Contents
Introduction
Expression, structure and characteristics of
PTEN
Molecular relationship between PPAR, SIRT and
PTEN/AKT through ROS production and DNA damage
Molecular relationship between WRN and PTEN/AKT
through ROS production and DNA damage
Catalytic activity of PTEN is modulated by ROS
Perspective
Introduction
Reactive oxygen species (ROS) are generated during
mitochondrial oxidative metabolism as well as in the cellular
response to pathogens, and act as signaling molecules and regulate
various physiological processes including proliferation,
differentiation, apoptosis and migration (1–3).
In addition, protein and lipid oxidation by ROS is proposed as a
crucial determinant of health. Many experiments have shown that ROS
also have a role in determining cellular senescence in mammalian
cells (4). Increased levels of
oxidative stress results in macromolecular damage and is implicated
in various disease states such as atherosclerosis, diabetes,
obesity and cancer (5).
One major source of endogenous ROS comes from
mitochondria during the process of oxidative phosphorylation to
produce energy in the form of ATP (6). In addition, ROS are produced by
intracellular membrane oxidases. Inflammation is a major source of
ROS at the sites of tissue. It is important for cells to neutralize
ROS before they can damage cellular macromolecules including DNA. A
major DNA lesion generated by excessive ROS is
8-oxo-2′-deoxyguanosine, which leads to a single- or double-strand
break when left unrepaired (7,8).
Persistent breaks can in turn lead to genomic instability. It is
widely believed that the accumulation of mutations is a main cause
of several aging processes. In addition, oxidative stress is known
to shorten telomeres a process likely leading to replicative
senescence and aging (9,10). Thus, an abnormal response to
increased levels of endogenous ROS would likely affect health and
aging. ROS modify the activity of several key enzymes, resulting in
the reorganization of the actin cytoskeleton, adhesion and
stimulation of cell migration. One mechanism by which ROS are
thought to exert effects is through the reversible regulation of
target molecules such as PKC; MAPK; phosphoinositide-3 kinase
(PI3K); tyrosine phosphatase; and phosphatase and tensin homologue
deleted on chromosome 10 (PTEN) (11). However, less is known in regards
to the initial regulation of signaling molecules by ROS. Cellular
ROS metabolism is tightly regulated by a variety of proteins
involved in the redox mechanism.
The proper functioning of several signaling pathways
relies on the action of several growth factors and cytokines, which
induce the generation of ROS (12). A number of studies have
demonstrated an antioxidant role for tumor-suppressor proteins,
activating the expression of antioxidant genes in response to
oxidative stress. Tumor-suppressor genes regulate diverse cellular
activities including DNA damage repair, cell cycle arrest, cell
proliferation, cell differentiation, migration and apoptosis
(13). PTEN is a
tumor-suppressor gene that is frequently deleted or mutated in a
variety of human cancers. It has been demonstrated that
upregulation of PTEN causes modulation of PI3K/AKT signaling to
reduce ROS generation in cells (14). In this review, the tumor
suppressor PTEN and its roles in oxidative stress are summarized
with a focus on the molecular links and interplay between ROS and
the signaling pathways.
Expression, structure and characteristics of
PTEN
Human genomic PTEN locus consists of nine
exons on chromosome 10q23.3, encoding a 5.5-kb mRNA that specifies
a 403-amino acid open reading frame (15,16). The translation product is a 53-kDa
protein with homology to tensin and protein tyrosine phosphatases
(PTPs). PTEN is ubiquitously expressed throughout early
embryogenesis (17). In addition,
peroxisome proliferator activated receptor γ (PPARγ) and tumor
suppressor p53 can transcriptionally upregulate PTEN
expression (18,19). Interestingly, rosemary extract
represses PTEN expression in K562 leukemic culture cells
(20). Methylation of the
PTEN promoter can result in transcriptional silencing of the
PTEN gene (21). Enzymatic
PTEN activity can be regulated by posttranslational regulation
including phosphorylation, acetylation and oxidation (22). The PTEN gene is the
frequently mutated and deleted tumor suppressor in human cancer.
Loss of heterozygosity studies have suggested that PTEN
plays an important role in advanced cancers (23). In addition, alteration of
PTEN in tumors is associated with a poor prognosis (24). PTEN heterozygous mice also
develop a number of cancers. In contrast, overexpression of
PTEN induces growth suppression by promoting G1 arrest
(25).
