Introduction
AML is an aggressive malignancy distinguished from
normal hematopoietic cells by key properties, including blocked
differentiation, enhanced self-renewal and increased proliferation.
Although AML treatment is among the most dose-intensive approaches
in the clinic, AML-related mortality remains high. Acute
erythroleukemia is an extremely rare subtype of AML, associated
with an extremely poor response to currently available therapeutic
agents as well as survival rate, compared to other subtypes
(1). Given the difficulties in
well-tolerance treatment, novel chemotherapeutic agents and more
effective therapies are urgently needed for the treatment of acute
erythroleukemia.
NF-κB, a member of the transcription factors family,
is widely expressed in almost every cell, while being crucial in
several signal transduction pathways (2). A common form of NF-κB is a
heterodimer, consisting of p65 (RelA) and p50, present in the
cytoplasm of unstimulated cells as an inactive IκBα-bound
formation. It may act both as a pro- and an anti-apoptotic
transcription factor through the regulation of different target
genes, such as Bcl-2, Bcl-xL, JNK kinase and p53 (2,3).
Although NF-κB activation is often associated with increased
survival of cancer cells and resistance to chemotherapy, there is
also evidence that NF-κB activation sensitizes cells to apoptosis
(4,5).
Nucleoporin 88 (Nup88) is a non-FG nucleoporin found
exclusively on the cytoplasmic face of the nuclear pore complex
(NPC) that mediates nucleocytoplasmic trafficking of macromolecules
(6). In tumors, Nup88 staining is
prominent in the cytoplasm, often in granular dots (7,8).
Furthermore, its expression levels are highly correlated with the
metastasis and mortality rates of colon cancer (9). Additionally, Nup88 is reported to be
closely associated with CAN/Nup214, another nucleoporin and a
proto-oncogene involved in leukemia (10). A study carried out on
Drosophila reported that Drosophila larvae lacking
either Nup214 or Nup88 showed a notable cytoplasmic accumulation of
the NF-κB homologs (the NES-containing Dorsal and Dif) that
normally shuttle between the nucleus and the cytoplasm. The FG
repetition domain of Nup214 as well as the N-terminal β-propeller
domain of Nup88 bind directly to CRM-1/exportin-1, the receptor
exporting most proteins from the nucleus.
In the present study, the effects of oridonin on
OCIM2 cells were investigated. OCIM2 was found to be relatively
resistant to oridonin treatment. Oridonin inhibited OCIM2 growth
and induced OCIM2 cell apoptosis by upregulating p65 and its target
gene Bax and activating apoptosis executioners including
caspases-6, -9 and -3, while the downregulation of Nup214 and Nup88
may protect OCIM2 by regulating the nucleocytoplasmic transport of
p65.
Materials and methods
Reagents
Oridonin of more than 98% purity (empirical formula,
C20H28O6; molecular weight,
360.42), purchased from Sigma-Aldrich (St. Louis, MO, USA), was
initially dissolved in dimethyl sulfoxide (DMSO), stored at −20°C
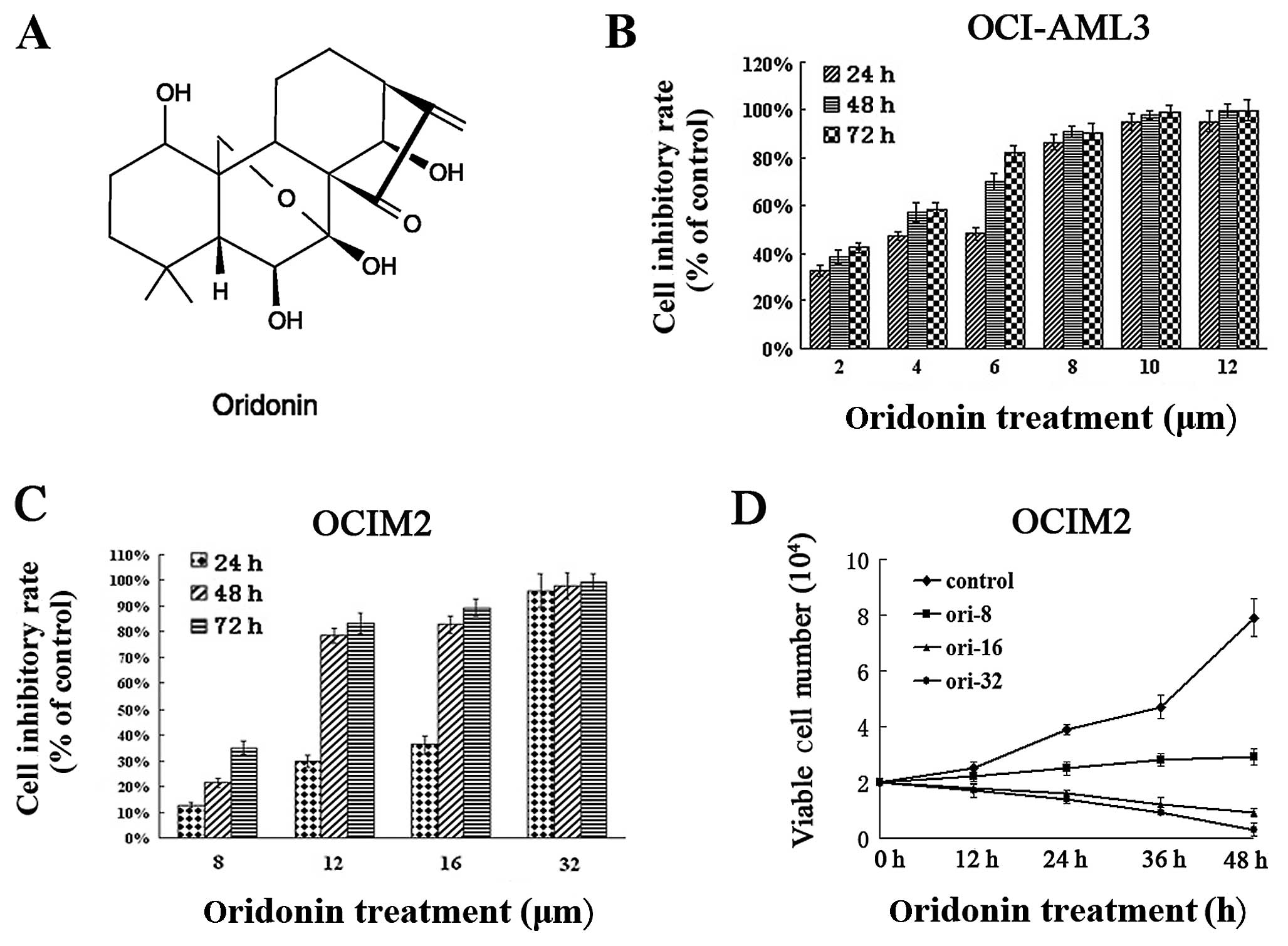
and thawed before use (Fig. 1A).
