Introduction
Inflammation is the biological response to harmful
stimuli, such as autoimmune diseases, pathogenic infections,
damaged cells and irritants; inflammation initiates the healing
process to prevent injury. Inflammation increases the expression of
inflammatory mediators, such as nitric oxide (NO) and prostaglandin
E2 (PGE2), which are regulated by NO synthase
(NOS) and cyclooxygenases (COXs), respectively, as well as
inflammatory cytokines, such as interleukin (IL)-1β, IL-6 and tumor
necrosis factor-α (TNF-α) (1,2).
These inflammatory regulators are required to regulate the cellular
pathways involved in protecting the organs (3–5).
However, excessive inflammatory response can lead to an
overexpression of pro-inflammatory factors, which can result in
severe inflammatory disorders (5–7).
Microglial cells are the resident macrophage-like
cells in the brain and have been proposed to play a major role in
host defense and tissue repair in the central nervous system (CNS).
In pathological conditions, activated microglial cells release
neurotoxic and pro-inflammatory mediators, including NO,
PGE2, reactive oxygen species (ROS) and pro-inflammatory
cytokines (8,9). Overproduction of these inflammatory
mediators and cytokines causes severe forms of various
neurodegenerative diseases such as Alzheimer's disease (AD),
Parkinson's disease (PD) and cerebral ischemia (10,11). Not surprisingly then, activated
microglial cells have been shown to be a major cellular source of
pro-inflammatory and/or cytotoxic factors that cause neuronal
damage in the CNS (10,12). Previous studies have also
demonstrated that a decrease in the number of pro-inflammatory
factors in microglial cells may attenuate the severity of these
disorders (9–12). Therefore, agents that attenuate
pro-inflammatory mediators and cytokines in microglial cells may
represent promising strategies to tackle brain injury and
neurodegenerative diseases.
Platycodon grandiflorus (P.
grandiflorus) A. DC., known as bellflower or balloon flower,
belongs to the species in the genus Platycodon L. of the
family Campanulaceae. This plant is an herbaceous perennial native
to Northeastern Asia and widely distributed in South Korea. The
root of P. grandiflorus, radix platycodi, has been
consumed as a food and has been used as a folk remedy for
conditions such as coughing, the common cold, bronchitis, asthma,
pulmonary tuberculosis and inflammation (13,14). Radix platycodi is abundant
in saponins (15,16). Several studies have reported that
various platycodon saponins exhibit strong anti-inflammatory
activity by blocking the generation of pro-inflammatory mediators
and cytokines through the inhibition of nuclear factor-κB (NF-κB)
and/or mitogen-activated protein kinase (MAPK) activation in
12-O-tetradecanoylphorbol 13-acetate (TPA) and
lipopolysaccharide (LPS)-treated macrophages (17-21). Although Jang et al
(22) have demonstrated the
anti-inflammatory effects of an aqueous extract of P.
grandiflorus on LPS-stimulated PGE2 synthesis, NO
generation and IL-8 production in BV2 microglial cells, the precise
anti-inflammatory mechanisms of action of saponins isolated from
radix platycodi (PGS) have not yet been elucidated in
microglial cells.
In the current study, we investigated the inhibitory
effects of PGS and the mechanisms by which PGS induces
anti-inflammatory effects by assessing the suppression of
anti-inflammatory mediator and cytokine production and expression
in an LPS-stimulated BV2 microglial cell model. The results
indicated that PGS inhibited the release of NO, PGE2,
IL-β and TNF-α, as well as their regulatory genes, which was
associated with the suppression of NF-κB translocation from the
cytosol to the nucleus. The regulation of NF-κB activity by PGS was
also associated with the inhibition of the phosphorylation of AKT
and MAPKs in an LPS-induced anti-inflammatory reaction. The data
from our study suggest that PGS may be a candidate for use in the
treatment of various neurodegenerative brain disorders.
Materials and methods
Cell culture and treatment with PGS
The BV2 murine cell line was maintained in
Dulbecco's modified Eagle's medium (Gibco-BRL, Grand Island, NY,
USA) supplemented with 10% fetal bovine serum (FBS), 100 U/ml of
penicillin and 100 mg/ml of streptomycin (Gibco-BRL) at 37°C in a
humidified incubator with 5% CO2. Confluent cultures
were passed by trypsinization. For the preparation of PGS, dried
samples of radix platycodi were purchased from Dongeui
University Hospital (Busan, Korea). The samples were extracted
twice with methanol by refluxing at 80°C for 2 h, and then the
methanol extract was suspended in water and partitioned
sequentially with n-hexane, chloroform, ethyl acetate and
n-butanol. Subsequently, the water-saturated
n-butanol fraction was evaporated to dryness in a vacuum.
The recovered crude saponins were loaded onto Diaion®
HP-20 MCI gel (Sigma-Aldrich Chemical Co., St. Louis, MO, USA), and
the sugar residues were then removed with 40% CH3OH. The
fractions were eluted with 60–80% CH3OH, collected, and
then dried to obtain PGS. Cells used in the experiments were washed
twice with warm DMEM and treated in serum-free medium for at least
4 h prior to the treatments. In all the experiments, the cells were
treated with various concentrations of PGS for the indicated
periods of time prior to exposure to 500 ng/ml LPS (Escherichia
coli 026:B6, Sigma-Aldrich Chemical Co.).
Cell viability assay
Cell viability was measured based on the formation
of blue formazan that was metabolized from colorless
3-(4,5-dimethylthi azol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich) by mitochondrial dehydrogenases, which are active
only in live cells. For this study, BV2 cells were plated in
24-well plates at a density of 2×105 cells/well for 24
h, and then washed. The cells were incubated with various
concentrations of PGS in the presence or absence of LPS (500 ng/ml)
for 24 h, and then incubated in 0.5 mg/ml of MTT solution. Three
hours later, the supernatant was removed, and the formation of
formazan was measured at 540 nm using an enzyme-linked
immunosorbent assay (ELISA) plate reader (Dynatech MR-7000;
Dynatech Laboratories, Chantilly, VA, USA).
NO production
Concentrations of NO in the culture supernatants
were determined by measuring the levels of nitrite, which is a
major stable product of NO, using Griess reagent (Sigma-Aldrich).
For this study, cells (5×105 cells/ml) were treated with
PGS in the presence or absence of LPS in 24-well plates for 24 h,
and then 100 μl of each culture medium were mixed with an equal
volume of Griess reagent [1% sulfanilamide/0.1%
N-(1-naphthyl)-ethylenediamine dihydrochloride/2.5%
H3PO4]. Nitrite levels were determined using
an ELISA plate reader at 540 nm, and nitrite concentrations were
calculated by referencing a standard curve generated by known
concentrations of sodium nitrite.
RNA isolation and reverse
transcription-polymerase chain reaction (PCR)
Total RNA was isolated using TRIzol reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA). Total RNA (1.0
μg) obtained from the cells was reverse transcribed using Moloney
murine leukemia virus (M-MLV) reverse transcriptase (Promega,
Madison, WI, USA) to produce DNA. The inducible NO synthase (iNOS),
COX-2, IL-1β and TNF-α genes were amplified from the cDNA by PCR.