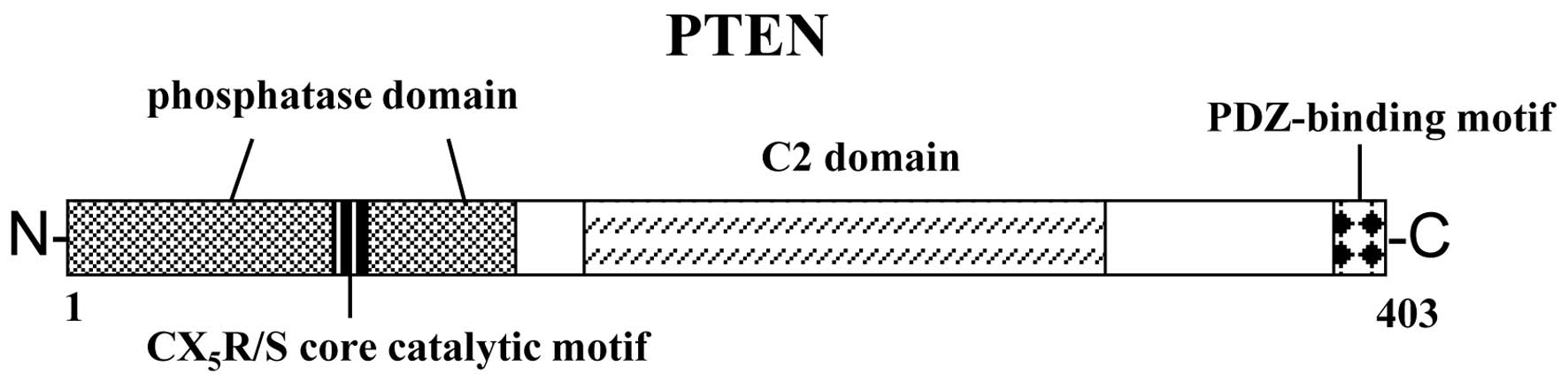
The schematic structure of the PTEN protein is shown
in Fig. 1. The PTEN protein
consists of N-terminal phosphatase, and C-terminal C2 and PDZ
(PSD-95, DLG1 and ZO-1) binding domains. The PTEN
CX5R(S/T) motif resides within an active site that
surrounds the catalytic signature with three basic residues, which
are critical for PTEN phosphatase activity. The structure provides
PTEN with its preference for acidic phospholipid substrates such as
phosphatidylinositol 3,4,5-triphosphate (PIP3). The PTEN lipid
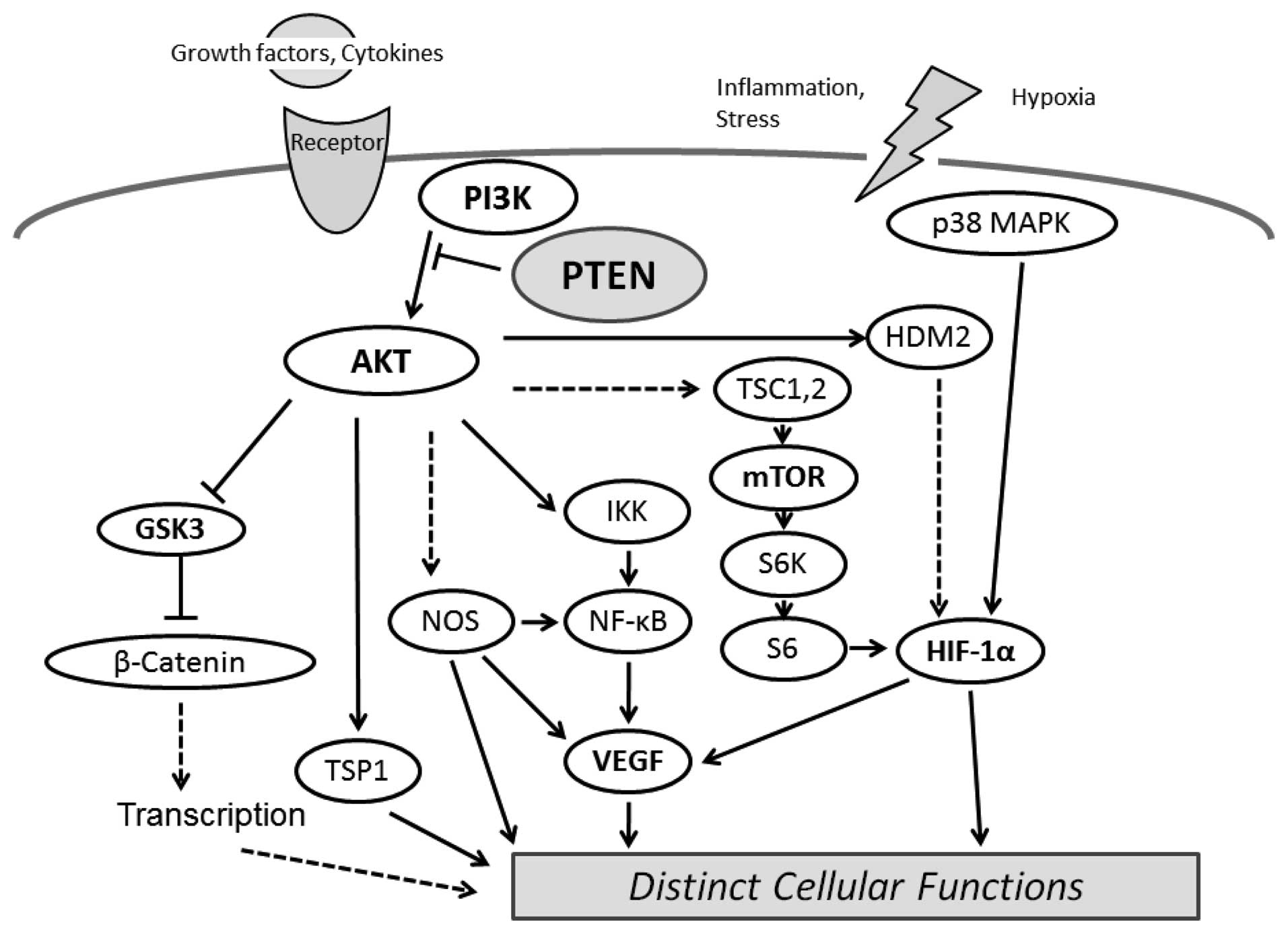
phosphatase activity regulates the AKT kinase pathway through
modulation of PIP3 levels, regulating cell proliferation, apoptosis
and migration. In addition, AKT activation leads to
hypoxia-inducible factor-1 (HIF-1) stabilization, whereas PTEN
attenuates hypoxia-mediated HIF-1 stabilization (26). HIF-1 is a critical element for the
transcriptional regulation of genes important for adaptation to low
oxygen conditions. The instability of mutant PTEN and the reduction
of HIF1 degradation have been shown to involve protein interactions
(27).
PIP3 is the major second messenger of the PI3K
pathway that mediates receptor tyrosine kinase signaling to the
survival kinase AKT. PTEN negatively regulates the activity of
PI3K/AKT signaling through converting PIP3 to phosphatidylinositol
4,5-bisphosphate (PIP2). Increased levels of PIP3 at the membrane
cause PH domain-containing proteins such as AKT to co-localize,
resulting in kinase-mediated phosphorylation and activation
(28). Activated AKT
phosphorylates target proteins involved in cell survival, cell
cycling, angiogenesis and metabolism (Fig. 2). PTEN acts as a regulator for
maintaining basal levels of PIP3 contrary to those signaling
activation. Increased proliferation, survival and motility are the