RPMI-1640 medium was purchased from Gibco-BRL (Gaithersburg, MD,
USA). Fetal bovine serum (FBS) was purchased from Shijiqing
(HangZhou, China). DMSO, trypan blue, Hoechst 33258 and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT)
were purchased from Sigma-Aldrich. RNA TRIzol reagents were
purchased from Invitrogen Life Technologies (Carlsbad, CA, USA) and
the RT-PCR kit was obtained from Toyobo Biologics (Osaka, Japan).
Primary antibody caspases-3, -6 and Bax were purchased from Cell
Signaling Technology (Danvers, MA, USA). Primary antibody of Nup88
was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). Primary antibodies of p65 and caspase-9 were purchased from
Proteintech Group (Chicago, IL, USA). Nup214 was purchased from
Abcam (Abcam, San Francisco, CA, USA). Horseradish peroxidase
(HRP)-labeled secondary antibodies and γ-tubulin antibody were
purchased from Jackson ImmunoResearch (Jackson, WY, USA). ECL
western blotting detection reagents and analysis system was
purchased from Pierce Biotechnology, Inc. (Rockford, IL, USA).
Cell lines and culture
The OCI-AML3 and OCIM2 were kindly provided by MD
Minden (Ontario Cancer Institute, Toronto, ON, Canada). OCI/AML3
(FAB M4) was obtained from a patient with NPM mutation and OCIM2
(FAB-M6) from a patient with erythroleukemia. The cells were grown
in RPMI-1640 and supplemented with 10% FBS. Cultures were
maintained in a humidified atmosphere with 5% CO2 at
37°C.
Cell proliferation assay
The anti-proliferative effect of oridonin on the
OCIM2 and OCI-AML3 cells was determined using MTT assay, according
to the Mosmann method. The OCIM2 and OCI-AML3 cells
(1×104/well) were exposed to varying concentrations of
oridonin (0–32 μmol/l) for 24, 48 and 72 h on 96-well
plates. Thereafter, 20 μl MTT solutions (5 mg/ml in PBS)
were added to each well. Subsequent to 4-h incubation, the
supernatant was discarded and 150 μl of DMSO was added. Once
the blue crystals were dissolved, the optical density (OD) was
measured at 490 nm using a 96-well multiscanner autoreader (BioTek
Instruments, Winooski, VT, USA). The inhibition of cell
proliferation was determined using the formula: inhibition of cell
proliferation (%) = [1−OD of the experimental samples/OD of the
control)] ×100% (n=3, mean ± SD). The half maximal inhibitory
concentration (IC50) was defined as the oridonin
concentration that causes 50% inhibition of cell proliferation.
Trypan blue dye exclusion assay
To assess the cytotoxicity of oridonin on OCIM2, the
cells were treated with varying concentrations of oridonin (0, 8,
16 and 32 μmol/l) for 12, 24, 36 and 48 h, and were then
stained using the Trypan Blue Staining Cell Viability Assay
(Beyotime, Haimen, China). Trypan blue staining was evaluated under
the microscope and cells were counted using a hemocytometer. Cells
that took up trypan blue were counted as dead cells.
Flow cytometry for apoptosis
Immunofluorescence flow cytometry was employed to
evaluate the apoptosis-inducing effect of oridonin on the OCIM2
cells. The cells seeded on 6-well plates (5×105/ml) were
treated with varying concentrations of oridonin (0, 2, 8 and 16
μmol/l) for 24 h. The cells were harvested and washed with
cold PBS, and then resuspended in 100 μl binding buffer.
Subsequently, phosphatidyl serine was detected on the surface of
apoptotic cells using Annexin V/FITC and a PI apoptosis detection
kit, following the manufacturer’s instructions (Bender MedSystems,
Inc., Burlingame, CA, USA). The number of cells in apoptosis was
evaluated by FACS flow cytometry (BD Biosciences, San Diego, CA,
USA). A population of at least 10,000 cells was analyzed.
Hoechst 33258 staining assay
Nuclear fragmentation was visualized by Hoechst
33258 staining of apoptotic nuclei. Cells treated with varying
concentrations of oridonin (0, 2, 8 and 16 μmol/l) for 24 h
were collected and washed, and then fixed for 10 min prior to
deposition onto polylysine-coated coverslips. Subsequently, the
samples were permeabilized with 0.25% Triton X-100 for 5 min and
stained with 1 μg/ml Hoechst 33258 for 30 min at 37°C. The
slides were mounted with glycerol-PBS, and the images were
visualized and captured using a FV500 confocal microscope (Olympus,
Tokyo, Japan).