PCR amplification was carried out for 26 cycles under the following
cycling conditions: denaturation at 95°C for 30 sec, annealing at
60°C for 30 sec and extension at 72°C for 1 min. A final extension
cycle was then performed at 72°C for 10 min. A sample of each
amplified product was subjected to 1.0% agarose gel electrophoresis
and stained with ethidium bromide (EtBr; Sigma-Aldrich) and
visualized using ultra violet (UV) illumination. The glyceraldehyde
3-phosphate dehydrogenase (GAPDH) housekeeping gene transcript was
used as the control. Primer sequences used in the RT-PCR analysis
are presented in Table I.
 | Table IDetails of the primer pairs used in
this study. |
Table I
Details of the primer pairs used in
this study.
| Gene | Primer
sequence |
|---|
| iNOS | F: 5′-ATG TCC GAA
GCA AAC ATC AC-3′ |
| R: 5′-TAA TGT CCA
GGA AGT AGG TG-3′ |
| COX-2 | F: 5′-CAG CAA ATC
CTT GCT GTT CC-3′ |
| R: 5′-TGG GCA AAG
AAT GCA AAC ATC-3′ |
| IL-1β | F: 5′-ATG GCA ACT
GTT CCT GAA CTC AAC T-3′ |
| R: 5′-TTT CCT TTC
TTA GAT ATG GAC AGG AC-3′ |
| TNF-α | F: 5′-ATG AGC ACA
GAA AGC ATG ATC-3′ |
| R: 5′-TAC AGG CTT
GTC ACT CGA ATT-3′ |
| GAPDH | F: 5′-CGT CTT CAC
CAC CAT GGA GA-3′ |
| R: 5′-CGG CCA TCA
CGC CAC AGT TT-3′ |
Protein extraction and western blot
analysis
Cells were washed 3 times with PBS, and total cell
lysates were lysed in extraction buffer [25 mM of Tris-Cl (pH 7.5),
250 mM of NaCl, 5 mM of ethylenediaminetetra acetic acid (EDTA), 1%
Nonidet P-40 (NP-40), 0.1 mM of sodium orthovanadate, 2 μg/ml of
leupeptin and 100 μg/ml of phenylmethylsulfonyl flouride (PMSF)]
containing protease inhibitor cocktail tablets (Roche Diagnostics,
Mannheim, Germany). In a parallel experiment, cytoplasmic and
nuclear proteins were extracted using NE-PER® Nuclear
and Cytoplasmic Extraction Reagents (Pierce Biotechnology,
Rockford, IL, USA) according to the manufacturer's instructions.
Protein concentration was determined using a Bio-Rad protein assay
kit (Bio-Rad, Hercules, CA, USA). For western blot analysis, an
equal amount of protein was subjected to electrophoresis on sodium
dodecyl sulfate (SDS)-polyacrylamide gels and transferred onto
nitro-cellulose membranes (Schleicher & Schuell Bioscience,
Inc., Keene, NH, USA) by electroblotting. The blots were probed
with the desired primary antibodies for 1 h, incubated with the
diluted enzyme-linked secondary antibodies (Amersham Co., Arlington
Heights, IL, USA), and visualized using the enhanced
chemiluminescence (ECL) method according to the recommended
procedure (Amersham Co.). The primary antibodies were purchased
from BD Biosciences (San Jose, CA, USA), Santa Cruz Biotechnology
Inc. (Santa Cruz, CA, USA), Cell Signaling Technology, Inc.
(Danvers, MA, USA) and Oncogene Science (Cambridge, MA, USA)
(Table II). Actin and lamin B
were used as the internal controls for cytosolic and nuclear
fractions, respectively.
| Table IIList of antibodies used in this
study. |
Table II
List of antibodies used in this
study.
| Antibody | Dilution | Product no. | Type of antibody,
supplier |
|---|
| iNOS | 1:1,000 | 610333 | Rabbit polyclonal,
BD Biosciences |
| COX-2 | 1:500 | SC-1999 | Mouse monoclonal,
Santa Cruz Biotechnology |
| TNF-α | 1:1,000 | 3707S | Rabbit polyclonal,
Cell Signaling Technology, Inc. |
| IL-1β | 1:1,000 | SC-7884 | Rabbit polyclonal,
Santa Cruz Biotechnology |
| NF-κB p65 | 1:500 | SC-109 | Mouse monoclonal,
Santa Cruz Biotechnology |
| AKT | 1:500 | SC-8312 | Rabbit polyclonal,
Santa Cruz Biotechnology |
| p38 MAPK | 1:500 | SC-728 | Rabbit polyclonal,
Santa Cruz Biotechnology |
| ERK | 1:2,000 | SC-535 | Rabbit polyclonal,
Santa Cruz Biotechnology |
| JNK | 1:500 | 9252S | Rabbit polyclonal,
Cell Signaling Technology, Inc. |
| p-AKT | 1:500 | 9271S | Rabbit polyclonal,
Cell Signaling Technology, Inc. |
| AKT | 1:500 | SC-8312 | Rabbit polyclonal,
Santa Cruz Biotechnology |
| p-p38 MAPK | 1:500 | 9211S | Rabbit polyclonal,
Cell Signaling Technology, Inc. |
| p38 MAPK | 1:500 | sc-535 | Rabbit polyclonal,
Santa Cruz Biotechnology |
| p-ERK | 1:500 | 9106S | Mouse monoclonal,
Cell Signaling Technology, Inc. |
| ERK | 1:500 | SC-154 | Rabbit polyclonal,
Santa Cruz Biotechnology |
| p-JNK | 1:500 | 9255S | Mouse monoclonal,
Cell Signaling Technology, Inc. |
| JNK | 1:500 | 9252S | Rabbit polyclonal,
Cell Signaling Technology, Inc. |
| Lamin B | 1:1,000 | NA12 | Mouse monoclonal,
Oncogene Science |
| Actin | 1:1,000 | SC-1616 | Goat polyclonal,
Cell Signaling Technology Inc. |
Cytokine assays
The concentrations of TNF-α and IL-1β were
determined using commercially available ELISA kits (R&D
Systems, Minneapolis, MN, USA) according to the manufacturer's
instructions. Briefly, BV2 cells (5×105 cells/ml) were
plated in 24-well plates and pre-treated with the indicated
concentrations of PGS for 1 h prior to treatment with 500 ng/ml of
LPS. After 24 h of treatment, fluid samples were diluted 1:10,000
in a Tris-buffered saline solution (pH 8.0) containing 1% bovine
serum albumin (BSA) and 0.5% Tween-20. They were then incubated
overnight in plates at 4°C, after which the plates were washed 5
times. Standard TNF-α and IL-1β were added to each plate in serial
dilutions, and a standard curve was constructed from which the
concentrations of TNF-α and IL-1β were obtained. The absorbance
values were determined with an ELISA microplate reader operating at
450 nm. Three ELISA experiments were conducted, with each sample
tested in duplicate as previously described (23).
Immunofluorescence analysis
For the detection of NF-κB p65 translocation, cells
were grown on glass coverslips for 24 h and then treated with 500
ng/ml LPS. The cells were either pre-treated or not with PGS for 1
h. The cells were fixed with 3.7% paraformaldehyde, treated with
0.2% Triton X-100, and blocked with 2% BSA. Cells were then
sequentially incubated with anti-NF-κB p65 antibody,
FITC-conjugated donkey anti-rabbit IgG and DAPI solution. They were
then examined under a fluorescence microscope (Carl Zeiss, Jena,
Germany).
Statistical analysis
Data values represent the means ± SD. Statistical
significance was determined using an analysis of variance followed
by the Student's t-test. A value of p<0.05 was considered to
indicate a statistically significant difference.
