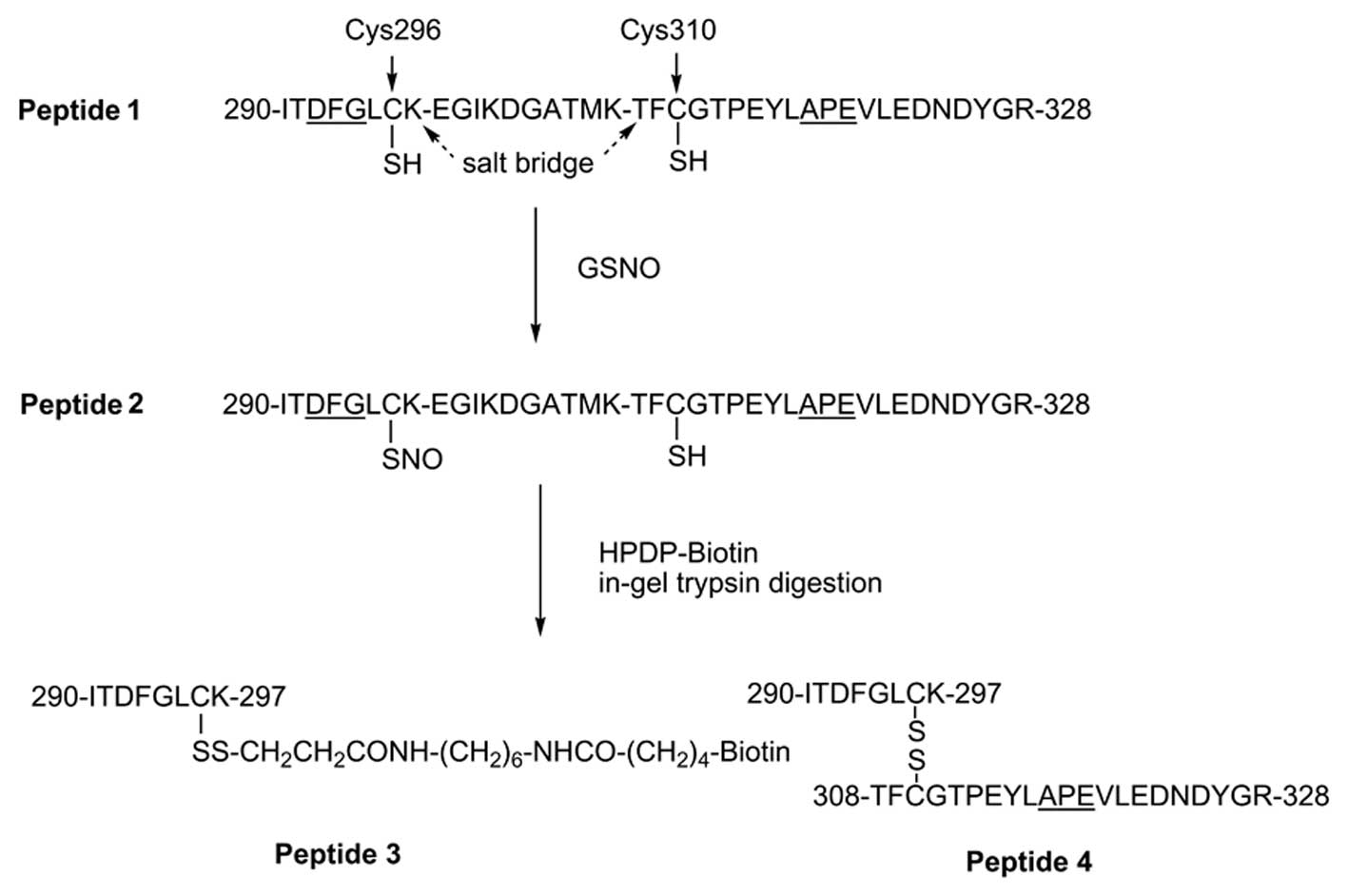
Introduction
Metabolic alterations that are produced by critical
illness such as burn trauma are associated with a
hypermetabolic/inflammatory state, increased protein catabolism
(with resulting muscle wasting) and insulin resistance. Muscle
wasting can lead to muscle weakness that can result in
hypoventilation, prolongation of dependence on mechanical
ventilation, prolonged rehabilitation and even death (1–4).
Insulin resistance is a well established state in critically ill
patients and is considered to play a key role in the metabolic
derangements and muscle wasting. Binding of insulin to its receptor
(IR) activates IR tyrosine kinase, which then phosphorylates IR
substrates (IRSs). Phosphorylation of IRS1 and IRS2 transduces the
signal from IR to phosphatidylinositol-3-kinase (PI3-kinase)
(4,5). Post-translational modifications
(PTMs) of the insulin signaling system are considered to be major
disease-dependent events that regulate glucose transport via GLUT-4
translocation and protein synthesis (6–12).
Akt1/PKBα is a critical downstream mediator
of the IR/IRS/PI3-kinase pathway of the insulin signaling system
(13–17). Akt1/PKBα consists of three
structural features: the N-terminal pleckstrin homology (PH)
domain, a large central kinase domain and a short C-terminal
hydrophobic motif. High specific binding of the PH domain with
membrane lipid products of PI3-kinase recruits Akt1/PKBα to
the plasma membrane where phosphorylations of Thr308
(pThr308, kinase domain) and Ser473
(pSer473, hydrophobic motif) occur. Phosphorylation of
Thr308 partially stimulates kinase activity; however,
additional phosphorylation of Ser473 is required for
full activity. Activation is associated with a disordered to
ordered transition of a specific αC helix of
Akt1/PKBα via an allosteric mechanism. A salt bridge between
the side-chain of Lys297 and the phosphate group of
pThr308 in this αC helix contributes to an
ordered activation segment from 292DFG to
APE319(18–21). Reversible dephosphorylations of
Thr308 and Ser473 by protein phosphatase 2A
(PP2A) and PH domain leucine-rich repeat protein phosphatase
(PHLPPα) also occur in the Akt1/PKBα
activation/deactivation cycle (22–25).
In addition to the role of reversible
phosphorylation/dephosphorylation in the regulation of
Akt1/PKBα activity, this kinase is also reversibly
inactivated by S-nitrosylation under conditions that result in
persistently increased production of nitric oxide; such as after
burn injury (13,26–29). Thiol titration and NMR data
indicate that a disulfide bond (Cys60-Cys77)
exists in the kinase PH domain (30). A second disulfide bond in the
critical kinase activation loop
(Cys297-Cys311) has been reported to be
associated with dephosphorylation under oxidative stress in
vitro(31). In addition, it
has been shown that when Cys224 of Akt1/PKBα is
mutated to a Ser residue, the kinase becomes resistant to NO
donor-induced S-nitrosylation and inactivation; suggesting that
this residue is a major S-nitrosylation acceptor site (28). In vivo S-nitrosylations of
the insulin receptor β and Akt1/PKBα result in reductions in
their kinase activities (27).
These data suggest that the redox status of Akt1/PKBα,
regulated by NO, is a second factor in the PTM that modulates
kinase activity (via dynamic conformational changes) and thus
GLUT-4 trafficking and protein synthesis. Nevertheless, to date,
published data on the reversible phosphorylation(s) and
S-nitrosylation(s) relevant to Akt1/PKBα activation,
conformation and regulation have not provided conclusive
information concerning their interrelationships nor critical
S-nitrosylation sites involved in the kinase
activation/deactivation cycle.
Recent technical developments have made it feasible
to study the molecular details of these important processes. These
techniques include: i) sensitive and site-specific procedures for
the detection of S-nitrosylation based upon nano-LC interfaced with
tandem MS (32,33); ii) the Biotin-Switch method for
qualitative discrimination of the thiol state between free,
disulfide bonded and S-nitroylated cysteine residues under
carefully defined conditions (34–39). Potential problems related to
quantification with this technique have been discussed previously
(33); and iii) highly specific
anti-Akt1/ PKBα mAbs that can be used to immunoprecipitate
quantities of protein that are sufficient to yield SDS-PAGE bands
with Coomassie brilliant blue R-250 staining which are compatible
with tandem MS analysis.
Burn injury-associated impairments in IRS1 signaling
and attenuated IR-IRS-PI3K-Akt/PKB activation have been the major
focuses of our research team (9,26,29,33). Significantly reduced
phosphorylations of Ser473 and Thr308, as
well as decreased Akt/PKB kinase activity were observed after burn
injury [55% total body surface area (TBSA), day 3] and insulin
stimulation (26). However, the
interrelationship between impaired kinase activity and the loop
disulfide bond (31) reported
under oxidative stress remains unclear. In the present study we
investigated the interaction between S-nitrosylation and
phosphorylation at Cys296-Lys297 and
Thr308-Phe309-Cys310 in the kinase
loop at the proteomic level.
Specifically, the following issues need to be
studied: i) the ability of Cys296 to chemically quench
elevated levels of free radicals, mainly nitric oxide; ii) loop
conformational changes associated with two types of PTMs; iii)
quantitative proteomics of Akt1/PKBα by stable isotope labeling in
mice. In this study, we obtained MS/MS sequence data to
characterize the thiol states of Cys296 in the kinase
activity loop of Akt1/PKB. These measurements were possible despite
the extremely low level of nitrosylated protein (at the
10−15 pmol level, the chance of positive hits is ∼25%
with lysates prepared from 25 mg of soleus muscle). The biochemical
role of S-nitrosylation at Cys296 was characterized as
an intermediate state which reduces the kinetic barrier to form the
disulfide bond with Cys310 within the activity loop.