Western blot analysis
Western blot analysis was used to evaluate the
levels of Bax, caspases-3, -6, -9, Nup88, Nup214 and p65 in the
OCIM2 cells exposed to oridonin. The OCIM2 cells (1×106)
were seeded and treated in vitro with different
concentrations of oridonin (0, 2, 8 and 16 μmol/l) for 24 h.
Cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.5; 150 mM
NaCl; 1% NP-40; 0.5% sodium deoxycholate; 0.1% SDS) containing
complete protease inhibitor cocktail (Calbiochem, San Diego, USA).
Nuclear and cytosolic extracts were prepared according to the
manufacturer’s instructions (Beyotime). The sample lysates (80
μg of protein/lane) were prepared, and the proteins were
separated by 10% polyacrylamide gel. The proteins were
electro-transferred onto nitrocellulose (NC) membranes and the
blots were incubated with a blocking solution (5% skimmed milk in
Tris-buffered saline Tween-20 buffer) for 2 h at room temperature.
The membranes were then incubated using the dilutions
(1:500–1:1,000) of the primary antibodies. The primary antibodies
were washed and incubated with secondary antibodies for 2 h at room
temperature. The bound antibodies were visualized using a
supersignal chemiluminescence detection (ECL) kit (Pierce
Biotechnology, Inc., Rockford, IL, USA). The signals were detected
using a chemiluminescence detection system (Bio-Rad, Hercules, CA,
USA). The results were expressed as the relative density of the
protein normalized to γ-tubulin. Enhancement or inhibition rates
were estimated by comparison with the control (100%).
Quantitative real-time RT-PCR
analysis
Total RNA was isolated from each group of OCIM2
cells treated with 0, 2, 8 and 16 μmol/l oridonin for 24 h,
according to the manufacturer’s instructions. Total RNA (2
μg) was reverse transcribed into cDNA, according to the
manufacturer’s instructions using the TOYOBO kits. The analysis was
carried out using SYBR-Green PCR master mix. Real-time RT-PCR
amplification was performed in an ABI Prism 7900HT Sequence
Detection System (Applied Biosystems, Foster City, CA, USA).
Samples were run in triplicates in three independent experiments.
All the threshold cycle (CT) values were normalized with β-actin in
the same master reaction. The 2−ΔΔCT (ΔΔ threshold
cycle) method of analysis was used to relatively quantify the
transcriptional levels of studied genes. Specific primers were used
for human Nup88, forward: 5′-GGA GCT TGC TTT GAA ACT GG-3′ and
reverse: 5′-ATT TCC CGC AGA CTT TTC CT-3′; Nup214, forward: 5′-AGT
CCT CAG TCT TGC CCT CA-3′ and reverse: 5′-CGA TTG TTG GCT AGG GTG
TT-3′; β-actin, forward: 5′-GCC CAG TCC TCT CCC AAG TC-3′ and
reverse: 5′-GGC ACG AAG GTC ATC ATT C-3′.
Immunofluorescence staining
An immunofluorescence staining assay was performed
to evaluate the oridonin-induced enhancement and translocation
effect of p65 on OCIM2 cells. The OCIM2 cells were exposed to
varying concentrations of oridonin. After 24 h, the cells were
fixed with 4% paraformaldehyde for 10 min and permeabilized with
0.25% Triton X-100 for 5 min. The cells were then washed twice with
PBS, blocked in 3% bovine serum albumin and then deposited onto
polylysine coated coverslips. The rabbit polyclonal anti-p65
antibody was diluted to 1:100 and non-immunoreactive IgG was added
as the negative control. Subsequent to washing, the cells were
incubated with Cy3-labeled goat anti-rabbit secondary antibody
(Pierce Biotechnology, Rockford, IL, USA), diluted in PBS for 2 h
and stained with Hoechst 33258 (10 μg/ml) for 10 min. The
image was visualized and captured using a FV500 confocal microscope
(Olympus). The p65 levels were estimated as the mean fluorescence
intensity after subtracting those of the negative control
cells.
Statistical analysis
Data were presented as the mean ± SD of at least
three independent experiments. The t-tests were used to determine
the differences between the treated samples and the controls.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of oridonin on the proliferation
and growth of AML cells
Initially, the effect of oridonin on the
proliferation of AML cells was examined. OCI-AML3 and OCIM2 were
cultured in varying concentrations of oridonin, while the
inhibitory effects of oridonin were examined by MTT, indicating the
mitochondrial activity of the cells. Results in Fig. 1 show that oridonin had a dose- and
time-dependent anti-proliferative effect on the OCI-AML3 (Fig. 1B), as well as on the OCIM2 cells
(Fig. 1C), subsequent to 24-, 48-
and 72-h exposure. The number of viable cells decreased with the
increasing concentration of oridonin over the exposure. The
IC50 value of OCIM2 at 24 h was 15.07±1.34
μmol/l, while that of OCI-AML3 was only 3.27±0.23
μmol/l. As the period of exposure increased, the
IC50 values gradually decreased. The IC50
values in OCIM2 cells after 48 and 72 h were 10.12±1.24 and
8.83±0.94 μmol/l, respectively. The values were much lower
(3.18±0.42 and 3.13±0.23 μmol/l) in OCI-AML3 cells.
The anti-proliferative effects of oridonin on OCIM2
cells were also examined, using trypan dye exclusion. As shown in
Fig. 1D, oridonin suppressed the
vitality of OCIM2 within 48 h, in a dose-dependent manner. These
results showed that oridonin suppressed the proliferation of the
tested AML cell lines, however, the oridonin concentration exerting
its effect on the OCIM2 cells, was much higher compared to the
concentration in OCI-AML3 cells.