main cellular effects associated with increased PIP3 levels and
also contribute to its tumorigenic effects. Dysregulation of the
PI3K/AKT pathway has been identified in many malignant cancers
(29). PTEN exerts its
tumor-suppressive effect by dephosphorylating PIP3, thereby
negatively regulating AKT activation and the survival pathway. As
elevated levels of PIP3 confer an advantage to cancer cells,
inactivating mutations in the PTEN gene may be common in
tumors.
Molecular relationship between PPAR, SIRT
and PTEN/AKT through ROS production and DNA damage
Peroxisome proliferator-activated receptors (PPARs)
are transcription factors that belong to the nuclear hormone
receptor superfamily. The responsive elements in genome DNA are
found in various genes that are involved in lipid metabolism and
energy homeostasis including lipid transport (30). The expression is modulated by
aging. Three subtypes PPARα, PPARδ (also known as PPARβ) and PPARγ,
have been identified. Distinct activation of PPARs may account for
the specific biological activity of the three PPAR subtypes. Fatty
acid can interact with PPARs, which induces upregulation of the
expression of enzymes necessary for their removal through
mitochondrial oxidation (31).
Then, lipid overload results in an increased production of ROS
(32). Since PPARs play an
essential role in mitochondrial biogenesis and respiration, and
detoxification of ROS, defects in this pathway are likely in turn
to contribute to mitochondrial impairment. For example, the PPARγ
signaling pathway improves many of the mitochondrial
insufficiencies (33). PPARγ
activation reduces the proliferation of endometrial cells via
regulation of PTEN. In addition, knockdown of PTEN with siRNA
abrogated the effects of PPARδ on cellular senescence and on
generation of ROS (34).
