Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic,
progressive, lethal disease of unknown etiology and pathogenesis.
It continues to be associated with considerable morbidity and
mortality (1). The recent
paradigm suggests that IPF is driven by chronic epithelial
micro-injury and a subsequent deregulated repair process, leading
to abnormal mesenchymal cell activation and proliferation and
excess extracellular matrix (ECM) accumulation (1,2). A
determinant role in ECM deposition is played by myofibroblasts
(3), which are α-smooth muscle
actin (α-SMA)-expressing fibroblasts and the main source of type
I/III collagen, fibronectin and fibrogenic cytokines in fibrotic
foci. However, the origin of fibroblasts in these fibrotic foci has
not yet been fully elucidated. It was once believed that the
migration, proliferation and activation of resident mesenchymal
cells are the main sources of fibroblasts; however, emerging
evidence has indicated that myofibroblasts may also be derived
through the process of epithelial-mesenchymal transition (EMT)
(4–7). Through this process, epithelial
cells lose certain characteristics, such as apical-basolateral
polarization, specialized cell-cell junctional structures and
epithelial markers, and undergo cytoskeletal reorganization,
ultimately acquiring the morphological and functional features of
mesenchymal-like cells (8–10).
The abnormal activation of EMT programs has been associated with an
abnormal wound healing process and tissue fibrosis, cancer invasion
and metastasis (8–10).
EMT is an orchestra which is delicately modulated by
several signaling molecules, including transforming growth factor-β
(TGF-β), epidermal growth factor (EGF), hepatocyte growth factor
(HGF), fiborblast growth factor (FGF) and integrin-linked kinase
(ILK) (7). Among these, TGF-β1 is
a multifunctional cytokine that regulates cell proliferation,
migration, or differentiation and is the master switch of EMT
(11). TGF-β1 is directly or
indirectly recognized by 3 heterogenic cell surface receptors
(types I, II and III) (12). Both
the type I TGF-β1 receptor (TβRI) and type II receptor (TβRII) are
transmembrane serine/threonine kinases. Ligand binding to TβRII and
TβRI induces the recruitment and phosphorylation of receptor-Smads
(R-Smads, Smad2 and Smad3). Activated Smad2/3 heterodimerize with
the Co-Smad (Smad4) to form a transcriptionally active complex
which translocates to the nucleus and modulates the expression of
TGF-β target genes (12,13).
EMT has been extensively investigated in
vitro and data have shown that many different types of cells
undergo EMT-like changes in response to TGF-β1 stimulation
(14,15). The transition process of lung
fibroblasts in vitro is mainly characterized by the
downregulation or complete loss of epithelial markers, such as
E-cadherin, cytokeratin, aquaporin 5 and prosurfactant protein
(pro-SP) B, and the acquisition of new mesenchymal proteins,
including vimentin, α-SMA, fibroblast specific protein 1 (FSP1),
type I collagen and fibronectin (14,15). However, EMT-associated tissue
fibrosis in vivo is a more complex and controversial
phenomenon (16) and studies on
the reverse process of EMT, mesenchymal-epithelial transition
(MET), are limited. In this study, we hypothesized that the EMT
program is activated in fibrotic lung tissue. Part of the injured
alveolar epithelial cells undergo complete EMT-like changes and
phenotypically resemble fibroblasts. Primarily cultured fibroblasts
from fibrotic lung tissue are partly from epithelial cells. Kinase
inhibitors targeting the EMT process may induce a possible MET
process in these epithelial cell-derived fibroblasts. In detail, we
examined the expression level of the TGF-β1 signaling network and
found that TGF-β1, TβRI/II/III, Smad2/3/4 and Snail1/2 were
significantly upregulated in the fibrotic lung tissue. We then
examined the possibility of MET in cultured fibroblasts from
fibrotic lung tissue using specific inhibitors of TβRI, Rho kinase
(ROCK), p38 mitogen-activated protein kinase (p38 MAPK) and c-Jun
NH-terminal kinase (JNK), and found that blocking TGF-β1 and other
kinase signals failed to induce the MET process.