Oridonin in a relatively high
concentration induced OCIM2 cell-apoptosis
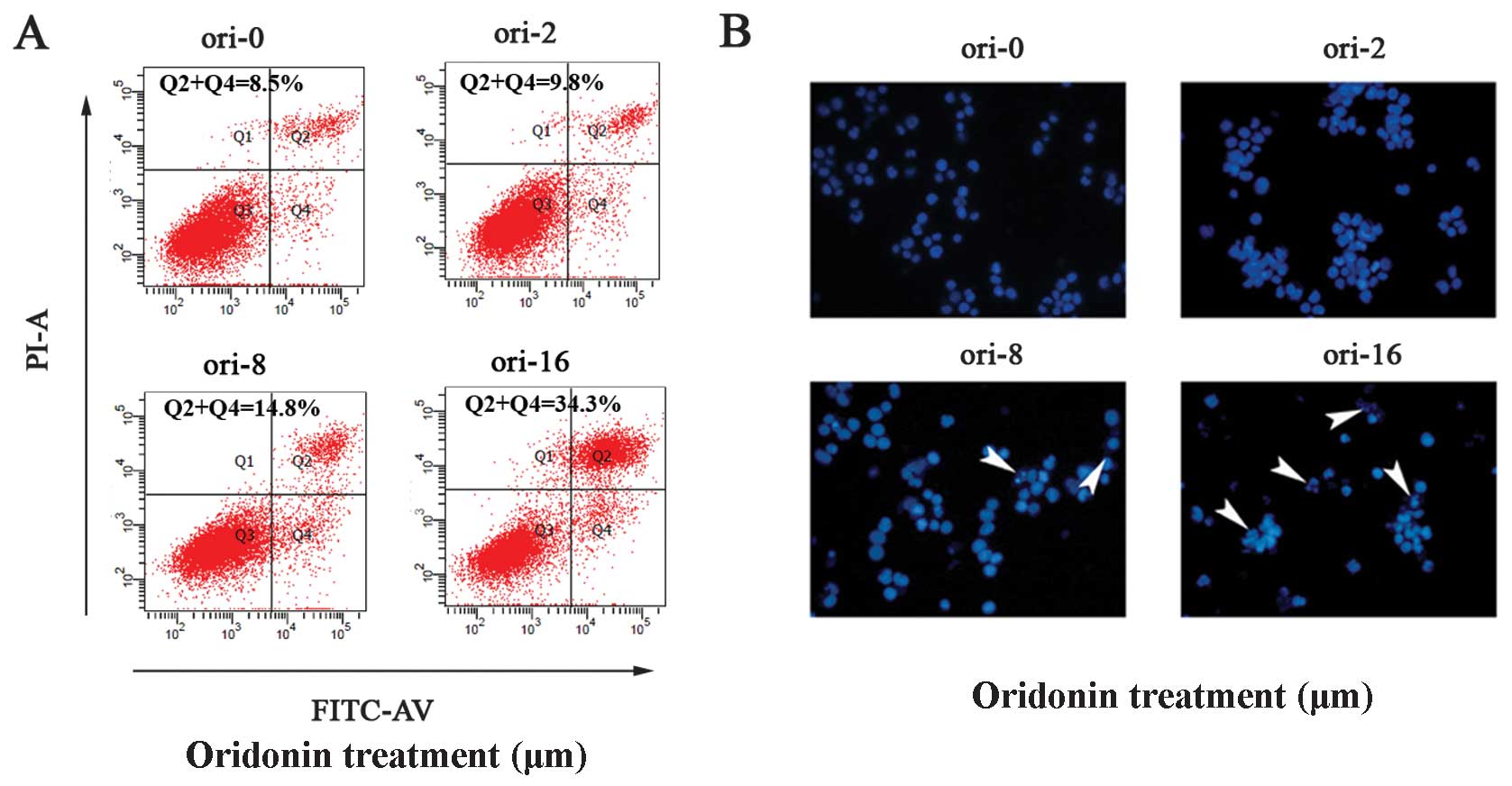
We investigated whether oridonin led to OCIM2
cell-apoptosis using the Annexin V/PI dual stain method. Annexin V
binds to those cells that express phosphatidylserine on the outer
layer of the cell membrane, a characteristic feature of cells
entering apoptosis. To check this, OCIM2 cells were treated with
different concentrations of oridonin for 24 h and then stained with
Annexin V-FITC and PI. Results in Fig. 2A show a dose-dependent increase in
Annexin V-positive cells, indicating the onset of apoptosis in
oridonin-treated cells.
In addition, the effect of oridonin on OCIM2 cell
apoptosis was observed subsequent to Hoechst 33258 staining, under
laser scanning confocal microscope. Nuclei in the control cells
were regular in shape. These cells decreased in size and darkened
subsequent to oridonin treatment, while the nuclei presented
chromatin condensation and marginalization or nuclear beading. The
number of apoptotic cells increased with the concentration, while
more typical morphological features appeared (8 and 16
μmol/l) (Fig. 2B).
