Introduction
Nitric oxide (NO) is synthesized by stereospecific
oxidation of terminal guanidine nitrogen of L-arginine by the
action of the NO synthase (NOS) (1). The synthesis of NO can be blocked by
inhibition of the NOS active site with guanidine-substituted
analogues of L-arginine, such as asymmetric dimethylarginine (ADMA)
(2,3), which is an important risk factor for
endothelial dysfunction.
Numerous studies have revealed that a high level of
ADMA is associated with increased renal oxidative stress (ROS)
(4,5) and activated the oxidant-responsive
transcription factor nuclear factor-κB (NF-κB), which enhanced
cytokine expression in human endothelial cells (6). Furthermore, in hepatic stellate
cells, treatment with ADMA significantly increased the
intracellular ROS production and activated NF-κB. The effects of
ADMA on the level of transforming growth factor-β (TGF-β) mRNA
could be markedly attenuated by pre-treatment with antioxidant
pyrrolidine dithiocarbamate (7).
We, and others, have demonstrated that actin cytoskeleton modulates
ADMA-induced NF-κB nuclear translocation in previous studies, which
plays a critical role on endothelial cell dysfunction (8,9).
ADMA can also increase endothelial permeability, which may involve
the p38 MAPK and NADPH signal pathway (10).
ADMA is considered an independent mortality and
cardiovascular risk factor in chronic kidney disease (CKD) patients
(11–13). Matsumoto et al (14) indicated that high plasma levels of
ADMA are associated with decreased number of peritubular
capillaries, increased tubulointerstitial fibrosis, TGF-β
expression and proteinuria levels in five-sixths subtotal
nephrectomy (Nx) rats. These data and similar observations of other
authors suggest that ADMA may contribute to the progression of CKD
(14–16). Furthermore, ADMA exposure induced
glomerular and vascular fibrosis as evidenced by the elevated
deposits of collagens I, III and fibronectin, suggesting that in
pathophysiological conditions of endothelial dysfunction, the
exaggerated endogenous synthesis of ADMA could contribute to CKD
progression by extracellular matrix synthesis (17). It was accepted that TGF-β was
related to renal fibrosis. However, the precise molecular
mechanisms underlying ADMA-induced TGF-β expression is not
explicit.
In addition, the current experimental research uses,
primarily, high concentration (10–500 μmol/l) and short-term (12–24
h) incubation conditions to study the cellular effects of ADMA.
However, the level is lower (1–10 μmol/l) and the affliction
proceed during the whole period in the high blood level ADMA
diseases, such as CKD, cardiovascular disease (CVD), hypertension,
and diabetes. The present study was designed to determine the
effects of long-term low-dose ADMA on the TGF-β expression in
endothelial cells and to investigate the underlying molecular
mechanisms.
Materials and methods
Cell culture
Primary human renal glomerular endothelial cells
(HRGECs) and primary human umbilical vein endothelial cells
(HUVECs) were purchased from ScienCell Research Laboratories (San
Diego, CA, USA). Both were cultured in endothelial cell medium
(ECM) (ScienCell Research Laboratories) containing 5% fetal bovine
serum (FBS), 1% endothelial cell growth supplement (ECGS) and 1%
penicillin/streptomycin solution (P/S).
The medium of HRGECs was changed every two days.
Actively growing HRGECs at the third to the fifth passage were used
for experiments.
HUVECs were used in the long-term low-dose ADMA
stimulation. ADMA was replaced every 48 h starting at the fourth
passage until the ninth passage. After reaching confluence (between
8–9 days), endothelial cells were trypsinized and seeded at a
density of 2,500 cells/cm2 per 25 cm2 flasks.
Following detachment with trypsin telomerase activity, mRNA and
protein levels of TGF-β expression were analyzed.
Western blot analysis
Cells were lysed with RIPA lysis buffer. Protein
concentrations were determined using a BCA assay, soluble proteins
were separated on 12% SDS-polyacrylamide gels and transferred to
polyvinylidene difluoride membranes. Nonspecific membrane binding
was blocked for 1 h at room temperature with 5% BSA (or milk) in
phosphate-buffered saline containing 0.05% Tween-20. Membranes were
incubated overnight at 4°C with primary antibodies (1:50; Beijing
Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China).
After washing, membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies for 1–2 h at room
temperature in blocking buffer. Signals generated by the
chemiluminescent substrate were captured by ECL plus reagent (Sun
Bio Corp., Beijing, China), and the images were analyzed by
densitometry using Labworks Image Acquisition and Analysis Software
(Ultra-Violet Products, Ltd., Cambridge, UK). Protein bands were
quantified by densitometry using the analysis software Image.
Quantitative real-time PCR
Total-RNA was isolated from cells using the TRIzol
reagent (Invitrogen, USA) and was quantified by analysis of
absorbance at 260 nm. Relative gene expression levels were
calculated using the comparative threshold-cycle method of
quantitative PCR, with data normalized to GAPDH and expressed
relative to untreated controls. Real-time PCR was performed using
the BioEasy SYBR-Green I Real-Time PCR kit (Bioer Technology Co.,
Ltd, China), according to the manufacturer’s instructions. The
primers used to amplify TGF-β were: 5′-GCCAGAGTGGTTATCTTTTGATG-3′
and 5′-AGTGTGTTATCCCTGCTGTCAC-3′. The total volume used in PCR was
20 μl. All experiments were performed in triplicate.
