Introduction
Hepatitis B virus (HBV) infection is a major risk
factor for the development of severe liver diseases including
hepatocellular carcinoma (HCC) (1). Hepatitis B virus X protein (HBx) is
a 17-kDa protein encoded by the 3.2-kb HBV genome (2). Previous studies have indicated that
HBx is oncogenic and can regulate HBV replication and transcription
(3). One study was performed
using a plasmid carrying a greater-than-unit-length HBV genome
(payw1.2) (4) and a HBx-deficient
plasmid containing a stop codon at amino acid position 7
(payw1.2.7) (5) transfected into
HepG2 cells, respectively. Results have shown that the absence of
HBx can induce a 65% reduction in HBV replication (6), and trans-complementation of HBx for
the HBx-deficient plasmid can restore replication to wild-type
levels (6–8). Another advanced study performed with
the hydrodynamic injection of an HBx-deficient plasmid into mice
showed the same results in vivo (6,9,10).
These results demonstrated that HBx has an important role in
modulating HBV replication.
However, to date, the mechanisms of HBx-dependent
HBV replication are not very clear. HBV covalently closed circular
DNA (cccDNA), the main replicative intermediate of HBV, is the
template for transcription of all viral RNAs including pregenomic
RNA (pgRNA) (11). Nuclear
cccDNA, predominately from relaxed circular DNA (rcDNA) (12), is organised by histone and
non-histone proteins into a viral minichromosome chromatin-like
structure (11,13,14). Changes in the nucleosome (the
basic unit of chromatin) structure and DNA-histone contacts may
result in the remodelling of minichromosomes (14). Chromatin remodelling is closely
associated with histone modifications, especially with
modifications of histone H3 and H4. These histone modifications may
influence the structure of nucleosomes directly, and provide DNA
binding sites for other proteins (13,15). Previous studies have shown that
HBx can regulate HBV replication and transcription. Multiple signal
transduction pathways and proteins may be involved in HBx-dependent
HBV replication and transcription (16,17). These signalling mediators may have
as their terminal target chromatin remodelling (18). Thus, it is hypothesized that the
mechanisms of HBx-dependent HBV replication may involve the
chromatin remodelling pathway.
Our previous study demonstrated that acetylation,
methylation and phosphorylation of cccDNA-bound histone H3 occurs
in HepG2 cells that are replicating wild-type HBV genome and that
these histone modifications are associated with HBV replication
(19). Our present study was
designed to investigate the mechanism of HBx-dependent HBV
replication through the pathway of chromatin remodelling. We
established two in vitro replication models by transfecting
human hepatoma HepG2 cells with the linear full-length HBV genome
(wild-type) or the HBx-deficient mutant HBV DNA (HBx mutant) and
investigated the regulation of HBx on replication, transcription
and antigen secretion, and in particular, on the methylation,
phosphorylation and acetylation of histone H3 bound to the cccDNA
in chromatin during HBV replication in HepG2 cells.
Materials and methods
Plasmid
The plasmid pUC-HBV1.0, which contains full-length
wild-type HBV genome and HindIII/SapI and
SacI/SapI restriction sites, was constructed as
previously described (19). The
HBx mutant plasmid pUC-HBV1.0.X7 that contains a stop codon
(CAA-UAA) at amino acid 7 of HBx was derived from plasmid
pUC-HBV1.0 by site-directed mutagenesis (5,8).
Briefly, mutagenic primers were designed using primer design
software developed by Stratagene. The forward primer was
5′-CTAGGCTGTGCTGCTAACTGGATCCTGCG-3′ (mutated
nucleotides underlined) and the reverse primer was 5′-CGCA
GGATCCAGTTAGCAGCACAGCCTAG-3′. Using plasmid pUC-HBV1.0 as a
template, the mutant products were amplified by the high-fidelity
enzyme Premix PrimeSTAR® HS (Takara) through polymerase
chain reaction (PCR). The PCR products were then digested with
DpnI enzyme (Fermentas), transformed into competent DH-5α
cells, which were plated on LB plates containing ampicillin (100
μg/ml). Four white colonies selected randomly were prepared, and
the plasmid DNA was extracted and digested with HindIII and
SacI enzymes (Takara). The plasmid, whose digested products
were determined to be correct, was then sequenced (Takara) to
confirm the mutation. All plasmids were prepared and purified using
the Endotoxin-Free Plasmid Maxi kit (Tiangen Biotech, Co.,
Ltd.).
Cell culture and DNA transfection
Human hepatoma HepG2 cells were cultured in 6-well
plates (Gibco) with high glucose DMEM containing 10% fetal calf
serum (Hyclone) under 5% CO2 at 37°C. The linear
full-length HBV genome and the HBx mutant HBV DNA were released
from plasmids pUC-HBV1.0 and pUC-HBV1.0.X7, respectively, by
SapI enzyme (MBI) digestion and gel-purified by a DNA gel
extraction kit (Promega). HBV DNA was transiently transfected into
HepG2 cells using PolyJet™ reagent (SignaGen Laboratories).
Briefly, HepG2 cells were seeded at a density of 1.0×106
cells in 6-well plates. Twenty-four hours later, cells at 70–80%
confluence were transfected with HBV DNA (1.5 μg) and PolyJet™
reagent (4 μl). HepG2 cells were transfected with the linear
full-length HBV genome (wild-type) or the HBx-deficient mutant HBV
DNA (HBx mutant). A green fluorescent protein (GFP) expression
vector (0.5 μg) was included in each transfection to assess
transfection efficiency. After transfection, the cell culture
medium was changed daily. The mean transfection efficiency was
approximately 40%. A negative control with no plasmid transfected
into the HepG2 cells was set up in each independent experiment.
Immunoprecipitation (IP) and western blot
analysis detection of HBx
Cells were harvested at 48 and 96 h
post-transfection and lysed in radioimmunoprecipitation assay
(RIPA) buffer (50 mM Tris, 150 mM NaCl, 1% NP-40, 0.5% sodium
deoxycholate and 0.1% SDS). Following centrifugation to remove
cellular debris, the supernatants were incubated with Protein G
Plus-Agarose (Santa Cruz Biotechnology, Inc.) and rabbit anti-HBx
polyclonal antibody (Abcam) for immunoprecipitation. The
precipitated complexes were subjected to sodium dodecyl
sulphate-polyacrylamide gel electrophoresis and transferred to
polyvinylidene fluoride (PVDF) membranes. After blocking with 5%
bovine serum albumin (Sigma) for 1 h, the membrane was incubated
overnight at 4°C with rabbit anti-HBx polyclonal antibody
(1:1,000), followed by incubation with an anti-rabbit secondary
antibody conjugated to horseradish peroxidase (1:2,000; Pierce).
The bound antibodies were visualised using an ECL chemiluminescence
system (6,20).
Analysis of secreted HBV antigens
Culture supernatants collected from transfected
cells at different time points were clarified by centrifugation at
3,000 rpm for 15 min and stored at −20°C until used. Hepatitis B
surface antigen (HBsAg) and hepatitis B e-antigen (HBeAg) were
detected by an enzyme-linked immunosorbent assay (ELISA) kit
(Shanghai, KeHua) according to the manufacturer’s instructions. The
absorbance of the contents in each well was determined at the
wavelength of 450 nm. Positive and negative control sera were
included in each assay. The results were expressed as mean optical
density (OD) values [mean ± standard deviation (SD)].
