Introduction
Liver fibrosis, resulting from chronic damage to the
liver, is the progressive accumulation of fibrillar extracellular
matrix (ECM) in the liver which eventually leads to cirrhosis and
liver failure. Generally, the main causes of liver fibrosis include
alcohol abuse, nonalcoholic steatohepatitis (NASH) and especially,
the chronic hepatitis virus infection, such as hepatitis B virus
(HBV) and C (HCV) (1). The
progression of fibrosis is a dynamic process where a potential
population of fibrogenic cells in the liver, such as portal
fibroblasts, mesenchymal cells derived from the bone marrow,
hepatocytes and biliary epithelial cells are involved. Hepatic
stellate cells (HSCs), the most important contributor cell type,
are the main ECM-producing cells in liver fibrosis following
activation into fibrogenic myofibroblast-like cells. Activated HSCs
express many ECM proteins, including collagen, α-smooth muscle
actin (α-SMA), transforming growth factor-β (TGF-β), matrix
metalloproteinase (MMP) and tissue inhibitors of metalloproteinases
(TIMP), which all contribute to liver fibrosis.
The activation of HSCs is regulated by several
cytokines and growth factors. The platelet-derived growth factor-B
(PDGF-BB) is the most prominent factor in HSCs proliferation and
liver fibrosis development (2).
Generally, PDGF-BB exerts its effects by binding to its receptor
(PDGFR-β), inducing receptor dimerization and
tyrosine-autophosphorylation. The activated phosphorylated receptor
recruits the signal transduction molecules, initiating various
intracellular signaling pathways (3–5).
Recently, the expression of PDGF-BB and its receptor (PDGFR-β) has
been shown to be increased in both experimental fibrosis in rats
and human fibrotic liver (6,7),
with a weak expression presented in the normal liver. Moreover,
HSCs may perpetuate their proliferative status by active secretion
of PDGF in a paracrine or autocrine manner (8). Mechanisms regulating the
PDGF-B/PDGFR-β signal transduction have recently begun to be
elucidated (9,10).
According to current clinical studies, chronic HBV
infection is strongly associated with the development of fibrosis,
cirrhosis and hepatocellular carcinoma (HCC), which may be caused
by the interaction between HBV or the viral proteins and HSCs,
directly, or indirectly. There is increasing evidence that the
intrahepatic accumulation of the HBV encoded x antigen (HBxAg)
correlates with the severity of chronic liver disease (CLD), as
well as with the development of fibrosis and cirrhosis (11,12). Certain studies also reported that
the x protein expression in hepatocytes leads to paracrine
activation and proliferation of HSCs (13). However, the possible role of the x
protein and other viral proteins in the development of liver
fibrosis, remains unknown. Thus, the role of HBV proteins in the
process of liver fibrosis needs to be explored intensively.
Moreover, according to several clinical data, the expression of
PDGF-BB in liver tissues or serum level in chronic HBV patients
reflects the degree of liver damage and the degree of hepatic
fibrosis (14–16). There may be some type of
correlation between the HBV viral proteins and the PDGF-B/PDGFR-β
signaling pathway, which may lead to the activation of HSCs.
Therefore, the aim of the present study was to
observe the effect of HBV and its antigen components on the
proliferation of HSCs and the expression of PDGF-BB and PDGFR-β, in
order to clarify whether HBV or virus gene products are able to
promote HSCs proliferation through PDGF-B/PDGFR-β signaling pathway
and further broaden our understanding of hepatic fibrogenesis.
Materials and methods
Purification of HBV Dane particles by
sucrose density gradient ultracentrifugation
Aliquots (6 ml) of 10, 20, 30, 40, 50, 60% (w/w)
sucrose in a solution containing 120 mM NaCl, 12 mM Tris·HCl, 1 mM
EDTA·Na2 (pH 8.0) were carefully layered in a 40-ml
ultracentrifuged tube and left at room temperature for 6 h.
Concentrated supernatant of HepG2.2.15, 4 ml, was layered on the
sucrose gradient mentioned above, and ultracentrifugation was
performed at 120,000 x g for 24 h at 4°C with the Beckman SW32Ti
Rotor. Finally, the density of 43.5 to 44.5% sucrose fraction was
collected, and a number of HBV Dane particles collected at this
density were confirmed by electron micrographs (17). The concentrated HBV was then
resuspended in fetal bovine serum (FBS) and stored at −80°C until
use. The copies of HBV were determined by real-time PCR.
Plasmid construction
The fragments of HBV preS, e, c and x, obtained by
PCR from the pHBV1.2 (complete genome of HBV isolate 57-1 subtype
adw, a kind gift by Professor Lai Wei, Peking University, and the
GenBank accession number AY518556.1) (18) respectively, were subcloned into
the efficient vector, pCAGGSP7, containing the β-actin promoter
(19). The positive clones were
named pHBV-S, pHBV-E, pHBV-C and pHBV-X, respectively. DNA
sequencing was used to verify the plasmids, and the expression of
these plasmids was further confirmed by indirect immunofluorescence
in Vero cells.
Cell culture and transfection
The human hepatocellular carcinoma cell line HepG2
and the human HSC line LX-2 were grown in Dulbecco’s modified
Eagle’s medium (DMEM, with 2 mM glutamine, 10% (FBS), 100 U/l
penicillin and 100 μg/ml streptomycin) at 37°C in a 5%
CO2 incubator. HepG2.2.15, an HBV (serotype ayw,
genotype D) stably transfected cell line, was also maintained in
10% FBS DMEM supplemented with 200 μg/ml G418 (Sigma).
To express the 4 kinds of HBV proteins, HepG2 cells
were transfected with the plasmid of pHBV-S, pHBV-E, pHBV-C, pHBV-X
and pCAGGSP7 (as the control), respectively, using FuGENE HD
transfection reagent (Roche), following the manufacturer’s
instructions. The cells were cultured with DMEM containing 10% FBS
and 600 μg/ml of G418 (Sigma). After selection, single cell
clones stably transfected with the plasmids mentioned above were
obtained, and named HepG-s, HepG-e, HepG-c, HepG-x and
HepG-control, respectively. The expression of viral protein was
evaluated by western blotting.
In vitro co-culture system
To elucidate the relationship between HBV proteins
and liver fibrosis, an in vitro co-culture system of HepG-s,
HepG-e, HepG-c, HepG-x, HepG2.2.15 and HepG-control cells with LX-2
cells was established according to a previous method with certain
modifications (20). In the
cell-to-cell non-contacted co-culture system, the cell line, which
stably expressed the viral protein, was separated from LX-2 cells
using a 0.4 μm transwell membrane (Corning). Briefly, the
same number of HepG-s, HepG-e, HepG-c, HepG-x, HepG2.2.15 and
HepG-control cells (as a negative control) were plated into the
upper chamber of a transwell insert. The same numbers of LX-2 cells
were plated on the lower chamber. After 24 h co-culture in DMEM
supplemented with 10% FBS, the confluence was achieved. The LX-2
cells were washed with phosphate-buffered saline (PBS) and
co-cultured for another 24 h in serum-free DMEM. Finally, the LX-2
cells and supernatants were collected for the following
experiments.