Electrophoretic mobility shift assay
(EMSA)
Ten micrograms of nuclear extract were incubated
with 1 μg of poly(dI-dC) in a binding buffer (10 mM Tris-HCl, pH
7.5, 50 mM NaCl, 0.5 mM dithiothreitol, 10% glycerol, 20 μl final
volume) for 15 min at room temperature. Then, end-labeled
double-stranded oligonucleotides containing an NF-κB site (30,000
cpm each) were added and the reaction mixtures were incubated for
15 min at room temperature. The DNA-protein complexes were resolved
in 5% native polyacrylamide gel electrophoresis in low ionic
strength buffer (0.25X Tris borate/EDTA). The oligonucleotide used
for the gel shift analysis was NF-κB 5′-AGTTGAGGGGACTTTCCCAGGC-3′.
The sequence motifs within the oligonucleotides are underlined.
Immunofluorescence
Cells grown on coverslips were fixed in 3.7%
paraformaldehyde/PBS for 30 min and permeabilized with 0.1% Triton
X-100 for 10 min. Cells were incubated with fluorescein
isothiocyanate-phalloidin (FITC-phalloidin) for 60 min at room
temperature to localize F-actin filaments. Single plain images of
the cells were obtained by confocal laser scanning microscopy
(Leica TCS SP5, Mannheim, Germany).
Statistical analysis
Data are expressed as the means ± SD. Comparisons
between more than two conditions were performed with the one-way
ANOVA test followed by the LSD and SNK test (western blot analysis
and real-time PCR data). P<0.05 was considered to indicate
statistically significant differences. All tests were performed
with SPSS version 17.0.
Results
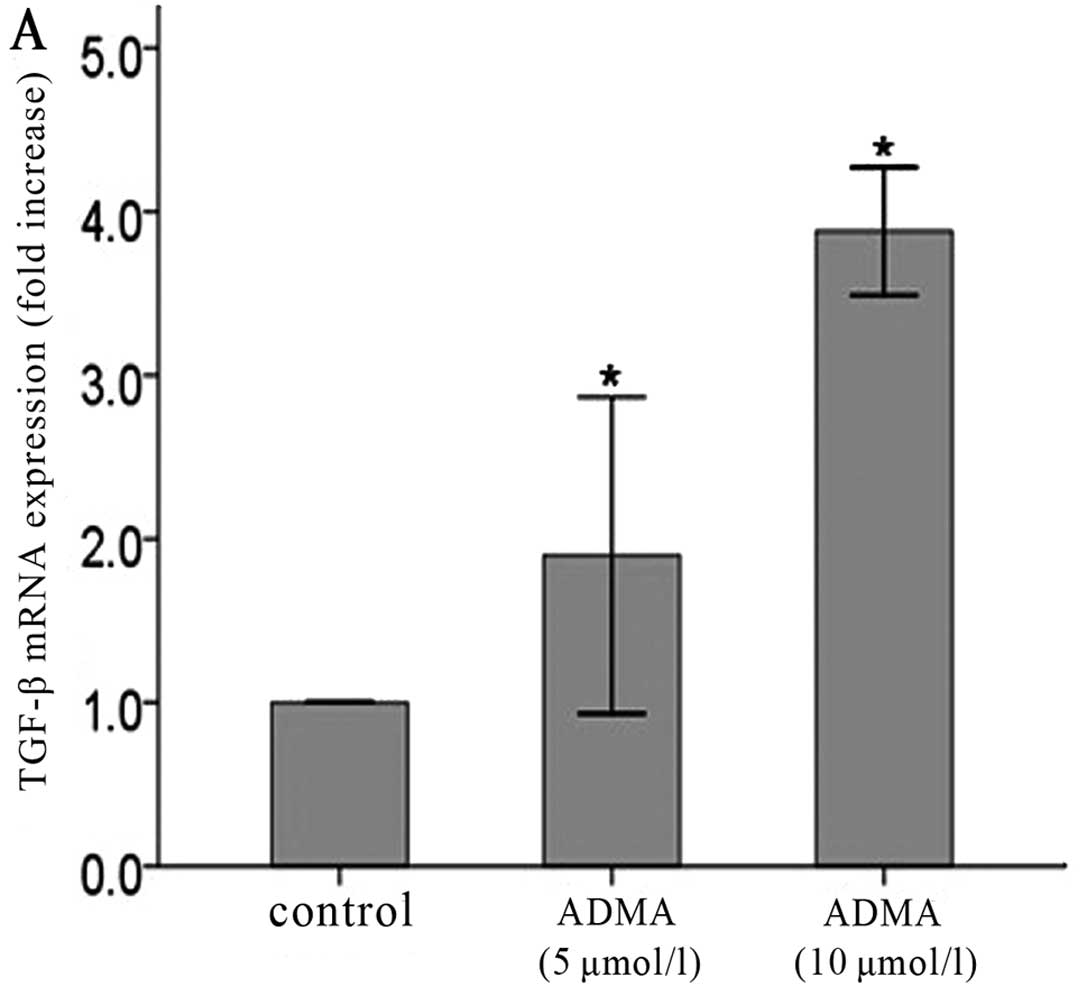
Long-term low-dose ADMA increases TGF-β
production in HUVECs
HUVECs were cultured until the ninth passage and
incubated in the presence of different low-dose concentrations of
ADMA (5 and 10 μmol/l), which were replaced every 48 h starting
from the fourth passage. Both protein (Fig. 1A, C and E) and mRNA (Fig. 1B, D and F) levels of TGF-β were
determined at the fifth, seventh, and ninth passage. In each
passage (P5, P7, P9), both concentrations of ADMA (5 and 10 μmol/l)
significantly increased TGF-β production compared with untreated
cells. More obvious effects were observed in the higher
concentration.