Furthermore, ligand-activated PPARδ upregulates the expression of
PTEN then suppresses the PI3K/AKT pathway. Activated PPARδ also
confers resistance to induced senescence by the upregulation of
PTEN, which reduces ROS generation in vascular cells. It has been
reported that manganese superoxide dismutase is induced by
resveratrol, which may activate PPARα, β and γ, via nuclear
translocation and activation of SIRT1 (35). Mammalian SIRT1 (known as yeast
homolog Sir2) is a deacetylase implicated in longevity and diverse
metabolic processes (36). The
enzymatic activity may be regulated by cellular energy, and SIRT1
may serve as a key regulator for obesity related to antioxidant
status. PTEN is acetylated on Lys-402, which is in the
COOH-terminal PDZ domain-binding motif, indicating a potential role
of acetylation in regulating PTEN function (37). The SIRT1 deacetylase is mainly
responsible for PTEN deacetylation. Thus, SIRT1 deficiency enhances
the acetylation level of target molecules such as PTEN. SIRT1 may
be a useful therapeutic targets for age-related diseases including
metabolic disorders. Overexpression of SIRT1 in cancer tissue,
compared with normal tissue, has been demonstrated, suggesting that
SIRT1 may act as a tumor promoter (38). Actually, SIRT1 promotes
carcinogenesis of hepatocellular carcinoma through the PI3K/AKT
signaling pathway. In addition, SIRT1 decreases PTEN acetylation
and inactivates the function of the pathway in a
deacetylase-dependent manner (39).
Molecular relationship between WRN and
PTEN/AKT through ROS production and DNA damage
Werner syndrome, which is characterized by the early
onset of premature age-associated pathologies including elevated
cancer incidence, is a rare autosomal disease caused by the lack of
a functional nuclear RecQ-like DNA helicase, the WRN protein
(40). Diploid fibroblasts have a
short lifespan and extensive genomic instability. The WRN protein
is a helicase involved in DNA repair, replication, transcription
and telomere maintenance. Repair mechanisms of DNA damage,
including oxidative DNA damage at telomeres require the presence of
WRN, which plays a critical role in optimizing double-strand break
repair mechanisms due to its end-processing helicase and
exonuclease activities. It was subsequently found that, in addition
to a 3′-5′ helicase activity, the WRN protein also possesses a
3′-5′ exonuclease activity (41).
Withdrawal of the WRN protein by knockdown through RNA interference
produces and accelerates cellular senescence phenotype and DNA
damage responses (42). The
depletion of the WRN protein also leads to increased HIF-1 complex
stabilization and activation (43). HIF-1 is also implicated in the
molecular mechanisms of ageing. HIF-1 activation in the absence of
WRN involves the generation of mitochondrial ROS. Studies indicate
that the WRN protein regulates HIF-1 activation by affecting