Materials and methods
Subjects and lung tissue procurement
Lung tissue was obtained from 5 IPF patients (5
males; mean age, 58.6±4.7 years) with histological evidence of
usual interstitial pneumonia at the time of surgical lung biopsy.
The diagnosis of IPF was derived according to the standards
accepted by the American Thoracic Society/European Respiratory
Society (17). For the controls,
histologically normal lung tissue was obtained from 3 patients (3
males; mean age, 20.8±2.8 years) with primary spontaneous
pneumothorax at the time of thoracoscopy with stapling of any air
leaks.
This study was approved by the Ethics Committee of
Beijing Chaoyang Hospital of Capital Medical University, Beijing,
China and written informed consent was obtained according to
institutional guidelines from all investigated subjects.
Cells and reagents
The explant culture method was used for the culture
of primary fibroblasts. In brief, lung specimens were minced with
scissors into pieces and washed with phosphate-buffered saline
(PBS). Subsequently, 5–10 pieces were transferred into culture
flasks (Corning Inc., Corning, NY, USA) with high glucose
Dulbecco's modified Eagle's medium (DMEM), containing 10% fetal
bovine serum (FBS), penicillin 100 U/ml and streptomycin 100 mg/ml.
Tissues were then incubated in a humidified 5% CO2
incubator at 37°C. The medium were changed every 5 to 6 days.
Approximately 3–4 weeks later, a monolayer of fibroblast-like cells
fully covered the bottom of the flask. Th explant tissue was then
removed and the cells were trypsinized and resuspended in
supplemented DMEM for subculture. All reagents were purchased from
HyClone (Logan, UT, USA). Cells at passages 4–8 (3 cell lines) were
used for all the experiments. The identification and purity of the
cultured primary lung fibroblasts were confirmed by morphological
observation as well as immunostaining with vimentin, fibronectin
and collagen I/III. The chemical inhibitors, SB203580, SP600125,
Y27632 and SB431542 (Biotrend, Cologne, Germany) were aliquoted
after reconstitution and frozen at −20°C.
Antibodies
The following antibodies were used in
immunofluorescence and western blot analyses: mesenchymal cell
markers included rabbit monoclonal anti-α-SMA, rabbit monoclonal
anti-vimentin, mouse polyclonal anti-collagen III, rabbit
polyclonal anti-collagen I, rabbit polyclonal anti-fibronectin
antibodies; epithelial cell marker, rabbit polyclonal
anti-E-cadherin antibodies; TGF-β1 system antibodies included mouse
monoclonal anti-TGF-β1 (active form), rabbit monoclonal
anti-TβRI/II, mouse monoclonal anti-TβRIII, rabbit monoclonal
anti-(phospho)Smad2/3, rabbit monoclonal anti-Snail and rabbit
monoclonal anti-phospho AKT antibodies. All of the antibodies were
obtained from Abcam (Cambridge, MA, USA) and were used according to
the manufacturer recommendations.
RNA purification and real-time
RT-PCR
Total RNA was isolated from the fibrotic and normal
lung tissue using the RNeasy MiniPrep kit (Tiangen Biotech,
Beijing, China) and 2 μg of RNA from each sample was
reverse-transcribed using the Omniscript RT kit (Tiangen Biotech)
using oligo(dT) primers (1 μM) at 37°C for 1 h. Real-time RT-PCR
was performed on an ABI PRISM 7500 instrument (Applied Biosystems,
Foster, CA, USA) using SYBR-Green PCR reagents (Tiangen Biotech).