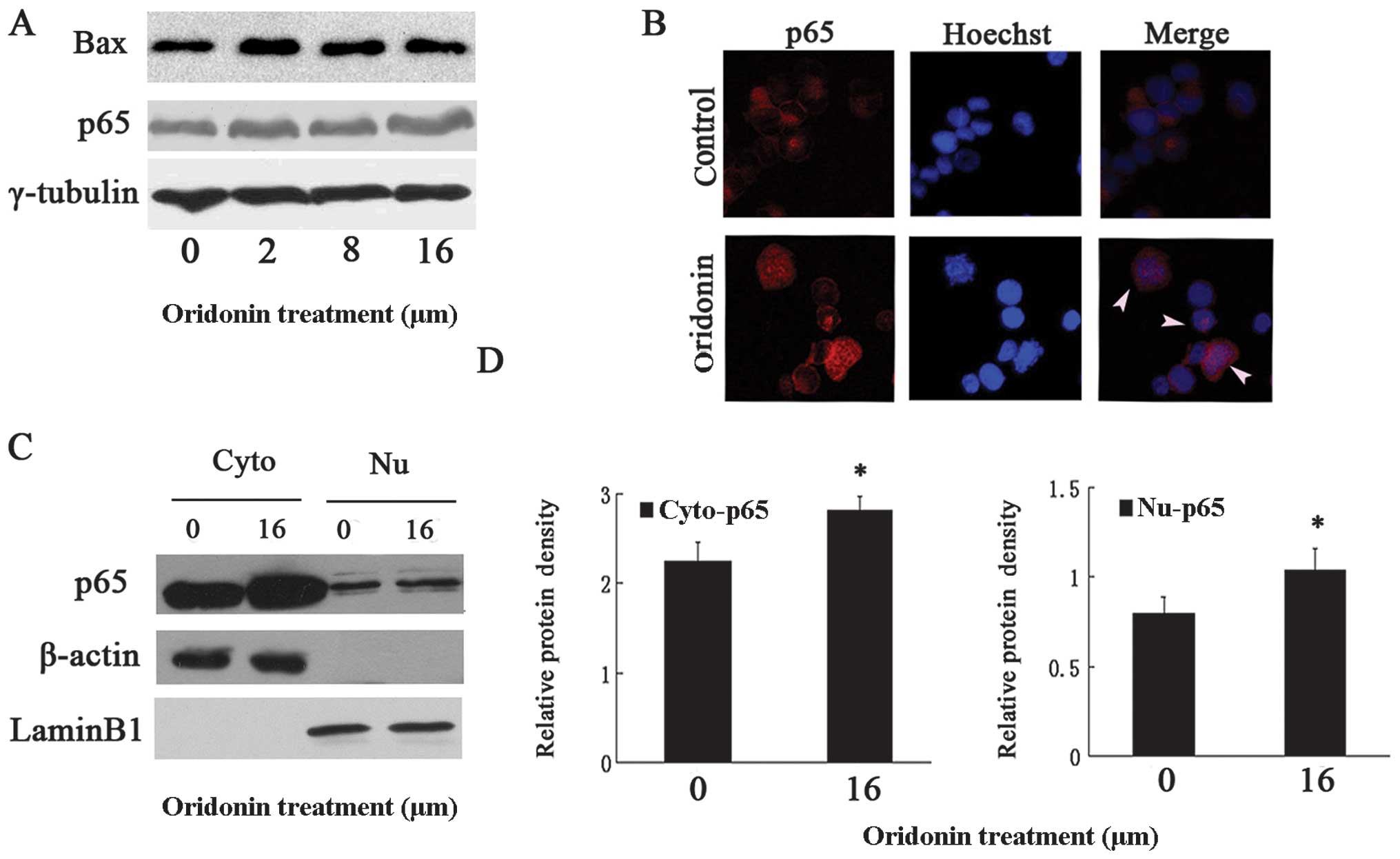
Oridonin-induced activated NF-κB was
partially blocked in the cytoplasm of OCIM2 cells
Since studies have indicated (4,5)
that the NF-κB pathway was involved in oridonin-induced apoptosis,
we examined whether or not NF-κB was activated by oridonin. OCIM2
cells were treated with varying concentrations of oridonin, while
their protein extracts were monitored for p65 expression, using
immuno-blotting. Results in Fig.
4A show that the total p65 increased in a dose-dependent manner
(P<0.05 vs. untreated control). While in the inactive state the
p65 subunit was retained in the cytoplasm, in an activated NF-κB
the p65 subunit containing the transactivation domain was
translocated into the nucleus. Therefore, to confirm that oridonin
translocated p65 from the cytoplasm to the nucleus,
immunofluorescence staining was used. The results in Fig. 4B clearly show that the p65 subunit
of NF-κB translocated to the nucleus in the oridonin-treated group
and especially in the typically apoptotic OCIM2 cells. In addition,
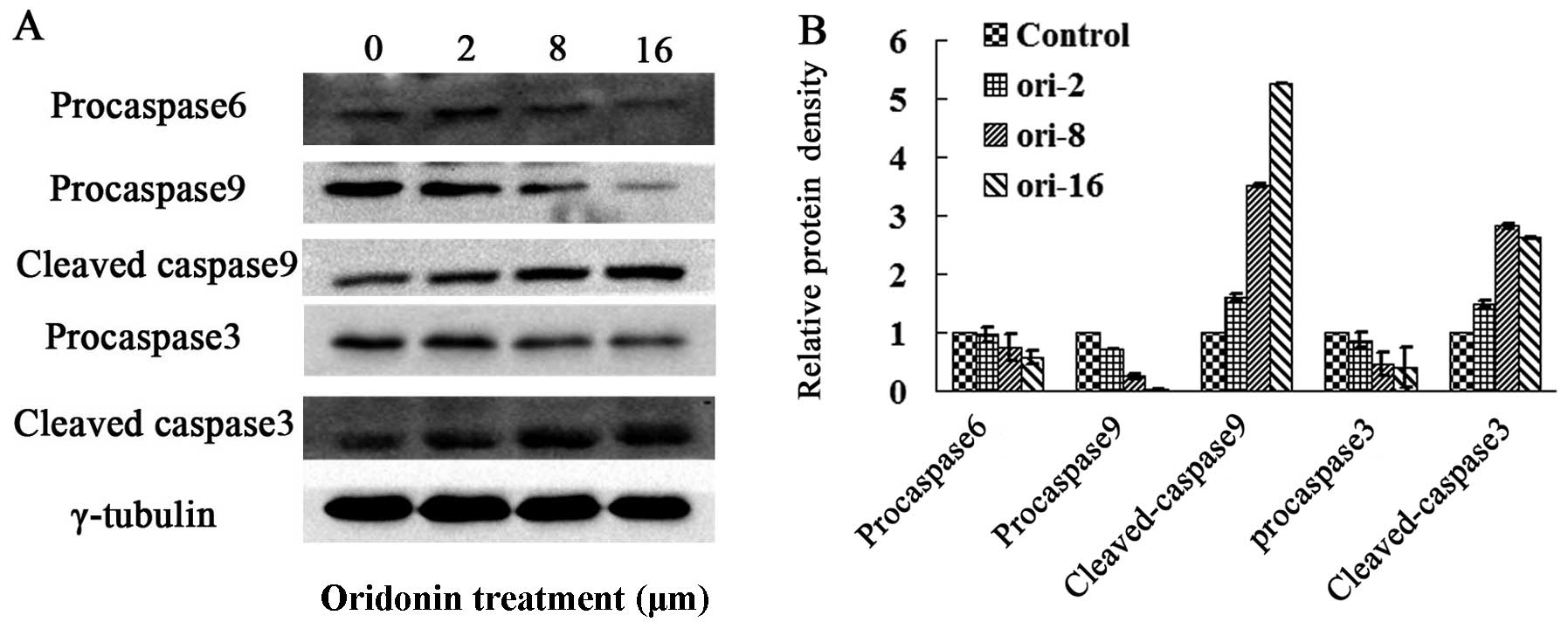
subsequent to oridonin treatment the protein level of Bax increased
accordingly (Fig. 4A). The
downstream caspase effectors of Bax, such as caspases-9 and -3 were
detected by western blot analysis. As shown in Fig. 3, oridonin significantly induced
the activation of caspases-9 and -3 while decreasing in the protein
level of pro-caspases-9 and -3 and increasing in the protein levels
of cleaved forms in OCIM2 cells. In addition, oridonin treatment
downregulated the protein level of pro-caspase-6, while indicating
an increased immunofluorescence p65 intensity in the cytoplasm. To
confirm a partially-retained p65 in the cytoplasm, the nuclear and
cytosolic extracts of untreated and oridonin-treated cells were
prepared and examined for p65 expression. Consistent with the
results in Fig. 4B, results in
Fig. 4C show that the cytoplasm
p65 content increased in the oridonin-treated OCIM2 cells. As shown
in Fig. 4D, there were
statistically significant differences in p65 expression in the
cytoplasm and nucleus in the oridonin-treated and control
groups.
Partial translocation of p65 might be
regulated by Nup88 and Nup214
OCIM2 cells were markedly more resistant to oridonin