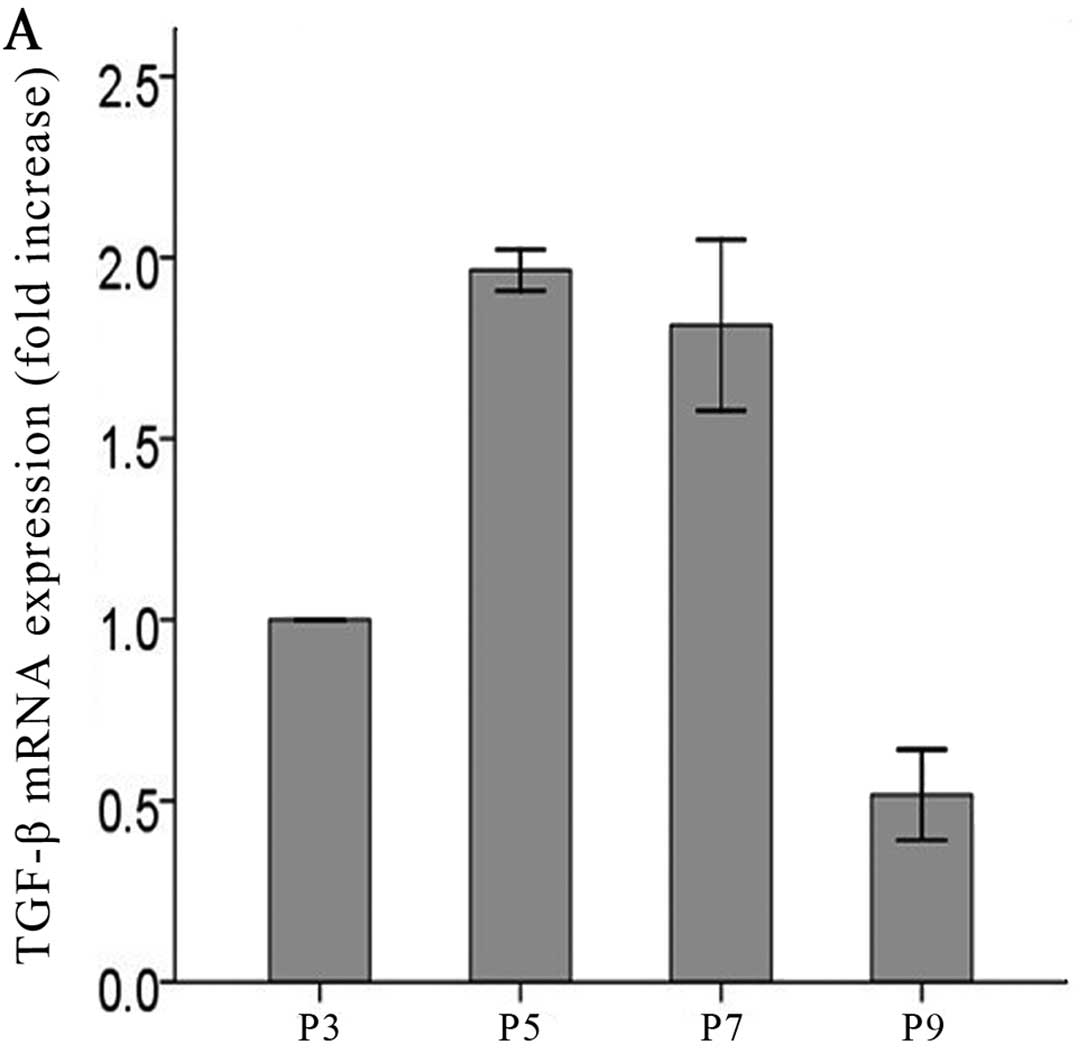
Long-term low-dose ADMA increases TGF-β
production in HUVECs in a time-dependent manner
To investigate the correlation between TGF-β
expression and ADMA, HUVECs were cultured until the ninth passage
and incubated in the presence of low-dose of ADMA (5 and 10
μmol/l), which was replaced every 48 h starting from passage four.
Both protein (Fig. 2A, C and E)
and mRNA (Fig. 2B, D and F)
levels of TGF-β were determined at the third, fifth, seventh, and
ninth passage. The results showed that both the mRNA and protein
levels of TGF-β were increased in a time-dependent manner (Fig. 2C–F) in the cells exposed to
low-dose ADMA. The time-dependent manner was absent in control
cells without ADMA stimulation (Fig.
2A and B).