Southern blot analyses
At 48 and 96 h post-transfection, capsid-associated
HBV DNA was extracted as described previously (8). Transfected cells were washed with
cold phosphate-buffered saline (PBS) and lysed in 1% NP lysis
buffer [50 mM Tris (pH 7.4), 1 mM EDTA, 1% NP-40, and 100 mM NaCl].
After centrifugation for 1 min at 12,000 rpm at 4°C, the
supernatants were treated with 100 μg/ml DNase I (Promega) for 30
min at 37°C and then 0.5 mg/ml proteinase K at 50°C for 2 h. Viral
DNA released from lysed cores were extracted with
phenol/chloroform, precipitated with ethanol, and dissolved in
Tris-EDTA. Nuclear HBV cccDNA was extracted as described (15,21). Simply, transfected cells were
lysed in cell lysis buffer [50 mM Tris-HCl (pH 8.0), 1 mM EDTA,
0.2% NP-40, and 150 mM NaCl]. After centrifugation for 10 min at
12,000 rpm at 4°C, the precipitate was resuspended in nuclear lysis
buffer (6% SDS, 100 mM NaOH) and incubated for 30 min at 37°C. The
lysates were then neutralised with potassium acetate (pH 4.8) and
centrifuged for 10 min at 12,000 rpm at 4°C. Nucleic acids were
purified by phenol/chloroform extraction and ethanol precipitation.
The replicative intermediates of capsid-associated HBV DNA and
nuclear HBV cccDNA were detected by Southern blotting. Ten
micrograms of capsid-associated HBV DNA and HBV cccDNA was
separated on a 1% agarose gel, transferred onto a positively
charged nylon membrane, and hybridised with a
32P-labelled full-length HBV DNA probe (Takara). The
membrane was then washed and exposed to film at −80°C (22).
Quantitative analysis of
capsid-associated HBV DNA
At 24, 48, 72 and 96 h after transfection,
capsid-associated HBV DNA in HepG2 cells was extracted and
quantitated by real-time PCR using TaqMan probes. The primers were
5′-AGAAACAACA CATAGCGCCTCAT-3′ (forward) and 5′-TGCCCCATGCTG
TAGATCTTG-3′ (reverse). The TaqMan probe was 5′-FAM-
TGTGGGTCACCATATTCTTGGG-TAMER-3′ (6). The cycling parameters, performed
with an Applied Biosystems 7300 sequence detection system, were as
followed: 95°C for 30 sec, then 40 cycles of 95°C for 5 sec and
60°C for 31 sec. The plasmid pUC-HBV1.0 was diluted over a range of
109–103 copies and used as a standard. The
results were expressed as the number of DNA copies/cell (mean ±
SD).
Quantitative analysis of HBV cccDNA
HBV cccDNA was quantified by real-time PCR as
previously described (15). HBV
cccDNA extracted from transfected HepG2 cells was treated with
Plasmid-Safe DNase (Epicentre) at 37°C for 1 h to remove the open
circular duplex HBV DNA and single-strand HBV DNA. The primers were
5′-CTCCCCGTCTGTGCCTTCT-3′ (forward) and 5′-GCCCCAAAGCCACCCAAG-3′
(reverse). The probes were 5′-GTTCACGGTGGTCTCCATGCAA CGT-FAM-3′ and
5′-ROX-AGGTGAAGCGAAGTGCACAC GGACC-PO4-3′ (15,19). Real-time PCR experiments were
performed with a LightCycler (Roche) as followed: pre-denaturation
for 10 min at 95°C, then 45 cycles of 10 sec at 95°C, 5 sec at
58°C, 10 sec at 63°C, and 20 sec at 72°C. Standard curves were
prepared as described in the quantitative analysis of
capsid-associated HBV DNA by real-time PCR. The results were
expressed as the number of DNA copies/cell (mean ± SD).
Quantitative analysis of HBV pregenomic
RNA (pgRNA)
For pgRNA analysis, total cellular RNA was extracted
with TRIzol reagent (Invitrogen) from transfected HepG2 cells at
different time points post-transfection. RNA concentration and
purity were determined by ultraviolet spectrometry. The RNA samples
were treated with RNase-Free DNase (Promega) at 37°C for 30 min,
and reverse transcribed into cDNA using PrimeScript® RT
reagent kit (Takara). Each cDNA was quantified by real-time PCR
using SYBR® Premix Ex Taq™ II kit (Takara). The specific
primers for pgRNA were 5′-GCCTTAGAGTCTCCTGAGCA-3′ (forward) and
5′-GAGGGAGTTCTTCTTCTAGG-3′ (reverse) (21), and for GAPDH were
5′-GAAGGTGAAGGTCGGAGTC-3′ (forward) and 5′-GAAGATGGTGATGGGATTTC-3′
(reverse). Amplification of GAPDH cDNA was used to normalise the
RNA samples.
cccDNA chromatin immunoprecipitation
(ChIP) assays
ChIP assays were performed with an EZ-Magna ChIP
assay kit (no. 17-408; Millipore) according to the manufacturer’s
specifications. Briefly, at 24, 48, 72 and 96 h post-transfection,
HepG2 cells were cross-linked by incubation with 1% formaldehyde
for 10 min at room temperature, which was terminated with 10X
glycine by incubation at room temperature for 5 min. The collected
cells were washed twice with cold PBS and lysed with cell lysis
buffer by incubation on ice for 15 min. The cell pellet was then
resuspended in nuclear lysis buffer and sonicated to generate
300–400 bp DNA fragments. After centrifugation, 50 μl of each of
the supernatants (1×106 cell equivalents) was diluted
1:10 with ChIP dilution buffer and a 1% volume of the mixture was
taken as input. The chromatin was then subjected to
immunoprecipitation for 14–16 h at 4°C with anti-H3 (no. 06-755),
anti-acetylated histone H3 at lysines 9 and 14 (no. 06-599),
anti-monomethylated histone H3 at lysine 4 (no. 07-436),
anti-phosphorylated histone H3 at serine 10 (no. 04-817; all were
from Millipore) antibodies, and 20 μl of fully suspended protein A
magnetic beads. Immunoprecipitation with the relevant nonspecific
immunoglobulin G (IgG) was included in each experiment as a
negative control. After reversal of the cross-linking, DNA from the
antibody-bound and input fractions was isolated and treated with
plasmid-safe DNase at 37°C for 1 h. Purified ChIP cccDNA and input
DNA were then analysed by PCR and real-time PCR using
cccDNA-selective primers and probes (15). HBV cccDNA-selective primers were
HBV P23 (5′-CTGAATCCCGCGGACGACCC-3′) (1443–1462), and P24
(5′-ACCCAAGGCACAGCTTGGAGG-3′) (1891–1871), which were specific to
the HBV precore-core promoter region to distinguish cccDNA from
rcDNA (25,26). The PCR reaction was performed with
the high-fidelity enzyme Premix PrimeSTAR® HS as
follows: 35 cycles of 10 sec at 98°C, 5 sec at 60°C and 1 min at
72°C. The PCR products were analysed by electrophoresis. ChIP
cccDNA and input DNA were absolutely quantified by real-time PCR as
described above. Results were expressed as the percentage of input
DNA.