Results
Effect of PGS on NO and PGE2
production in LPS-stimulated BV2 microglial cells
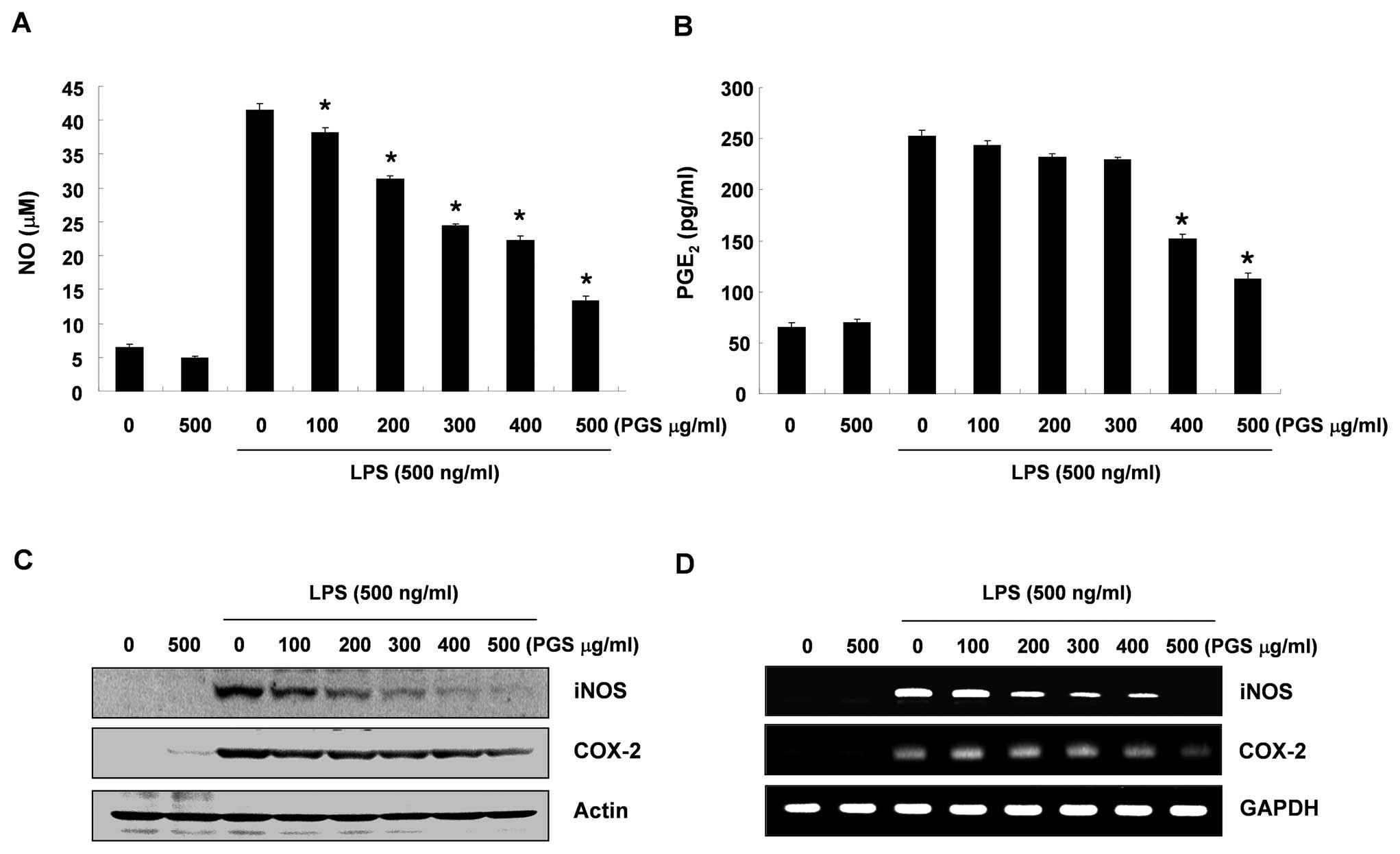
According to the NO detection assay using Griess
reagent, treatment with LPS alone markedly induced NO production by
the cells when compared to that generated by the control. However,
pre-treatment with PGS significantly repressed the levels of NO
production in the LPS-stimulated BV2 microglial cells in a
concentration-dependent manner (up to 500 μg/ml) (Fig. 1A). The amount of PGE2
present in the culture medium also increased after 24 h of exposure
to LPS alone; however, a marked repression was observed after PGS
was administered in a concentration-dependent manner (Fig. 1B).
Effect of PGS on iNOS and COX-2
expression in LPS-stimulated BV2 microglial cells
We performed RT-PCR and western blot analysis to
determine whether the inhibition of NO and PGE2
production by PGS in the LPS-stimulated BV2 cells is associated
with the decreased levels of iNOS and COX-2, which produce NO and
PGE2 as key mediators of inflammation, respectively. As
demonstrated in Fig. 2C, the
levels of iNOS and COX-2 proteins were markedly upregulated
following 24 h of exposure to LPS; however, PGS significantly
inhibited iNOS and COX-2 protein expression in the LPS-stimulated
BV2 microglial cells in a concentration-dependent manner.
Subsequently, in order to investigate whether PGS suppresses the
LPS-mediated induction of iNOS and COX-2 via a pre-translational
mechanism, the effects of PGS on iNOS and COX-2 mRNA expression
were evaluated. RT-PCR analyses indicated that the reduced iNOS and
COX-2 mRNA levels correlated with the corresponding reduction in
protein levels (Fig. 2D). These
results suggest that PGS-induced reductions in the expression of
iNOS and COX-2 inhibited NO and PGE2 production.
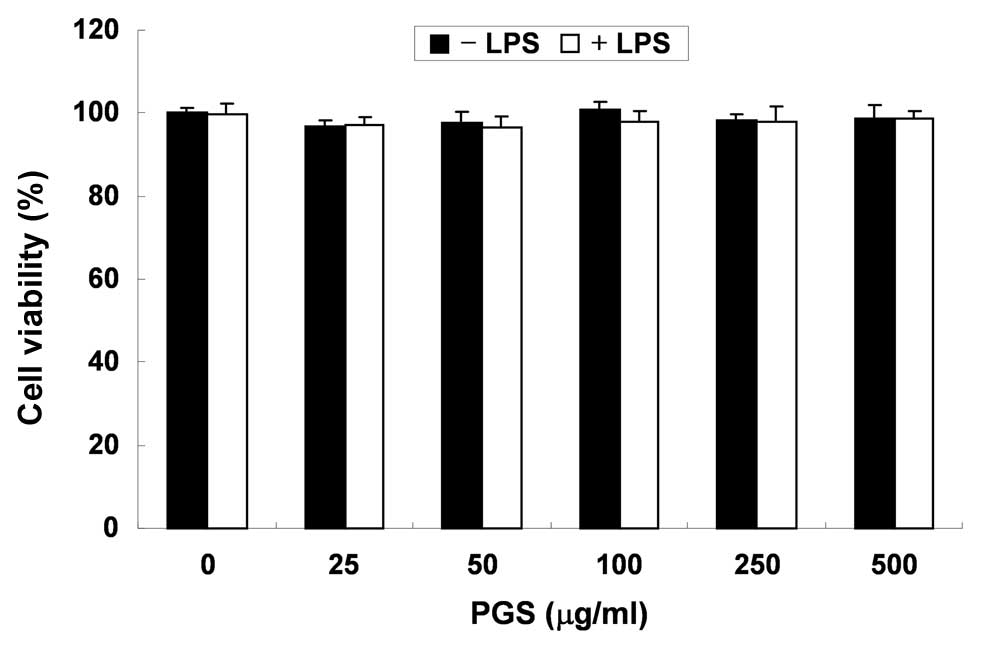
In order to exclude the possibility that the
inhibition of NO and PGE2 production was due to
cytotoxicity caused by PGS treatment, MTT assays were performed in
BV2 microglial cells treated with PGS for 24 h in the presence or
absence of LPS (500 ng/ml). As demonstrated in Fig. 2, at the concentrations (100–500
μg/ml) used to inhibit NO and PGE2 production, PGS alone
did not affect cell viability. Furthermore, co-treatment with PGS
and LPS did not demonstrate any cytotoxic effects. These results
clearly indicated that the inhibition of NO and PGE2
production in LPS-stimulated BV2 cells was not due to the cytotoxic
effects of PGS.