This occurs simultaneously with dephosphorylation of
pThr308 after burn injury. The facts that no other
disulfide bonds associated with Cys296 were detected
suggest that they may be thermodynamically forbidden; due to
geometry and/or dihedral strain. The data obtained with soleus
muscle from burned and sham-treated rats indicates that NO-mediated
formation of the Cys296-Cys310 disulfide bond
(which likely downregulates kinase activity) plays a reciprocal
role with formation of a Lys297-pThr308 salt
bridge (which upregulates kinase activity) during
disease-associated reversible activation/deactivation
processes.
Materials and methods
Chemicals
Acetonitrile (ACN, LC-MS Chromasolv), formic acid
(FA), glacial acetic acid, LC-MS grade water, dithiothreitol (DTT),
iodoacetic acid, iodoacetamide, [Glu1]-fibrinopeptide B,
methyl methanethiolsulfonate (MMTS), S-nitrosoglutathione (GSNO),
sodium L-ascorbate, neocuproine, N,N-dimethylformamide (DMF),
dimethyl sulfoxide (DMSO) were obtained from Sigma Chemical Co.
(St. Louis, MO). SDS-PAGE Ready gels (4-15% Tris-HCl, cat. no.
161-1122), Laemmli sample buffer (cat. no. 161-0737) and Coomassie
brilliant blue R-250 (no. 161-0436) were obtained from Bio-Rad.
Trypsin profile IGD kits (cat. no. PP0100) were obtained from
Sigma. Anti-Akt1/PKBα monoclonal antibody (cat. no. 05-798;
lot, 26860) and inactive Akt1/PKBα (cat. no. 14-279) were
purchased from Upstate (Charlottesville, VA, USA). Streptavidin
agarose CL-4B (cat. no. 85881) was a product of Fluka (Milwakee,
WI, USA). HPDP-Biotin (cat. no. 21341) and Iodoacetyl-LC-Biotin
(cat. no. 21333) were purchased from Pierce (Rockford, IL,
USA).
Mapping of cysteine residues in inactive
Akt1/PKBα
Inactive Akt1/PKBα (10 μg, 0.18 nmol, in 10
μl stock solution) was transferred to a siliconized Eppendorf tube
(0.6 ml) containing Laemmli sample buffer (2X, 10 μl, pH was
adjusted to 8.0) and DDT (2 μl, 20 nmol, PBS, pH 8.0), and the
solution was kept at 95°C for 5 min. Freshly prepared
Iodoacetyl-LC-Biotin (15 μl, 55 nmol, in DMF) was added to the
denatured protein solution followed by stirring for an additional
15 min at room temperature. The resulting biotinylated
Akt1/PKBα was purified by SDS-PAGE and stained with
Coomassie brilliant blue R-250. The protein bands were excised (∼1
mm size) and digested (Akt1/PKBα: trypsin 25, overnight at
37°C) with a Trypsin Profile IGD kit according to the
manufacturer's instructions. The biotinylated peptide mixture was
captured by gentle stirring with streptavidin agarose CL-4B (30 μl
packed) at room temperature for 1 h (final vol, 100 μl). The
streptavidin beads were washed with PBS (0.5 ml ×3), followed by
water/ acetonitrile (ACN 10%, 0.5 ml ×3). Biotinylated peptides
were released from the streptavidin beads with formic acid (70%,
100 μl) at room temperature for 15 min with brief vortexing. The
supernatant containing biotinylated peptides was transferred to a
new vial and the formic acid was evaporated with a SpeedVac. The
biotinylated peptide mixture was resuspended in water/acetonitrile
(ACN, 2%, with 0.1% FA, 70 μl), and the aliquots (10 μl) were
injected into a Waters CapLC-tandem quadrupole time-of-fight mass
spectrometry (Q-TOF) system.
Identification of disulfide bonds in
inactive Akt1/PKBα
Inactive Akt1/PKBα (10 μg, 0.18 nmol, in 10
μl stock solution) was transferred into a siliconized Eppendorf
tube (0.6 ml) containing Laemmli sample buffer (2X, 10 μl, pH 8.0)
and iodoacetamide (2 μl, 20 nmol, PBS, pH 8.0). The mixture was
maintained at 95°C for 5 min and then stirred at room temperature
for an additional 15 min. The Akt1/PKBα was purified by
SDS-PAGE and stained with Coomassie brilliant blue R-250. The
protein bands were processed as above.
Identification of NO acceptor sites in
inactive Akt1/PKBα
Three samples of inactive Akt1/PKBα (10 μg,
0.18 nmol, in 10 μl stock solution) were treated with GSNO (250
nmol, 50 μl PBS, pH 8.0, 200-fold excess/thiol group) for 1 h at
room temperature in the dark in siliconized Eppendorf tubes (0.6
ml). Separation of Akt1/PKBα and GSNO was achieved by two
successive acetone/water precipitations (0.3 ml, 70% ACN) at −40°C
for 10 min. The supernatants (containing GSNO) were removed by
centrifugation at 14,000 × g for 2 min. The kinase pellets were
resuspended in blocking buffer (100 μl, 20 mM Tris-HCl, pH 7.7,
2.5% SDS, 20 mM MMTS, 1 mM EDTA, 0.1 mM neocuproine) at room
temperature for 1 h with gentle stirring (1 mm ID ×5 mm bar).
Excess MMTS was removed by acetone (100%, 0.3 ml) precipitation (as
above), and the protein pellets were resuspended in PBS (50 μl, pH
8.0). Freshly prepared iodoacetic acid (5 μl, 2 mM in PBS, pH 8.0),
HPDP-Biotin (5 μl, 2 mM in DMSO), Iodoacetyl-LC-Biotin (5 μl, 2 mM
in DMF) and sodium ascorbate (20 μl, 5 mM, PBS) were added to the
three vials containing nitrosylated Akt1/PKBα, respectively.
The reaction mixtures were stirred at room temperature for 15 min
(iodoacetic acid and Iodoacetyl-LC-Biotin) or 1 h for the
thiol-disulfide exchange reaction. Aliquots of SDS sample buffer
(2X, with 5% 2-mercaptoethanol, 50 μl) were added to the protein
solutions, and the mixtures were incubated at 95°C for 5 min. The
derivatized proteins were processed as above. Carboxymethyl
cysteine (CMC)-containing peptides, were neutralized with FA (5 μl)
and sequenced via parent ion discovery trigged by the CMC immonium
ion (134.02±0.05 mDa) as reported previously (33). Biotinylated peptides were
sequenced with data-dependent acquisition after capture with
streptavidin agarose beads. Ten-microliter aliquots of each final
solution were injected into the CapLC-Q-TOF system.
Analysis of the
Cys296-Cys310 disulfide bond formation in
Akt1/PKBα after treatment with S-nitrosoglutathione
Inactive Akt1/PKBα (10 μg, 10 μl, 0.18 nmol)
and freshly prepared GSNO (5 μl, 250 nmol, PBS, pH 8.0) were
stirred in an Eppendorf tube (0.6 ml) in the dark at room
temperature for 1 h. Separation of Akt1/PKBα and GSNO was
performed with acetone/water (70%) as above. The kinase pellet was
resuspended in PBS (10 μl), and SDS sample buffer (10 μl with
iodoacetamide, 20 nmol) was added. The cysteine alkylation was
performed at room temperature for 15 min. The protein samples were
separated with SDS-PAGE Ready gels and digested as above. Aliquots
of the final solution (10 μl) were injected into the CapLC-Q-TOF
system.
Measurement of the free and disulfide
bonded Cys296 in Akt1/PKBα from soleus muscle of burned
rats
Soleus muscle lysates from rats with third degree
burn (40% TBSA) were prepared as previously described (29,33). The lysates (∼10 mg/ml total
proteins) were diluted to ∼3-5 mg protein/ml protein with PBS, and
filtered through 0.22-μm membranes. Immunoprecipitation was
performed as follows. Anti-Akt1/PKBα mAb (clone AW24, 5 μg;
Upstate) and prewashed protein G agarose beads (50 μl, packed) were
kept at 4°C (100 μl of PBS) for 1 h under gentle stirring. Without
washing the beads, the soleus lysates (5 ml) were added and
stirring was continued for an additional 90 min. Non-specific
proteins were removed by washing with PBS (3X), Laemmli sample
buffer (50 μl, pH 8) containing HPDP-Biotin (400 μM) was added and
the mixtures were maintained at 95°C for 5 min. The procedures for
SDS-PAGE separation and in-gel trypsin digestion were the same as
described above.
The burn injury protocol was approved by the
Committee on Research Animal Care and Use of the Massachusetts
General Hospital (MGH). The MGH animal care facility is accredited
by the Association for Assessment and Accreditation of Laboratory
Animal Care.