Detection of LX-2 cells proliferation by
flow cytometry
In order to detect the LX-2 cell proliferation, 2
measures were employed: i) LX-2 cells were co-cultured with the
stable transfected cell line, respectively, for 48 h as described
above. ii) LX-2 cells were cultured in complete medium for 24 h,
serum-starved for 16 h and were then added the series of diluted
concentrations of HBV (final concentration of HBV of each well was
5×106, 5×105, 5×104,
5×103, 5×102 and 0 copies/ml). After
incubation of 12 h, the cell cycle was detected with an FITC BrdU
Flow kit (BD Pharmingen) according to the manufacturer’s
instructions. Briefly, the pretreated LX-2 cells were labeled with
BrdU for 45 min, washed, and fixed and permeabilized with BD
Cytofix/Cytoperm buffer. After repeated incubation on ice, washed
and centrifugated cells were treated with DNase to expose BrdU
epitope for 1 h at 37°C, The cells were washed and stained with
fluorescent anti-BrdU for 20 min at room temperature, washed again
and centrifugated. Staining buffer containing 7-Aminoactinomycin D
(7-AAD) (1 ml; BD Pharmingen) was added to each tube to resuspend
the cells. Finally, the cells were analyzed by flow cytometry
(Becton-Dickinson, San Jose, CA, USA). Acquired multiparameter data
were analyzed using CellQuest software. With the combination of
BrdU and 7-AAD, 2-color flow cytometric analysis permits the
enumeration and characterization of cells that are actively
synthesizing DNA (BrdU Incorporation) in terms of their cell cycle
position (i.e., G0/1, S or G2/M phases defined by 7-AAD staining
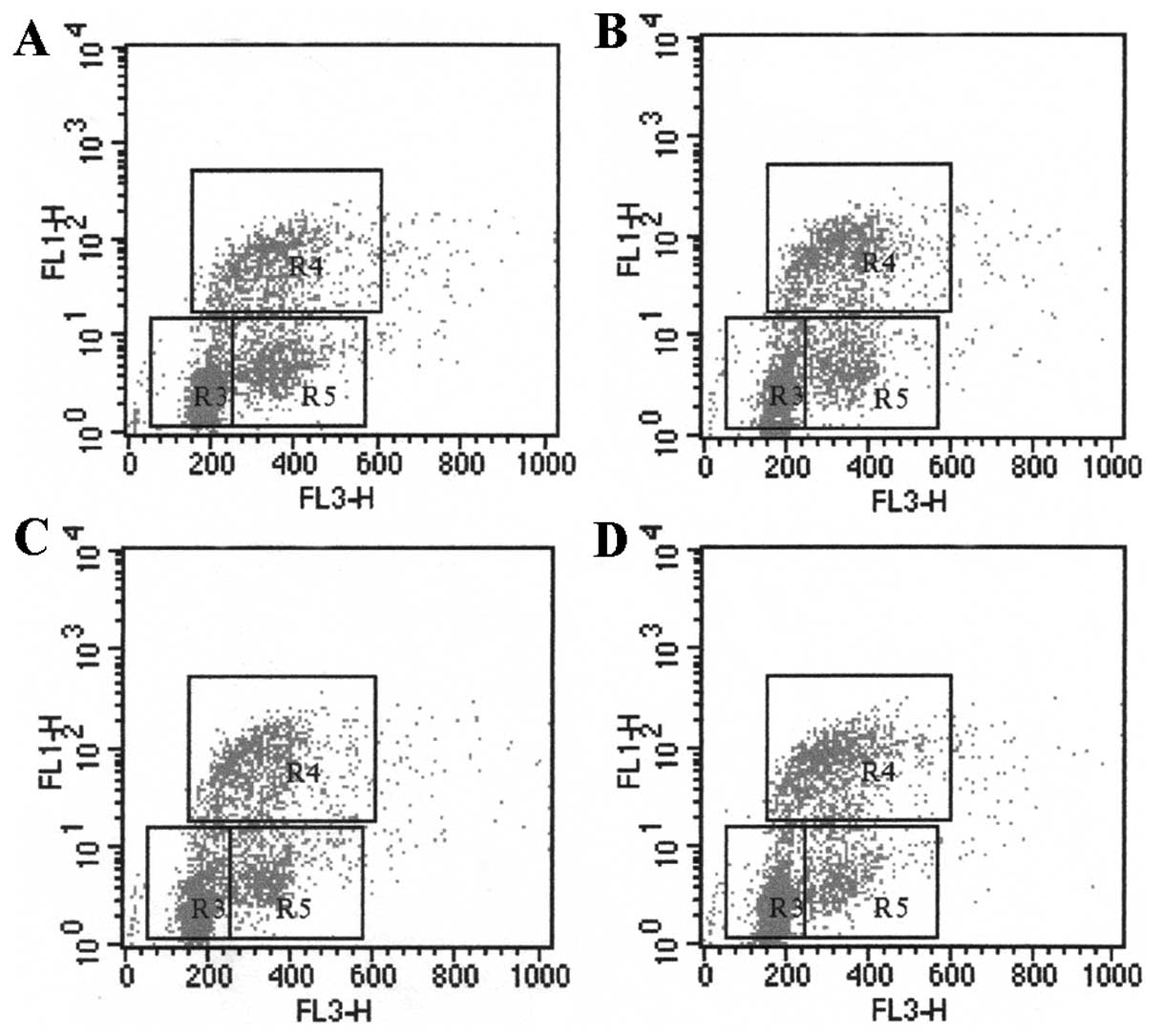
intensities). As shown by the region gates applied to the 7-AAD vs.
BrdU dot plot, flow cytometric analysis of cells stained with the
reagents allowed the discrimination of cell subsets that were in
the G0/G1 phase (R3), S-phase (R4), G2+M phase (R5) (21).
Detection of the mRNA of PDGF-B, PDGFR-β,
α-SMA, collagen-I by real-time PCR
Total RNA was extracted with TRIzol (Invitrogen) and
was reverse-transcribed by random hexamer primer. Then quantitative
real-time PCR analysis was performed using the GeneAmp 7500 system
and SYBR® Green (Applied Biosciences). All expression
data were normalized to GAPDH. Each reaction contained: 10
μl of SYBR-Green, 1 μl sense and antisense specific
primer respectively and 1 μl of cDNA matrix, in a final
volume of 20 μl. All reactions were repeated 3 times. PCR
products were obtained after 10 min at 95°C, followed by 45 cycles
of 10 sec at 95°C, 5 sec at 60°C and 10 sec at 72°C. The primers
used for amplification are shown in Table I. The relative quantity of the
products was expressed as a fold-induction of the target gene
compared with the control primers according to the formula
2−ΔΔCT (22).
 | Table IPrimers for real-time PCR. |
Table I
Primers for real-time PCR.
| Gene | Accession | Primer
sequence |
|---|
| PDGF-B | NM_033016.2 | F:
5′-tgatctccaacgcctgct-3′
R: 5′-tcatgttcaggtccaactcg-3′ |
| PDGFR-β | NM_002609.3 | F:
5′-tctgggaccagcagtctttc-3′
R: 5′-cctccaggaagtcctccttac-3′ |
| α-SMA | NM_001613.2 | F:
5′-ctgttccagccatccttcat-3′
R: 5′-tcatgatgctgttgtaggtggt-3′ |
| Collagen-I | NM_000088.3 | F:
5′-cagcgctggtttcgactt-3′
R: 5′-ccatcgtgagccttctcttg-3′ |
| GAPDH | NM_002046.3 | F:
5′-agccacatcgctcagacac-3′
R: 5′-gcccaatacgaccaaatcc-3′ |
Detection of PDGF-BB by ELISA
To quantify the expression levels of PDGF-BB in the
co-culture supernatant, ELISA Systems (R&D Systems) was
performed according to the manufacturer’s instructions.