Statistical analysis
The data presented from at least 3 separate
experiments were expressed as the means ± SD. Statistical
comparisons of the continuous variables between the 2 groups were
performed using the nonparametric Wilcoxon rank-sum test (SPSS 19.0
software). P-values of <0.05 were assigned to indicate
statistically significant results.
Results
HBx is required for enhancement of HBV
replication in HepG2 cells
To detect the effect of HBx on HBV replication,
equivalent number of HepG2 cells were transfected with the linear
full-length HBV genome (wild-type) or the HBx mutant.
Capsid-associated HBV DNA was extracted from HepG2 cells at 24, 48,
72 and 96 h post-transfection and quantified by real-time PCR. The
levels of capsid-associated HBV DNA from the wild-type
HBV-transfected cells were 39.1±2.9 copies/cell at 48 h which
declined to 10.5±1.4 copies/cell at 96 h, while levels from the HBx
mutant-transfected cells were 36.6±2.5 copies/cell at 48 h which
decreased to 4.9±1.1 copies/cell at 96 h (Fig. 1A). Between 24 and 48 h, when HBV
replication reached peak levels, the levels of capsid-associated
DNA in the HBx mutant-transfected cells were slightly affected by
the lack of HBx (P>0.05); however, the levels of
capsid-associated HBV DNA were significantly reduced at 72 and in
particular at 96 h, which showed a 50–70% reduction (P<0.05) as
compared to those levels in the wild-type HBV-transfected cells.
The results of the Southern blotting by which replicative
intermediates of capsid-associated DNA were detected with a
32P-labelled full-length HBV DNA probe were consistent
with those of the real-time PCR quantitation of capsid-associated
DNA (Fig. 1B). A sensitive
IP/western blot assay was used to detect the expression of HBx in
transfected HepG2 cells. HBx was below the limit of detection by
IP/western blot assay in the HBx mutant-transfected cells, but was
detected at both 48 and 96 h in the wild-type HBV-transfected
cells, with the expression level of HBx at 96 h much lower than
that at 48 h (Fig. 1C). Together,
these results demonstrate that HBx is required for the enhancement
of HBV replication in HepG2 cells.
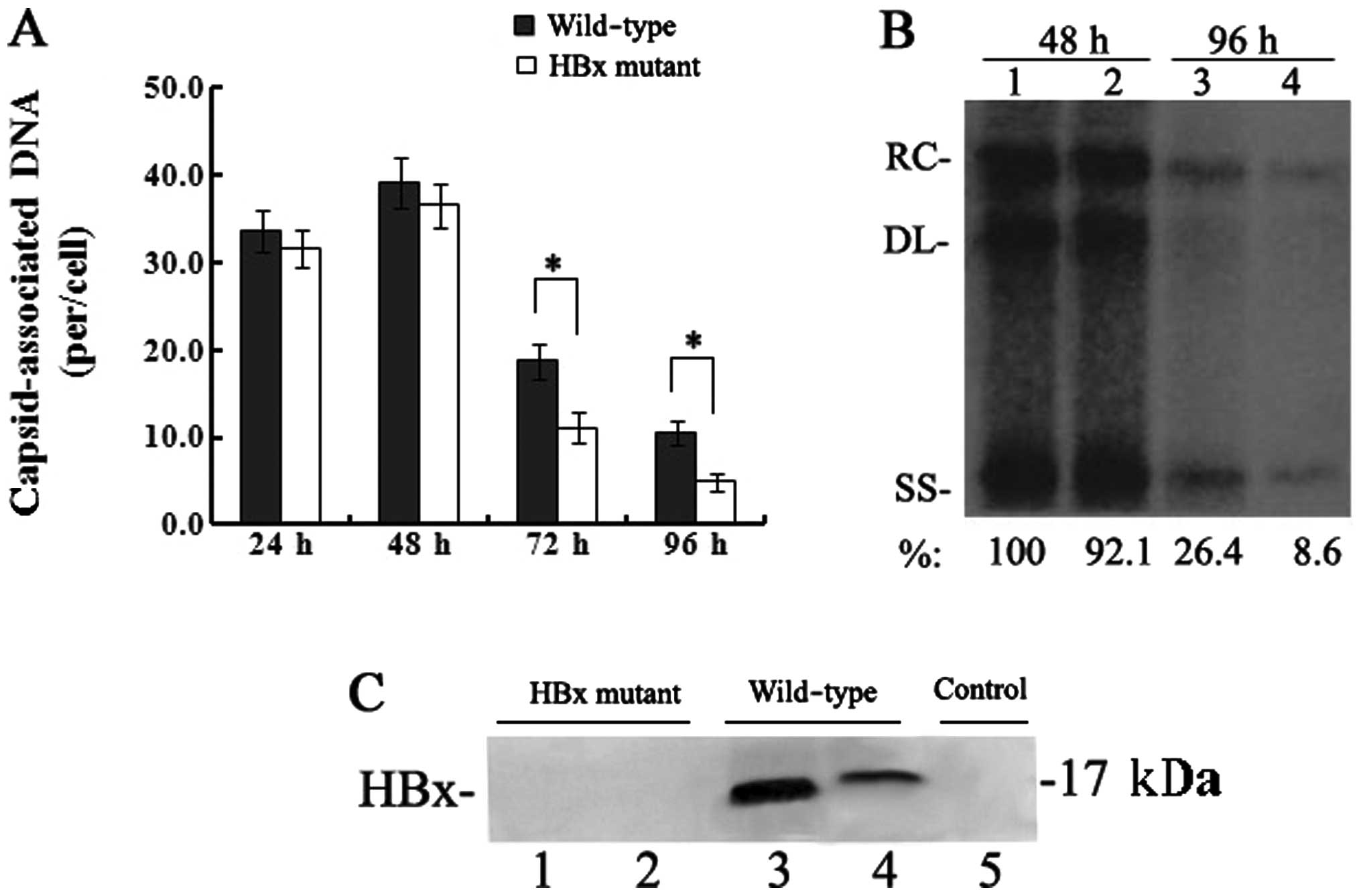 | Figure 1Effect of HBx on HBV replication in
transfected HepG2 cells. (A) Quantification of capsid-associated
HBV DNA by real-time PCR at 24, 48, 72 and 96 h post-transfection.