compared to other AML cells, albeit oridonin had the potential to
induce OCIM2 cell-apoptosis. In addition, p65 was observed to be
retained in the cytoplasm of oridonin-treated OCIM2 cells, while
the nucleocytoplasmic transport of p65 was reported to be
selectively mediated by the Nup88/214 complex. We hypothesized that
oridonin downregulated Nup88 as well as Nup214, while possibly
inhibiting the translocation of p65 from the cytoplasm to the
nucleus. To confirm the effect of oridonin on the Nup88 and Nup214
expressions, immunoblotting was employed on the OCIM2 cells treated
with varying concentrations of oridonin as well as on their protein
extracts, with a view to detect Nup88 and Nup214 expression.
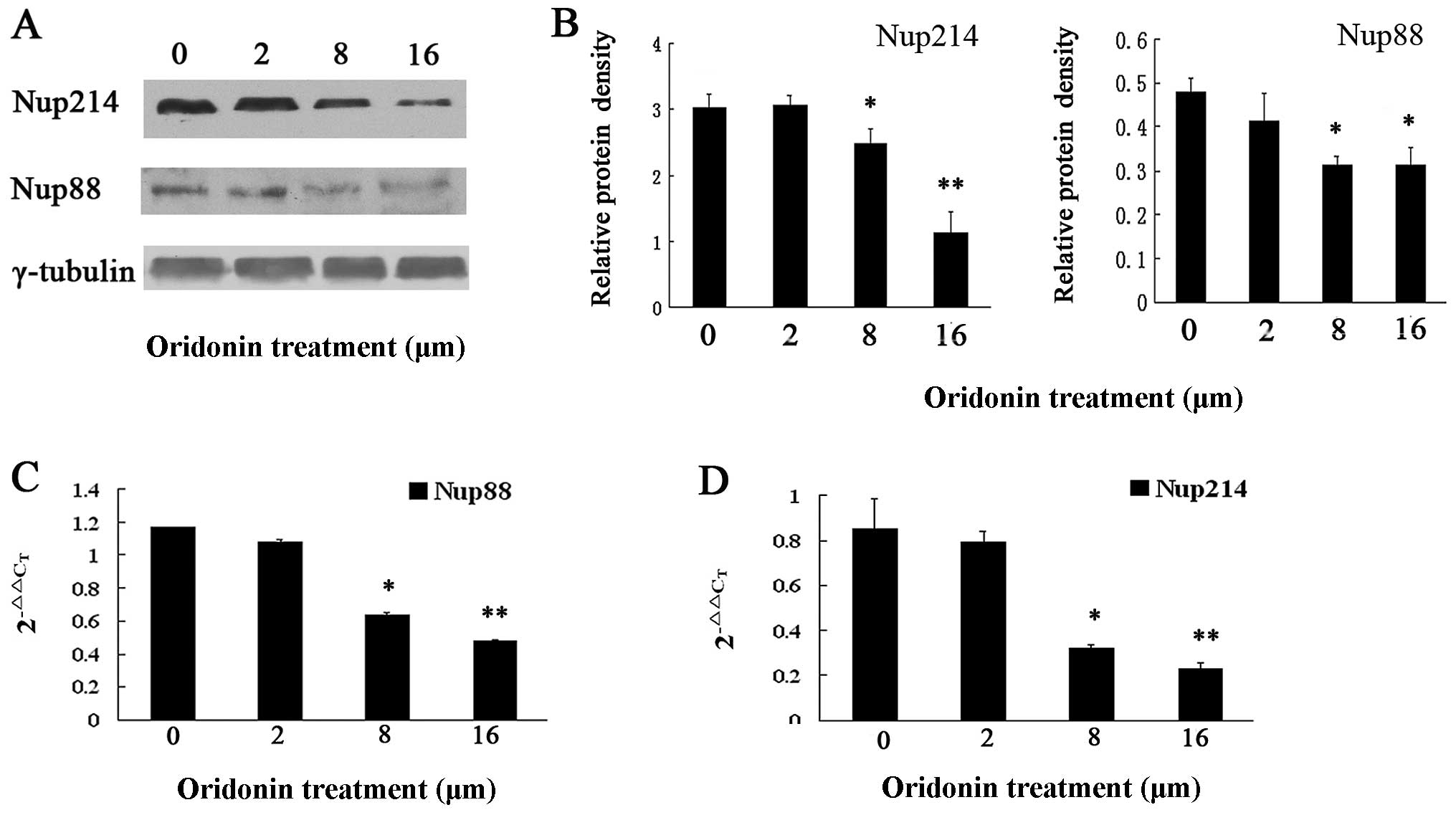
Results in Fig. 5A show that the
Nup88 and Nup214 expressions decreased in a dose-dependent
manner.
To determine whether oridonin affected the
transcriptional expression of Nup88 and Nup214, total RNA was
extracted from untreated and oridonin-treated OCIM2 cells and
Nup88, while Nup214 mRNA was analyzed by quantitative real-time
PCR. Consistent with the results of immunoblotting, results shown
in Fig. 5 demonstrate that the
2−ΔΔCT of Nup88 (Fig.
5C) and Nup214 (Fig. 5D) were
significantly decreased in the oridonin-treated, compared to the
untreated cells.
Discussion
Oridonin, an active anti-cancer diterpenoid, is a
well-known and widely used traditional Chinese medicament. Previous
reports have demonstrated that oridonin had significant inhibitory
effects on leukemia cells, including NB4, HL-60, U937, kausima-1,
K562 and primary AML, both in vitro and in vivo
(11–14). Although Zhou et al
(12) found that oridonin-induced
AML cell apoptosis (8,21) through targeting the AML1-ETO (AE)
fusion protein that is crucial in leukemogenesis, data regarding
the potential of oridonin to induce apoptosis in erythroleukemia,
as well as the precise underlying molecular mechanisms are scarce.
In the present study, oridonin was found to have the potential to
inhibit cell proliferation in the AML cell lines OCI-AML3 and
OCIM2, when increasing treatment concentration. Subsequent to
Hoechst 33258 staining, oridonin-treated OCIM2 cells demonstrated
typical morphological changes, including nuclear chromatin
condensation and a crescent shape. The number of apoptotic cells