Ninety-six-well polysty-rene microplates were pre-coated with
recombinant human PDGFRβ/Fc chimera and 100 μl standard,
control and culture supernatant was added to each well. Cells were
incubated for 2 h, aspirated and washed 4 times. Then, 200
μl of conjugate was added to the cells, incubated for 2 h
and washed 4 times. A 200 μl substrate solution was added to
each well and the cells were incubated for 30 min. Finally, a stop
solution was added to each well and the absorbance at 450 nm was
determined on an ELISA reader. We repeated the experiment 3
times.
Western blotting
To detect the expression of PDGFR-β, and
phos-PDGFR-β, LX-2 cells were lysed in RIPA lysis buffer (50 mmol/l
Tris-HCl (pH 7.4), 1% NP-40, 0.25% sodium deoxycholate, 150 mmol/l
NaCl, 0.1% SDS, 1 mmol/l EDTA, 1 mmol/l PMSF, 1 mmol/l
Na3VO4, and 1 mmol/l NaF). Protein
concentrations were determined using the Bio-Rad protein assay kit
(Bio-Rad, Hercules, CA USA). After boiling for 5 min, the lysate
were separated on 10% SDS-PAGE polyacrylamide gel. Proteins were
transferred to nitrocellulose membranes which were blocked in
Tris-buffered saline with Tween-20 (TTBS) containing 5% non-fat
dried milk. The membranes were incubated with primary antibody
against PDGFR-β, or phospho-PDGFR-β and β-actin at 4°C overnight.
After rinsing three times in TTBS, the membranes were incubated
with secondary antibody conjugated with horseradish peroxidase
(HRP) at room temperature for 1 h and then developed using a
chemiluminescence detection kit (Amersham Biosciences), according
to the manufacturer’s instructions and exposed to X-ray film. The
relative expression of these proteins was determined by
densitometric scanning and calculating the ratios of each protein
to β-actin bands, which were expressed constitutively.
Statistical analysis
Statistical analysis of the results was performed by
one-way ANOVA, the Newman-Keuls test, the Mann-Whitney test, and
the unpaired Student’s t-test when appropriate. Differences were
considered to be significant at P<0.05.
Results
Proliferation of LX-2 cells after
incubation with HBV or viral proteins
The cell cycle was analyzed by using BrdU and 7-AAD
to measure the proliferation of LX-2 cells stimulated by HBV or
viral proteins. After incubation with HBV, the cell number in
S-phase of LX-2 cells increased with different concentrations of
HBV (Fig. 1 and Table IIA). The proliferation rate of
LX-2, stimulated with 5×104, 5×105 and
5×106 copies/ml HBV, was 30.42±1.58%, 34.12±2.35% and
35.11±2.05%, respectively. HBV had a significant effect on the cell
proliferation as compared with that of 0 copies/ml of HBV
(25.50±2.98%, P<0.05), indicating that HBV particles promote
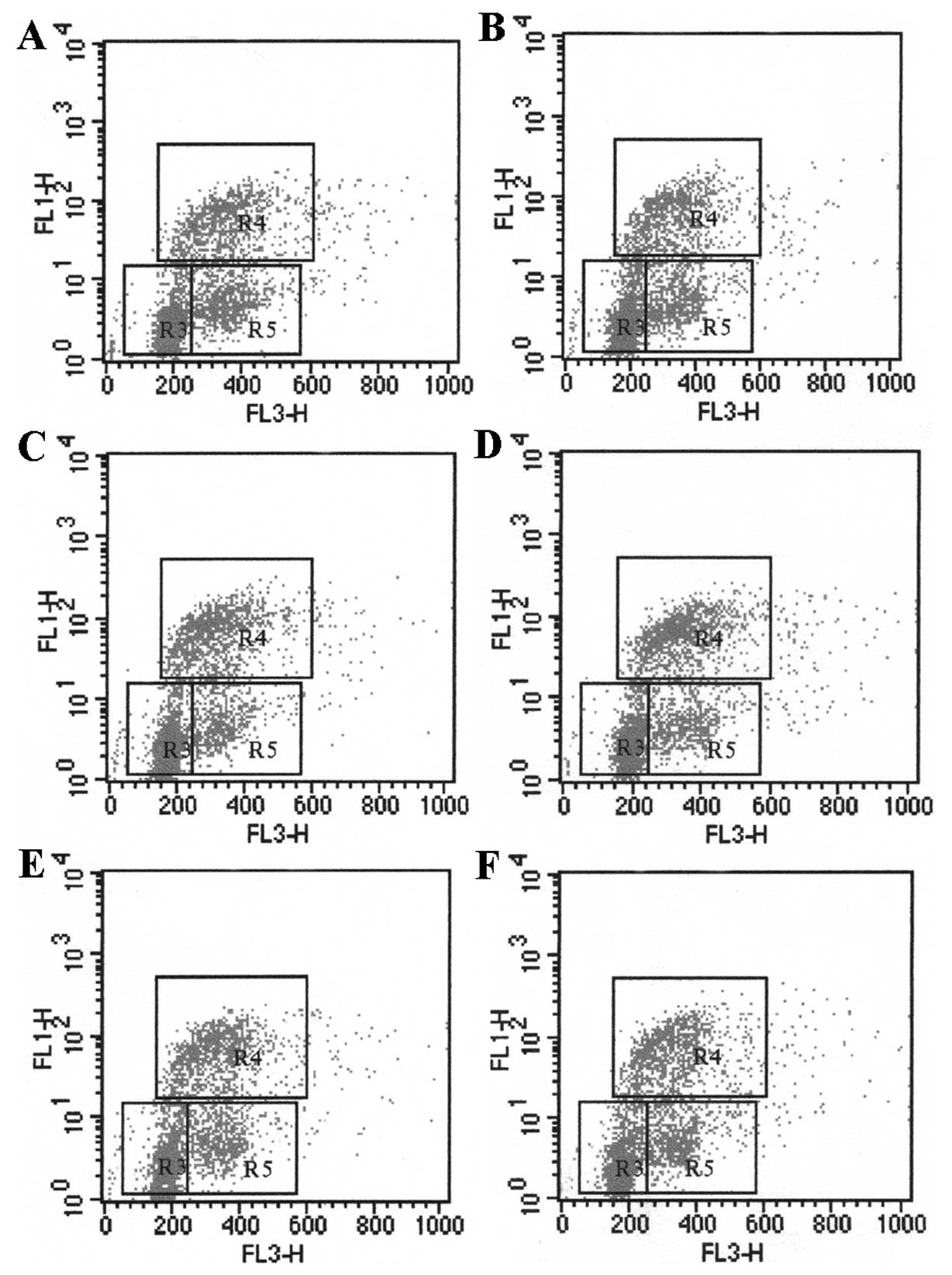
LX-2 proliferation in a dose-dependent pattern (Table IIA). In the co-culture system, the
proliferation of LX-2 cells in S-phase significantly increased up
to 36.44±2.45% and 35.34±2.85% after incubation with HepG-x or
HepG-c cells, which express x or c protein of HBV, compared with
HepG-control cell (P<0.05). However, HepG-e and HepG-s had no
obvious effect on the LX-2 proliferation. The results indicated
that the HBV x and c proteins promote LX-2 proliferation (Fig. 2 and Table IIB).