Results are expressed as the number of capsid-associated DNA copies
per cell (mean ± SD) from 3 independent experiments. Statistical
significance is designated with asterisks above the brackets. (B)
Southern blot analysis of capsid-associated viral DNA replicative
intermediates at 48 and 96 h after transfection. Signal intensity
of the single-stranded (SS) band underneath the double-stranded
linear (DL) HBV DNA band was quantified with Quantity One Analysis
software (Bio-Rad). The band corresponding to the DL HBV DNA was
not included in the quantitative analysis, as this DNA might be
partially derived from transfected input DNA. Lanes 1 and 3, the
wild-type HBV genome; lanes 2 and 4, HBx mutant HBV DNA. The number
at the bottom of each lane represents the relative levels of HBV
DNA replicative intermediates, with those levels detected in the
wild-type HBV-transfected cells at 48 h set to 100%, and levels
measured in the HBx mutant-transfected cells are compared to those
in the wild-type HBV-transfected cells from three independent
experiments; RC, relaxed circular; DL, double-stranded linear; and
SS, single-stranded forms. (C) Representative IP/western blotting.
A negative control with no plasmid transfected into HepG2 cells
(lane 1); HepG2 cells transfected with the linear HBx-deficient
mutant HBV DNA (HBx mutant) and the full-length HBV genome
(wild-type) were harvested at 48 and 96 h, and analysed by
IP/western blotting for HBx protein. Lanes 1 and 3, 48 h; lanes 2
and 4, 96 h. |
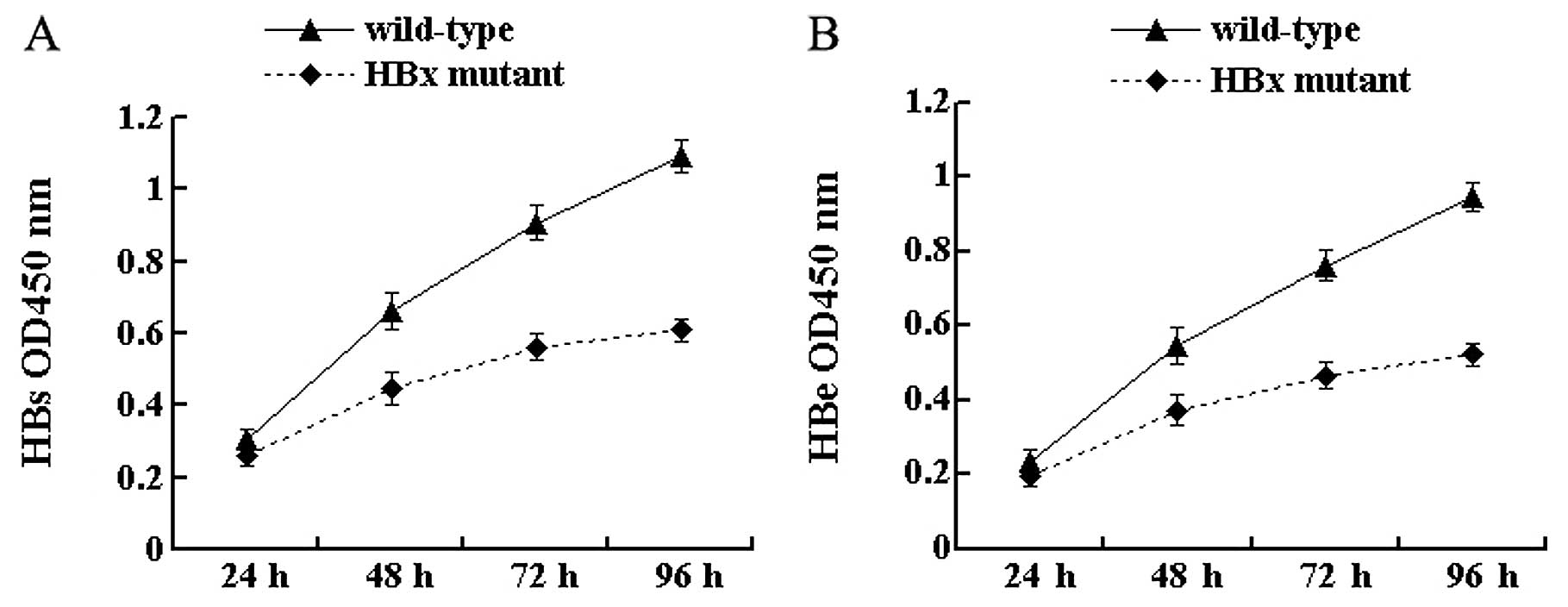
HBx affects the secretion of the HBV
antigen
To investigate the effect of HBx on antigen
secretion, the secretion of HBsAg and HBeAg from cell culture
supernatants was tested by ELISA at 4 time-points. The results
showed that the secretion of HBV antigens was detected in cells
transfected with both wild-type and HBx mutant HBV DNA. At 24 h
after transfection, there was no significant difference in the
secretion of HBsAg and HBeAg between the 2 types of HBV DNA
(P>0.05) (Fig. 2). With the
extension of time after transfection, differences gradually
appeared. In particular, at 96 h the secretion of HBsAg and HBeAg
from HBx mutant-transfected cells was 55.7% (P<0.05) (Fig. 2A) and 55.2% (P<0.05) (Fig. 2B), respectively, of that from
wild-type HBV-transfected cells. These results indicate that the
lack of HBx reduces the secretion of viral antigens in HepG2
cells.
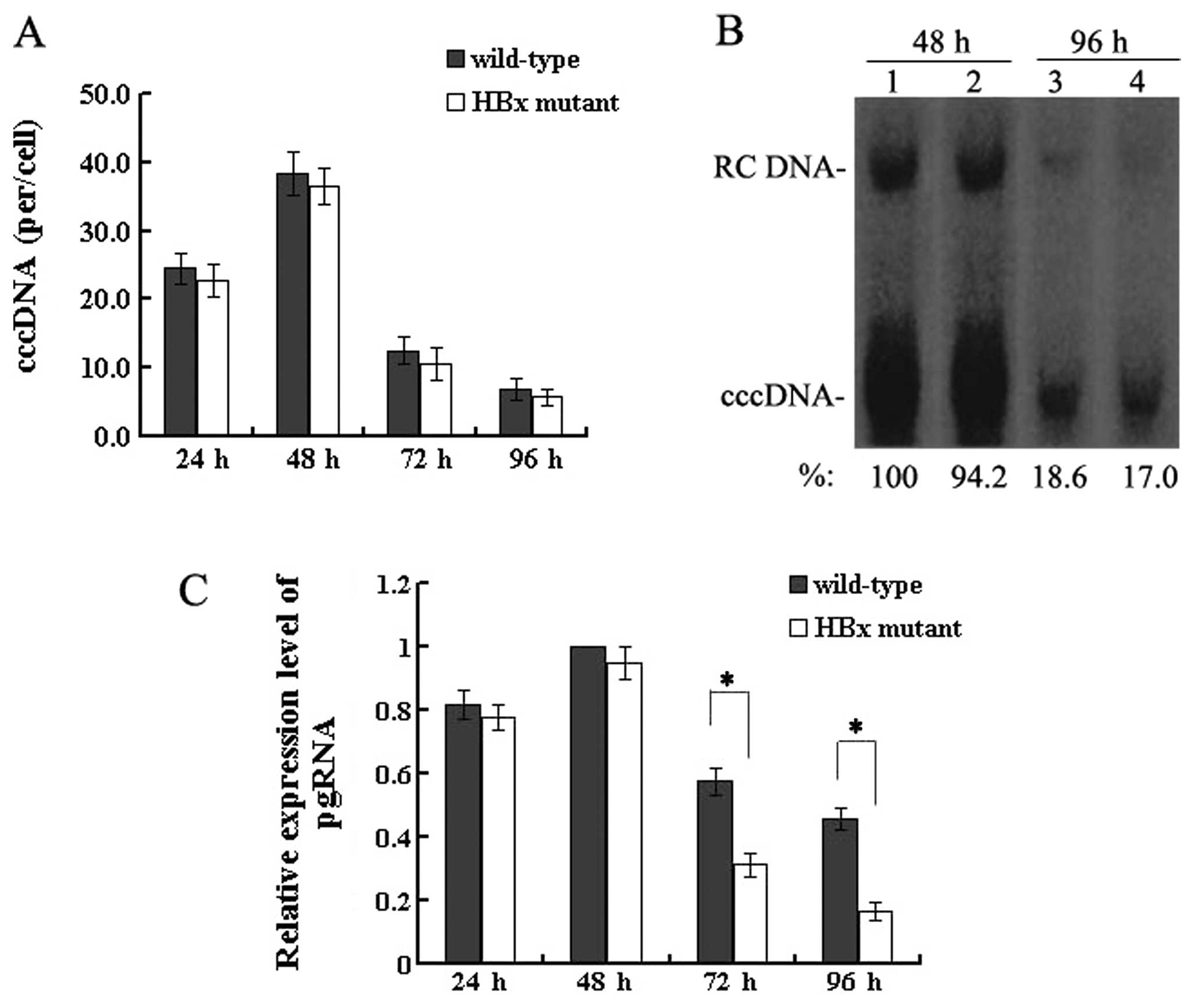
HBx does not affect HBV cccDNA formation,
but upregulates the transcription of pgRNA
The HBV cccDNA extracted from HepG2 cells at 24, 48,
72 and 96 h post-transfection was purified after treatment with
plasmid-safe DNase and quantitatively analysed by real-time PCR.