LC-MS/MS analysis
All experiments were performed using a Waters
CapLC-Q-TOFmicro system (Waters Corporation, Milford,
MA, USA) as previously described (32,33). An analytical column (75 mm ID ×150
mm, C18 PepMap300, 5 mm, LC Packings) was used to connect the
stream select module of the CapLC with the voltage supply adapter
for ESI. Peptide mixtures were loading onto the precolumn (C18
resin) at a flow rate of 15 μl/min. Dead volume from the CapLC
injector to the precolumn was measured to be ∼1.5 μl. After washing
with mobile phase C (auxiliary pump, 0.1% formic acid in water/ACN,
2% ACN) for 2 min, the trapped peptides were back-washed from the
precolumn onto the analytical column using the 10-position stream
switching valve. Freshly prepared mobile phases A and C were
sonicated under vacuum for ∼25 min, and mobile phase B was treated
in this way for 5 min. The mobile phases were degassed every week,
and the CapLC pumps were wet primed for 20 cycles. A linear
gradient was used to elute the peptide mixture from mobile phase A
(0.1% FA in water/ACN, 2% ACN) to mobile phase B (0.1% FA in ACN).
The gradient was segmented as follow: isocratic elution with 2% B
for 3 min, increasing B from 2 to 70% (3-40 min), isocratic elution
with 70% B (40-45 min) and decreasing B from 70 to 2% (over 2 min).
The injector syringe (25 μl) was washed with degassed mobile phase
A, and the injection volume was set as full loop mode (10 μl). The
gradient flow rate was set at 1.5 μl/min before the 16/1 Nanotee
splitter and the pressure drop from the analytical column was ∼800
psi. The pressure drop (or the flow splitting ratio) was adjusted
and maintained with 20 μm ID capillary tubing at the waste outlet
position of the Nanotee splitter. The gradient flow rate was ∼95
nl/min. The electrospray voltage was set to ∼3,000 V to obtain an
even ESI plume at the beginning of the gradient (high water
content). As a routine sensitivity check, the PicoTip Emitter
position and other parameters were adjusted to achieve ∼45
counts/sec for the capillary tubing background peak (m/z 429).
Sample cone and extraction cone voltages were set at 45 and 3 V,
respectively. The instrument was operated in positive ion mode with
the electrospray source maintained at 90°C. The instrument was
calibrated with synthetic human [Glu1]-fibrinopeptide B
(100 fmol/μl in acetonitrile/water, 10:90, 0.1% formic acid, v/v)
at an infusion rate of 1 μl/min in TOF MS/MS mode. The peptide was
selected at m/z 785.8 and focused into the collision cell
containing argon gas at ∼3×10−5 Torr; the collision
energy was set at 35 V. Instrument resolution for the
[Glu1]-fibrinopeptide B parent ion, m/z 785.84, was
found to be 5,250 FWHM. All data were acquired and processed using
MassLynx 4.1 software. For parent ion discovery triggered by the
CMC immonium ion (134.02±0.03 Da), the survey low and high
collision energies were set at 5 and 30 V, respectively. MS survey
data were collected in continuum mode over the m/z 100–1,200 range.
Data-dependent acquisition (DDA) was set from 450 to 1,500 m/z for
the biotinylated peptides. Scan time was in the range of 1.9–3.8
sec (depending upon sample conditions), and the inter-scan delay
was 0.1 sec. MS to MS/MS switch criteria were dependent upon the
reporter ion intensity (5 counts/sec) and detection window (2.3 Da,
charge status). The instrument was switched from MS/MS back to MS
after 5 sec without intensity restriction.
Evaluation of the S-nitrosylated cysteine
site
Confirmations of the S-nitrosylation sites were
performed by the following three step procedure. i) For parent ion
discoveries by continuum MS survey, the peptide mass tolerance was
0.2 Da for the CMC immonium ion. Under these conditions, only a few
false positive ions were observed and these were eliminated
manually from the expected CMC parent ion list. ii) The positively
discovered parent ions were analyzed with PepSeq of MassLynx V4.1
software; oxidation of methionine was searched as a variable
modification. iii) For peptides, with MS/MS scores <35, manual
interpretations of candidate parent ions were performed with the
following procedure: continuum MS/MS spectra were smoothed, the
upper 80% was centroided and cysteine residues were confirmed with
three different thiol-specifically derivatized y ions. Cysteine
residue monoisotopic mass C3H5NOS = 103.01 Da
was replaced with CMC residue monoisotopic mass
C5H7NO3S = 161.01 Da, HPDP-Biotin
derivatized adduct residue monoisotopic mass
C22H37N5O4S3
= 531.20 Da and Iodoacetyl-LC-Biotin derivatized adduct residue
monoisotopic mass
C21H35N5O4S2 = 485.21
Da, respectively.
Results and Discussion
It has been reported that NO production is elevated
by stressors such as burn injury and in patients with type 2
diabetes (29–41). It has also been shown that the
Cys297-Cys311 disulfide bond in the critical
kinase activation loop of Akt1/PKBα may be formed in
association with dephosphorylation under oxidative stress in
vitro(31). Thus, we
hypothesized that reversible S-nitrosylation at either
Cys296 or Cys310 in the kinase active loop
may be a second PTM factor which complements reversible
phosphorylation at Thr308 in the regulation of kinase
activity and we sought to determine how S-nitrosylation interacts
with phosphorylation during the Akt1/PKBα activation cycle
(22). To address these issues,
GSNO was used as the only NO donor in a model S-nitrosylation
system to randomly target the seven cysteine residues of the kinase
at pH 8. Vicinal Cys296 and Cys310 take
advantage of the pKa for dissociation of the thiol to thiolate, and
these electron-rich thiolate groups can lead to formation of an
intradomain disulfide bond. Under these conditions, intracellular
free cysteine residues, and cysteines at the kinase surface without
interactions or located in hydrophobic environments (i.e. high
pKa), are unlikely to be affected by GSNO. In contrast,
Cys296 and Cys310, which may have low pKa
values due to weak interactions with vicinal residues inside the
loop, are potential S-nitrosylation sites as predicted from the 3D
structure of the kinase (19). NO
donors, such as thioredoxin and thiol/disulfide oxidoreductases
were excluded from the system to prevent possible interferences
(42,43); however, a small amount of
2-mercaptoethanol (∼0.05% v/v) was necessary to prevent oxygen
effects.
The simple, but well-defined, S-nitrosylation
reaction model was used to probe for particular NO acceptor sites
in human Akt1/PKBα (inactive, 89% pure containing
2-mercaptoethanol and EGTA; Upstate) in three steps. i) Mapping of
all cysteine residues with DTT reduction, Iodoacetyl-LC-Biotin
alkylation and affinity capture provided relative MS ionization
efficacies and charge states. ii) Detection of disulfide bonds with
and without GSNO, provided an understanding of NO-mediated
disulfide bond formation. The concentrations of the NO donor used
here were similar to the levels used in reported studies (35–37). iii) MS/MS pinpointed the
S-nitrosylated sites with three different thiol-specific
derivatives. As indicated above, false-negatives may occur with the
Biotin-Switch method (33),
whereas false-positives are more common with the other methods;
however, thiolether derivatives can be identified with MS/MS data.
The findings of these studies were used to study the biological
consequences of S-nitrosylation of Akt1/PKBα in soleus
muscle from burned rats. This in vivo system was used
because soleus muscle is an insulin-sensitive tissue with high
levels of IRS-1.
A base peak intensity (BPI) nano-LC chromatogram of
all seven affinity captured cysteine residues that were
biotinylated with Iodoacetyl-LC-Biotin is shown (Fig. 1A). Cysteine residue monoisotopic
mass of C3H5NOS = 103.01 Da was replaced with
derivatized Cys residue monoisotopic mass of
C21H35N5O4S2
= 485.21 Da. The relative simplicity of the nano-LC chromatogram
indicates the high purification efficacy for removing
non-biotinylated tryptic peptides from streptavidin agarose beads.
Three predominate TOF MS tryptic parent ions were identified; m/z
639.79 (T41, M+2H+ = 639.83) eluting at 50.5 min, m/z
1088.49 (T9, M-CH4+2H+ = 1088.03) eluting at
51.5 min and m/z 924.67 (T44, M+3H+ = 924.43) eluting at
53 min are doubly and triply charged tryptic peptides containing
Cys296, Cys310 and Cys60,
respectively. Fig. 1B shows the
parent ions co-eluting at ∼53 min as well as the charge state
assignments. Parent ions m/z 924.67 (T44, M+3H+ =
924.43) and m/z 1386.51 (T44, M+2H+ = 1386.14) are
triply and doubly charged ions from the same tryptic peptide,
308TFCGTPEYLAPEVLEDNDYGR328, which contains
Cys310. Parent ion m/z 1266.09 (T58, M+3H+ =
1266.41) is triply charged and derived from the peptide,
437YFDEEFTAQMTITPPDQDDSMECVDSER465, which
contains Cys460. Parent ion m/z 815.87 (T11,
M+2H+ = 815.93) is doubly charged from the peptide,
77CLQWTTVIER86, which contains
Cys77. Parent ion m/z 1088.49 resulted from
CH4 neutral loss from m/z 1096.48. Fig. 1C shows TOF MS parent ions that
co-eluted at ∼50.8 min; chromatographic peak tailing the most
intense peak at 50.5 min. Parent ions m/z 731.33 (T9,
M+3H+ = 731.03) and m/z 1096.46 (T9, M+2H+ =
1096.04) are triply and doubly charged ions from the same tryptic
peptide, 49ESPLNNFSVAQCQLMK64, which contains
Cys60. Parent ion m/z 639.79 (T41, M+2H+ =
639.83) is a doubly charged ion from the tryptic peptide,
290ITDFGLCK297, which contains
Cys296. Fig. 1D shows
the TOF MS parent ions that co-eluted at ∼53.5 min. Parent ion m/z
829.00 (T45, M+3H+ = 829.05) is triply charged and
derived from the tryptic peptide,
329AVDWWGLGVVMYEMMCGR346, which contains
Cys344. Parent ion m/z 872.70 (T32, M+3H+ =
872.43) is triply charged and derived from the tryptic peptide,
223LCFVMEYANGGELFFHLSR241, which contains
Cys224. No doubly charged T58, T45 or T32 ions were
observed. It is clear that the ionization efficacies for the
peptides containing Cys296 (M+2H+),
Cys310 (M+2H+ and M+3H+),
Cys60 (M+2H+ and M+3H+) and
Cys77 (M+2H+) are much higher than for the
triply charged peptides containing Cys460
(M+3H+), Cys334 (M+3H+) and Cys224
(M+3H+) under the same conditions.