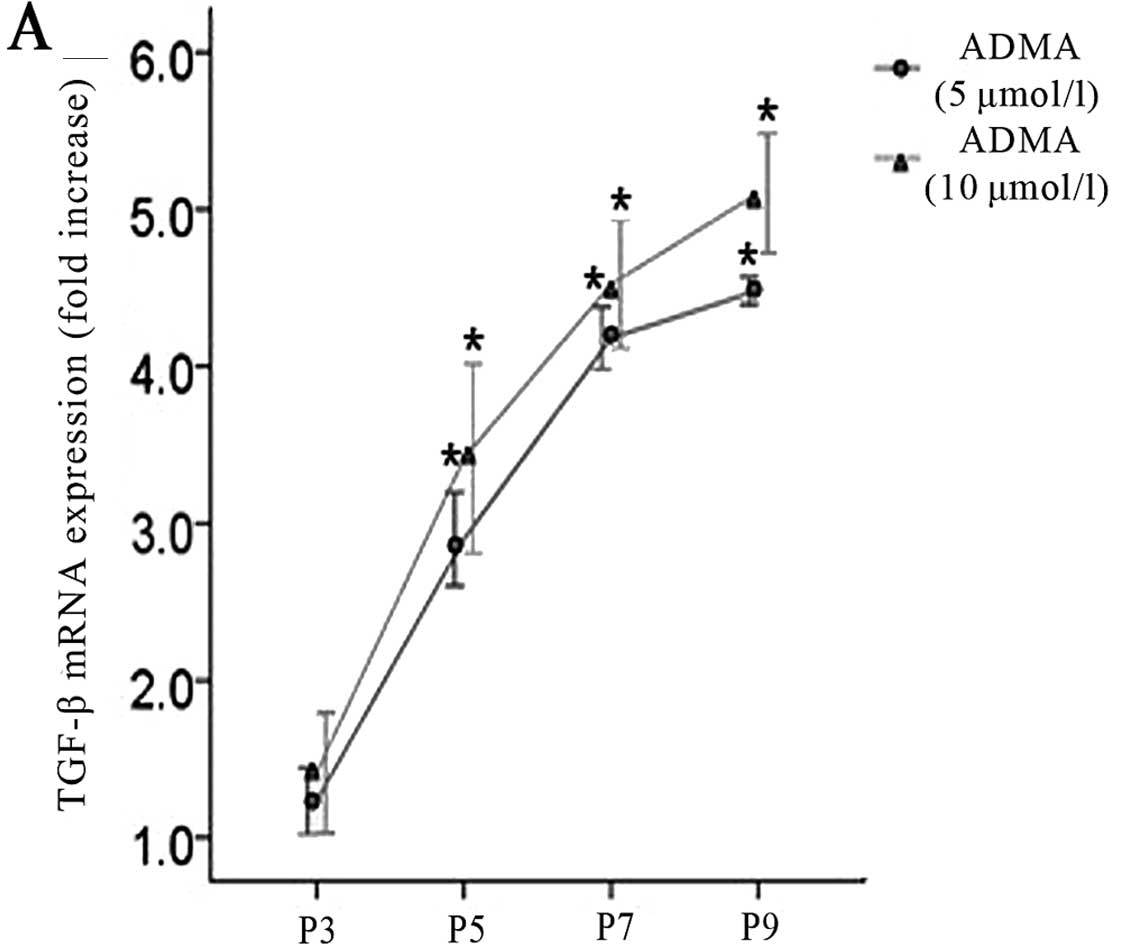
Since TGF-β, a pleiotropic cytokine, is also present
in control cells, and is involved in the effects of cell apoptosis,
the following formula was used for correction:
Relative TGF-β expression = TGF-β expression in
ADMA-stimulated (5 and 10 μmol/l) cells/TGF-β expression in control
cells.
In Fig. 3, the
changes in TGF-β production in endothelial cells are shown as a
function of passage number. Treatment of cultured endothelial cells
with ADMA (5 and 10 μmol/l) significantly increased TGF-β
production compared with untreated cells in a time-dependent
manner.
Reorganization of actin filaments
inhibits ADMA-induced stress fiber formation and TGF-β expression
in HRGECs
To address the role of actin cytoskeleton in the
mechanism of ADMA-induced NF-κB activation in HRGECs, we firstly
evaluated the effect of reorganizing actin filaments on
NF-κB-dependent reporter gene activity. We used the four types of
agents which can prevent the formation of actin stress fibers
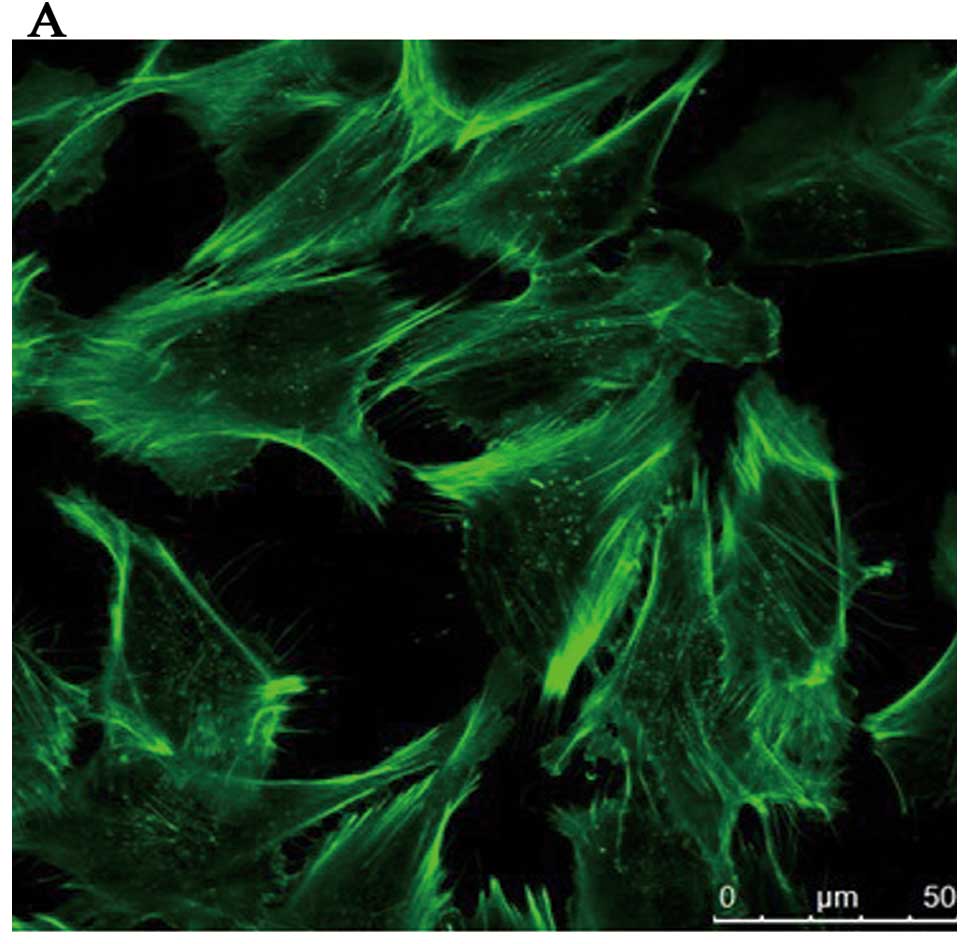
induced by ADMA. Analysis by immunofluorescence microscopy of
HRGECs showed that the cells of the control group were polygonal in
shape and F-actin was found mostly in the cortical regions
comprising the cortical actin bands (Fig. 4A), which rearranged into stress
fibers after treatment with ADMA (Fig. 4B). Pre-treatment with 5 μM Cyto D,
the prototypic actin depolymerizing drug, induced obvious
destabilization of the actin filaments and then prevented the
formation of stress fibers induced by ADMA (Fig. 4E). Similar effects were observed
with the presence of SB 203580, the p38 MAPK inhibitor (Fig. 4C), apocynin, the NADPH oxidase
inhibitor (Fig. 4D), and Jas, the
prototypic actin stabilizing drug (Fig. 4F).