The level of cccDNA in wild-type HBV-transfected cells was 38.4±3.1
copies/cell at 48 h which decreased to 6.9±1.5 copies/cell at 96 h,
and the level in the HBx mutant-transfected cells was 36.5±2.7
copies/cell at 48 h which declined to 5.8±1.2 copies/cell at 96 h
(Fig. 3A). When HBV replication
reached a peak between 24 and 48 h or when levels of both nuclear
cccDNA and HBV replication were reduced severely at 72 and 96 h,
the mean copies of cccDNA per HepG2 cell were similar with the 2
types of HBV genome. The results of the real-time PCR quantitation
of HBV cccDNA were confirmed by Southern blotting by which nuclear
cccDNA was detected (Fig. 3B).
Altogether, these results demonstrate that HBx is not required for
HBV cccDNA formation.
Since HBx is not required for the formation of HBV
cccDNA, the effect of HBx on a downstream step, pgRNA
transcription, was investigated. At 4 time-points
post-transfection, the extracted RNA was reverse transcribed and
quantified by real-time PCR. These results showed that levels of
pgRNA were slightly lower between 24 and 48 h after transfection in
the HBx mutant DNA (P>0.05). However, differences gradually
appeared after this time. In particular, at 96 h, the levels of
pgRNA were reduced by 50–70% in the absence of HBx, compared to
levels in the cells transfected with wild-type HBV DNA (P<0.05)
(Fig. 3C). Taken together, these
findings indicate that HBx upregulates pgRNA transcription without
affecting the formation of HBV cccDNA.
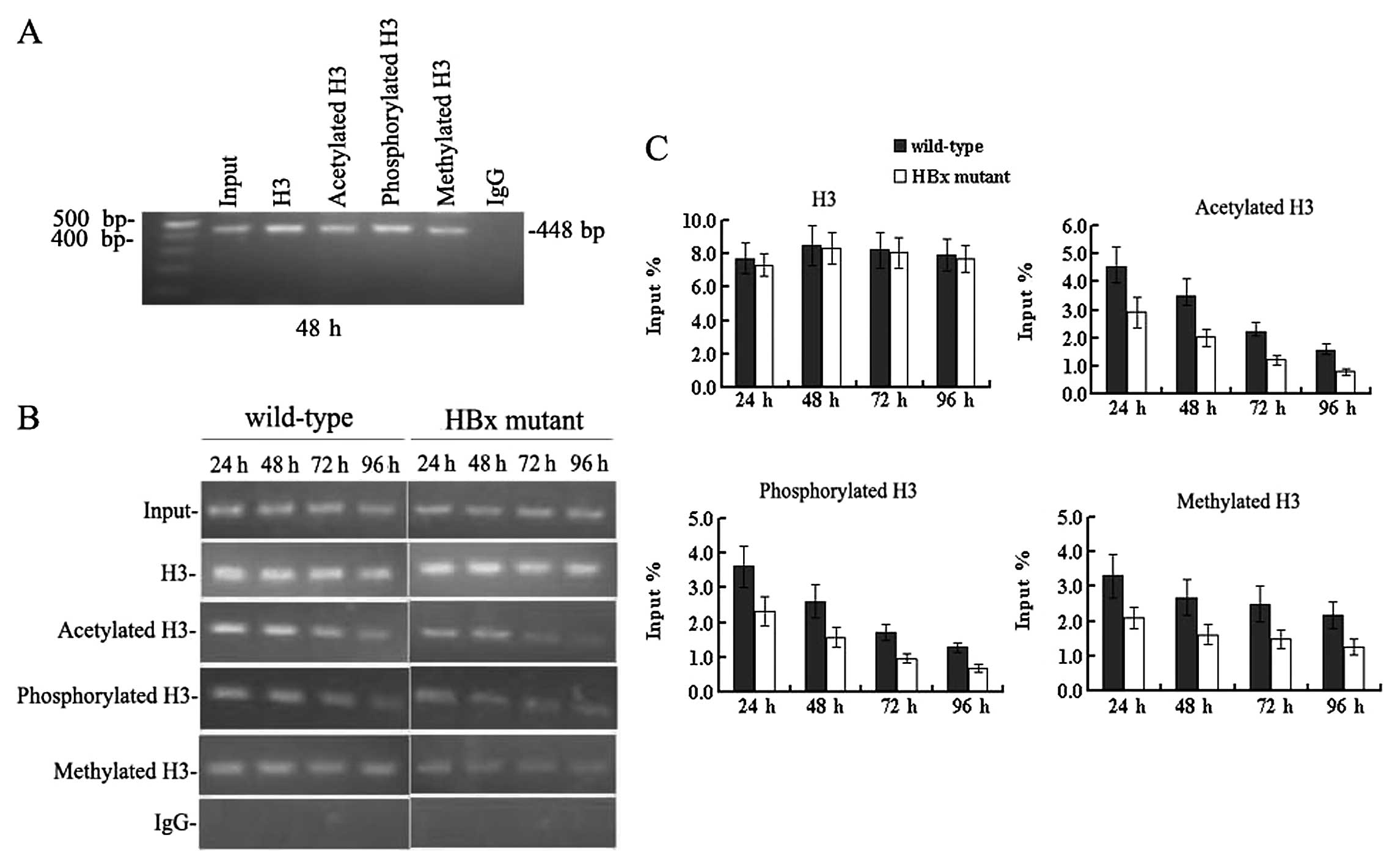
HBx regulates remodelling of the
minichromosome related to HBV replication
Since HBx has proven to be crucial for HBV
replication and transcription, we sought to study whether HBx
affects remodelling of the minichromosome related to HBV
replication. Immunoprecipitated cccDNA from HepG2 cells at 24, 48,
72 and 96 h after transfection was amplified by PCR and quantified
by real-time PCR. In cells transfected with the wild-type HBV
genome (Fig. 4), the cccDNA-bound
histone H3 was highly acetylated, hypermethylated and
hyperphosphorylated simultaneously between 24 and 48 h
post-transfection when the level of HBV replication reached a peak.