mitochondrial ROS production. As mentioned above, activation of the
PI3K/AKT pathway leads to HIF-1 stabilization, whereas PTEN
attenuates HIF-1 stabilization. Mutation of PTEN can also
destabilize HIF-1. Mice lacking the helicase domain of the WRN
homologue exhibit many phenotypic features of Werner syndrome,
including a shorter mean lifespan compared to wild-type animals
(44). Vitamin C supplementation
rescues the shorter mean lifespan of WRN-mutant mice and
reverses several related abnormalities (45). The deletion of AKT homologues
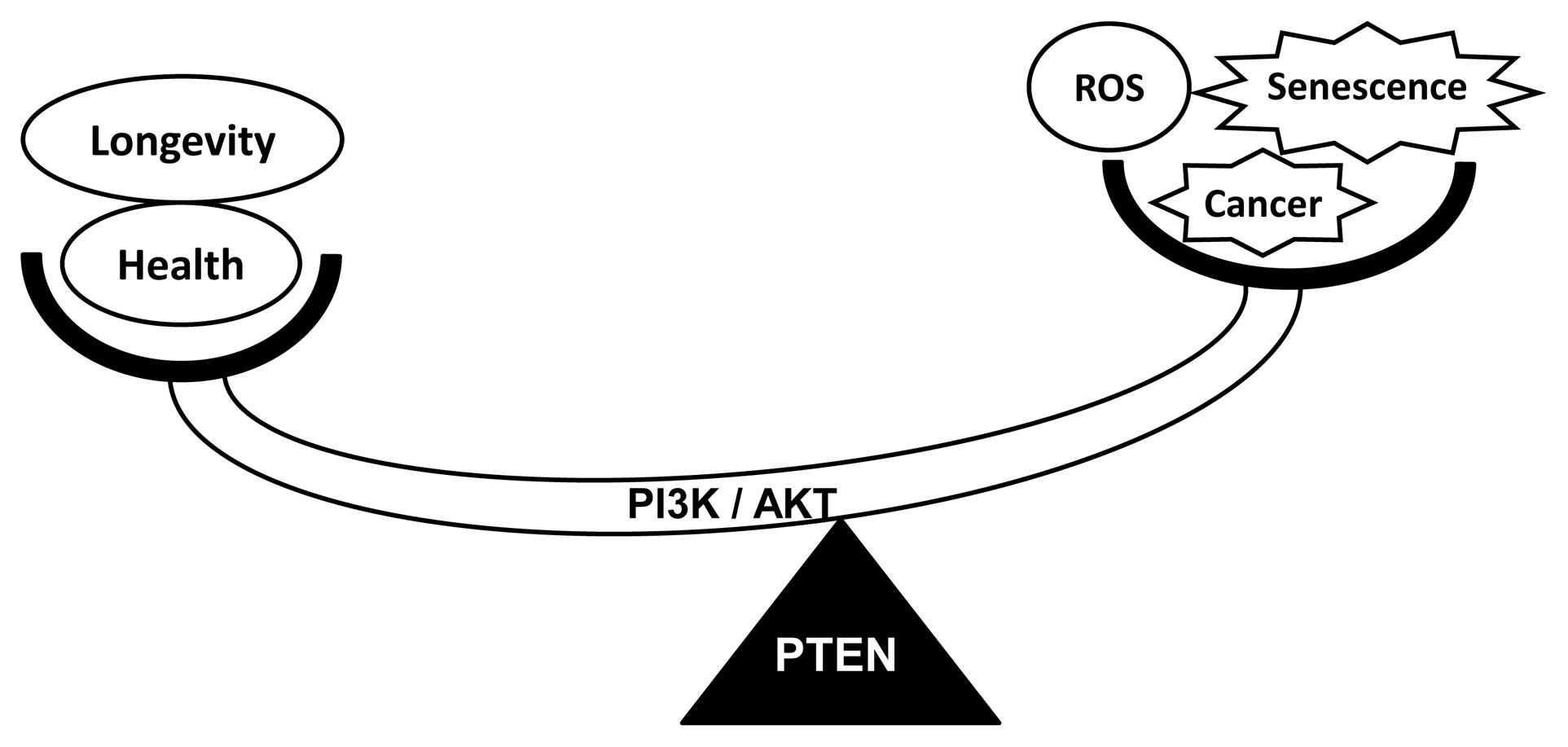
protects against age-dependent defects, raising the possibility
that modulation of the PTEN/AKT pathway protects against premature
aging syndromes. Downregulation of the PI3K/AKT pathway by PTEN
expression may protect against age-dependent DNA damage and cancer
(Fig. 3), including the premature
disorders observed in Werner syndrome.
Catalytic activity of PTEN is modulated by
ROS
ROS directly interact with signaling molecules to
modulate the signaling in a variety of cellular processes such as
proliferation and survival. In fact, the catalytic activity of PTEN
is modulated by ROS, and cellular PTEN phosphatase activity is
inhibited by oxidative stress (46). PTEN inactivation then causes an
increase in cellular PIP3 levels; PIP3 accumulation occurs at the
plasma membrane and activation of the downstream PIP3 target
including AKT, indicating a functional role for elevated
intracellular ROS. In addition, activated PI3K/AKT signaling causes
increased expression of several genes related to cell survival.
Endogenous oxidant production in macrophages inactivates a fraction
of the cellular PTEN, and this is also associated with an
oxidant-dependent activation of downstream signaling (47). It has been reported that ROS
levels are increased in retinal pigment epithelium cells in
association with phosphorylation and inactivation of PTEN (48). PTEN phosphorylated inactivation
and the consequent AKT activation in cells are inhibited by
antioxidant treatment. ROS mediate PTEN inactivation but do not
affect PTEN expression. On the other hand, mitochondrial PTEN
protein levels are decreased by N-acetylcysteine and increased by
H2O2 (49).
The increase in mitochondrial PTEN may further promote ROS
production in cells. However, exposure of purified PTEN to
H2O2 results in inactivation of PTEN in a
time- and concentration-dependent manner (50). Analysis of various cysteine
mutants has indicated that the essential Cys residue in the active
site of PTEN specifically forms a disulfide bond during oxidation
(51). The uncontrolled
generation of ROS may contribute to cell proliferation by
inhibiting PTEN function. In fact, inactivation of PTEN is
implicated in the tumorigenesis of prostate cancer. This may have
implications for the carcinogenic processes in which PIP3 and
oxidants are produced.