| Table IIProliferation of LX-2 cells after
incubation with HBV. |
Table II
Proliferation of LX-2 cells after
incubation with HBV.
| A, Proliferation of
LX-2 cells after incubation with HBV (n=5) |
|
| Concentration of
HBV (copies/ml) | S-phase cell
(%) | P-value |
|
| 0 | 25.50±2.98 | |
|
5×104 | 30.42±1.58 | |
|
5×105 | 34.12±2.35 | <0.05a |
|
5×106 | 35.11±2.05 | <0.05a |
|
| B, Proliferation of
LX-2 cells after incubation with HBV proteins (n=5) |
|
| Co-culture with
viral protein | S-phase cell
(%) | P-value |
|
| HepG-control | 25.32±2.78 | |
| HepG-s | 25.35±1.88 | |
| HepG-e | 26.84±3.05 | |
| HepG-c | 35.34±2.85 | <0.05b |
| HepG-x | 36.44±2.45 | <0.05b |
| HepG2.2.15 | 30.94±2.60 | <0.05b |
HBV or HBV-x or HBV-c protein upregulated
mRNA levels of Collagen-I, α-SMA PDGF-B and PDGFR-β in LX-2
cells
After incubation with different concentrations of
HBV for 12 h, total mRNA from LX-2 cells was extracted with TRIzol.
mRNA levels of collagen-I, α-SMA, PDGF-B and PDGFR-β were detected
by real-time PCR, respectively. The increased mRNA levels of
collagen-I and α-SMA were observed in 5×105 and
5×106 copies/ml HBV concentration groups (Fig. 3). Both collagen-I and α-SMA showed
an increase approximately 2 times greater than that in the control
group (P<0.05). The mRNA levels of PDGF-B and PDGFR-β in LX-2
cells were also upregulated significantly in the 5×105
and 5×106 copies/ml groups, compared with the control
group (P<0.05). However, there were no apparent changes in mRNA
levels of the parameters mentioned above in HBV concentration of
5×102−5×104 copies/ml (Fig. 3).
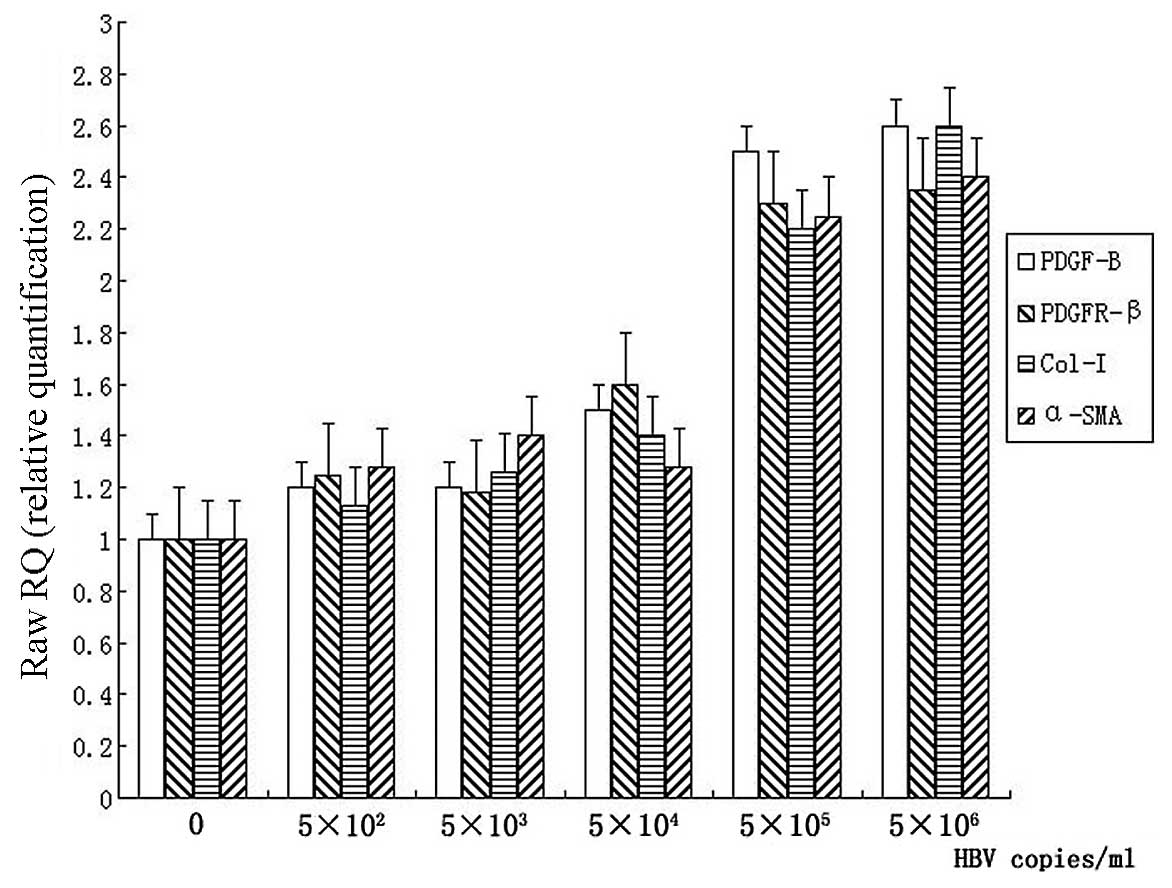 | Figure 3Detection of the mRNA levels of
PDGF-B, PDGFR-β, collagen-I (Col-I) and α-SMA in LX-2 cells. LX-2
cells were cultured in complete medium for 24 h, serum starved for