Levels of cccDNA-bound acetylated, mono-methylated and
phosphorylated histone H3 peaked at 4.53±0.71, 3.31±0.62 and
3.62±0.59, in units of % input DNA, respectively. At 72 h, when the
HBV replication declined, the levels of cccDNA-bound acetylated and
phosphorylated histone H3 were sharply reduced compared to those at
48 h (all P<0.05). Both the acetylation and phosphorylation of
cccDNA-bound H3 histone decreased by 72 h to 2.24±0.31 and
1.71±0.22 in units of % input DNA, respectively (all P<0.05).
However, levels of cccDNA-bound methylated H3 did not change
appreciably at 72 h, and the values in units of % input DNA were
2.69±0.52 and 2.49±0.51 at 48 and 72 h, respectively (P>0.05)
(Fig. 4B and C). These results
indicate that the acetylation and phosphorylation of cccDNA-bound
histones H3 are dynamic, but the methylation of cccDNA-bound
histone H3 is relatively stable at lysine 4. Acetylation,
methylation and phosphorylation of cccDNA-bound histone H3
paralleled HBV replication in HepG2 cells.
Next we investigated the epigenetic changes in cells
transfected with the HBx mutant HBV genome. cccDNA-bound histone H3
was not only rapidly hypo-acetylated but also hypo-phosphorylated
and hypomethylated (Fig. 4B and
C). At 48 h post-transfection, cccDNA-bound acetylated,
phosphorylated and methylated histone H3 were reduced by 40–50% in
the absence of HBx compared to those levels in cells transfected
with the wild-type HBV DNA (all P<0.05). The acetylation,
methylation, and phosphorylation of cccDNA-bound H3 were 2.01±0.32,
1.61±0.28 and 1.58±0.29 in units of % input DNA, respectively. At
96 h, levels of the above-mentioned modifications of histone H3
were very low. These results were in agreement with the reduction
observed in HBV replication (Fig. 1A
and B), pgRNA transcription (Fig.
3C) and antigen secretion (Fig.
2). Together, these findings demonstrate that HBx can affect
the methylation, phosphorylation and acetylation of cccDNA-bound
histone H3 during HBV replication in HepG2 cells. In other words,
HBx can regulate the remodelling of the minichromosomes related to
HBV replication in HepG2 cells. Considering that epigenetic
modifications of cccDNA-bound H3 histone parallel HBV replication,
it is now possible to state that HBx may regulate viral replication
through the pathway of chromatin remodelling.
Discussion
In our study, the HBx-deficient plasmid
pUC-HBV1.0.X7 was successfully constructed by site-directed
mutagenesis, and two in vitro replication models by
transfecting HepG2 cells with the linear full-length HBV genome
(wild-type) or the HBx-deficient mutant HBV DNA (HBx mutant) were
established successfully. We found that although the formation of
HBV cccDNA was not affected by HBx, there was a dramatic reduction
in HBV replication, pgRNA transcription and antigen secretion in
the absence of HBx compared to levels in cells transfected with the
wild-type HBV genome. In addition, the levels of cccDNA-bound
methylated, phosphorylated and acetylated histone H3 decreased
sharply in HBx mutant HBV DNA. These results suggest that HBx is
required for the enhancement of HBV replication and transcription.
HBx modulates not only the status of acetylation but also the
methylation and phosphorylation of cccDNA-bound histone H3 related
to HBV replication in HepG2 cells.
Although it is not completely clear why HBV cccDNA
formation is not affected by HBx, our study has demonstrated that
HBx plays an importance role in regulating methylation and
phosphorylation in addition to acetylation of cccDNA-bound histone
H3 during HBV replication and elucidated the mechanism of
HBx-dependent HBV replication through the pathway of chromatin
remodelling. It has been shown that HBx does not directly bind to
DNA sequences but is recruited onto the chromatin through its
ability to interact with various cellular partners and proteins
(23). The recruitment of HBx
onto the cccDNA parallels the dynamic changes of cccDNA-bound
acetylated H3 (21). HBx favours
the aceylation of histone H3 bound to cccDNA and modulates HBV
replication and transcription (15,24). However, our present study has
shown that the methylation, phosphorylation and acetylation of
cccDNA-bound histone H3 paralleled HBV replication in HepG2 cells.
The cccDNA-bound histone H3 was highly acetylated, hypermethylated
and hyperphosphorylated when the level of HBV replication reached a
peak; while the levels of cccDNA-bound acetylated and
phosphorylated histone H3 were also reduced when the HBV
replication declined, although the levels of cccDNA-bound
methylated H3 did not change appreciably. But why acetylation and
phosphorylation of cccDNA-bound histone H3 decreased over time
apart from methylation remains obscure. The difference in HBV
replication between the groups of wild-type and HBx-mutant appeared
only at 72 h post-transfection whereas the difference in epigenetic
modifications was already detected 24 h post-transfection. The
possible interpretations are that the absence of HBx may change
chromatin structure and DNA-histone contacts, resulting in
remodelling of minichromosomes; the decline of HBV replication may
be associated with the early decrease in histone modifications. HBx
modulates not only the status of acetylation but also the
methylation and phosphorylation of histone H3 bound to the cccDNA
during HBV replication. Therefore, one of the mechanisms of
HBx-dependent HBV replication may be that HBx influences epigenetic
modifications, leading to chromatin remodelling.
Histone lysine methylation is closely related to
gene transcription, which has a different effect at different
amino-terminal residues. Methylation of lysines 9 and 27 in histone
H3 correlates with gene repression, whereas methylation of lysines
4, 36 and 79 in histone H3 correlates with activation (27,28). Our study suggested that HBx
modulates the status of hisone H3 methylation. The possible
explanation from some studies is that SET and MYND
domain-containing protein 3 (SMYD3) is one of the
methyltransferases for H3 lysine 4 (29,30). RNA polymerase II is recruited to
the promoter region of SMYD3 gene by HBx, leading to the
enhancement of the expression of SMYD3 and cellular activities of
HMTs at H3 lysine 4 (30,31). Histone modifications, as well as
modifications of the DNA, can influence chromatin structure, induce
the remodelling of chromatin and consequently result in gene
silencing (27). HBx can increase
the activities of total DNA methyltransferases (DNMTs) by
upregulation of DNMT1, DNMT3A1 and DNMT3A2, and can selectively
promote regional hypermethylation of specific tumour-suppressor
genes (32). In the cytoplasm,
HBx increases DNMT1 by activating the Ras signalling pathway
(32,33) and/or by inhibiting p53 function
(32,34). HBx interacts with DNMT3A to
trigger epigenetic modifications at different loci, thus regulating
the transcription of target genes. For example, HBx recruits DNMT3A
to the promoter region of ML1F and IL4R, inducing inhibition of
ML1F and IL4R and regional hypermethylation. In contrast, HBx
separates DNMT3A from the promoter of IGFBP-3 and CDH6, resulting
in activation of IGFBP-3 and CDH6 and downregulation of DNA
methylation (35).