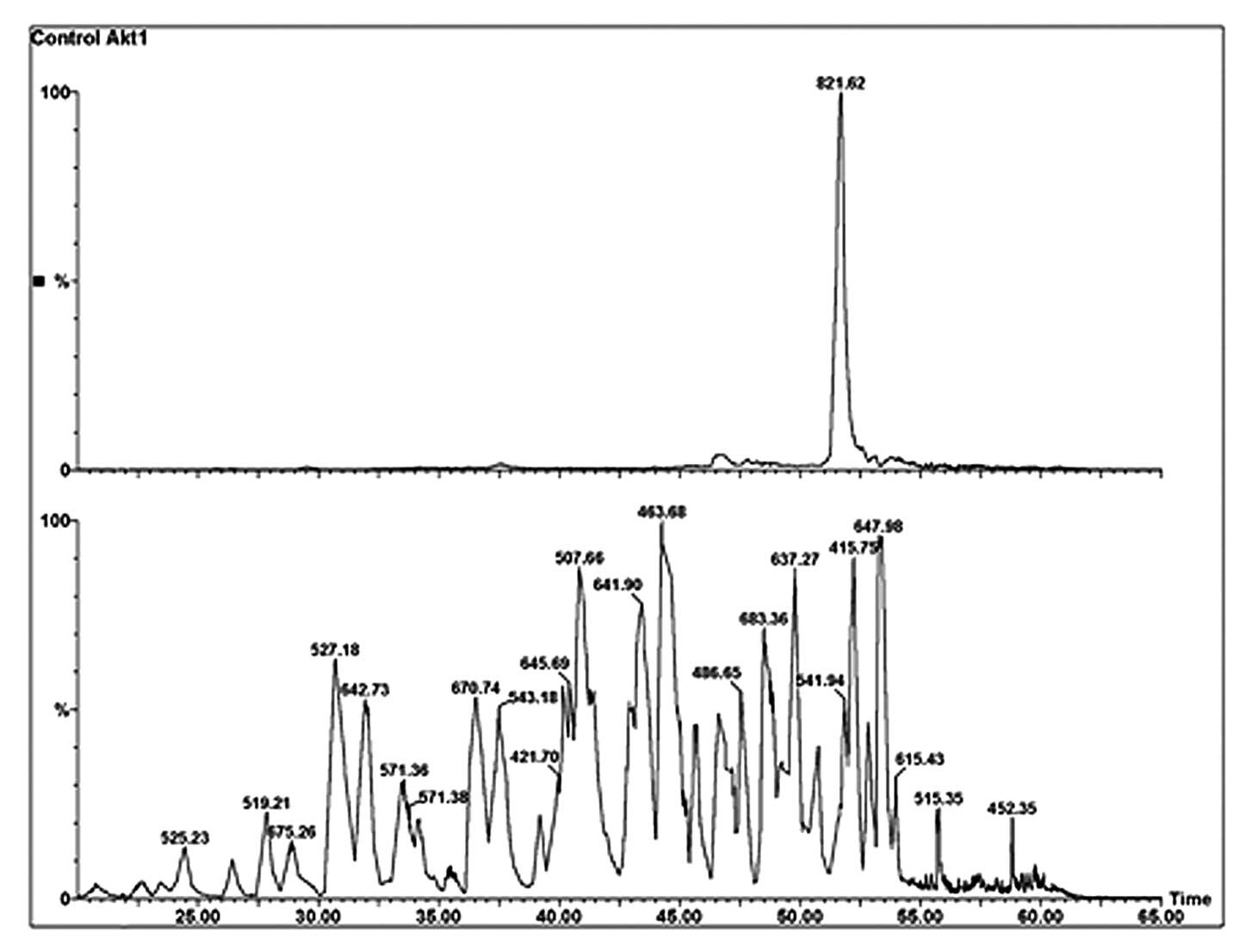
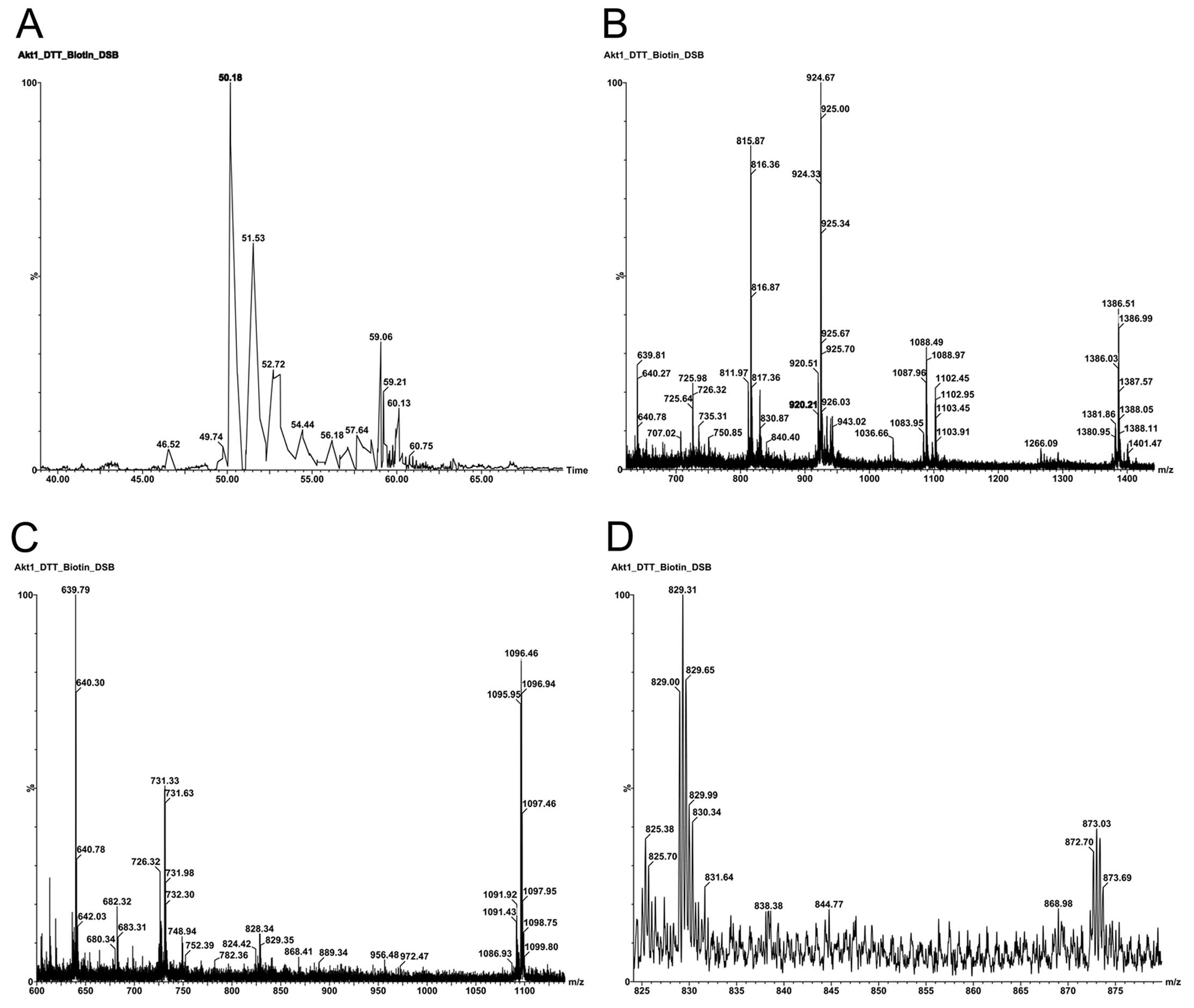 | Figure 1.Mapping of cysteine residues in
inactive Akt1/PKBα. (A) Base peak intensity (BPI) nano-LC
chromatogram of affinity capture of all seven cysteine residues
that were biotinylated with Iodoacetyl-LC-Biotin. Sample
preparation: see materials and methods section for details. Column
conditions: 75 mm ID ×150 mm, C18 PepMap300, 5 mm, under linear
gradient conditions at a flow rate 95 nl/min. (B) TOF MS analysis
of parent ions co-eluted at retention time of ∼53 min. Parent ions
m/z 924.67 and 1386.51 are triply and doubly charged ions from the
same tryptic peptide
308TFCGTPEYLAPEVLEDNDYGR328 which contains
Cys310. Parent ion m/z 1266.09 is a triply charged ion
from the tryptic peptide,
437YFDEEFTAQMTITPPDQDDSMECVDSER465, which
contains Cys460. Parent ion m/z 815.87 is a doubly
charged ion derived from the tryptic peptide,
77CLQWTTVIER86, which contains
Cys77. The parent ion at m/z 1088.49 results from
CH4 neutral loss from m/z 1096.46 as shown in C. (C) TOF
MS analysis of parent ions co-eluting at retention time of ∼50.8
min. Parent ions m/z 731.33 and 1096.46 are triply and doubly
charged ions from the same tryptic peptide,
49ESPLNNFSVAQCQLMK64, which contains
Cys60. Parent ion m/z 639.79 is doubly charged and is
derived from tryptic peptide, 290ITDFGLCK297,
which contains Cys296. (D) TOF MS analysis of parent
ions co-eluting at retention time of ∼53.5 min. Parent ion m/z
829.00 is triply charged and derived from tryptic peptide,
329AVDWWGLGVVMYEMMCGR346, which contains
Cys344. Parent ion m/z 872.70 is triply charged and
derived from tryptic peptide,
223LCFVMEYANGGELFFHLSR241, which contains
Cys224. |
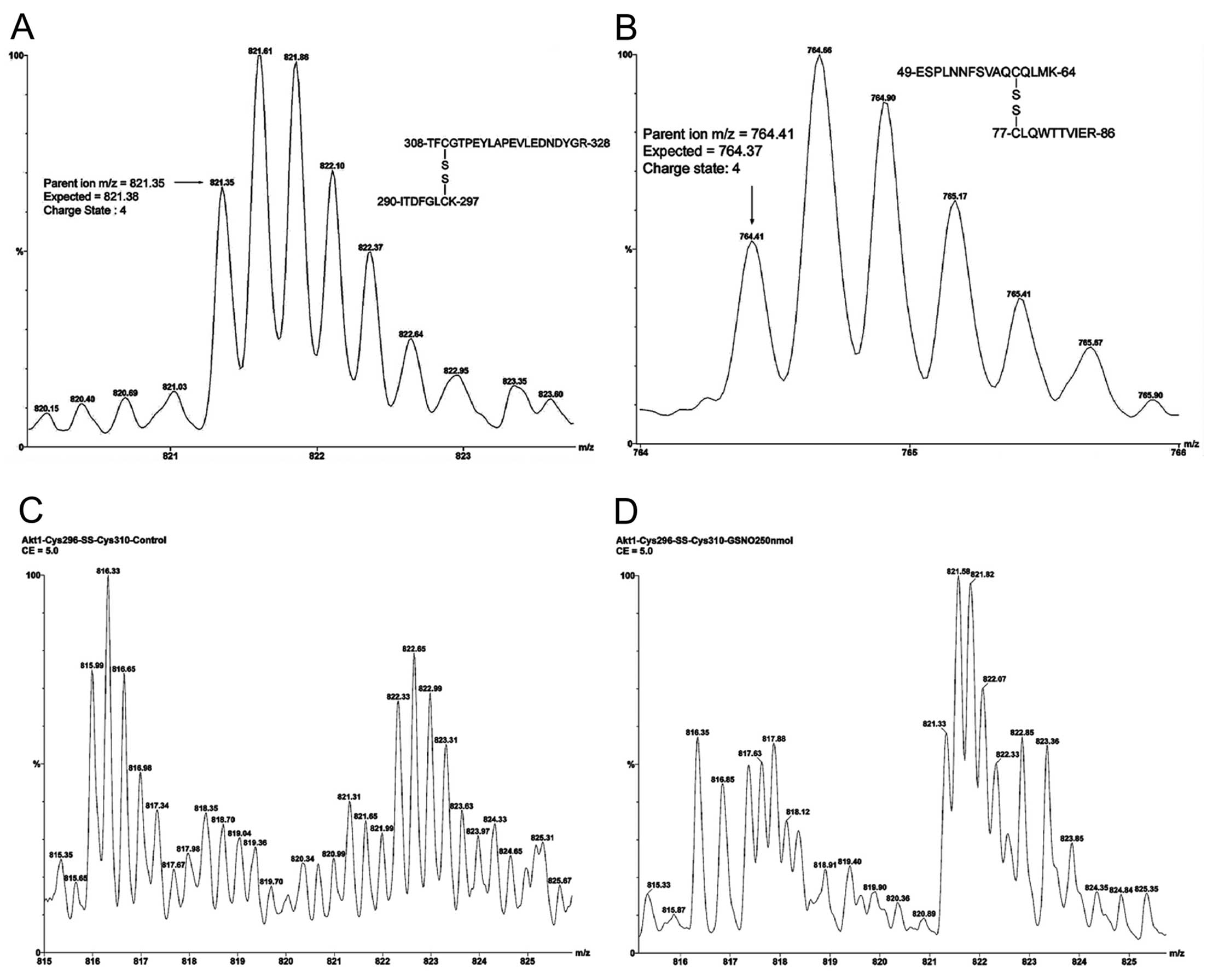
When Akt1/PKBα was treated with GSNO without
cleavage of disulfide bonds and the free cysteine residues were
alkylated with iodoacetamide, two intradomain disulfide bonds were
identified: Cys60-Cys77 in the PH domain and
Cys296-Cys310 in the kinase active loop. The
monoisotopic parent ion with m/z 821.35, shown in Fig. 2A, represents two tryptic peptides
containing the Cys296-Cys310 disulfide bond
in the kinase loop. The isotopic peaks at m/z 821.61 and m/z 821.35
are attributed to the M+1 and M+0 ions. A mass difference of 0.26
Da (expected 0.25 Da) indicated four positive charges: two at
N-terminals and two at side chains of the C-terminals of the
dipeptides. The expected quadruply charged disulfide bond linked
Cys296 and Cys310-containing peptides
(T41-SS-T44, M+4H+) were calculated to be m/z 821.38
[(894.45 + 2387.06 + 4)/4]. The monoisotopic parent ion with m/z
764.41, shown in Fig. 2B,
represents the two tryptic peptides containing the
Cys60-Cys77 disulfide bond in the PH domain.