16 h. Following incubation with 0, 5×102,
5×103, 5×104, 5×105 or
5×106 copies/ml of HBV for 12 h, total mRNA was
extracted and mRNA levels of PDGF-B, PDGFR-β, collagen-I and α-SMA
were detected by real-time PCR. All the values were normalized to
GAPDH (n=5). |
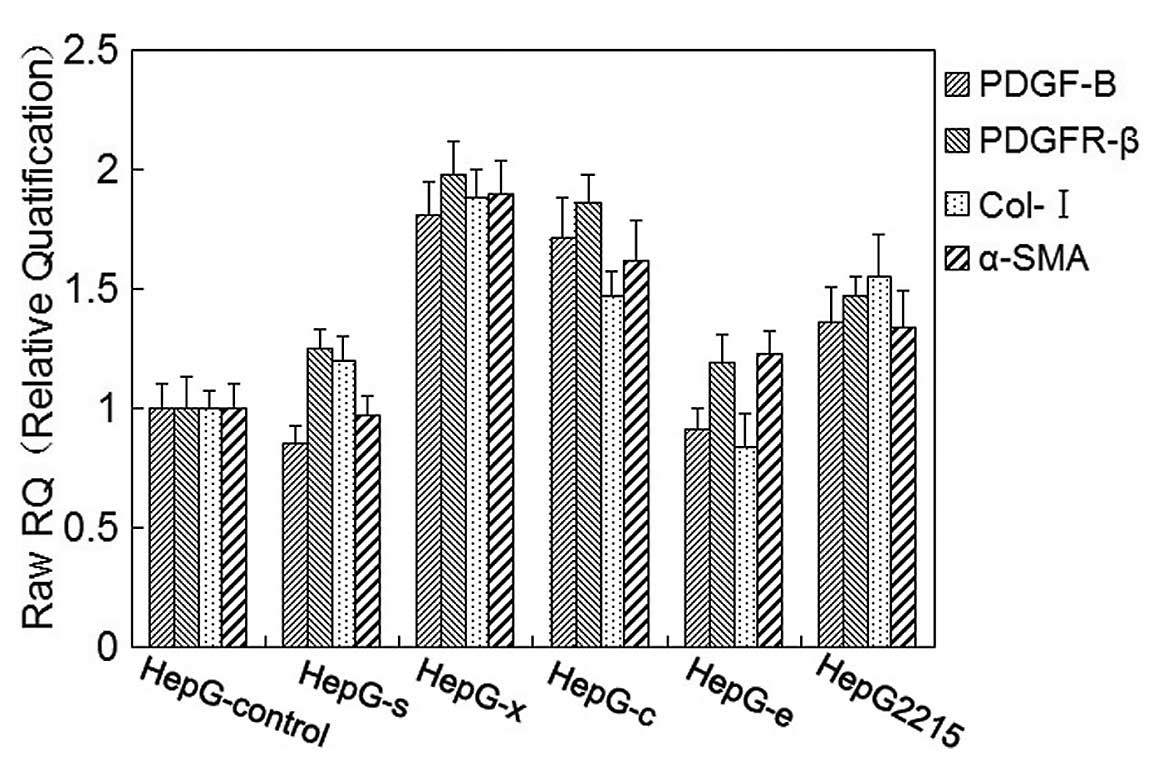
Meanwhile, mRNA levels of collagen-I, α-SMA, PDGF-B
and PDGFR-β in LX-2 cells were also detected by real-time PCR after
co-culture with HepG-s, HepG-x, HepG-c, HepG-e, HepG2.2.15 and
HepG-control cells, respectively, for 48 h. Increased mRNA levels
of collagen-I, α-SMA, PDGF-B and PDGFR-β were observed in LX-2
cells after being co-cultured with HepG-x or HepG-c, and they were
approximately 1.5–2 times higher than the control group. In
co-culture with HepG2.2.15, mRNA levels of collagen-I, α-SMA,
PDGF-B and PDGFR-β in LX-2 cells slightly increased and there were
no significant difference as compared with control group
(p>0.05) (Fig. 4). There were
no effects of HepG-s or HepG-e on mRNA levels of collagen-I, α-SMA,
PDGF-B and PDGFR-β in LX-2 cells.
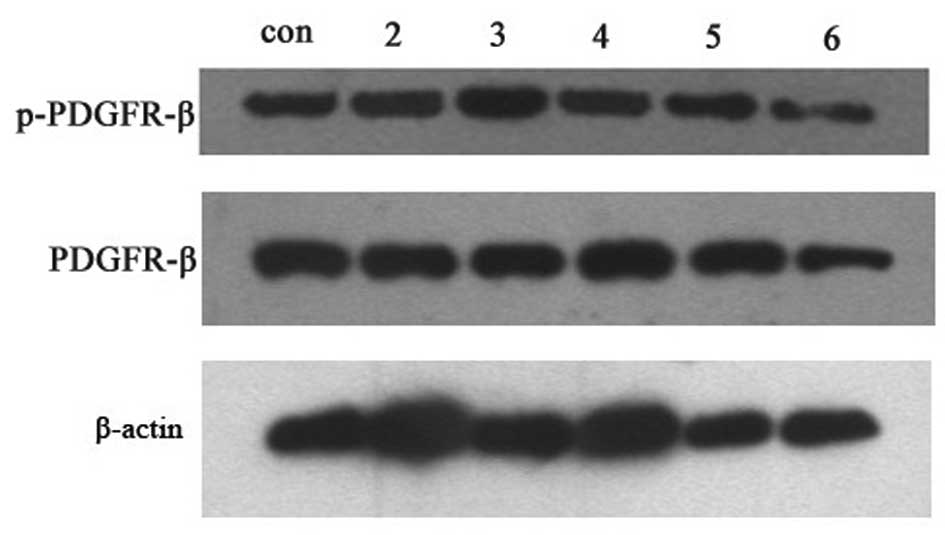
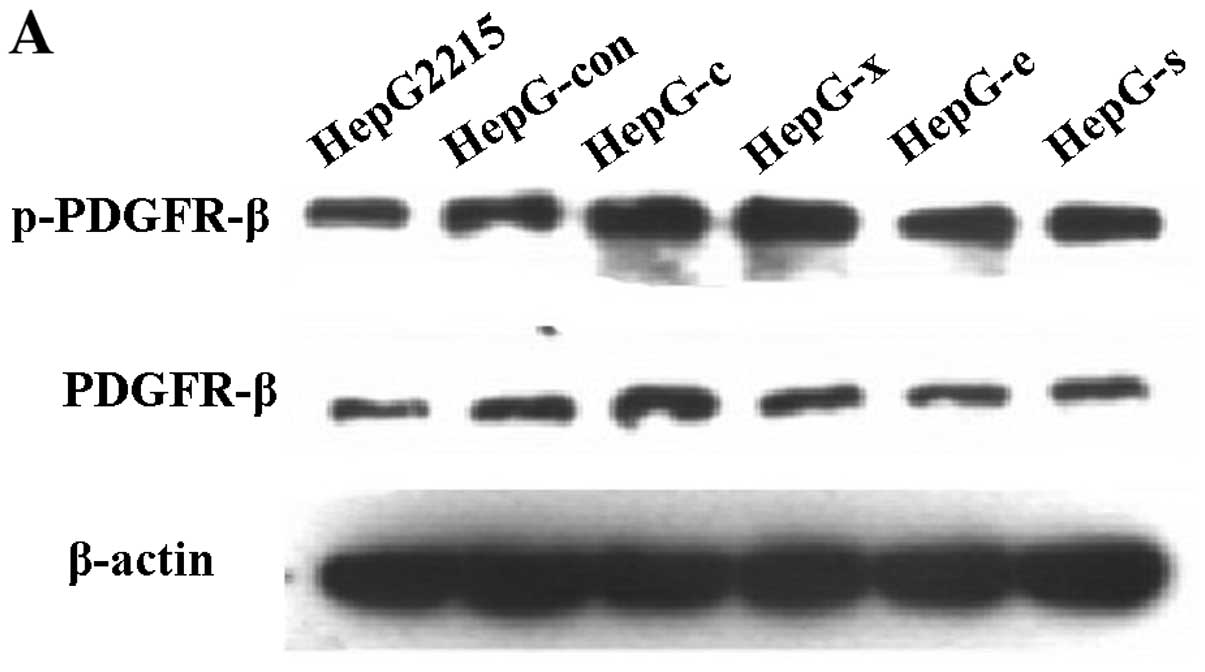
HBV or HepG-c or HepG-x upregulated the
expression levels of the phos-PDGFR-β protein in LX-2 cells
After incubation with HBV at concentration of 0
copies/ml, 5×102−5×106 copies/ml for 12 h or
co-cultured with HepG-s, HepG-x, HepG-c, HepG-e, HepG-control and
HepG2.2.15 cells for 48 h, respectively, LX-2 cells were collected
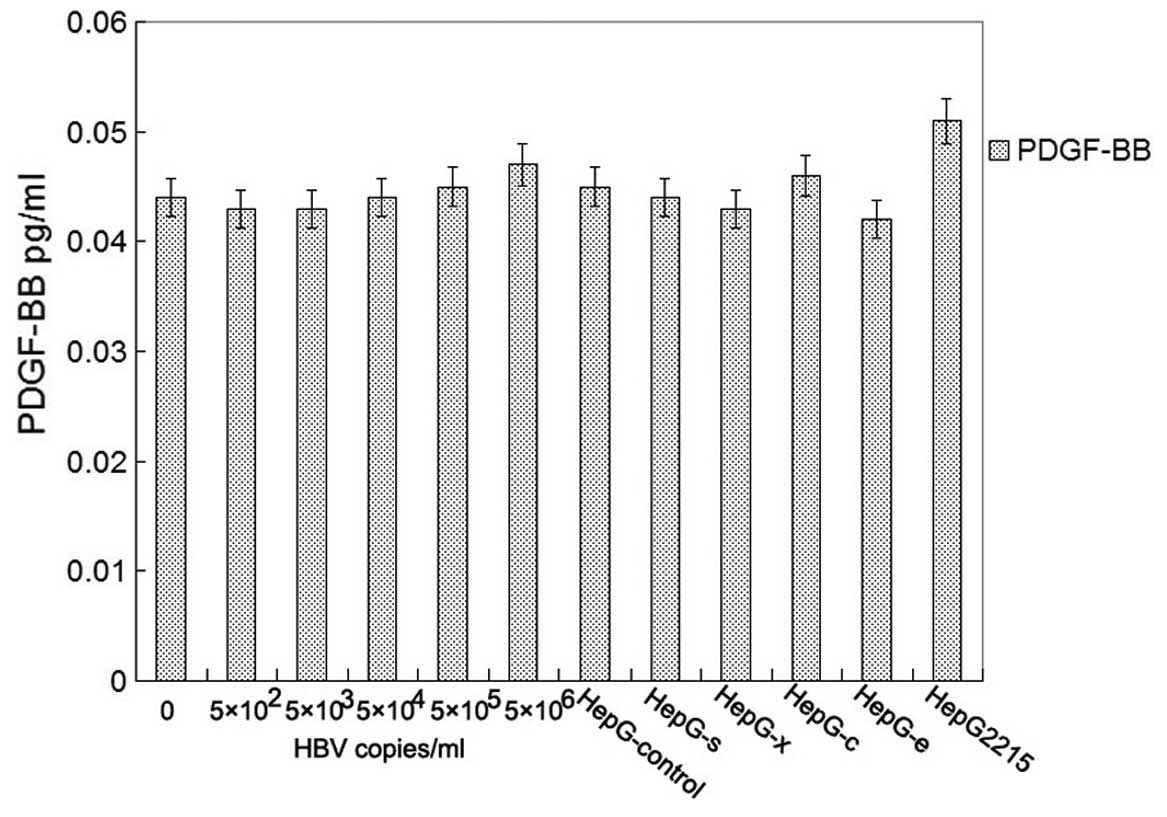
for detection PDGFR-β and phospho-PDGFR-β by western blotting. One
milliliter of culture supernatants were collected for detection of
PDGF-BB by ELISA. Unfortunately, there was no detectable level of
PDGF-BB in all of groups (Fig.
5). The relative expression levels of the proteins were
determined by densitometric scanning and calculating the ratios of