Since NF-κB is an essential regulator of TGF-β
transcription, we determined if the effects of reorganization of
the actin filaments on NF-κB activity are reproduced on
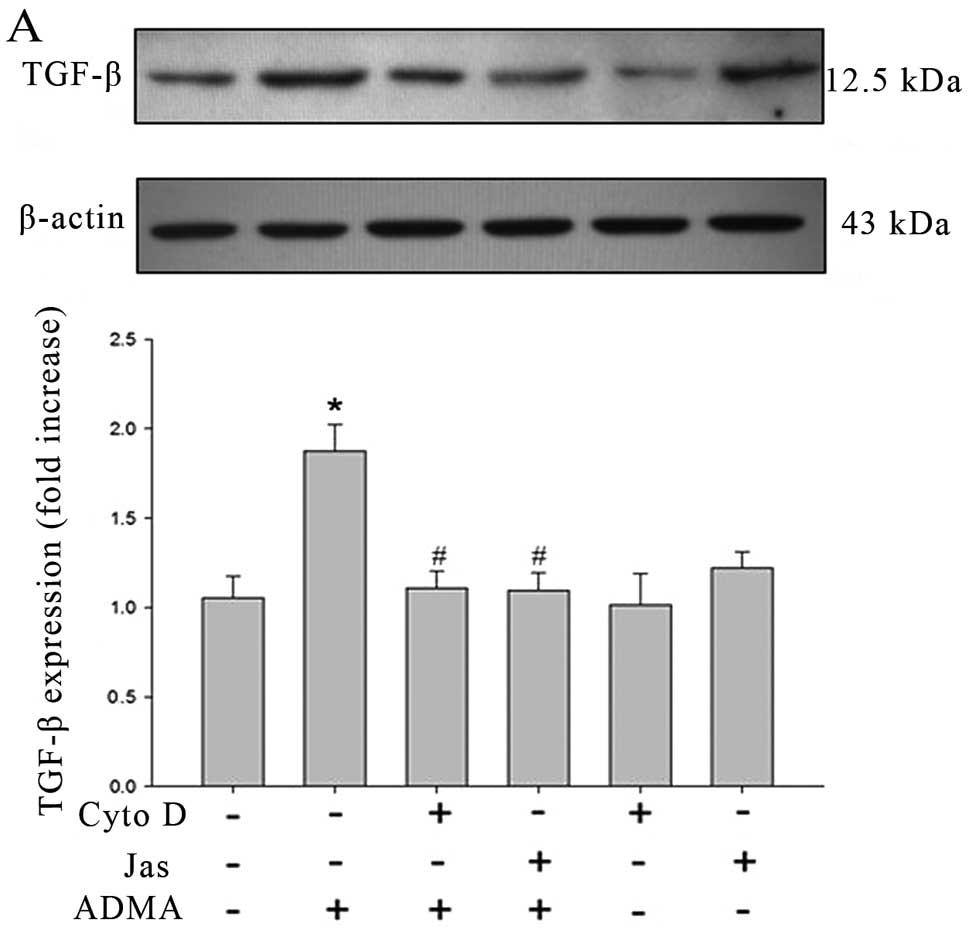
ADMA-induced TGF-β expression in HRGECs. Western blot analysis
showed that HRGECs exposed to ADMA (100 μmol/l) expressed much
higher levels of TGF-β than control cells and the increased TGF-β
protein was significantly inhibited in cells pre-treated with Cyto
D (5 μmol/l) or Jas (1 μmol/l) for 30 min (Fig. 5A). However, the drugs per
se have no significant effect on TGF-β expression. The similar
inhibition of ADMA-induced TGF-β expression was also observed from
the pre-treatment of p38 MAPK inhibitor and NADPH oxidase inhibitor
(Fig. 5B).