Perspective
It is accepted that one of the hallmarks of aging is
the accumulation of DNA damage caused by ROS, which are produced
during normal metabolism and inflammatory reactions. ROS are known
to induce genomic alterations such as point mutations and
deletions, inhibit tumor-suppressor genes and/or activate
oncogenes. It has been shown that another tumor suppressor p53 also
promotes ROS production and participates in the induction of
apoptosis (52). ROS are known to
be critical for senescence, and the p53 target genes that increase
ROS may also play an important role in senescence induction. As ROS
can regulate PTEN, the role of PTEN in modulating ROS may form part
of a feedback loop. The induction of ROS is likely to play a role
in cell growth inhibitory responses such as the induction of
senescence. Premature senescence has a compensatory role in
apoptosis in the absence of the tumor suppressor PTEN through the
AKT/ROS/p53 pathway. Furthermore, increasing ROS can enhance
insulin signaling to attenuate the development of insulin
resistance. Enhanced ROS-dependent insulin signaling is
attributable to the oxidation and inhibition of PTEN. Given the
importance of PTEN in regulating metabolism, there may be much
evidence linking PTEN to diabetes and senescence. A better
understanding of the intricate relationships between ROS and aging
via the PTEN pathway may provide clues to ROS-mediated aging
pathways for new drug discovery. Future experiments may determine
which pathways are responsible for the redox imbalance noted in the
human PTEN mutant.
Abbreviations:
|
PTEN
|
phosphatase and tensin homologue
deleted on chromosome 10;
|
|
PIP3
|
phosphatidylinositol
3,4,5-triphosphate;
|
|
PIP2
|
phosphatidylinositol
4,5-bisphosphate;
|
|
PI3K
|
phosphoinositide-3 kinase;
|
|
PPAR
|
peroxisome proliferator-activated
receptor;
|
|
ROS
|
reactive oxygen species;
|
|
HIF-1
|
hypoxia-inducible factor-1;
|
|
mTOR
|
mammalian target of rapamycin;
|
|
PTP
|
protein tyrosine phosphatase.
|
Acknowledgements
This study was supported by
grants-in-aid from the Ministry of Education, Culture, Sports,
Science and Technology of Japan. In addition, this study was
supported, in part, by a grant from SHIN-EI Pharmaceutical Co.,
Ltd.
References
|
1
|
Al-Gubory KH, Fowler PA and Garrel C: The
roles of cellular reactive oxygen species, oxidative stress and
antioxidants in pregnancy outcomes. Int J Biochem Cell Biol.
42:1634–1650. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang Y, Du Y, Le W, Wang K, Kieffer N and
Zhang J: Redox control of the survival of healthy and diseased
cells. Antioxid Redox Signal. 15:2867–2908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Scatena R: Mitochondria and cancer: a
growing role in apoptosis, cancer cell metabolism and
dedifferentiation. Adv Exp Med Biol. 942:287–308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leonarduzzi G, Sottero B, Testa G, Biasi F
and Poli G: New insights into redox-modulated cell signaling. Curr
Pharm Des. 17:3994–4006. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Roberts CK and Sindhu KK: Oxidative stress
and metabolic syndrome. Life Sci. 84:705–712. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ballard JW: Drosophila simulans as
a novel model for studying mitochondrial metabolism and aging. Exp
Gerontol. 40:763–773. 2005. View Article : Google Scholar
|
|
7
|
Shubassi G, Robert T, Vanoli F, Minucci S
and Foiani M: Acetylation: a novel link between double-strand break
repair and autophagy. Cancer Res. 72:1332–1335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Murray JM, Stiff T and Jeggo PA: DNA
double-strand break repair within heterochromatic regions. Biochem
Soc Trans. 40:173–178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Babizhayev MA, Vishnyakova KS and Yegorov
YE: Telomere-dependent senescent phenotype of lens epithelial cells
as a biological marker of aging and cataractogenesis: the role of
oxidative stress intensity and specific mechanism of phospholipid
hydroperoxide toxicity in lens and aqueous. Fundam Clin Pharmacol.
25:139–162. 2011. View Article : Google Scholar
|
|
10
|
Saretzki G and Von Zglinicki T:
Replicative aging, telomeres, and oxidative stress. Ann NY Acad
Sci. 959:24–29. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li ZY, Yang Y, Ming M and Liu B:
Mitochondrial ROS generation for regulation of autophagic pathways
in cancer. Biochem Biophys Res Commun. 414:5–8. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Azad N, Rojanasakul Y and Vallyathan V:
Inflammation and lung cancer: roles of reactive oxygen/nitrogen
species. J Toxicol Environ Health B Crit Rev. 11:1–15. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Asai T, Liu Y, Bae N and Nimer SD: The p53
tumor suppressor protein regulates hematopoietic stem cell fate. J
Cell Physiol. 226:2215–2221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu J, Tian W, Ma X, et al: The molecular
mechanism underlying morphine-induced Akt activation: roles of
protein phosphatases and reactive oxygen species. Cell Biochem
Biophys. 61:303–311. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Okumura N, Yoshida H, Kitagishi Y,
Murakami M, Nishimura Y and Matsuda S: PI3K/AKT/PTEN signaling as a
molecular target in leukemia angiogenesis. Adv Hematol.