Effects of PGS on the LPS-induced
production of pro-inflammatory cytokines in BV2 microglial
cells
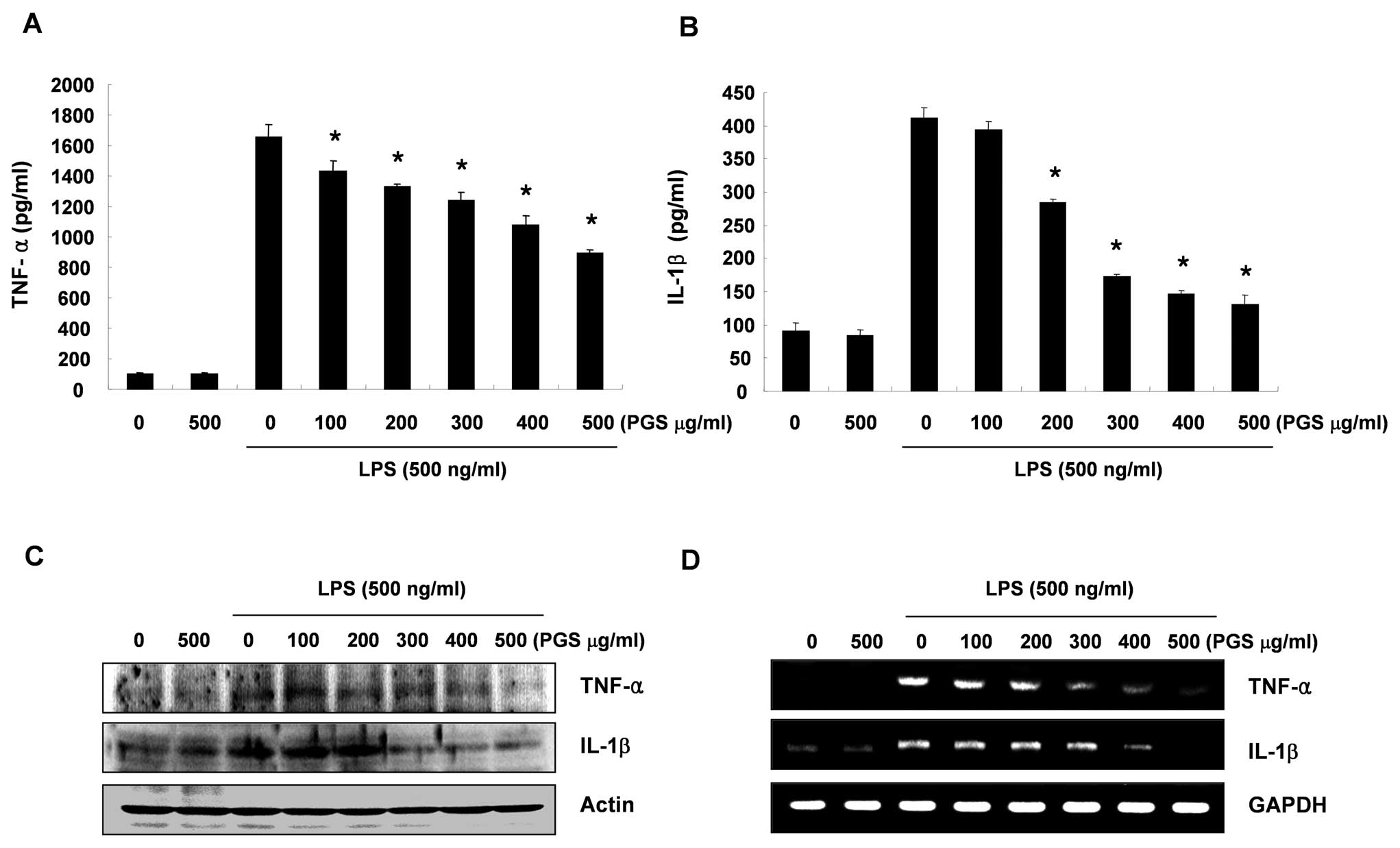
To determine the effects of PGS on LPS-induced
inflammatory-associated cytokine production, BV2 cells were treated
with various concentrations of PGS in the presence or absence of
LPS (500 ng/ml) for 24 h. The production of TNF-α and IL-1β induced
by LPS was evaluated by ELISA. As shown in Fig. 3A, the levels of TNF-α were
markedly increased in the culture medium of LPS-stimulated BV2
microglial cells; however, pre-treatment with PGS resulted in a
significant decrease in the release of TNF-α in a
concentration-dependent manner. In addition, a similar tendency was
also observed as regards IL-1β production (Fig. 3B).
Effects of PGS on the LPS-induced
expression of pro-inflammatory cytokines in BV2 microglial
cells
Since PGS markedly suppressed the production of
TNF-α and IL-1β in the LPS-treated BV2 cells, we investigated
whether the inhibitory effects of PGS on the levels of TNF-α and
IL-1β expression are associated with the inhibitory
effects on the release of TNF-α and IL-1β. As shown in Fig. 3D, the increased IL-1β and TNF-α
mRNA levels as a result of exposure to LPS decreased in a
concentration-dependent manner following treatment with PGS. In a
parallel experiment, using western blot analysis, the elevated
protein levels of IL-1β and TNF-α resulting from LPS treatment were
also decreased following treatment with PGS (Fig. 3C). These results suggest that PGS
is effective in suppressing pro-inflammatory cytokine production by
altering the transcriptional levels of IL-1β and TNF-α in activated
microglial cells.
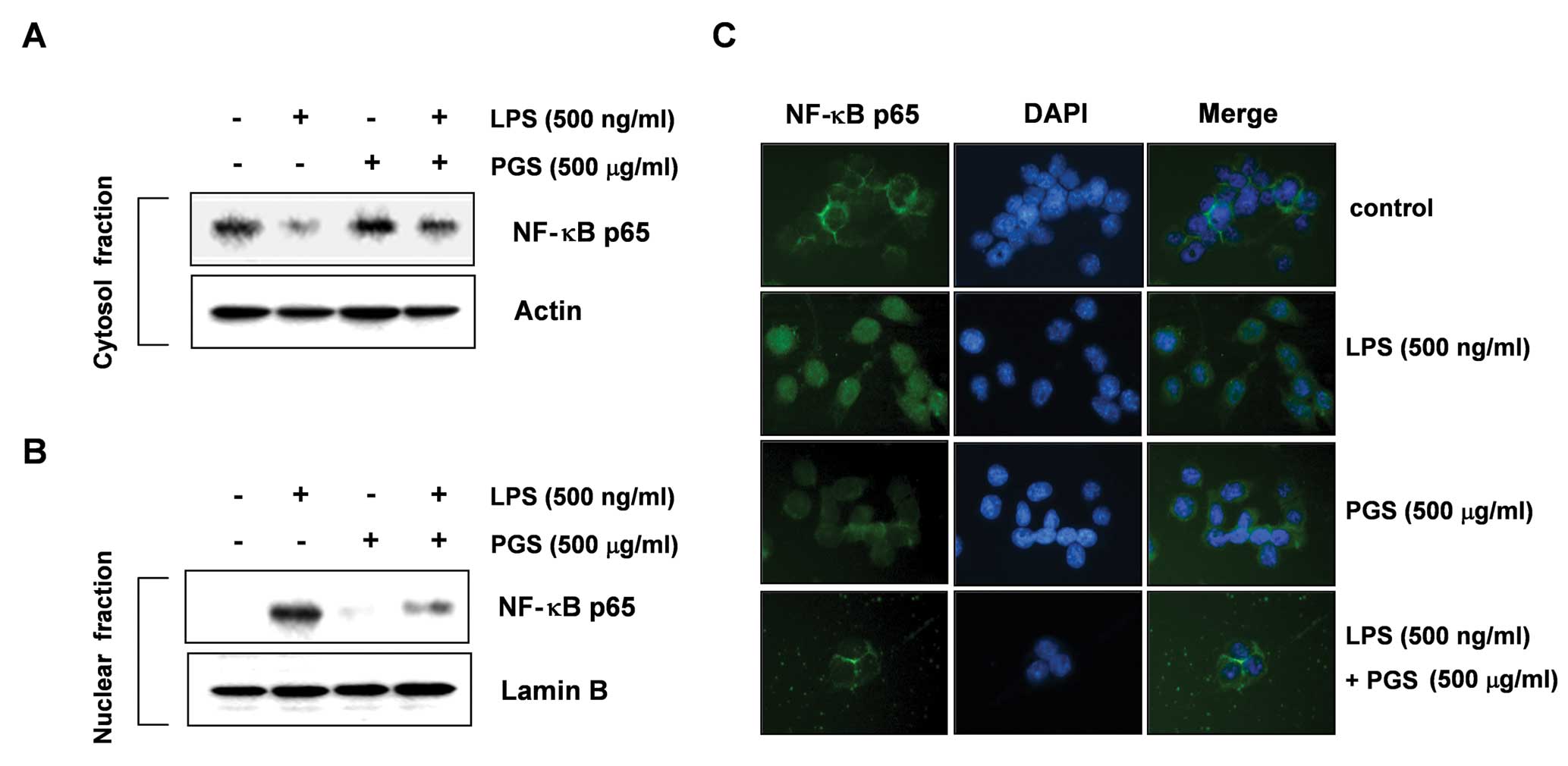
Effect of PGS on LPS-induced NF-κB
translocation in BV2 microglial cells
To further characterize the mechanisms through which
PGS inhibits pro-inflammatory responses, we investigated whether
PGS prevents the translocation of the p65 subunit of NF-κB to the
nucleus. The immunoblot analysis results presented in Fig. 4A and B show that the amount of
NF-κB p65 in the nucleus was markedly increased following exposure
to LPS; however, the LPS-induced p65 level in the nuclear fractions
was reduced as a result of pre-treatment with PGS. Furthermore, we
wished to confirm the inhibition of LPS-induced NF-κB activation by
PGS by immunofluorescence microscopy assay. As shown in Fig. 4C, similar results were observed.