Reorganization of actin filaments
prevents ADMA-induced NF-κB DNA binding activity
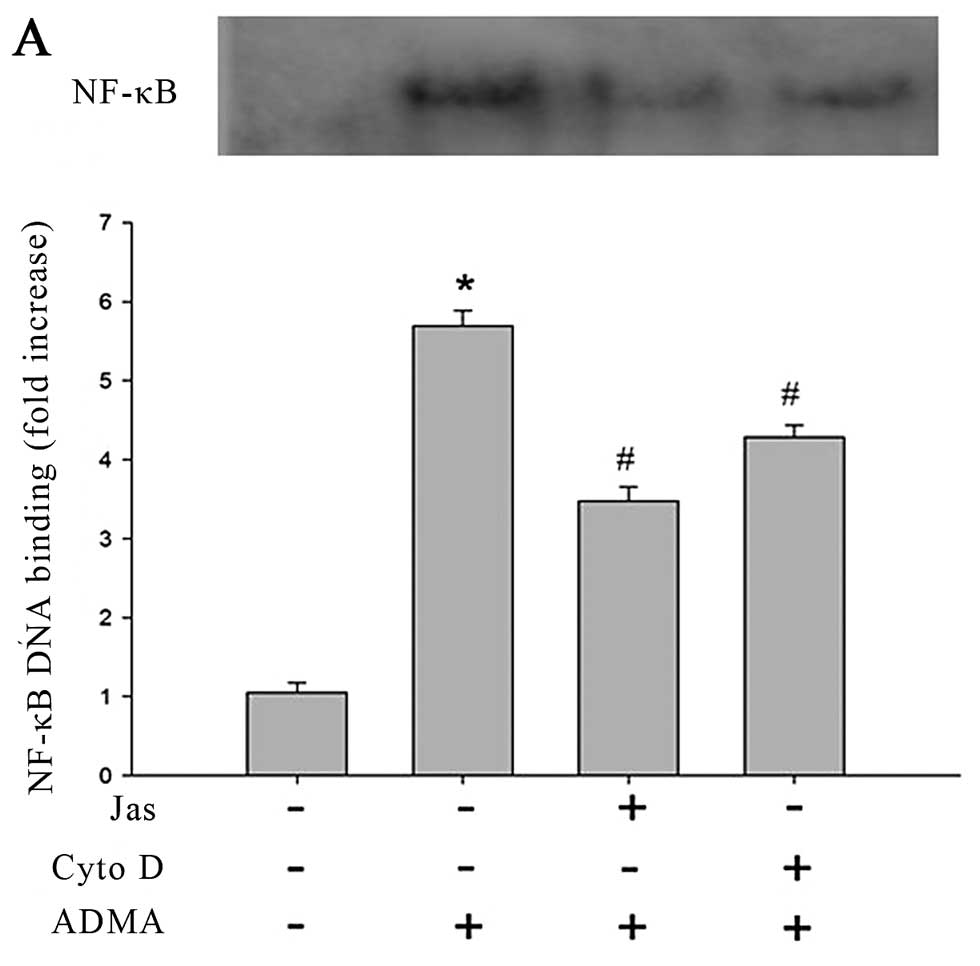
The results of EMSA revealed that the control group
showed a faint shift, while the cells exposed to ADMA (100 μmol/l)
produced a strong shift. In subsequent experiments, it was found
that both Cyto D and Jas reduced the DNA binding of NF-κB in
response to ADMA challenge (Fig.
6A). The similar inhibition of ADMA-induced NK-κB nuclear
translocation was also observed from the cells pretreated by p38
MAPK inhibitor and NADPH oxidase inhibitor (Fig. 6B).
Discussion
The main purpose of this study was to demonstrate
that ADMA, the endogenous NOS inhibitor, can induce TGF-β
expression in endothelial cells, and that cytoskeleton organization
may be involved in modulating ADMA-induced NF-κB activation and the
ensuing TGF-β expression in HRGECs.
NF-κB is a ubiquitously expressed family of
transcription factors controlling varied biological effects ranging
from inflammatory, oxidative stress, and stress-induced responses
to cell fate decisions such as proliferation, differentiation,
tumorigenesis, and apoptosis (18–20). Fazal et al (9) revealed the existence of actin
cytoskeleton-dependent and -independent pathways that may be
involved in a stimulus-specific manner to facilitate NF-κB nuclear
import and thereby ICAM-1 expression in endothelial cells. We have
already confirmed that stabilization and destabilization of the
actin filaments prevented ADMA-induced NF-κB activation and the
ensuing cytokine production in HUVECs (8). However, it was confirmed that the
actin organization responses to ADMA challenge and the subsequent
NF-κB activation are cell type specific (21), and, to date, how cytoskeleton
responds to ADMA stimulation and the role of cytoskeleton
reorganization in ADMA-induced NF-κB activation in HRGECs has yet
to be investigated. In the present study, we demonstrated that ADMA
significantly enhanced the stress fiber formation and active DNA
binding of NF-κB in HRGECs, and such effects were inhibited by
pre-treatment with actin depolymerizing or stabilizing drug.
Previous studies have demonstrated that the p38 MAPK
and NADPH pathways may be involved in the cytoskeleton formation.
Accumulating evidence suggests that ROS are important regulators of
the actin cytoskeletal dynamics and cellular motility, and an
important source of ROS within endothelial cells is the
non-phagocytic NADPH oxidase (22). It has been demonstrated that NADPH
oxidase activation and the ensuing ROS production associated with
actin or with the F-actin binding protein moesin (23–25), dramatically increasing the speed
of actin polymerization. Furthermore, phosphorylation of Hsp27, the
downstream effector of p38 (26),
leads to dissociation of multimers and promotes actin
polymerization (27). Inhibiting
p38 can inhibit stress fiber formation in adherent cells and lead
to a decrease in cell permeability (28–30). We have already confirmed that ADMA
increases endothelial permeability, which may involve the p38 MAPK
and NADPH oxidase pathway in HUVECs (10). In the experiments presented here,
it has been demonstrated that the inhibition of the p38 MAPK
pathway and NADPH oxidase activation can inhibit the ADMA-induced
NK-κB activation via decreasing cytoskeleton reorganization in
HRGECs, suggesting the cytoskeleton is involved in modulating the
NF-κB nuclear transcription.