increased with the increased drug dose, while nuclei broke into
fragments and formed apoptotic bodies. Flow cytometry-detected
apoptosis showed a gradually increasing population of apoptotic
cells. Based on these findings, a series of experiments were
carried out to further determine potential apoptotic signaling
pathways.
NF-κB regulates genes involved in cell survival,
proliferation and apoptosis (15). Activation of NF-κB mediates cell
survival signals in most tumor cells, acting simultaneously as a
pro-apoptotic factor under certain conditions (4,5).
Results in the present study have shown that NF-κB was activated by
oridonin in a dose-dependent manner, while the p65 translocated to
the nucleus in the oridonin-treated group, especially in the
typically apoptotic OCIM2 cells, indicating that NF-κB promoted
apoptosis in this study. Furthermore, oridonin treatment with
varying concentrations was found to have upregulated the Bax
expression, in accordance with the increasing NF-κB level.
Members of the Bcl-2 family, such as Bax, being one
of the target genes of NF-κB are crucial in the mitochondria
pathway. The pro-apoptotic member Bax translocated to the
mitochondria and integrated into the outer mitochondrial membrane,
facilitating the release of cytochrome c into the cytosol
(16) and activating caspase-9,
and subsequently cleaving and activating downstream effector
proteases, such as caspase-3, resulting in apoptosis. In
particular, caspase-3 has been shown to have a pivotal role in the
terminal and execution phases of apoptosis induced by diverse
stimuli (17). To elucidate
whether Bax was involved in oridonin-induced apoptosis, caspase-9
expression and its downstream caspase were detected in
oridonin-treated OCIM2 cells. The present study indicated that
oridonin treatment with varying concentrations cleaved and
activated caspase-9. Subsequently, caspase-3 was activated with an
increase in the cleaved caspase-3 protein, and a decrease in the
protein levels of the pro-caspase-3 form. In addition, western blot
analysis demonstrated that the pro-caspase-6 decreased, thus
oridonin activated caspase-6. However, most of the p65 was found to
have been retained in the cytoplasm, possibly generating a markedly
higher resistance to oridonin in OCIM2 cells.