These results suggest that PGS inhibits NF-κB activation in BV2
microglial cells by suppressing IκB degradation and the nuclear
translocation of NF-κB.
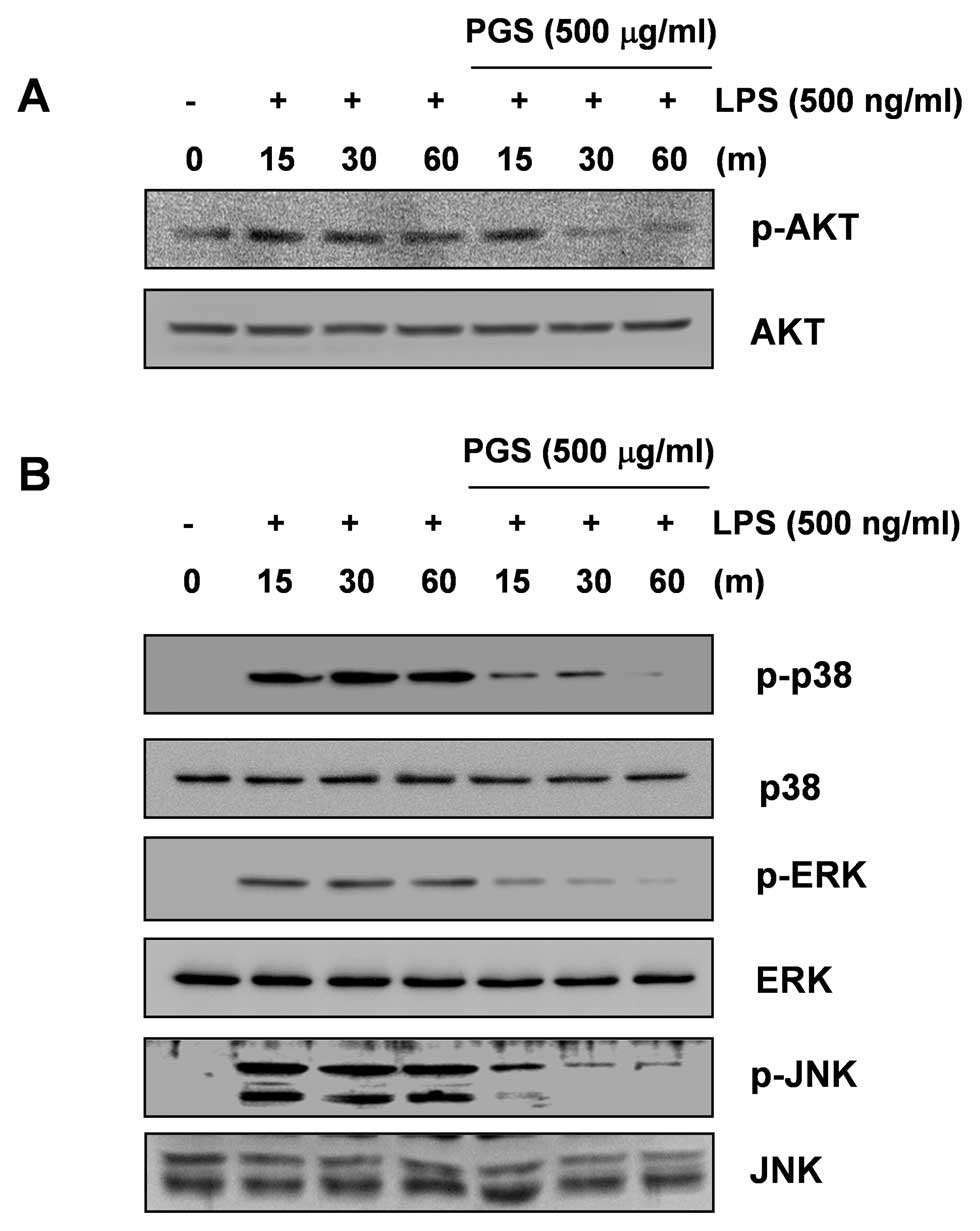
Inhibition of LPS-induced AKT activation
by PGS in LPS-stimulated BV2 microglial cells
Previous studies have indicated that the
phosphoinositide 3-kinase (PI3K)/AKT signaling molecule triggers
NF-κB activation through IκB degradation (24,25). Therefore, we determined the
activation levels of AKT at various time points following the
stimulation of BV2 cells with LPS and the effect of PGS on
LPS-induced AKT activation. As shown in Fig. 5A, although the amount of
non-phosphorylated AKT was unaffected by either PGS or LPS
treatment, the phosphorylation of AKT showed a marked increase
within 15 min following stimulation with LPS. However,
pre-treatment with PGS resulted in significant blockage of
LPS-induced AKT phosphorylation. These results indicate that the
inhibition of pro-inflammatory mediator and cytokine expression by
PGS in LPS-stimulated BV2 microglial cells is associated with the
inactivation of the PI3K/AKT signaling pathway.
Inhibition of LPS-induced MAPK activation
by PGS in LPS-stimulated BV2 microglial cells
To further elucidate the molecular targets of PGS in
further upstream signaling pathways, we examined the effect of PGS
on the activity of MAPKs, such as extracellular signal-regulated
kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 MAPK, which
regulate the induction of several genes encoding inflammatory
factors. As indicated in Fig. 5B,
the stimulation of BV2 cells with LPS induced the rapid activation
of ERK, JNK and p38 MAPK, with the peak levels of each
phosphorylated MAPK occurring 15 to 60 min after the addition of
LPS without altering their unphosphorylated forms. However,
pre-treatment with PGS significantly inhibited MAPK phosphorylation
in LPS-stimulated BV2 microglial cells. These findings suggest that
PGS is capable of disrupting MAPK signal transduction pathways
activated by LPS in BV2 microglial cells, which subsequently
prevents the production of pro-inflammatory mediators and
cytokines.
Discussion
In this study, in order to evaluate the cellular and
molecular mechanisms by which PGS exerts its anti-inflammatory
effects, the inhibitory effects of PGS on the production of
LPS-induced pro-inflammatory mediators and cytokines in BV2
microglial cells were investigated. The present results demonstrate
that PGS significantly exhibits anti-inflammatory activities via
the attenuation of pro-inflammatory factors in LPS-treated BV2
microglial cells. The inhibitory effects were also mediated through
the inhibition of the NF-κB, PI3K/AKT and MAPK signaling pathways.
Therefore, on the basis of the anti-inflammatory effects of PGS,
P. grandiflorus may be a possible therapeutic candidate for
the treatment of neurodegenerative diseases.