2012:8430852012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Okumura N, Yoshida H, Kitagishi Y,
Nishimura Y and Matsuda S: Alternative splicings on p53, BRCA1 and
PTEN genes involved in breast cancer. Biochem Biophys Res Commun.
413:395–399. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Croushore JA, Blasiole B, Riddle RC, et
al: Ptena and ptenb genes play distinct roles in zebrafish
embryogenesis. Dev Dyn. 234:911–921. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Teresi RE, Shaiu CW, Chen CS, Chatterjee
VK, Waite KA and Eng C: Increased PTEN expression due to
transcriptional activation of PPARgamma by Lovastatin and
Rosiglitazone. Int J Cancer. 118:2390–2398. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harikumar KB and Aggarwal BB: Resveratrol:
a multitargeted agent for age-associated chronic diseases. Cell
Cycle. 7:1020–1035. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yoshida H, Okumura N, Kitagishi Y,
Nishimura Y and Matsuda S: Ethanol extract of rosemary repressed
PTEN expression in K562 culture cells. Int J Appl Boil Pharm
Technol. 2:316–322. 2011.
|
|
21
|
Mueller S, Phillips J, Onar-Thomas A, et
al: PTEN promoter methylation and activation of the PI3K/Akt/mTOR
pathway in pediatric gliomas and influence on clinical outcome.
Neuro Oncol. 14:1146–1152. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Singh G and Chan AM: Post-translational
modifications of PTEN and their potential therapeutic implications.
Curr Cancer Drug Targets. 11:536–547. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tysnes BB and Mahesparan R: Biological
mechanisms of glioma invasion and potential therapeutic targets. J
Neurooncol. 53:129–147. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu X, Qin X, Fei M, et al: Combined
phosphatase and tensin homolog (PTEN) loss and fatty acid synthase
(FAS) overexpression worsens the prognosis of Chinese patients with
hepatocellular carcinoma. Int J Mol Sci. 13:9980–9991. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Weng LP, Brown JL and Eng C: PTEN
coordinates G(1) arrest by down-regulating cyclin D1 via its
protein phosphatase activity and up-regulating p27 via its lipid
phosphatase activity in a breast cancer model. Hum Mol Genet.
10:599–604. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carver DJ, Gaston B, Deronde K and Palmer
LA: Akt-mediated activation of HIF-1 in pulmonary vascular
endothelial cells by S-nitrosoglutathione. Am J Respir Cell Mol
Biol. 37:255–263. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li YM, Zhou BP, Deng J, Pan Y, Hay N and
Hung MC: A hypoxia-independent hypoxia-inducible factor-1
activation pathway induced by phosphatidylinositol-3 kinase/Akt in
HER2 overexpressing cells. Cancer Res. 65:3257–3263.
2005.PubMed/NCBI
|
|
28
|
Howes AL, Arthur JF, Zhang T, et al:
Akt-mediated cardiomyocyte survival pathways are compromised by G
alpha q-induced phosphoinositide 4,5-bisphosphate depletion. J Biol
Chem. 278:40343–40351. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Castaneda CA, Cortes-Funes H, Gomez HL and
Ciruelos EM: The phosphatidyl inositol 3-kinase/AKT signaling
pathway in breast cancer. Cancer Metastasis Rev. 29:751–759. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fruchart JC: Peroxisome
proliferator-activated receptor-alpha (PPARalpha): at the
crossroads of obesity, diabetes and cardiovascular disease.