The quadruply charged state is calculated as m/z 764.66 (M+1) -
764.41 (M+0) = 0.25 which indicates four positive proton charges.
The quadruply charged disulfide bond linked Cys60 and
Cys77 containing peptides (T9-SS-T11, M+4H+) are
calculated as m/z 764.37 [(1806.86 + 1246.63 + 4)/4]. Without GSNO
treatment, only the Cys60-Cys77 disulfide
bond was detected. The mass accuracies for the two measurements
were found to be 36 ppm (Cys296-Cys310
disulfide bond linked dipeptides) and 78 ppm
(Cys60-Cys77 disulfide bond linked
dipeptides). The impact of GSNO on
Cys296-Cys310 disulfide bond formation is
demonstrated in Fig. 2C and D.
The S-nitrosylation reaction without GSNO (Fig. 2C) shows the triply charged tryptic
peptide, 308TFCGTPEYLAPEVLEDNDYGR328,
[carboxyamidomethyl cysteine (CAM) derivative] containing
Cys310 at m/z 815.99 (expected monoisotopic parent ion,
816.03). The observed M+1 isotopic peak was at m/z 816.33. The
difference between the isotopic M+1 and M+0 peak of 0.34 Da
indicates three proton charges. In contrast, the triply charged
ions at m/z 821.31 and 821.65 (difference = 0.31 Da) do not
represent the quadruply charged Cys296-Cys310
dipeptides in Fig. 2C. The triply
charged Cys310-containing peptide was found to be
totally absent with GSNO treatment as shown in Fig. 2D. The doubly charged ions at m/z
816.35 and 816.85 (difference = 0.50 Da) are not related to the
triply charged tryptic peptide
308TFCGTPEYLAPEVLEDNDYGR328 (CAM derivative)
containing Cys310 at m/z 815.99 as shown in Fig. 2C. In contrast, the ions at m/z
821.33 and 821.58 (difference = 0.25 Da) are indeed from quadruply
charged Cys296-Cys310-linked dipeptides.
Since quadruply charged Cys296-Cys310-linked
dipeptides are formed at the expense of triply charged
Cys310-containing peptide after GSNO treatment, it is
obvious that S-nitrosylation and disulfide bond formation occur
simultaneously in the kinase loop.
We next sought to determine which cysteine residue
is the NO acceptor that initializes
Cys296-Cys310 disulfide bond formation. There
are three possibilities for the two cysteine residue thiol states:
single S-nitrosothiol, double S-nitrosothiols and nitroxyl
disulfide. The last case (nitroxyl disulfide) can be ruled out from
the list, since the expected net mass increases of 28 Da (NO - 2H =
30 - 2 Da) were not observed for the corresponding dipeptides. The
second case, double S-nitrosothiols of Cys296 and
Cys310, may occur if both pKa values are acidic inside
the kinase loop. The Biotin-Switch method was used to identify the
S-nitrosothiol within the loop under gentle reaction conditions
(GSNO 250 nmol, 1 h). In addition, two other thiol-specific
reagents, iodoacetic acid and Iodoacetyl-LC-Biotin (leaving
molecule: HI, fast and quantitative), were evaluated.