COXs are the enzymes that catalyze the conversion of
arachidonic acid to prostaglandin H2 (PGH2)
and PGH2 is the precursor of a variety of biologically
active mediators, such as PGE2, prostacyclin and
thromboxane A2. COXs exist as two major isozymes: COX-1,
a constitutive COX and COX-2, an isoform that is induced during the
response to a number of stimulants and is activated at the site of
inflammation (26–28). Previous studies have indicated
that LPS, a microbe-derived ligand, significantly activates
microglial cells and induces the COX-2 gene, leading to the
synthesis of PGE2 (26,28). A number of studies have also
reported that COX-2 is associated with cytotoxicity in brain
diseases since the inhibition of COX-2 induction and/or activity
reduces brain injury following ischemia and slows the progression
of AD, PD and cerebral ischemia (29,30). In addition, NO has been shown to
be an important regulatory molecule for diverse physiological
functions, including vasodilation, neural communication and host
defense (31,32). In mammalian cells, NO is
synthesized from three different isoforms of NOS: endothelial NOS
(eNOS), neuronal NOS (nNOS) and iNOS. It has been reported that
iNOS is not usually expressed in the brain, but activated
microglial cells are a major cellular source of iNOS in the brain.
The excessive release of NO by activated microglial cells
correlates with the progression of neurodegenerative disorders
(33,34). Collectively, inflammatory
mediators, including PGE2 and NO, are responsible for
much of the neuronal damage (35,36). In the present study, PGS
significantly suppressed LPS-stimulated PGE2 and NO
production in BV2 microglial cells, which appears to be due to the
transcriptional suppression of both COX-2 and iNOS (Fig. 1). This inhibition occurred in a
concentration-dependent manner and PGS did not exhibit any
cytotoxic effects (Fig. 2).
The neuro-inflammatory response in activated
microglial cells produces elevated levels of pro-inflammatory
cytokines, including TNF-α and IL-1β (37,38). These cytokines have been shown to
induce neuronal cell damage; therefore, suppressing their
production is important for the prevention of neurode-generative
diseases (39,40). TNF-α is primarily produced by
activating monocytes, macrophages and T cells, and it exerts
various pro-inflammatory effects. The major producers of TNF-α in
the brain are microglial cells, and they may play a role in several
pathological conditions in the brain (41,42). IL-1β is also a potent
pro-inflammatory cytokine that acts through the IL-1 receptors
found on numerous cell types, including neurons and microglial
cells. Moreover, IL-1β has been shown to be an important mediator
of neuroimmune interactions that participate directly in
neurodegeneration (43,44). Thus, the inhibition of cytokine
production or function serves as a key mechanism in the control of
neurodegeneration. In the current study, treatment with PGS prior
to exposure to LPS significantly attenuated the production of IL-1β
and TNF-α in BV2 microglial cells and this was associated with the
downregulation of their transcriptional activities (Fig. 3). Taken together, our results
indicate that PGS at non-toxic concentrations may be a promising
candidate for the treatment of neurodegenerative diseases caused by
microglial cell activation in the brain.
The excessive production of pro-inflammatory
components in over-activated microglial cells may be a risk factor
for initiating neurodegenerative disease through a number of cell
signaling pathways. Among them, the nuclear transcriptional factor,
NF-κB, is a key regulator of inflammation due to its ability to
induce the transcription of pro-inflammatory genes, which are
modulated by the binding of NF-κB to specific promoter regions
(40,45). NF-κB is usually located in the
cytoplasm where it is complexed with the inhibitory IκB (IκB)
protein. In response to pro-inflammatory stimuli, IκB is
phosphorylated and subsequently degraded, and NF-κB is released and
translocated to the nucleus where it promotes the expression of
inflammation-associated genes that are involved in the production
of pro-inflammatory cytokines and enzymes (40,46). In addition, the involvement of the
PI3K/AKT signaling pathway in inflammatory mediator expression in
microglial cells through NF-κB activation has been demonstrated
(25,47,48). Furthermore, it is well known that
the blockade of NF-κB transcriptional activity and the PI3K/AKT
signaling pathway in microglial cells can also suppress the
expression of iNOS, COX-2 and pro-inflammatory cytokines, such as
IL-1β, IL-6 and TNF-α (38,46,49). Therefore, modulating the NF-κB and
PI3K/AKT signaling pathways is considered a promising strategy for
the treatment of several neuropathological disorders. Our results
indicated that PGS inhibited the LPS-induced nuclear translocation
of the p65 subunit of NF-κB in BV2 microglial cells (Fig. 4), suggesting that PGS inhibits the
expression of pro-inflammatory genes by suppressing LPS-induced
NF-κB activity. Furthermore, PGS markedly attenuated AKT activation
in LPS-stimulated BV2 microglial cells (Fig. 5A), indicating that PGS inhibits
LPS-induced NF-κB activation by inactivating the PI3K/AKT signaling
pathway. Therefore, the inhibition of the NF-κB and PI3K/AKT
signaling pathways in microglial cells as a result of PGS may
result in the downregulation of pro-inflammatory mediators and
cytokines, resulting in an anti-inflammatory effect.
MAPKs, including ERK, JNK and p38 MAPK, mediate
important signaling responses in the immune and inflammatory
systems, as well as in the regulation of cellular activities,
including mitosis, cell proliferation and survival and gene
expression. In general, the JNK and p38 MAPK pathways are activated
by pro-inflammatory cytokines, such as IL-1β and TNF-α, microbial
endotoxins (e.g., LPS), or cellular stress; however, the ERK
pathway is activated by mitogenic stimuli (50,51). The activation of the p38 MAPK
pathway is crucial for a number of immune and inflammatory
response-related functions in macrophages (51,52). Furthermore, the expression of
TNF-α and IL-1β are strongly regulated by p38 MAPK (53,54). Therefore, further experiments were
performed to determine whether PGS regulates MAPK activation to
induce anti-inflammatory effects in LPS-stimulated BV2 cells. Our
results revealed that LPS induced the phosphorylation of all three
classes of MAPKs in BV2 microglial cells, and that PGS markedly
inhibited their phosphorylation (Fig.
5B). However, the amounts of non-phosphorylated ERK, JNK and
p38 MAPK were unaffected by treatment with PGS and LPS, indicating
that PGS diminished the LPS-mediated pro-inflammatory response via
the inhibition of MAPK activation in BV2 microglial cells.