Histone H3 serine 10 phosphorylation is important
for transcriptional activation and chromosome condensation
occurring during mitosis, meiosis, apoptosis and DNA damage
(27). Our study has shown that
HBx modulates the status of histone H3 phosphorylation. HBx can
also promote nuclear protein serine phosphorylation and increases
pgRNA encapsidation and HBV DNA synthesis, which may be
attributable to the activation of the HBx-induced signal
transduction pathways including the core protein serine kinases
(36). A rapid phosphorylation of
histone H3 at serine 10 is induced by the early response genes
c-fos and c-myc when the Ras mitogen-activated protein kinase
(MAPK) signalling pathway is stimulated (37,38). Modifications of histones are quite
complex and can interplay with each other due to the cross-talk
between them (39). The
activation of Aurora-kinase-B-mediated phosphorylation of H3 serine
10 may serve as a ‘phos-methyl switch’, leading to the separation
of HP1 from heterochromatin while maintaining H3K9 methylation
during mitosis (39,40). Acetylation of histones at lysines
9 and 14 can serve as a prelude to transcriptional activation,
whereas methylation of histones at lysine 9 can lead to gene
silencing and formation of heterochromatin. These modifications may
influence the ability of serine 10 to be phosphorylated and vice
versa (41).
Histone acetyltransferases (HATs) and histone
deacetylases (HDACs) are responsible for the steady-state balance
of acetylation modification of histones, although this balance can
be affected by HBx protein. Cellular histone acetytransferases
p300, CBP, PCAF/GCN5, and the histone deactylases HDAC1 and hSirtl
are recruited with different kinetics onto cccDNA in HBV
replication (13,21). A previous study demonstrated that
there is no HBx recruited onto cccDNA in cells replicating the HBx
mutant HBV virus (21).
cccDNA-bound histones are rapidly hypoacetylated in the absence of
HBx, and the recruitment of p300 is severely impaired while the
recruitment of the histone deacetylases HDAC1 and hSirtl is
increased and occurs earlier (21). Our study also demonstrated that
HBx modulates the status of histone H3 acetylation. The possible
explanation is that HBx affects the expression of important
cellular genes resulting from its transcriptional transactivation
and transrepression properties. HBx can directly interact with the
acetyltransferase p300/CBP complex in coordination to enhance the
activity of CREB to promote transcription, leading to activation of
the acetylated histone state of the target cellular genes (18,42). HBx recruits HDAC1 to the promoter
of IGFBP-3 and induces the formation of the Sp1/HDAC1 complex,
resulting in deacetylation of Sp1 and inhibition of the
transcription of IGFBP-3 (43).
In summary, HBx plays a pivotal role in HBV
replication and transcription. Specifically, HBx affects not only
the status of acetylation but also methylation and phosphorylation
of cccDNA-bound histone H3 during HBV replication in HepG2 cells.
HBx may modulate HBV replication through the pathway of
minichromosome remodelling related to HBV replication. Further
study is required to ascertain whether HBx modulates HBV
replication by affecting the recruitment of other histone-modifying
enzymes bound to the cccDNA in addition to HATs and HDACs. In this
regard, our research provides experimental evidence for elucidating
the mechanism of HBx-dependent HBV replication through the pathway
of chromatin remodelling and identified the HBx protein as a new
target for antiviral treatment at the level of cccDNA.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (grant no. 30872249) and key
program of Medical Science of Chongqing Health Bureau (grant no.
2010-1-37).
Abbreviations:
|
cccDNA
|
covalently closed circular DNA
|
|
ChIP
|
chromatin immunoprecipitation
|
|
CREB
|
cAMP-response element protein
|
|
DNMTs
|
DNA methyltransferases
|
|
DL
|
double-stranded linear
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
GFP
|
green fluorescence protein
|
|
HBV
|
hepatitis B virus
|
|
HBx
|
hepatitis B virus X protein
|
|
HCC
|
hepatocellular carcinoma
|
|
HBsAg
|
hepatitis B surface antigen
|
|
HBeAg
|
hepatitis B e-antigen
|
|
HATs
|
histone acetyltransferases
|
|
HDACs
|
histone deacetylases
|
|
HMTs
|
histone methyltransferases
|
|
IP
|
immunoprecipitation
|
|
IgG
|
immunoglobulin G
|
|
MAPK
|
mitogen-activated protein kinase
|
|
OD
|
optical density
|
|
pgRNA
|
pregenomic RNA
|
|
PVDF
|
polyvinylidene fluoride
|
|
PBS
|
phosphate-buffered saline
|
|
rcDNA
|
relaxed circular DNA
|
|
RIPA
|
radioimmunoprecipitation assay
|
|
SS
|
single-strand
|
|
SD
|
standard deviation
|
References
|
1
|
Dienstag JL: Hepatitis B virus infection.
N Engl J Med. 359:1486–1500. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Seeger C and Mason WS: Hepatitis B virus
biology. Microbiol Mol Biol Rev. 64:51–68. 2000. View Article : Google Scholar
|
|
3
|
Murakami S: Hepatitis B virus X protein: a
multifunctional viral regulator. J Gastroenterol. 36:651–660. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scaglioni PP, Melegari M and Wands JR:
Posttranscriptional regulation of hepatitis B virus replication by
the precore protein. J Virol. 71:345–353. 1997.PubMed/NCBI
|
|
5
|
Melegari M, Scaglioni PP and Wands JR:
Cloning and characterization of a novel hepatitis B virus x binding
protein that inhibits viral replication. J Virol. 72:1737–1743.
1998.PubMed/NCBI
|
|
6
|
Keasler VV, Hodgson AJ, Madden CR and
Slagle BL: Enhancement of hepatitis B virus replication by the
regulatory X protein in vitro and in vivo. J Virol. 81:2656–2662.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bouchard MJ, Wang LH and Schneider RJ:
Calcium signaling by HBx protein in hepatitis B virus DNA
replication. Science. 294:2376–2378. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tang H, Delgermaa L, Huang F, et al: The
transcriptional transactivation function of HBx protein is
important for its augmentation role in hepatitis B virus
replication. J Virol. 79:5548–5556. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang PL, Althage A, Chung J and Chisari
FV: Hydrodynamic injection of viral DNA: a mouse model of acute
hepatitis B virus infection. Proc Natl Acad Sci USA.