Findings of this study show that the inhibition of
NOS by ADMA increased the expression of TGF-β in HUVECs after the
repeated addition of ADMA. ADMA is considered an independent
mortality and cardiovascular risk factor in CKD patients (11–13), possibly contributing to the
progression of CKD (14–16). Evidence has implicated TGF-β as a
major causative agent in the pathogenesis of tubulointerstitial
fibrosis in CKD (31,32). Recently, blockade of NOS or NO
deficiency was reported to promote cardiac or renal fibrosis,
respectively, via induction of TGF-β (33,34), thus suggesting the pathological
role for the ADMA-elicited NO reduction in fibrosis in various
aggressive disorders. Mihout et al (17) demonstrated that high levels of
ADMA induced renal TGF-β production associated with increased renal
oxidative stress in uninephrectomized mice, dimethylarginine
dimethylaminohydrolase (DDAH) ameliorated the renal dysfunction by
reducing ADMA accumulation (14),
while the precise molecular mechanisms were not explicit. It is
known that ADMA increases TGF-β expression via the NO/NF-κB pathway
in hepatic stellate cells, while there is no report on ADMA-induced
NF-κB activity and the ensuing TGF-β expression in endothelial
cells. Our study demonstrated that ADMA-induced TGF-β expression is
associated with NF-κB activity, but whether other pathways are also
involved in the effect remains to be confirmed in future
studies.
TGF-β, a pleiotropic cytokine, regulates cell
proliferation, differentiation, and apoptosis, and plays a key role
in development and tissue homeostasis. Hence, in untreated cells,
TGF-β also expressed along with the growth of cells. The results
presented here show that in the control cells without ADMA
stimulation, TGF-β expression was not time-dependent. We analyzed
the relative TGF-β expression using the corrected formula mentioned
previously to exclude the influencing factor with the normal cell
apoptosis and other cell homeostasis. The experiment demonstrated
that long-term low-dose ADMA increased TGF-β production in both
mRNA and protein levels in HUVECs in a time-dependent manner.
Moreover, in this investigation, more obvious TGF-β
production was observed in the higher ADMA concentration. Hence we
can assume that ADMA accumulated in the progression of CKD,
associated with the higher blood concentration, induced the TGF-β
production in endothelial cells along with the extension of
stimulation time, which could cause the renal fibrosis.
The dose of ADMA used in previous in vitro
studies was usually relatively high (100 μmol/l), however the blood
level of ADMA is 1–10 μmol/l reported in patients with chronic
renal disease. We demonstrated that long-term low-dose of ADMA
induces TGF-β expression in endothelial cells via NF-κB activation
and highlighted the cytoskeleton related pathways responsible.
To date, the technology of extracting HRGECs has not
been improved significantly. In our experiment, the cells failed to
grow at the fifth or sixth passage, hence the long-term stimulation
can not be implemented in HRGECs and we used HUVECs to replace
them. They have common endothelial cell characteristics and HUVECs
have also been studied extensively in kidney research. Whether
long-term low-dose ADMA challenge induces cytokine expression in
HRGECs remains to be studied.
Furthermore, while there is emerging evidence that
the reorganization of cytoskeleton in endothelial cells can be
protectable for endothelial dysfunction, it remains to be
determined whether it can protect the renal fibrosis in further
in vivo studies.
In summary, the present study demonstrated that
long-term low-dose ADMA induces TGF-β expression in endothelial
cells at both the gene and protein level, which may be involved in
the development of renal fibrosis. Actin cytoskeleton may be
involved in modulated ADMA-induced NF-κB activation and the ensuing
TGF-β expression in HRGECs.
Acknowledgements
The present study was financially supported by the
Beijing Municipal Science and Technology Commission Funds
(D09050704310903), the Collaborative Project Funds on Fundamental
and Clinical Research of Capital Medical University (10JL26) and
the Specialized Research Fund for the Doctoral Program of Higher
Education (SRFDP) (20101107120003).
References
|
1
|
Boger RH: Asymmetric dimethylarginine, an
endogenous inhibitor of nitric oxide synthase, explains the
‘L-arginine paradox’ and acts as a novel cardiovascular risk
factor. J Nutr. 134(Suppl 10): S2842–S2853. 2004.
|
|
2
|
Cooke JP: Does ADMA cause endothelial
dysfunction. Arterioscler Thromb Vasc Biol. 20:2032–2037. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boger RH: Asymmetric dimethylarginine
(ADMA) and cardiovascular disease: insights from prospective
clinical trials. Vasc Med. 10(Suppl 1): S19–S25. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Palm F, Onozato ML, Luo Z and Wilcox CS:
Dimethylarginine dimethylaminohydrolase (DDAH): expression,
regulation, and function in the cardiovascular and renal systems.
Am J Physiol Heart Circ Physiol. 293:H3227–H3245. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Teerlink T, Luo Z, Palm F and Wilcox CS:
Cellular ADMA: regulation and action. Pharmacol Res. 60:448–460.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scalera F, Borlak J, Beckmann B, et al:
Endogenous nitric oxide synthesis inhibitor asymmetric dimethyl
L-arginine accelerates endothelial cell senescence. Arterioscler
Thromb Vasc Biol. 24:1816–1822. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li JC, Chang L, Lu D, Jiang DJ and Tan DM:
Effect of asymmetric dimethylarginine on the activation of hepatic
stellate cells and its mechanism. Zhong Nan Da Xue Xue Bao Yi Xue
Ban. 32:427–432. 2007.(In Chinese).
|
|
8
|
Guo WK, Zhang DL, Wang XX, Zhang Y, Zhang
QD and Liu WH: Actin cytoskeleton modulates ADMA-induced NF-kappaB
nuclear translocation and ICAM-1 expression in endothelial cells.
Med Sci Monit. 17:BR242–BR247. 2011.PubMed/NCBI
|
|
9
|
Fazal F, Minhajuddin M, Bijli KM, McGrath
JL and Rahman A: Evidence for actin cytoskeleton-dependent and
-independent pathways for RelA/p65 nuclear translocation in
endothelial cells. J Biol Chem. 282:3940–3950. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang LY, Zhang DL, Zheng JF, Zhang Y,
Zhang QD and Li WH: Apelin-13 passes through the ADMA-damaged
endothelial barrier and acts on vascular smooth muscle cells.