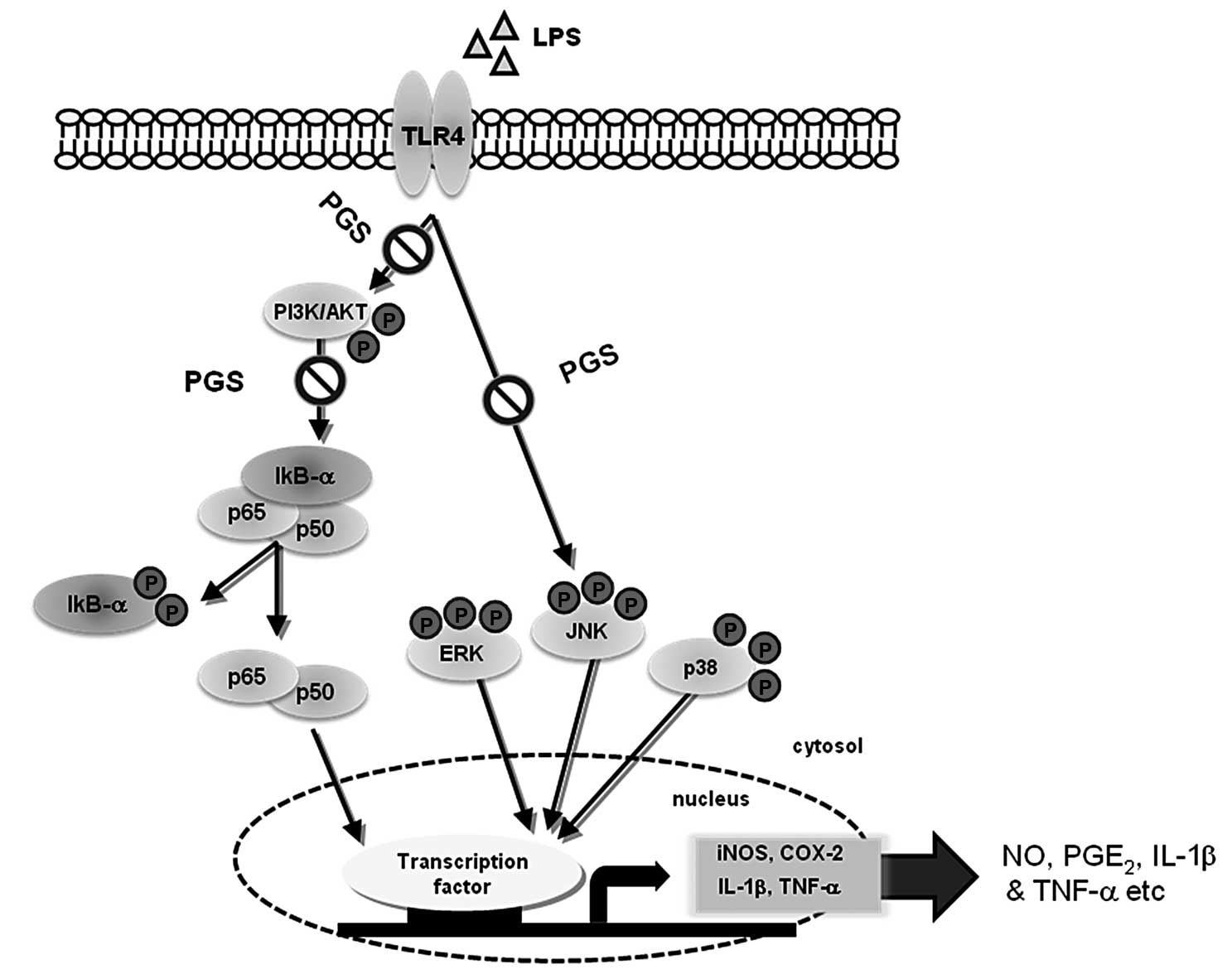
In conclusion, we found that PGS significantly
attenuated the release of neurotoxic pro-inflammatory mediators,
including NO and PGE2, which are regulated by iNOS and
COX-2 and pro-inflammatory cytokines, TNF-α and IL-1β, in
LPS-stimulated microglial cells (Fig.
6). The inhibitory effects of PGS were accompanied by the
attenuation of NF-κB activity through the prevention of NF-κB
translocation from the cytoplasm to the nucleus, which was
associated with the inactivation of the PI3K/AKT signaling pathway.
In addition, the levels of phosphorylated MAPKs were significantly
downregulated by pre-treatment with PGS in LPS-stimulated
microglial cells. These results indicate that PGS exerts its
anti-inflammatory effects by inhibiting NF-κB activation and the
PI3K/AKT and MAPK signaling pathways. The data presented in this
study suggest that PGS may ultimately prove useful in the treatment
of inflammatory diseases and several neurodegenerative diseases
that are associated with microglial cell activation.
Acknowledgements
This study was supported by the
R&D program of MKE/KEIT (10040391, Development of Functional
Food Materials and Device for Prevention of Aging-Associated Muscle
Function Decrease) and the Blue-Bio Industry Regional Innovation
Center (RIC08-06-07) at Dongeui University as an RIC program under
the Ministry of Knowledge Economy, Busan, Republic of Korea.
References
|
1
|
Knowles RG and Moncada S: Nitric oxide
synthases in mammals. Biochem J. 298:249–258. 1994.PubMed/NCBI
|
|
2
|
O'Banion MK: Cyclooxygenase-2: molecular
biology, pharmacology, and neurobiology. Crit Rev Neurobiol.
13:45–82. 1999.PubMed/NCBI
|
|
3
|
Abrams J: Beneficial actions of nitrites
in cardiovascular disease. Am J Cardiol. 77:31C–37C. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hou YC, Janczuk A and Wang PG: Current
trends in the development of nitric oxide donors. Curr Pharm Des.
5:417–441. 1999.PubMed/NCBI
|
|
5
|
Rock RB and Peterson PK: Microglia as a
pharmacological target in infectious and inflammatory diseases of
the brain. J Neuroimmune Pharmacol. 1:117–126. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kaur C and Ling EA: Antioxidants and
neuroprotection in the adult and developing central nervous system.
Curr Med Chem. 15:3068–3080. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Deng YY, Lu J, Ling EA and Kaur C: Role of
microglia in the process of inflammation in the hypoxic developing
brain. Front Biosci (Schol Ed). 3:884–900. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perry VH and Gordon S: Macrophages and
microglia in the nervous system. Trends Neurosci. 92:273–274. 1988.
View Article : Google Scholar
|
|
9
|
Napoli I and Neumann H: Microglial
clearance function in health and disease. Neuroscience.
158:1030–1038. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eikelenboom P and van Gool WA:
Neuroinflammatory perspectives on the two faces of Alzheimer's
disease. J Neural Transm. 111:281–294. 2004.PubMed/NCBI
|
|
11
|
Loane DJ and Byrnes KR: Role of microglia
in neurotrauma. Neurotherapeutics. 7:366–377. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu B and Hong JS: Role of microglia in
inflammation-mediated neurodegenerative diseases: mechanisms and
strategies for therapeutic intervention. J Pharmacol Exp Ther.
304:1–7. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Germida JJ, Siciliano SD, Freitas JR and
Seib AM: Diversity of root-associated bacteria associated with
field-grown canola (Brassica napus L.) and wheat
(Triticum aestivum L.). FEMS Microbiol Ecol. 26:43–50. 1998.
View Article : Google Scholar
|
|
14
|
Lee JY, Hwang WI and Lim ST: Antioxidant
and anticancer activities of organic extracts from Platycodon
grandiflorum A. De Candolle roots. J Ethnopharmacol.
93:409–415. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ishii H, Tori K, Tozyo T and Yoshimura Y:
Structures of polygalacin-D and -D2, platycodin-D and -D2, and
their monoacetates, saponins isolated from Platycodon
grandiflorum A. DC., determined by carbon-13 nuclear magnetic
resonance spectroscopy. Chem Pharm Bull. 26:674–677. 1978.
View Article : Google Scholar
|
|
16
|
Fu WW, Shimizu N, Dou DQ, Takeda T, Fu R,
Pei YH and Chen YJ: Five new triterpenoid saponins from the roots
of Platycodon grandiflorum. Chem Pharm Bull (Tokyo).
54:557–560. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim YP, Lee EB, Kim SY, Li D, Ban HS, Lim
SS, Shin KH and Ohuchi K: Inhibition of prostaglandin E2 production
by platycodin D isolated from the root of Platycodon
grandiflorum. Planta Med. 67:362–364. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang C, Schuller Levis GB, Lee EB, Levis
WR, Lee DW, Kim BS, Park SY and Park E: Platycodin D and D3
isolated from the root of Platycodon grandiflorum modulate
the production of nitric oxide and secretion of TNF-alpha in
activated RAW 264.7 cells. Int Immunopharmacol. 4:1039–1049.
2004.PubMed/NCBI
|
|
19
|
Yoon YD, Kang JS, Han SB, Park SK, Lee HS,
Kang JS and Kim HM: Activation of mitogen-activated protein kinases
and AP-1 by polysaccharide isolated from the radix of Platycodon
grandiflorum in RAW 264.7 cells. Int Immunopharmacol.
4:1477–1487. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ahn KS, Noh EJ, Zhao HL, Jung SH, Kang SS
and Kim YS: Inhibition of inducible nitric oxide synthase and
cyclooxygenase II by Platycodon grandiflorum saponins via
suppression of nuclear factor-kappaB activation in RAW 264.7 cells.
