakiJT LaitinenThe basic
secretagogue compound 48/80 activates G proteins indirectly via
stimulation of phospholipase D-lysophosphatidic acid receptor axis
and 5-HT1A receptors in rat brain sectionsBr J
Pharmacol147596606200610.1038/sj.bjp.070667116415902
|
|
26.
|
SY KimSH KimHY ShinEffects of Prunella
vulgaris on mast cell-mediated allergic reaction and
inflammatory cytokine productionExp Biol Med
(Maywood)2329219262007
|
|
27.
|
LJ WalshG TrinchieriHA WaldorfD WhitakerGF
MurphyHuman dermal mast cells contain and release tumor necrosis
factor alpha, which induces endothelial leukocyte adhesion molecule
1Proc Natl Acad Sci USA8842204224199110.1073/pnas.88.10.4220
|
|
28.
|
KD StoneC PrussinDD MetcalfeIgE, mast
cells, basophils, and eosinophilsJ Allergy Clin Immunol125Suppl
2S73S80201010.1016/j.jaci.2009.11.01720176269
|
|
29.
|
SJ GalliJR GordonBK WershilCytokine
production by mast cells and basophilsCurr Opin
Immunol3865872199110.1016/S0952-7915(05)80005-61793528
|
|
30.
|
PS ThomasTumour necrosis factor-alpha: the
role of this multifunctional cytokine in asthmaImmunol Cell
Biol79132140200110.1046/j.1440-1711.2001.00980.x11264706
|
|
31.
|
JA MicanN AroraPR BurdDD MetcalfePassive
cutaneous anaphylaxis in mouse skin is associated with local
accumulation of interleukin-6 mRNA and immunoreactive interleukin-6
proteinJ Allergy Clin
Immunol90815824199210.1016/0091-6749(92)90107-D1430707
|
|
32.
|
M KarinNF-kappaB as a critical link
between inflammation and cancerCold Spring Harb Perspect
Biol1a000141200910.1101/cshperspect.a00014120066113
|
|
33.
|
C DongRJ DavisRA FlavellMAP kinases in the
immune responseAnnu Rev
Immunol205572200210.1146/annurev.immunol.20.091301.13113311861597
|