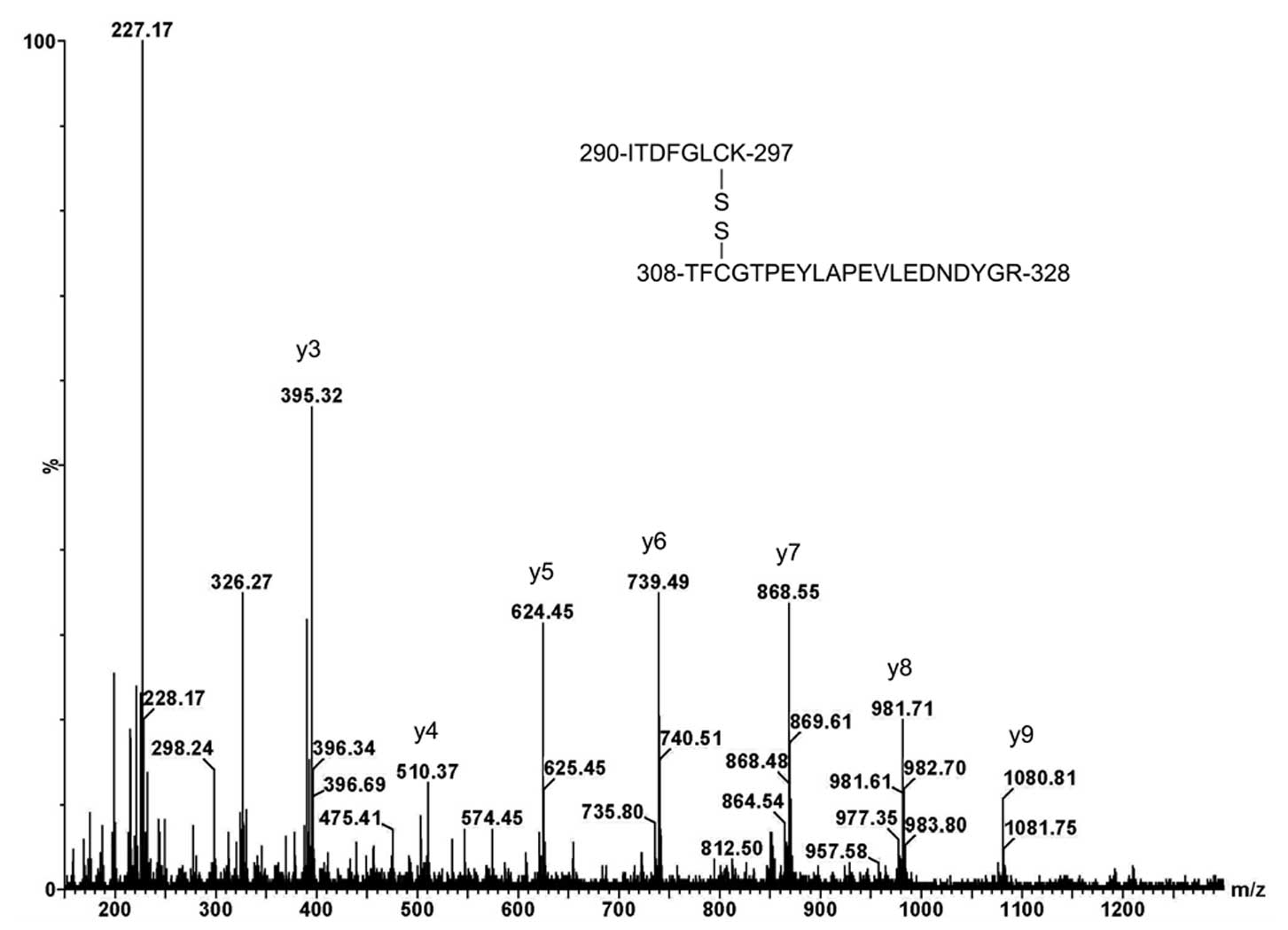
Table I shows the
expected results of Cys296 S-nitrosylation in the kinase
loop with the three different chemical modifications. The resulting
S-nitrosylated Cys was reduced with ascorbate and then derivatized
with iodoacetic acid to afford the CMC derivative (the Cys residue
with a monoisotopic mass C3H5NOS = 103.01 Da
was replaced by the CMC residue with a monoisotopic mass
C5H7NO3S = 161.01 Da) for sequence
analysis. The CMC derivative of the y2 ion of the doubly charged
tryptic peptide, 290ITDFGLCK297, was
confirmed at m/z 308.17 (expected 308.13 = 161.01 + 145.10+ 2.02).
The Cys HPDP-Biotin adduct (Cys residue monoisotopic mass
C3H5NOS = 103.01 Da was replaced with the
adduct residue monoisotopic mass
C22H37N5O4S3
= 531.20 Da) was used for sequence analysis. The corresponding y2
ion of the Biotin-HPDP derivatized,
290ITDFGLCK297, was confirmed at m/z 678.29
(expected 678.32 = 531.20 + 145.10 + 2.02). The Cys
Iodoacetyl-LC-Biotin adduct (Cys residue monoisotopic mass
C3H5NOS = 103.01 Da was replaced with adduct
residue monoisotopic mass
C21H35N5O4S2
= 485.21 Da) was used for peptide sequence analysis. The
corresponding y2 ion of Iodoacetyl-LC-Biotin derivatized,
290ITDFGLCK297 was confirmed at m/z 632.38
(expected 632.33 = 485.21 + 145.10 + 2.02). Since the y2 ions of
296Cys-Lys297 produced with the three
different derivatization procedures were unambiguously observed it
is likely that Cys296 is a favorable S-nitrosylation
site under the conditions used. Although studies with mutated
Akt1/PKBα (Cys224) indicated that
Cys224 is a major S-nitrosylation acceptor site in
vitro(28), the biological
role of S-nitrosylated Cys224 in kinase regulation needs
to be further explored. In the current study it was determined that
significant S-nitrosylation of Cys224 is improbable,
since using the three alkylation approaches and trypsin digestion,
the levels of positive ionization of Cys224-containing
peptides were below the level of detection. This failure in
detection of S-nitrosylated Cys224 may be a
false-negative under our experimental conditions and clearly
warrants further investigation. Nevertheless, our findings clearly
demonstrate that S-nitrosylated Cys296 is directly
relevant to the kinase activation regulation cycle.
 | Table I.Characterization of the
thiol-specifically modified Akt1/PKBα peptide
290ITDFGLCK297. |
Table I.
Characterization of the
thiol-specifically modified Akt1/PKBα peptide
290ITDFGLCK297.
| Chemical
derivatives | Parent calc. | Parent found | y2 ion calc. | y2 ion found |
|---|
| CMC | 953.45 | 953.42 | 308.13 | 308.17 |
| HPDP-Biotin | 1323.64 | 1323.68 | 678.32 | 678.29 |
|
Acetyl-LC-Biotin | 1277.65 | 1277.58 | 632.33 | 632.38 |
One possible explanation for the kinetics of
Cys296-Cys310 disulfide bond formation in the
kinase loop may be that there is a high kinetic barrier without
GSNO. Due to its highly labile nature (44), S-nitrosylated Cys296,
which forms rapidly in the presence of GSNO, may function as an
intermediate state. Since this intermediate is likely to have a
lower kinetic barrier for Cys296-Cys310
disulfide bond formation, the overall speed of the reaction should
increase greatly. It has been reported that
trans-nitrosylation reactions between vicinal thiols can
occur and accelerate disulfide bond formation (45). The well characterized
Cys296-Cys310 disulfide bond can be used as a
signature peptide for detection of S-nitrosylation of
Cys296 after immunoprecipitation. The separation of
tryptic peptide mixtures with our nano-LC interfaced
Q-TOFmicro is demonstrated in Fig. 3 (bottom panel). The extracted mass
ion peak m/z 821.62, as shown in Fig.
3 (top panel), is the M+1 isotopic peak of the quadruply
charged dipeptides (the most intense isotopic peak due to a high
number of carbon atoms).