each protein to constitutively expressed β-actin bands. There were
no changes in PDGFR-β levels after treatment with any concentration
of HBV or viral protein (Figs. 6
and 7). However, phosphorylation
activity of PDGFR-β in LX-2 cells was increased after treatment
with HBV and the peak value was observed in group of
5×105 copies/ml HBV (Fig.
6) (P<0.05). Treatment with viral protein of HBV-c or HBV-x
could also upregulated the level of phospho-PDGFR-β in LX-2 cells
(Fig. 7) (P<0.05).
Discussion
HBV Dane particles, HBV protein c and x,
not e or s, promote HSCs proliferation
Chronic HBV infection has been recognized to
exacerbate liver fibrosis in patients. However, HSCs, activated by
a variety of host factors and/or viral proteins, is considered to
be the most important contributor to fibrosis progression and has
been investigated intensively. Generally, the activation process of
HSCs includes a loss of vitamin A droplets, an increased
proliferation rate, a phenotypic transition to a
myofibroblast-like, α-SMA positive cell and a dramatic increase in
the synthesis of extracellular matrix proteins. Activated HSCs are
the major collagen producing cells during hepatic fibrogenesis.
Although chronic HBV infection is one of the major causes of liver
fibrosis, the HBV-specific steps in HSCs proliferation and
pathogenesis of liver fibrosis remain unclear. In our previous
study, it was found that supernatants from the HepG2.2.15 culture
could promote LX-2 cell proliferation (23); however, the possible role of HBV
viral particles or HBV proteins in the development of liver
fibrosis remains unclear. It is necessary to identify which factors
of HBV are involved in the activation of HSCs. Thus the activation
of HSCs followed by HBV stimulation was characterized in the
present study.