Atherosclerosis. 205:1–8. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schreurs M, Kuipers F and van der Leij FR:
Regulatory enzymes of mitochondrial beta-oxidation as targets for
treatment of the metabolic syndrome. Obes Rev. 11:380–388. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ruggiero C, Ehrenshaft M, Cleland E and
Stadler K: High-fat diet induces an initial adaptation of
mitochondrial bioenergetics in the kidney despite evident oxidative
stress and mitochondrial ROS production. Am J Physiol Endocrinol
Metab. 300:E1047–E1058. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pipatpiboon N, Pratchayasakul W,
Chattipakorn N and Chattipakorn SC: PPARγ agonist improves neuronal
insulin receptor function in hippocampus and brain mitochondria
function in rats with insulin resistance induced by long term
high-fat diets. Endocrinology. 153:329–338. 2012.
|
|
34
|
Kim HJ, Ham SA, Kim MY, et al: PPARδ
coordinates angiotensin II-induced senescence in vascular smooth
muscle cells through PTEN-mediated inhibition of superoxide
generation. J Biol Chem. 286:44585–44593. 2011.
|
|
35
|
Das UN: A defect in the activity of Delta6
and Delta5 desaturases may be a factor predisposing to the
development of insulin resistance syndrome. Prostaglandins Leukot
Essent Fatty Acids. 72:343–350. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee J and Kemper JK: Controlling SIRT1
expression by microRNAs in health and metabolic disease. Aging.
2:527–534. 2010.PubMed/NCBI
|
|
37
|
Ikenoue T, Inoki K, Zhao B and Guan KL:
PTEN acetylation modulates its interaction with PDZ domain. Cancer
Res. 68:6908–6912. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Choi HN, Bae JS, Jamiyandorj U, et al:
Expression and role of SIRT1 in hepatocellular carcinoma. Oncol
Rep. 26:503–510. 2011.PubMed/NCBI
|
|
39
|
Qu Y, Zhang J, Wu S, Li B, Liu S and Cheng
J: SIRT1 promotes proliferation and inhibits apoptosis of human
malignant glioma cell lines. Neurosci Lett. 525:168–172. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
van Brabant AJ, Stan R and Ellis NA: DNA
helicases, genomic instability, and human genetic disease. Annu Rev
Genomics Hum Genet. 1:409–459. 2000.PubMed/NCBI
|
|
41
|
Li B, Reddy S and Comai L:
Sequence-specific processing of telomeric 3′ overhangs by the
Werner syndrome protein exonuclease activity. Aging. 1:289–302.
2009.
|
|
42
|
Szekely AM, Bleichert F, Nümann A, et al:
Werner protein protects nonproliferating cells from oxidative DNA
damage. Mol Cell Biol. 25:10492–10506. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Labbé A, Lafleur VN, Patten DA, et al: The
Werner syndrome gene product (WRN): a repressor of
hypoxia-inducible factor-1 activity. Exp Cell Res. 318:1620–1632.
2012.PubMed/NCBI
|
|
44
|
Massip L, Garand C, Turaga RV, Deschênes
F, Thorin E and Lebel M: Increased insulin, triglycerides, reactive
oxygen species, and cardiac fibrosis in mice with a mutation in the
helicase domain of the Werner syndrome gene homologue. Exp
Gerontol. 41:157–168. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Massip L, Garand C, Paquet ER, et al:
Vitamin C restores healthy aging in a mouse model for Werner
syndrome. FASEB J. 24:158–172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Barbieri SS, Ruggiero L, Tremoli E and
Weksler BB: Suppressing PTEN activity by tobacco smoke plus
interleukin-1beta modulates dissociation of
VE-cadherin/beta-catenin complexes in endothelium. Arterioscler
Thromb Vasc Biol. 28:732–738. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Leslie NR, Bennett D, Lindsay YE, Stewart
H, Gray A and Downes CP: Redox regulation of PI 3-kinase signalling
via inactivation of PTEN. EMBO J. 22:5501–5510. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim JW, Kang KH, Burrola P, Mak TW and
Lemke G: Retinal degeneration triggered by inactivation of PTEN in
the retinal pigment epithelium. Genes Dev. 22:3147–3157. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zu L, Zheng X, Wang B, et al: Ischemic
preconditioning attenuates mitochondrial localization of PTEN
induced by ischemia-reperfusion. Am J Physiol Heart Circ Physiol.
300:H2177–H2186. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lee SR, Yang KS, Kwon J, Lee C, Jeong W
and Rhee SG: Reversible inactivation of the tumor suppressor PTEN
by H2O2. J Biol Chem. 277:20336–20342. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Matsuda M, Takeshita K, Kurokawa T, et al:
Crystal structure of the cytoplasmic phosphatase and tensin homolog
(PTEN)-like region of Ciona intestinalis voltage-sensing
phosphatase provides insight into substrate specificity and redox
regulation of the phosphoinositide phosphatase activity. J Biol
Chem. 286:23368–23377. 2011.PubMed/NCBI
|
|
52
|
Maillet A and Pervaiz S: Redox regulation
of p53, redox effectors regulated by p53: a subtle balance.
Antioxid Redox Signal. 16:1285–1294. 2012. View Article : Google Scholar : PubMed/NCBI
|