The in vitro system allowed us to determine
conditions that are favorable for evaluation of S-nitrosylation of
Cys296 by MS/MS and was useful for studying the
mechanism of intradomain disulfide bond formation. The reason for
using inactive Akt1/PKBα (unphosphorylated) in these studies
was to find possible S-nitrosylation sites in relationship with the
following published data: i) Akt1/PKBα undergoes transient
phosphorylation/dephosphorylation which regulates the kinase
activity conformation cycle (22); ii) kinase disulfide bond
formation, Cys297-Cys311, and
dephosphorylation at pThr308 are induced simultaneously
by H2O2 oxidative stress in vitro(31); iii) high levels of nitric oxide
production occur both after burn injury (29,42) and in diabetic patients (43). Previous results from our
laboratory have indicated that there is S-nitrosylation at
Cys296 in rat soleus muscle (33). A parent ion at m/z 690.83
containing Cys296 (T41-T42:
290ITCFGLCKEGIK301) was observed with CAM
immonium trigged parent ion discovery; however, MS/MS sequencing
data were not obtained. As a continuation of these studies to
explore S-nitrosylation in the kinase active loop, large amounts of
rat soleus muscle lysate (∼3-5 mg/ml total proteins, 3 ml for each
experiment, day 4 after 40% TBSA, 3rd degree burn) were used. In
the present study, detailed MS/MS analyses of HPDP-biotinylated
free Cys296 peptide and
Cys296-Cys310 disulfide bound dipeptides of
Akt1/PKBα were performed with lysates of rat soleus muscle
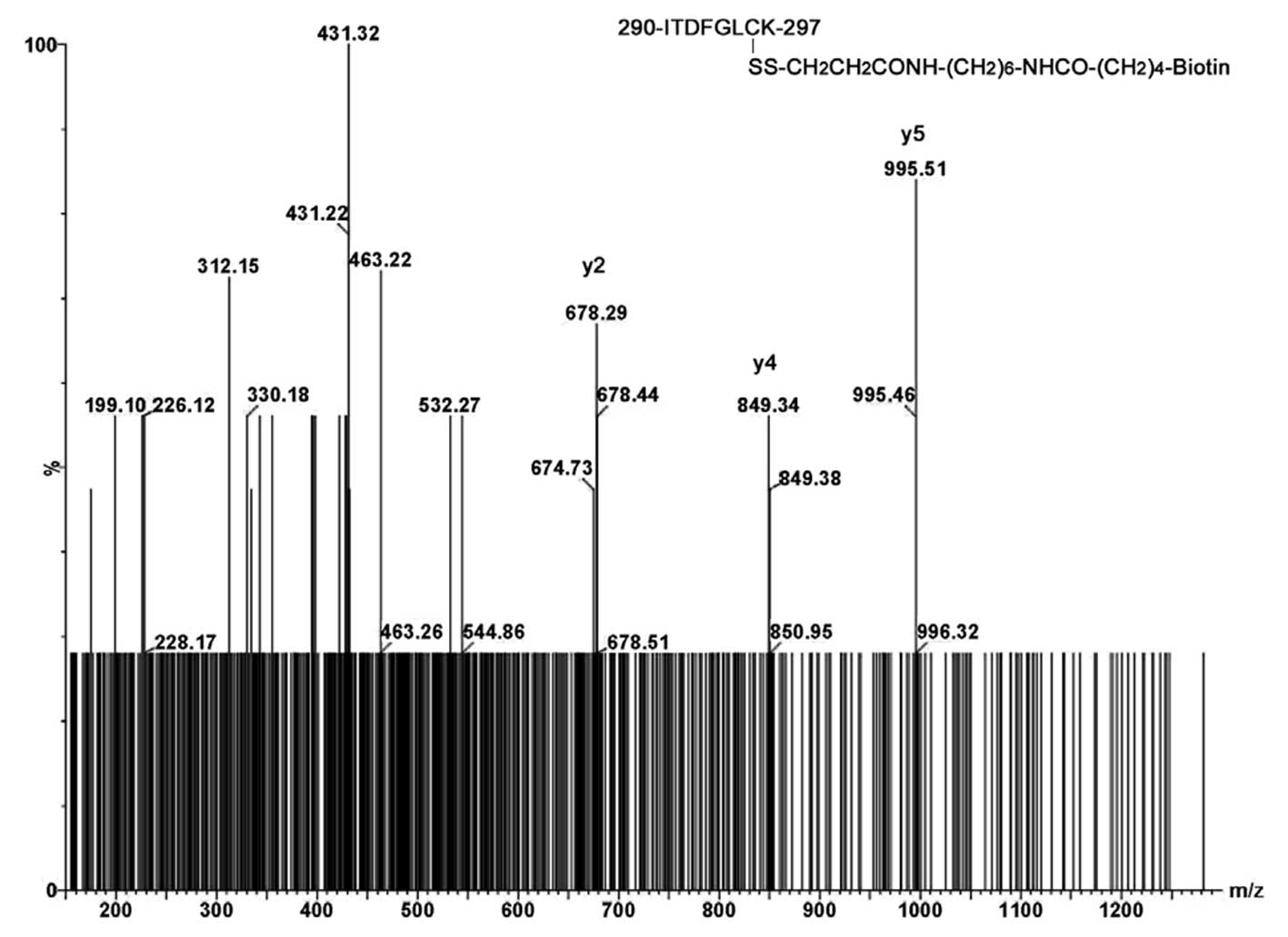
after burn injury. The tryptic parent ion derivatized from free
Cys296 after burn injury was observed at m/z 662.84
(M+2H+, expected 662.82) and the MS/ MS sequence data
are shown in Fig. 4. A low
sequence score of 18 was obtained from the parent ion with S/N = 3.
However, the critical diagnostic y2, y4 and y5 ions at m/z 678.29,
849.34 and 995.51 confirmed that trace amounts of free
Cys296 are indeed present after intradomain disulfide
bond formation induced by burn injury. In addition, partial
sequencing data for Cys296-Cys310
disulfide-linked dipeptides are shown in Fig. 5. The C-terminal y ion series of
Cys310-containing peptide,
308TFCGTPEYLAPEVLEDNDYGR328, was observed for
the quadruply charged parent ion (T41-SS-T44, M+4H+).
Cys296-Cys310 disulfide-linked dipeptides
were not observed in muscle lysates from sham-treated animals
(negative controls). The chance of obtaining the MS/MS sequence
using our in vivo experimental conditions is only ∼20–25%.
This indicates that one interpretable MS/MS outcome (score >25)
is expected in four or five independent experiments in which three
successive injections are performed. Nevertheless, these MS/MS data
for peptides containing free Cys296 and
Cys296-Cys310-linked dipeptides are
sufficient to verify our hypothesis that S-nitrosylation promotes
intradomain disulfide bond formation and dephosphorylation at
pThr308 after burn injury as illustrated in Fig. 6. Due to its high lability of
Cys296-SNO, direct identification of this species in
vivo was not possible.
S-nitrosylation of Akt1/PKBα is a key factor
for understanding the regulation of glucose transport and
downstream protein synthesis. A recent study demonstrated that
blockade of iNOS prevents the S-nitrosylations of Akt and IRS-1 and
results in insulin resistance in vivo(46). Although it is clear that two PTMs
of Akt1/PKBα, phosphorylation at Thr308 and
S-nitrosylation at Cys296, are critical for the
regulation of Akt1/PKBα activity under stress conditions,
there are still many unanswered questions concerning how reversible
phosphorylation/dephosphorylation and
S-nitrosylation/denitrosylation modulate Akt1/PKBα activity.
For example, it has been reported that the
Cys296-Cys310 disulfide bond is present only
when there is binding of substrate to the active kinase loop and
phosphorylation at Thr308(25); indicating that both disulfide bond
formation as well as phosphorylation of Thr308 are
important for kinase activity. In contrast, this disulfide bond was
not observed under similar conditions in two studies of the ternary
structure of the kinase (19,21); even though, oxidative stress was
shown to induce dephosphorylation of pThr308 and
disulfide bond formation in the kinase loop in an in vitro
study (31).
In summary, our data establish that
Cys296 is an important S-nitrosylation site in the
kinase loop of Akt1/PKBα under gentle reaction conditions:
i) iodoacetic acid as previously described; ii) the HPDP-Biotin
switch method; and iii) the Iodoacetyl-LC-Biotin method to ensure
indirect capture of Cys296-SNO which may be undetectable
with HPDP-Biotin. The corresponding derivatized y2 ions
(296Cys-Lys297) in the tryptic peptide
(Ile-Thr-Asp-Phe-Gly-Leu-Cys-Lys) were obtained with mass sequences
to eliminate false-positive discovery. Although no other
S-nitrosylated cysteine residues were detected, it is possible that
S-nitrosylations at Cys224, Cys344 and
Cys460 were missed due to very low ionizations (i.e.,
false-negative discoveries). As a consequence of S-nitrosylation at
Cys296, there is rapid disulfide bond formation with
vicinal Cys310 in the kinase loop, which alters kinase
substrate recognition (47) as
well as Akt-FOXO switch (48).
This affords a stable disulfide bond linked quadruply charged
parent ion at m/z 821.35 (M+4H+). Partial sequencing
data for Cys296-Cys310 linked dipeptides from
soleus muscle lysates indicated that burn injury is associated with
both dephosphorylation of pThr308 and disulfide bond
formation. These two types of PTMs may provide insights for
understanding negative cooperative effects on reduced Akt/PKB
kinase activity after burn injury as previously reported by our
laboratory (26). Although our
results have provided important mechanistic information,
quantitative measurements of Thr308/pThr308
and free Cys296/ SNO-Cys296/bound
Cys296 in patients with burn injury and type 2 diabetes
remain very challenging.
Abbreviations:
|
Akt1/PKBα
|
Akt1/protein kinase Bα;
|
|
CAM
|
carboxyamidomethyl cysteine;
|
|
CMC
|
carboxymethyl cysteine;
|
|
GSNO
|
S-nitrosoglutathione;
|
|
PH
|
pleckstrin homology;
|
|
PTM
|
post-translational modification;
|
|
Q-TOF
|
tandem quadrupole time-of-fight mass
spectrometry;
|
|
TBSA
|
total body surface area
|
Acknowledgements
This study was supported in part by
grants from the National Institutes of Health (NIGMS P50 GM21000)
and Shriners Hospital for Children.
References
|
1
|
Biolo G, Fleming RY, Maggi SP, Nguye TT,
Herndon DN and Wolfe RR: Inverse regulation of protein turnover and
amino acid transport in skeletal muscle of hypercatabolic patients.
J Clin Endocrinol Metab. 87:3378–3384. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bodine SC, Stitt TN, Gonzalez M, Kline WO,
Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC,
Glass DJ and Yancopoulos GD: Akt/mTOR pathway is a crucial
regulator of skeletal muscle hypertrophy and can prevent muscle
atrophy in vivo. Nat Cell Biol. 3:1014–1019. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bruning JC, Winnay J, Cheatham B and Kahn
CR: Differential signaling by insulin receptor substrate 1 (IRS-1)
and IRS-2 in IRS-1-deficient cells. Mol Cell Biol. 17:1513–1521.