Transport receptors recognizing nuclear import (NLS)
or export signals (NES) on the cargo proteins are vital for the
nucleocytoplasmic protein-transporting (6), while distinct nucleoporins determine
substrate specificity. Previous reports identified anarrower
specificity of Nup214 compared to CRM1-dependent export (18,19). A study on Drosophila
demonstrated that Drosophila members Only (mbo), an
ortholog of Nup88, selectively regulated the nuclear translocation
or export of dorsal, a member of the Rel protein family (20–22). Results in the present study showed
that oridonin downregulated Nup214 and Nup88 at the gene and
protein levels. As a result of the decreased Nup214 and Nup88, p65
did not translocate into the nucleus, since the nucleocytoplasmic
transporting of p65 depended on Nup214 and Nup88. These results
suggest that the downregulation of Nup214 and Nup88 protects OCIM2
cells by regulating the nucleocytoplasmic transport of p65.
Acknowledgements
This study was supported by a grant
from the National Natural Science Foundation of China (nos.
30871036 and 81070429).
References
|
1.
|
FP SantosCE Bueso-RamosF RavandiAcute
erythroleukemia: diagnosis and managementExpert Rev
Hematol3705718201010.1586/ehm.10.6221091147
|
|
2.
|
MS HaydenS GhoshShared principles in
NF-kappaB
signalingCell132344362200810.1016/j.cell.2008.01.02018267068
|
|
3.
|
KM RyanMK ErnstNR RiceKH VousdenRole of
NF-kappaB in p53-mediated programmed cell
deathNature404892897200010.1038/3500913010786798
|
|
4.
|
V BaudM KarinIs NF-kappaB a good target
for cancer therapy?Hopes and pitfalls Nat Rev Drug
Discov83340200910.1038/nrd278119116625
|
|
5.
|
ND PerkinsTD GilmoreGood cop, bad cop: the
different faces of NF-kappaBCell Death
Differ13759772200610.1038/sj.cdd.440183816410803
|
|
6.
|
K WeisNucleocytoplasmic transport: cargo
trafficking across the borderCurr Opin Cell
Biol14328335200210.1016/S0955-0674(02)00337-X12067655
|
|
7.
|
VE GouldA OrucevicH ZentgrafP GattusoN
MartinezA AlonsoNup88 (karyoporin) in human malignant neoplasms and
dysplasias: correlations of immunostaining of tissue sections,
cytologic smears, and immunoblot analysisHum
Pathol33536544200210.1053/hupa.2002.124785
|
|
8.
|
VE GouldN MartinezA OrucevicJ SchneiderA
AlonsoA novel, nuclear pore-associated, widely distributed molecule
overexpressed in oncogenesis and developmentAm J
Pathol15716051613200010.1016/S0002-9440(10)64798-011073820
|
|
9.
|
A EmterlingJ SkoglundG
ArbmanClinicopathological significance of Nup88 expression in
patients with colorectal
cancerOncology64361369200310.1159/00007029412759533
|
|
10.
|
M FornerodM OhnoM YoshidaIW MattajCRM1 is
an export receptor for leucine-rich nuclear export
signalsCell9010511060199710.1016/S0092-8674(00)80371-29323133
|
|
11.
|
F GaoQ TangP YangY FangW LiY WuApoptosis
inducing and differentiation enhancement effect of oridonin on the
all-trans-retinoic acid-sensitive and -resistant acute
promyelocytic leukemia cellsInt J Lab
Hematol32e114e122201010.1111/j.1751-553X.2009.01147.x
|
|
12.
|
GB ZhouSJ ChenZY WangZ ChenBack to the
future of oridonin: again, compound from medicinal herb shows
potent antileukemia efficacies in vitro and in vivoCell
Res17274276200710.1038/cr.2007.2117426700
|
|
13.
|
FH GaoF LiuW WeiOridonin induces apoptosis
and senescence by increasing hydrogen peroxide and glutathione
depletion in colorectal cancer cellsInt J Mol
Med29649655201222294162
|
|
14.
|
Z JiQJ TangJS ZhangY YangYF LiuYJ
PanOridonin-induced apoptosis in SW620 human colorectal
adenocarcinoma cellsOncol Lett213031307201122848306
|
|
15.
|
S GhoshM KarinMissing pieces in the
NF-kappaB
puzzleCell109SupplS81S96200210.1016/S0092-8674(02)00703-111983155
|
|
16.
|
V KirkinS JoosM ZornigThe role of Bcl-2
family members in tumorigenesisBiochim Biophys
Acta1644229249200410.1016/j.bbamcr.2003.08.00914996506
|
|
17.
|
NA ThornberryCaspases: key mediators of
apoptosisChem
Biol5R97R103199810.1016/S1074-5521(98)90615-99578633
|
|
18.
|
R BernadD EngelsmaH SandersonH
PickersgillM FornerodNup214-Nup88 nucleoporin subcomplex is
required for CRM1-mediated 60 S preribosomal nuclear exportJ Biol
Chem2811937819386200610.1074/jbc.M51258520016675447
|
|
19.
|
S HuttenRH KehlenbachNup214 is required
for CRM1-dependent nuclear protein export in vivoMol Cell
Biol2667726785200610.1128/MCB.00342-0616943420
|
|
20.
|
AE UvP RothN XylourgidisA WickbergR
CanteraC Samakovlismembers only encodes a Drosophila
nucleoporin required for rel protein import and immune response
activationGenes Dev1419451957200010921908
|
|
21.
|
P RothN XylourgidisN SabriA UvM FornerodC
SamakovlisThe Drosophila nucleoporin DNup88 localizes
DNup214 and CRM1 on the nuclear envelope and attenuates
NES-mediated nuclear exportJ Cell Biol1637017062003
|
|
22.
|
N XylourgidisP RothN SabriV TsarouhasC
SamakovlisThe nucleoporin Nup214 sequesters CRM1 at the nuclear rim
and modulates NFkappaB activation in DrosophilaJ Cell
Sci11944094419200610.1242/jcs.0320117032737
|