Peptides. 32:2436–2443. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fliser D, Kronenberg F, Kielstein JT, et
al: Asymmetric dimethylarginine and progression of chronic kidney
disease: the mild to moderate kidney disease study. J Am Soc
Nephrol. 16:2456–2461. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zoccali C, Bode-Boger S, Mallamaci F, et
al: Plasma concentration of asymmetrical dimethylarginine and
mortality in patients with end-stage renal disease: a prospective
study. Lancet. 358:2113–2117. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zoccali C: Traditional and emerging
cardiovascular and renal risk factors: an epidemiologic
perspective. Kidney Int. 70:26–33. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matsumoto Y, Ueda S, Yamagishi S, et al:
Dimethylarginine dimethylaminohydrolase prevents progression of
renal dysfunction by inhibiting loss of peritubular capillaries and
tubulointerstitial fibrosis in a rat model of chronic kidney
disease. J Am Soc Nephrol. 18:1525–1533. 2007. View Article : Google Scholar
|
|
15
|
Wagner L, Riggleman A, Erdely A, Couser W
and Baylis C: Reduced nitric oxide synthase activity in rats with
chronic renal disease due to glomerulonephritis. Kidney Int.
62:532–536. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ravani P, Tripepi G, Malberti F, Testa S,
Mallamaci F and Zoccali C: Asymmetrical dimethylarginine predicts
progression to dialysis and death in patients with chronic kidney
disease: a competing risks modeling approach. J Am Soc Nephrol.
16:2449–2455. 2005. View Article : Google Scholar
|
|
17
|
Mihout F, Shweke N, Bige N, et al:
Asymmetric dimethylarginine (ADMA) induces chronic kidney disease
through a mechanism involving collagen and TGF-beta1 synthesis. J
Pathol. 223:37–45. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Aggarwal BB: Nuclear factor-kappa B: the
enemy within. Cancer Cell. 6:203–208. 2004.
|
|
19
|
Hayden MS and Ghosh S: Signaling to
NF-kappaB. Genes Dev. 18:2195–2224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Karin M and Lin A: NF-kappa B at the
crossroads of life and death. Nat Immunol. 3:221–227. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wojciak-Stothard B, Torondel B, Tsang LY,
et al: The ADMA/DDAH pathway is a critical regulator of endothelial
cell motility. J Cell Sci. 120:929–942. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Moldovan L, Mythreye K,
Goldschmidt-Clermont PJ and Satterwhite LL: Reactive oxygen species
in vascular endothelial cell motility. Roles of NAD(P)H oxidase and
Rac1. Cardiovasc Res. 71:236–246. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li JM and Shah AM: Intracellular
localization and preassembly of the NADPH oxidase complex in
cultured endothelial cells. J Biol Chem. 277:19952–19960. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tamura M, Kai T, Tsunawaki S, Lambeth JD
and Kameda K: Direct interaction of actin with p47(phox) of
neutrophil NADPH oxidase. Biochem Biophys Res Commun.
276:1186–1190. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wientjes FB, Reeves EP, Soskic V,
Furthmayr H and Segal AW: The NADPH oxidase components p47(phox)
and p40(phox) bind to moesin through their PX domain. Biochem
Biophys Res Commun. 289:382–388. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gerthoffer WT and Gunst SJ: Invited
review: focal adhesion and small heat shock proteins in the
regulation of actin remodeling and contractility in smooth muscle.
J Appl Physiol. 91:963–972. 2001.PubMed/NCBI
|
|
27
|
McMullen ME, Bryant PW, Glembotski CC,
Vincent PA and Pumiglia KM: Activation of p38 has opposing effects
on the proliferation and migration of endothelial cells. J Biol
Chem. 280:20995–21003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lamalice L, Houle F, Jourdan G and Huot J:
Phosphorylation of tyrosine 1214 on VEGFR2 is required for
VEGF-induced activation of Cdc42 upstream of SAPK2/p38. Oncogene.
23:434–445. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garcia JG, Wang P, Schaphorst KL, et al:
Critical involvement of p38 MAP kinase in pertussis toxin-induced
cytoskeletal reorganization and lung permeability. FASEB J.
16:1064–1076. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Song C, Perides G, Wang D and Liu YF:
beta-Amyloid peptide induces formation of actin stress fibers
through p38 mitogen-activated protein kinase. J Neurochem.
83:828–836. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Okuda S, Languino LR, Ruoslahti E and
Border WA: Elevated expression of transforming growth factor-beta
and proteoglycan production in experimental glomerulonephritis.
Possible role in expansion of the mesangial extracellular matrix. J
Clin Invest. 86:453–462. 1990. View Article : Google Scholar
|
|
32
|
Tamaki K, Okuda S, Ando T, Iwamoto T,
Nakayama M and Fujishima M: TGF-beta 1 in glomerulosclerosis and
interstitial fibrosis of adriamycin nephropathy. Kidney Int.
45:525–536. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boffa JJ, Lu Y, Placier S, Stefanski A,
Dussaule JC and Chatziantoniou C: Regression of renal vascular and
glomerular fibrosis: role of angiotensin II receptor antagonism and
matrix metalloproteinases. J Am Soc Nephrol. 14:1132–1144. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tomita H, Egashira K, Ohara Y, et al:
Early induction of transforming growth factor-beta via angiotensin
II type 1 receptors contributes to cardiac fibrosis induced by
long-term blockade of nitric oxide synthesis in rats. Hypertension.
32:273–279. 1998. View Article : Google Scholar : PubMed/NCBI
|