To explore the direct interaction between HBV and
HSCs, purified HBV particles were obtained from the supernatants of
HepG2.2.15 cells cultured by sucrose density gradient
ultracentrifugation and plasmids expressing HBV proteins were
constructed. The roles of HBV or the viral proteins on the LX-2
cells proliferation were investigated with the co-cuture system. We
found that the cell number in S-phase of LX-2 cells was upregulated
after HBV incubation in a dose-dependent manner, and the peak value
was observed at 5×106 copies/ml HBV (Fig. 1 and Table 2A). Moreover, the cell number in
S-phase of LX-2 cells also increased after co-culture with HepG2
cells which were transfected with plasmids pHBV-C and pHBV-X, but
not pHBV-E or pHBV-S (Fig. 2 and
Table IIB). As we mentioned
above, HSCs convert into proliferating α-SMA-expressing and
collagen-producing cells after a fibrogenic stimulus. Thus, the
mRNA levels of α-SMA and collagen-I were further detected, and
showed an increase approximately 2 times greater than those in the
control group with the concentration of 5×106 copies/ml
(Fig. 3). Similarily, α-SMA and
collagen-I mRNA levels were also detected in the LX-2 cells
co-cultured with HepG-c and HepG-x (Fig. 4). The results mentioned above
indicated that HBV Dane particles, as well as the HBV viral protein
c or x could promote LX-2 proliferation, which was characterized
with the increased proliferation rate and metabolic changes.
Our results were consistent with other studies which
have reported that HCV viral proteins could directly induce HSC
proliferation and release inflammatory cytokines. Treatment with a
conditioned medium from Huh-7 cells expressing an HCV core protein
(24) or recombinant core protein
(25), led to the upregulation of
α-SMA and other cytokines in LX-2 cells. Moreover, it was reported
that the HBV x protein may lead to a paracrine activation and
proliferation of HSCs (13).
These data show that the viral protein may be able to stimulate
HSCs into an active status; however, the intracellular signaling
mechanisms of activation and perpetuation are under active
investigation.
The PDGF signal pathway involved in the
activation of HSCs
During liver fibrosis, activated HSCs proliferate
and deposit ECM proteins, a process that is driven by an array of
cytokines and growth factors. PDGF has been identified as the most
potent mitogen for HSCs, making it an attractive therapeutic target
for the treatment of liver fibrosis (26,27). Marra et al (8) have also demonstrated how HSCs
perpetuate their proliferative status by active secretion of PDGF
in an autocrine or paracrine manner. Moreover, PDGF receptors
(PFGFR) are highly upregulated on the cell surface of activated
HSCs during fibrosis (7). Of the
PDGF ligand/receptor systems, PDGF-BB signaling through PDGFR-β is
an important mediator in the initiation and progression of liver
fibrosis (28). Interestingly,
the PDGF-B protein overexpression in the livers of transgenic mice
was associated with an increased number of α-SMA-positive cells and
was also marked by an increase in the PDGFR-β transcription
(6). Thus, in order to
investigate the mechanisms of the proliferation of HSCs caused by
HBV infection, we address the role of paracrine PDGF-B/PDGFR-β
signaling further.
We detected the mRNA and protein levels of PDGF-B
and PDGFR-β from the LX-2 cells, respectively. The PDGF-B mRNA in
LX-2 cells inoculated with HBV particles was upregulated in a
dose-dependent manner and the highest level was observed in the
5×106 HBV copies/ml group (Fig. 3). Subsequently, a co-culture
system was used to detect the effects of viral proteins on LX-2
cells. We found that the HBV c and x antigen but not the large s or
e antigen promote the expression of PDGF-B mRNA in LX-2 cells
(Fig. 4). We did not detect the
expression of PDGF-BB by ELISA. Similarly, the mRNA and
phosphorylation level, not the protein level of PDGFR-β was also
upregulated when stimulated with HBV particles or HBV c and x. Our
results indicated that the HBV particles or virus proteins did not
promote the protein expression of PDGF-BB and PDGFR-β, but the
phosphorylation of PDGFR-β was upregulated, which indicated the
activation of PDGFR. Collectively, HSC proliferation mediated by
HBV or viral proteins via PDGFR-β phosphorylation and the
subsequent activation of the PDGF-BB signal pathway.
Several studies provide ample evidence of the
PDGF-B/PDGFR-β pathway being a strong stimulus of HSC
proliferation, which may be attenuated by anti-PDGF strategies. For
example, the tyrosine kinase inhibitor AG 1295 of PDGF inhibit HSCs
proliferation through reducing the phosphorylation of PDGFR-β and
the downstream signaling molecules ERK1/2 and Akt (29). Administration of A771726,
metabolite of leflunomide, markedly blunted the PDGFR-β expression
and phosphorylation in activated HSCs (30). Moreover, in culture-activated
HSCs, a soluble PDGF-B receptor was able to block the
phosphorylation of endogenous PDGF receptors and reduce the
proliferative activity of HSCs (31).
In conclusion, our data suggest that HBV Dane
particles, x and c protein may induce HSCs proliferation through
effecting on PDGF-B/PDGFR-β signal pathway which play an important
role of in liver fibrosis caused by HBV infection. HBV Dane
particles and x and c protein may upregulate the mRNA levels of
PDGF-B and PDGFR-β and promote the phosphorylation of PDGFR-β,
leading to the later auto-phosphorylation. Therefore, interference
with PDGF-B/PDGFR-β signal pathway may be a potential target for
antifibrotic therapies in liver disease.
Abbreviations:
|
HBV
|
hepatitis B virus;
|
|
HSCs
|
hepatic stellate cells;
|
|
PDGF
|
platelet-derived growth factor;
|
|
PDGFR-β
|
platelet-derived growth factor
receptor-β;
|
|
α-SMA
|
α-smooth muscle actin;
|
|
GAPDH
|
glyceraldehyde phosphate
dehydrogenase
|
Acknowledgements
We thank Dr Lijun Zhang and Dr Xia
Peng (School of Public Health, Fudan University) for their
assistance and comments during this study. This study was supported
by the National Natural Science Foundation of China (no. 30671854),
the National High Technology Research and Development Program of
China (863 Program, no. 2006AA02A410) and the Major State Basic
Research Development Program of China (973 Program, no.
2007CB512802).