Life Sci. 76:2315–2328. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chung JW, Noh EJ, Zhao HL, Sim JS, Ha YW,
Shin EM, Lee EB, Cheong CS and Kim YS: Anti-inflammatory activity
of prosapogenin methyl ester of platycodin D via nuclear
factor-kappaB pathway inhibition. Biol Pharm Bull. 31:2114–2120.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jang MH, Kim CJ, Kim EH, Kim MG, Leem KH
and Kim J: Effects of Platycodon grandiflorum on
lipopolysaccharide-stimulated production of prostaglandin E2,
nitric oxide, and interleukin-8 in mouse microglial BV2 cells. J
Med Food. 9:169–174. 2006.
|
|
23
|
Bae DS, Kim YH, Pan CH, Nho CW, Samdan J,
Yansan J and Lee JK: Protopine reduces the inflammatory activity of
lipopolysaccharide-stimulated murine macrophages. BMB Rep.
45:108–113. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Madrid LV, Wang CY, Guttridge DC,
Schottelius AJ, Baldwin AS Jr and Mayo MW: Akt suppresses apoptosis
by stimulating the transactivation potential of the RelA/p65
subunit of NF-kappaB. Mol Cell Biol. 20:1626–1638. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wei J and Feng J: Signaling pathways
associated with inflammatory bowel disease. Recent Pat Inflamm
Allergy Drug Discov. 4:105–117. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fiebich BL, Lieb K, Kammerer N and Hüll M:
Synergistic inhibitory effect of ascorbic acid and acetylsalicylic
acid on prostaglandin E2 release in primary rat microglia. J
Neurochem. 86:173–178. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liang X, Wu L, Wang Q, Hand T, Bilak M,
McCullough L and Andreasson K: Function of COX-2 and prostaglandins
in neurological disease. J Mol Neurosci. 33:94–99. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Scher JU and Pillinger MH: The
anti-inflammatory effects of prostaglandins. J Investig Med.
57:703–708. 2009.PubMed/NCBI
|
|
29
|
Levi G, Minghetti L and Aloisi F:
Regulation of prostanoid synthesis in microglial cells and effects
of prostaglandin E2 on microglial functions. Biochimie. 80:899–904.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Giovannini MG, Scali C, Prosperi C,
Bellucci A, Pepeu G and Casamenti G: Experimental brain
inflammation and neurode-generation as model of Alzheimer's
disease: protective effects of selective COX-2 inhibitors. Int J
Immunopathol Pharmacol. 16:31–40. 2003.
|
|
31
|
MacMicking J, Xie QW and Nathan C: Nitric
oxide and macrophage function. Ann Rev Immunol. 15:323–330. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mitchell JA, Larkin S and Williams TJ:
Cyclooxygenase-2: regulation and relevance in inflammation. Biochem
Pharmacol. 50:1535–1542. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mulrennan SA and Redington AE: Nitric
oxide synthase inhibition: therapeutic potential in asthma. Treat
Respir Med. 3:79–88. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cau SB, Carneiro FS and Tostes RC:
Differential modulation of nitric oxide synthases in aging:
therapeutic opportunities. Front Physiol. 3:2182012.PubMed/NCBI
|
|
35
|
Iadecola C and Ross ME: Molecular
pathology of cerebral ischemia: delayed gene expression and
strategies for neuroprotection. Ann NY Acad Sci. 835:203–217. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
del Zoppo G, Ginis I, Hallenbeck JM,
Iadecola C, Wang X and Feuerstein GZ: Inflammation and stroke:
putative role for cytokines, adhesion molecules and iNOS in brain
response to ischemia. Brain Pathol. 10:95–112. 2000.PubMed/NCBI
|
|
37
|
Boka G, Anglade P, Wallach D, Javoy-Agid
F, Agid Y and Hirsch EC: Immunocytochemical analysis of tumor
necrosis factor and its receptors in Parkinson's disease. Neurosci
Lett. 172:151–154. 1994.PubMed/NCBI
|
|
38
|
Hunot S, Dugas N, Faucheux B, Hartmann A,
Tardieu M, Debré P, Agid Y, Dugas B and Hirsch EC:
FcepsilonRII/CD23 is expressed in Parkinson's disease and induces,
in vitro, production of nitric oxide and tumor necrosis
factor-alpha in glial cells. J Neurosci. 19:3440–3447.
1999.PubMed/NCBI
|
|
39
|
De Nardin E: The role of inflammatory and
immunological mediators in periodontitis and cardiovascular
disease. Ann Periodontol. 6:30–40. 2001.PubMed/NCBI
|
|
40
|
Viscido A, Aratari A, Maccioni F, Signore
A and Caprilli R: Inflammatory bowel diseases: clinical update of
practical guidelines. Nucl Med Commun. 26:649–655. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sawada M, Kondo N, Suzumura A and
Marunouchi T: Production of tumor necrosis factor-alpha by
microglia and astrocytes in culture. Brain Res. 491:394–397. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Owens T: Identification of new therapeutic
targets for prevention of CNS inflammation. Expert Opin Ther
Targets. 6:203–215. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rothwell N, Allan S and Toulmond S: The
role of interleukin 1 in acute neurodegeneration and stroke:
pathophysiological and therapeutic implications. J Clin Invest.
100:2648–2652. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Suzumura A, Takeuchi H, Zhang G, Kuno R
and Mizuno T: Roles of glia-derived cytokines on neuronal
degeneration and regeneration. Ann NY Acad Sci. 1088:219–229. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lee JY, Jhun BS, Oh YT, Lee JH, Choe W,
Baik HH, Ha J, Yoon KS, Kim SS and Kang I: Activation of adenosine
A3 receptor suppresses lipopolysaccharide-induced TNF-alpha
production through inhibition of PI3-kinase/Akt and NF-kappaB
activation in murine BV2 microglial cells. Neurosci Lett. 396:1–6.
2006. View Article : Google Scholar
|
|
46
|
Mankan AK, Lawless MW, Gray SG, Kelleher D
and McManus R: NF-kappaB regulation: the nuclear response. J Cell
Mol Med. 13:631–643. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lee JW, Lee MS, Kim TH, Lee HJ, Hong SS,
Noh YH, Hwang BY, Ro JS and Hong JT: Inhibitory effect of
inflexinol on nitric oxide generation and iNOS expression via
inhibition of NF-kappaB activation. Mediators Inflamm.
2007:931482007.PubMed/NCBI
|
|
48
|
Lee YH, Jeon SH, Kim SH, Kim C, Lee SJ,
Koh D, Lim Y, Ha K and Shin SY: A new synthetic chalcone
derivative, 2-hydroxy-3′,5,5′-trimethoxychalcone (DK-139),
suppresses the Toll-like receptor 4-mediated inflammatory response
through inhibition of the Akt/NF-κB pathway in BV2 microglial
cells. Exp Mol Med. 44:369–377. 2012.PubMed/NCBI
|
|
49
|
Moon DO, Choi YH, Kim ND, Park YM and Kim
GY: Anti-inflammatory effects of beta-lapachone in
lipopolysaccharide-stimulated BV2 microglia. Int Immunopharmacol.
7:506–514. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Garrington TP and Johnson GL: Organization
and regulation of mitogen-activated protein kinase signaling
pathways. Curr Opin Cell Biol. 11:211–218. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ashwell JD: The many paths to p38
mitogen-activated protein kinase activation in the immune system.
Nat Rev Immunol. 6:532–540. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cuenda A, Rouse J, Doza YN, Meier R, Cohen
P, Gallagher TF, Young PR and Lee JC: SB 203580 is a specific
inhibitor of a MAP kinase homologue which is stimulated by cellular
stresses and interleukin-1. FEBS Lett. 364:229–233. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Barone FC, Irving EA, Ray AM, Lee JC,
Kassis S, Kumar S, Badger AM, Legos JJ, Erhardt JA, Ohlstein EH,
Hunter AJ, Harrison DC, Philpott K, Smith BR, Adams JL and Parsons
AA: Inhibition of p38 mitogen-activated protein kinase provides
neuroprotection in cerebral focal ischemia. Med Res Rev.
21:129–145. 2001. View Article : Google Scholar : PubMed/NCBI
|