1997.PubMed/NCBI
|
|
4
|
Carvalho E, Rondinone C and Smith U:
Insulin resistance in fat cells from obese Zucker rats - evidence
for an impaired activation and translocation of protein kinase B
and glucose transporter 4. Mol Cell Biochem. 206:7–16. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Araki E, Lipes MA, Patti ME, Bruning JC,
Haag B, Johnson RS and Kahn CR: Alternative pathway of insulin
signaling in mice with targeted disruption of the IRS-1 gene.
Nature. 372:186–190. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Khoury W, Klausner JM, Ben-Abraham R and
Szold O: Glucose control by insulin for critically ill surgical
patients. J Trauma. 57:1132–1138. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carter EA, Burks D, Fischman AJ, White M
and Tompkins RG: Insulin resistance in thermally-injured rats is
associated with post-receptor alterations in skeletal muscle, liver
and adipose tissue. Int J Mol Med. 14:653–658. 2004.PubMed/NCBI
|
|
8
|
Johan Groeneveld AB, Beishuizen A and
Visser FC: Insulin: a wonder drug in the critically ill? Crit Care.
6:102–105. 2002.PubMed/NCBI
|
|
9
|
Ikezu T, Okamato T, Yonezawa K, Tompkins
RG and Martyn JA: Analysis of thermal injury-induced insulin
resistance in rodents. J Biol Chem. 272:25289–25295. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
White MF: Insulin signaling in health and
disease. Science. 302:1710–1711. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Q, Carter EA, Ma BY, White M,
Fischman AF and Tompkins RG: Molecular mechanism(s) of burn-induced
insulin resistance in murine skeletal muscle: role of IRS
phosphorylation. Life Sci. 77:3068–3077. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ishiki M and Klip A: Minireview: recent
developments in the regulation of glucose transporter-4 traffic:
new signals, locations, and partners. Endocrinology. 146:5071–5078.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kaneki M, Shimizu N, Yamada D and Chang K:
Nitrosative stress and pathogenesis of insulin resistance. Antioxid
Redox Signal. 9:1–11. 2007.
|
|
14
|
Song G, Quyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Neels JG and Olefsky JM: Cell signaling. A
new way to burn fat. Science. 312:1756–1758. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tian R: Another role for celebrity: Akt
and insulin resistance. Circ Res. 96:139–140. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lawlor MA and Alessi DR: PKB/Akt: a key
mediator of cell proliferation, survival and insulin responses? J
Cell Sci. 114:2903–2910. 2001.PubMed/NCBI
|
|
18
|
Yang J, Cron P, Thompson V, Good VM, Hess
D, Hemmings BA and Barford D: Molecular mechanism for the
regulation of protein kinase B/Akt by hydrophobic motif
phosphorylation. Mol Cell. 9:1227–1240. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang J, Cron P, Good VM, Thompson V,
Hemmings BA and Barford D: Crystal structure of an activated
Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP.
Nat Struct Biol. 9:940–944. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang X, Begley M, Morgenstern KA, Gu Y,
Rose P, Zhao H and Zhu X: Crystal structure of an inactive Akt2
kinase domain. Structure. 11:21–30. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kumar CC and Madison V: Akt crystal
structure and Akt-specific inhibitors. Oncogene. 24:7493–7501.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fayard E, Tintignac LA, Baudry A and
Hemmings BA: Protein kinase B/Akt at a glance. J Cell Sci.
118:5675–5678. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brazil DP, Yang ZZ and Hemmings BA:
Advances in protein kinase B signaling: AKTion on multiple fronts.
Trends Biochem Sci. 29:233–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brazil DP, Park J and Hemmings BA: PKB
binding proteins: getting in on the Akt. Cell. 111:293–303. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang BX and Kim HY: Interdomain
conformational changes in Akt activation revealed by chemical
cross-linking and tandem mass spectrometry. Mol Cell Proteomics.
5:1045–1053. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sugita H, Kaneki M, Sugita M, Yasukawa T,
Yasuhara S and Martyn JA: Burn injury impairs insulin-stimulated
Akt/PKB activation in skeletal muscle. Am J Physiol Endocrinol
Metab. 288:E585–E591. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Carvalho-Filho MA, Ueno M, Hirabara SM,
Seabra AB, Carvalheira JB, de Oliveira MG, Velloso LA, Curi R and
Saad MJ: S-nitrosation of the insulin receptor, insulin receptor
substrate 1, and protein kinase B/Akt: novel mechanism of insulin
resistance. Diabetes. 54:959–967. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yasukawa T, Tokunaga E, Ota H, Sugita H,
Martyn JA and Kaneki M: S-Nitrosylation-dependent inactivation of
Akt/protein kinase B in insulin resistance. J Biol Chem.
280:7511–7518. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Carter EA, Derojas-Walker T, Tamir S,
Tannenbaum SR, Yu YM and Tompkins RG: Nitric oxide production is
intensely and persistently increased in tissue by thermal injury.
Biochem J. 304:201–204. 1994.PubMed/NCBI
|
|
30
|
Auguin D, Barthe P, Auge-Senegas MT, Stern
MH, Noguchi M and Roumestand C: Solution structure and backbone
dynamics of the Pleckstrin homology domain of the human protein
kinase B (PKB/Akt). Interaction with inositol phosphates. J Biomol
NMR. 28:137–155. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Murata H, Ihara Y, Nakamura H, Yodoi J,
Sumikawa K and Kondo T: Glotaredoxin exerts an antiapoptotic effect
by regulating the redox state of Akt. J Biol Chem. 278:50226–50233.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lu XM, Lu MY, Fischman AJ and Tompkins RG:
A new approach for sequencing human IRS1 phosphotyrosine-containing
peptides using CapLC-Q-TOF(micro). J Mass Spectrom. 40:599–607.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lu XM, Lu M, Tompkins RG and Fischman AJ:
Site-specific detection of S-nitrosylated PKBa/Akt1 from rat soleus
muscle using CapLC-Q-TOF(micro) mass spectrometry. J Mass Spectrom.
40:1140–1148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jaffrey SR, Erdjument-Bromage H, Ferris
CD, Tempst P and Snyder SH: Protein S-nitrosylation: a
physiological signal for neuronal nitric oxide. Nat Cell Biol.
3:193–197. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jaffrey SR and Snyder SH: The biotin
switch method for detection of S-nitrosylated proteins. Science
STKE. 86:1–9. 2001.
|
|
36
|
Greco TM, Hodara R, Parastatidis I,
Heijnen HFG, Dennehy MK, Liebler DC and Ischiropoulos H:
Identification of S-nitrosylation motifs by site-specific mapping
of the S-nitrosocysteine proteome in human vascular smooth muscle
cells. Proc Natl Acad Sci USA. 103:7420–7425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hao G, Derakhshan B, Shi L, Campagne F and
Gross SS: SNOSID, a proteomic method for identification of cysteine
S-nitrosylation sites in complex protein mixtures. Proc Natl Acad
Sci USA. 103:1012–1017. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kuncewicz T, Sheta EA, Goldknopf IL and
Kone BC: Proteomic analysis of S-nitrosylated proteins in mesangial
cell. Mol Cell Proteomics. 2:156–163. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Martinez-Ruiz A and Lamas S: Detection and
proteomic identification of S-nitrosylated proteins in endothelial
cells. Arch Biochem Biophys. 423:192–199. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gan HT and Chen JDZ: Roles of nitric oxide
and prostaglandins in pathogenesis of delayed colonic transit after
burn injury in rats. Am J Physiol Regul Integr Comp Physiol.
288:R1316–R1324. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Torres SH, De Sanctis JB, de L Briceno M,
Hernandez N and Finol HJ: Inflammation and nitric oxide production
in skeletal muscle of type 2 diabetic patients. J Endocrinol.
181:419–427. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Benhar M, Forrester MT and Stamler JS:
Nitrosative stress in the ER: a new role for S-nitrosylation in
neurodegenerative diseases. ACS Chem Biol. 1:355–358. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tannenbaum SR and White FM: Regulation and
specificity of S-nitrosylation and denitrosylation. ACS Chem Biol.
1:615–618. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hogg N: The biochemistry and physiology of
S-nitrosothiols. Ann Rev Pharmacol Toxicol. 42:585–600. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Arnelle DR and Stamler JS: NO+,
NO•, and NO− donation by S-nitrosothiols:
implications for regulation of physiological functions by
S-nitrosylation and acceleration of disulfide formation. Arch
Biochem Biophys. 318:279–285. 1995.
|
|
46
|
Carvalho-Filho MA, Ueno M, Carvalheira JB,
Velloso LA and Saad MJ: Targeted disruption of iNOS prevents
LPS-induced S-nitrosation of IRbeta/IRS-1 and Akt and insulin
resistance in muscle of mice. Am J Physiol Endocrinol Metab.
291:E476–E482. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wu WI, Voegtli WC, Sturgis HL, Dizon FP,
Vigers GP and Brandhuber BJ: Crystal structure of human AKT1 with
an allosteric inhibitor reveals a new mode of kinase inhibition.
PLos One. 5:e129132010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kloet DE and Burgering BM: The PKB/FOXO
switch in aging and cancer. Biochim Biophys Acta. 1813:1926–1937.
2011. View Article : Google Scholar : PubMed/NCBI
|