99:13825–13830. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Keasler VV, Hodgson AJ, Madden CR and
Slagle BL: Hepatitis B virus HBx protein localized to the nucleus
restores HBx-deficient virus replication in HepG2 cells and in vivo
in hydrodynamically-injected mice. Virology. 390:122–129. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Newbold JE, Xin H, Tencza M, Sherman G,
Dean J, Bowden S and Locarnini S: The covalently closed duplex form
of the hepadnavirus genome exists in situ as a heterogeneous
population of viral minichromosomes. J Virol. 69:3350–3357.
1995.PubMed/NCBI
|
|
12
|
Gao W and Hu J: Formation of hepatitis B
virus covalently closed circular DNA: removal of genome-linked
protein. J Virol. 81:6164–6174. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Levrero M, Pollicino T, Petersen J,
Belloni L, Raimondo G and Dandri M: Control of cccDNA function in
hepatitis B virus infection. J Hepatol. 51:581–592. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bock CT, Schwinn S, Locarnini S, Fyfe J,
Manns MP, Trautwei C and Zentgraf H: Structural organization of the
hepatitis B virus minichromosome. J Mol Biol. 307:183–196. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pollicino T, Belloni L, Raffa G, Pediconi
N, Squadrito G, Raimondo G and Levrero M: Hepatitis B virus
replication is regulated by the acetylation status of hepatitis B
virus cccDNA-bound H3 and H4 histones. Gastroenterology.
130:823–837. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bouchard MJ, Wang L and Schneider RJ:
Activation of focal adhesion kinase by hepatitis B virus HBx
protein: multiple functions in viral replication. J Virol.
80:4406–4414. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cougot D, Wu Y, Cairo S, et al: The
hepatitis B virus X protein functionally interacts with
CREB-binding protein/p300 in the regulation of CREB-mediated
transcription. J Biol Chem. 282:4277–4287. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harju S, McQueen KJ and Peterson KR:
Chromatin structure and control of beta-like globin gene switching.
Exp Biol Med (Maywood). 227:683–700. 2002.PubMed/NCBI
|
|
19
|
Gong Q, Chen S, Guo J, et al: Chromosome
remodeling related to hepatitis B virus replication in HepG2 cells.
DNA Cell Biol. 30:347–354. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Slagle BL, Lee TH, Medina D, Finegold MJ
and Butel JS: Increased sensitivity to the hepatocarcinogen
diethylnitrosamine in transgenic mice carrying the hepatitis B
virus X gene. Mol Carcinog. 15:261–269. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Belloni L, Pollicino T, De Nicola F, et
al: Nuclear HBx binds the HBV minichromosome and modifies the
epigenetic regulation of cccDNA function. Proc Natl Acad Sci USA.
106:19975–19979. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sambrook J and Russel DW: Molecular
Cloning: A Laboratory Manual. 3rd edition. Cold Spring Harbor
Laboratory Press; New York: pp. 138–140. pp. 151–153. 2001
|
|
23
|
Brechot C, Kremsdorf D, Soussan P, Pineau
P, Dejean A, Paterlini-Brechot P and Tiollais P: Hepatitis B virus
(HBV)-related hepatocellular carcinoma (HCC): molecular mechanisms
and novel paradigms. Pathol Biol (Paris). 58:278–287. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lucifora J, Arzberger S, Durantel D, et
al: Hepatitis B virus X protein is essential to initiate and
maintain virus replication after infection. J Hepatol. 55:996–1003.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pollicino T, Squadrito G, Cerenzia G, et
al: Hepatitis B virus maintains its pro-oncogenic properties in the
case of occult HBV infection. Gastroenterology. 126:102–110. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stoll-Becker S, Repp R, Glebe D, et al:
Transcription of hepatitis B virus in peripheral blood mononuclear
cells from persistently infected patients. J Virol. 71:5399–5407.
1997.PubMed/NCBI
|
|
27
|
Hake SB, Xiao A and Allis CD: Linking the
epigenetic ‘language’ of covalent histone modifications to cancer.
Br J Cancer. 90:761–769. 2004.
|
|
28
|
Strahl BD and Allis CD: The language of
covalent histone modifications. Nature. 403:41–45. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dillon SC, Zhang X, Trievel RC and Cheng
X: The SET-domain protein superfamily: protein lysine
methyltransferases. Genome Biol. 6:2272005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hamamoto R, Furukawa Y, Morita M, et al:
SMYD3 encodes a histone methyltransferase involved in the
proliferation of cancer cells. Nat Cell Biol. 6:731–740. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang L, He J, Chen L and Wang G: Hepatitis
B virus X protein upregulates expression of SMYD3 and C-MYC in
HepG2 cells. Med Oncol. 26:445–451. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Park IY, Sohn BH, Yu E, et al: Aberrant
epigenetic modifications in hepatocarcinogenesis induced by
hepatitis B virus X protein. Gastroenterology. 132:1476–1494. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rouleau J, MacLeod AR and Szyf M:
Regulation of the DNA methyltransferase by the Ras-AP-1 signaling
pathway. J Biol Chem. 270:1595–1601. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Peterson EJ, Bogler O and Taylor SM:
p53-mediated repression of DNA methyltransferase 1 expression by
specific DNA binding. Cancer Res. 63:6579–6582. 2003.PubMed/NCBI
|
|
35
|
Zheng DL, Zhang L, Cheng N, et al:
Epigenetic modification induced by hepatitis B virus X protein via
interaction with de novo DNA methyltransferase DNMT3A. J Hepatol.
50:377–387. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Melegari M, Wolf SK and Schneider RJ:
Hepatitis B virus DNA replication is coordinated by core protein
serine phosphorylation and HBx expression. J Virol. 79:9810–9820.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chadee DN, Hendzel MJ, Tylipski CP, Allis
CD, Bazett-Jones DP, Wright JA and Davie JR: Increased Ser-10
phosphorylation of histone H3 in mitogen-stimulated and
oncogene-transformed mouse fibroblasts. J Biol Chem.
274:24914–24920. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Min L, He B and Hui L: Mitogen-activated
protein kinases in hepatocellular carcinoma development. Semin
Cancer Biol. 21:10–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang GG, Allis CD and Chi P: Chromatin
remodeling and cancer, Part I: Covalent histone modifications.
Trends Mol Med. 13:363–372. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hirota T, Lipp JJ, Toh BH and Peters JM:
Histone H3 serine 10 phosphorylation by Aurora B causes HP1
dissociation from heterochromatin. Nature. 438:1176–1180. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nowak SJ and Corces VG: Phosphorylation of
histone H3: a balancing act between chromosome condensation and
transcriptional activation. Trends Genet. 20:214–220. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Herceg Z and Paliwal A: Epigenetic
mechanisms in hepatocellular carcinoma: how environmental factors
influence the epigenome. Mutat Res. 727:55–61. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shon JK, Shon BH, Park IY, et al:
Hepatitis B virus-X protein recruits histone deacetylase 1 to
repress insulin-like growth factor binding protein 3 transcription.
Virus Res. 139:14–21. 2009. View Article : Google Scholar : PubMed/NCBI
|