References
|
1.
|
G Gutierrez-ReyesMC Gutierrez-RuizD
KershenobichLiver fibrosis and chronic viral hepatitisArch Med
Res38644651200710.1016/j.arcmed.2006.10.00117613356
|
|
2.
|
K BreitkopfC RoeyenI SawitzaL WickertJ
FloegeAM GressnerExpression patterns of PDGF-A, -B, -C and -D and
the PDGF-receptors alpha and beta in activated rat hepatic stellate
cells
(HSC)Cytokine31349357200510.1016/j.cyto.2005.06.00516039137
|
|
3.
|
M PinzaniS MilaniH HerbstExpression of
platelet-derived growth factor and its receptors in normal human
liver and during active hepatic fibrogenesisAm J
Pathol14878580019968774134
|
|
4.
|
M PinzaniPDGF and signal transduction in
hepatic stellate cellsFront
Biosci7d1720d1726200210.2741/pinzani12133817
|
|
5.
|
F MarraM PinzaniR DeFrancoG LaffiP
GentiliniInvolvement of phosphatidylinositol 3-kinase in the
activation of extracellular signal-regulated kinase by PDGF in
hepatic stellate cellsFEBS
Lett376141145199510.1016/0014-5793(95)01261-07498528
|
|
6.
|
P CzochraB KlopcicE MeyerLiver fibrosis
induced by hepatic overexpression of PDGF-B in transgenic miceJ
Hepatol45419428200610.1016/j.jhep.2006.04.01016842882
|
|
7.
|
M PinzaniL GesualdoGM SabbahHE
AbboudEffects of platelet-derived growth factor and other
polypeptide mitogens on DNA synthesis and growth of cultured rat
liver fat-storing cellsJ Clin
Invest8417861793198910.1172/JCI1143632592560
|
|
8.
|
F MarraGG ChoudhuryM PinzaniHE
AbboudRegulation of platelet-derived growth factor secretion and
gene expression in human liver fat-storing
cellsGastroenterology1071110111719947926460
|
|
9.
|
K LehtiE AllenH Birkedal-HansenAn
MT1-MMPPDGF receptor-beta axis regulates mural cell investment of
the microvasculatureGenes
Dev19979991200510.1101/gad.129460515805464
|
|
10.
|
P LindahlM HellstromM KalenParacrine
PDGF-B/PDGF-Rbeta signaling controls mesangial cell development in
kidney glomeruliDevelopment1253313332219989693135
|
|
11.
|
MA FeitelsonHM ReisNL TufanB SunJ PanZ
LianPutative roles of hepatitis B x antigen in the pathogenesis of
chronic liver diseaseCancer
Lett2866979200910.1016/j.canlet.2008.12.01019201080
|
|
12.
|
GH GuoDM TanPA ZhuF LiuHepatitis B virus X
protein promotes proliferation and upregulates TGF-beta1 and CTGF
in human hepatic stellate cell line, LX-2Hepatobiliary Pancreat Dis
Int85964200919208517
|
|
13.
|
S Martin-VilchezP Sanz-CamenoY
Rodriguez-MunozThe hepatitis B virus X protein induces paracrine
activation of human hepatic stellate
cellsHepatology4718721883200810.1002/hep.2226518449922
|
|
14.
|
SM LouYM LiKM WangWM CaiHL WengExpression
of platelet-derived growth factor-BB in liver tissues of patients
with chronic hepatitis BWorld J
Gastroenterol10385388200414760763
|
|
15.
|
WM CaiBB ZhangHL WengThe diagnostic value
of eight serum indices for liver fibrosisZhonghua Gan Zang Bing Za
Zhi122192222004(In Chinese).
|
|
16.
|
BB ZhangWM CaiHL WengDiagnostic value of
platelet derived growth factor-BB, transforming growth
factor-beta1, matrix metalloproteinase-1, and tissue inhibitor of
matrix metalloproteinase-1 in serum and peripheral blood
mononuclear cells for hepatic fibrosisWorld J
Gastroenterol9249024962003
|
|
17.
|
M SeiferKH HeermannWH GerlichReplication
of hepatitis B virus in transfected nonhepatic
cellsVirology179300311199010.1016/0042-6822(90)90298-62219725
|
|
18.
|
XB PanJC HanY GaoL WeiHigh replicated
hepatitis B virus induces apoptosis of hepatocytesZhonghua Yi Xue
Za Zhi888408432008(In Chinese).
|
|
19.
|
N GaoW ChenQ ZhengCo-expression of
Japanese encephalitis virus prM-E-NS1 antigen with
granulocyte-macrophage colony-stimulating factor enhances humoral
and anti-virus immunity after DNA vaccinationImmunol
Lett1292331201010.1016/j.imlet.2009.12.023
|
|
20.
|
E HintermannM BayerJM PfeilschifterAD
LusterU ChristenCXCL10 promotes liver fibrosis by prevention of NK
cell mediated hepatic stellate cell inactivationJ
Autoimmun35424435201010.1016/j.jaut.2010.09.00320932719
|
|
21.
|
J KitagawaT HaraH TsurumiCell
cycle-dependent priming action of granulocyte colony-stimulating
factor (G-CSF) enhances in vitro apoptosis induction by cytarabine
and etoposide in leukemia cell linesJ Clin Exp
Hematop5099105201010.3960/jslrt.50.99
|
|
22.
|
P Melgar-LesmesG CasalsM PautaApelin
mediates the induction of profibrogenic genes in human hepatic
stellate
cellsEndocrinology15153065314201010.1210/en.2010-075420843995
|
|
23.
|
X LiuST ZhuH YouHepatitis B virus infects
hepatic stellate cells and affects their proliferation and
expression of collagen type IChin Med J
(Engl)12214551461200919567171
|
|
24.
|
S ClementS PascarellaS ConzelmannC
Gonelle-GispertK GuillouxF NegroThe hepatitis C virus core protein
indirectly induces alpha-smooth muscle actin expression in hepatic
stellate cells via interleukin-8J
Hepatol52635643201010.1016/j.jhep.2009.10.03520347177
|
|
25.
|
M CoenenHD NischalkeB KramerHepatitis C
virus core protein induces fibrogenic actions of hepatic stellate
cells via toll-like receptor 2Lab
Invest9113751382201110.1038/labinvest.2011.7821537327
|
|
26.
|
T GonzaloL BeljaarsM van de BovenkampLocal
inhibition of liver fibrosis by specific delivery of a
platelet-derived growth factor kinase inhibitor to hepatic stellate
cellsJ Pharmacol Exp
Ther321856865200710.1124/jpet.106.11449617369283
|
|
27.
|
S OgawaT OchiH ShimadaAnti-PDGF-B
monoclonal antibody reduces liver fibrosis developmentHepatol
Res4011281141201010.1111/j.1872-034X.2010.00718.x20880061
|
|
28.
|
E Borkham-KamphorstCR van RoeyenT
OstendorfJ FloegeAM GressnerR WeiskirchenPro-fibrogenic potential
of PDGF-D in liver fibrosisJ
Hepatol4610641074200710.1016/j.jhep.2007.01.02917397961
|
|
29.
|
H IwamotoM NakamutaS TadaR SugimotoM
EnjojiH NawataPlatelet-derived growth factor receptor tyrosine
kinase inhibitor AG1295 attenuates rat hepatic stellate cell
growthJ Lab Clin
Med135406412200010.1067/mlc.2000.10597410811056
|
|
30.
|
HF SiX LvA GuoH JiangJ LiSuppressive
effect of leflunomide on rat hepatic stellate cell proliferation
involves on PDGF-BB-elicited activation of three mitogen-activated
protein
kinasesCytokine422431200810.1016/j.cyto.2008.01.01718343153
|
|
31.
|
N KinnmanC FrancozV BarbuThe
myofibroblastic conversion of peribiliary fibrogenic cells distinct
from hepatic stellate cells is stimulated by platelet-derived
growth factor during liver fibrogenesisLab
Invest83163173200310.1097/01.LAB.0000054178.01162.E4
|