Reaction mixtures were incubated for 2 min at 94°C followed by 40
cycles of 15 sec at 94°C, 20 sec at 55°C and 35 sec at 68°C. For
each sample, gene expression was corrected against the β-actin mRNA
level and the comparative threshold cycle number (Ct) method was
used to assess the relative quantification of gene expression. The
fold-change of the target genes were calculated using the
2−ΔΔCT method. The primers used for real-time PCR are
shown in Table I.
 | Table IPrimers used for real-time RT-PCR. |
Table I
Primers used for real-time RT-PCR.
| Gene | Forward
sequences | Reverse
sequences |
|---|
| Smad4 |
5′-TGATGTCTGAGAAGATGTCC-3′ |
5′-GCATCGACAGAGACATACAG-3′ |
| TGF-β1 |
5′-GAGCCTGAGGCCGACTACTA-3′ |
5′-CGGAGCTCTGATGTGTTGAA-3′ |
| TβRII |
5′-CGGAGCTCTGATGTGTTGAA-3′ |
5′-AGCAACTGCAGCATCACCTC-3′ |
| Snail1 |
5′-CAACAGTAACAATAGGGCAG-3′ |
5′-GAGCTGCAGGACTCTAATCCA-3′ |
| Snail2 |
5′-CGGTGGGGTTGAGGATCT-3′ |
5′-TGGTTGCTTCAAGGACACAT-3′ |
| Smad2 |
5′-GTTGCAGTGAGGGCAAGAA-3′ |
5′-ATCCTAACAGAACTTCCGCC-3′ |
| β-actin |
5′-TGCTATCCAGGCTGTGCTAT-3′ |
5′-AGTCCATCACGATGCCAGT-3′ |
Protein extraction, SDS-PAGE and indirect
immunoblot analysis
Cells were harvested and lysed in
radioimmunoprecipitation assay (RIPA; Pierce, Rockford, IL, USA)
buffer supplemented with complete proteinase and phosphatase
inhibitor cocktails (Roche, Basel, Switzerland). Protein extracts
were clarified by centrifugation (12,000 rpm, 15 min) at 4°C and
the concentrations were determined using the bicinchoninic acid
assay kit (Pierce). Equal amounts (30 μg) of protein extracts were
then separated by 8–12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred onto polyvinylidene
difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The
membranes were then blocked with 5% non-fat dry milk or bovine
serum albumin (BSA) in TBS (10 mM Tris-HCl, pH 7.6; 150 mM NaCl;
0.1% Tween-20) and incubated with the indicated primary antibodies
for overnight at 4°C according to the manufacturer's instructions.
After washing, membranes were incubated for 1 h with appropriate
secondary, HRP-labeled antibodies (Proteintech, Chicago, IL, USA).
Finally, an enhanced chemiluminescence detection (ECL) buffer (KPL,
Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD, USA)
with ChemiDoc XRS (Bio-Rad, Hercules, CA, USA) was used for the
visualization of the protein bands. Relative protein levels were
determined by densitometry using Quantity One Software (Bio-Rad) or
ImageJ software and normalized by mouse anti-β-actin monoclonal
antibody (mAb) or mouse anti-GAPDH mAb (both from Abcam, Cambridge,
CA, USA) when the detected protein has a similar molecule weight
with β-actin.
Indirect immunofluorescence staining
Cells were seeded on glass coverslips and cultured
as described above. The slides were then rinsed once with ice-cold
PBS and fixed with 4% paraformaldehyde for 20 min at room
temperature. The cells were then permeabilized in PBS containing
0.1% Triton X-100. After rinsing in PBS and blocking with 5% BSA at
room temperature for 1 h, monolayers were incubated with the
appropriate primary antibody overnight at 4°C. After extensive
washing and blocking with 5% BSA at room temperature for 30 min,
the slides were incubated with appropriate secondary antibodies
conjugated to fluorescein isothiocyanate (FITC) or Texas Red (KPL)
at room temperature in a humidified chamber in the dark. The
coverslips were then overturned on a microscope slide containing
one drop of anti-fade solution with DAPI. Images were then taken at
room temperature using an Olympus inverted microscope (Olympus,
Tokyo, Japan).
Statistical analysis
Unless otherwise indicated, all experiments were
performed on 3 separate occasions, each time with triplicates. For
statistical evaluation, the results are presented as the means ±
SD. For comparisons between groups, we used the Mann-Whitney or
Kruskal-Wallis test with SPSS software. A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
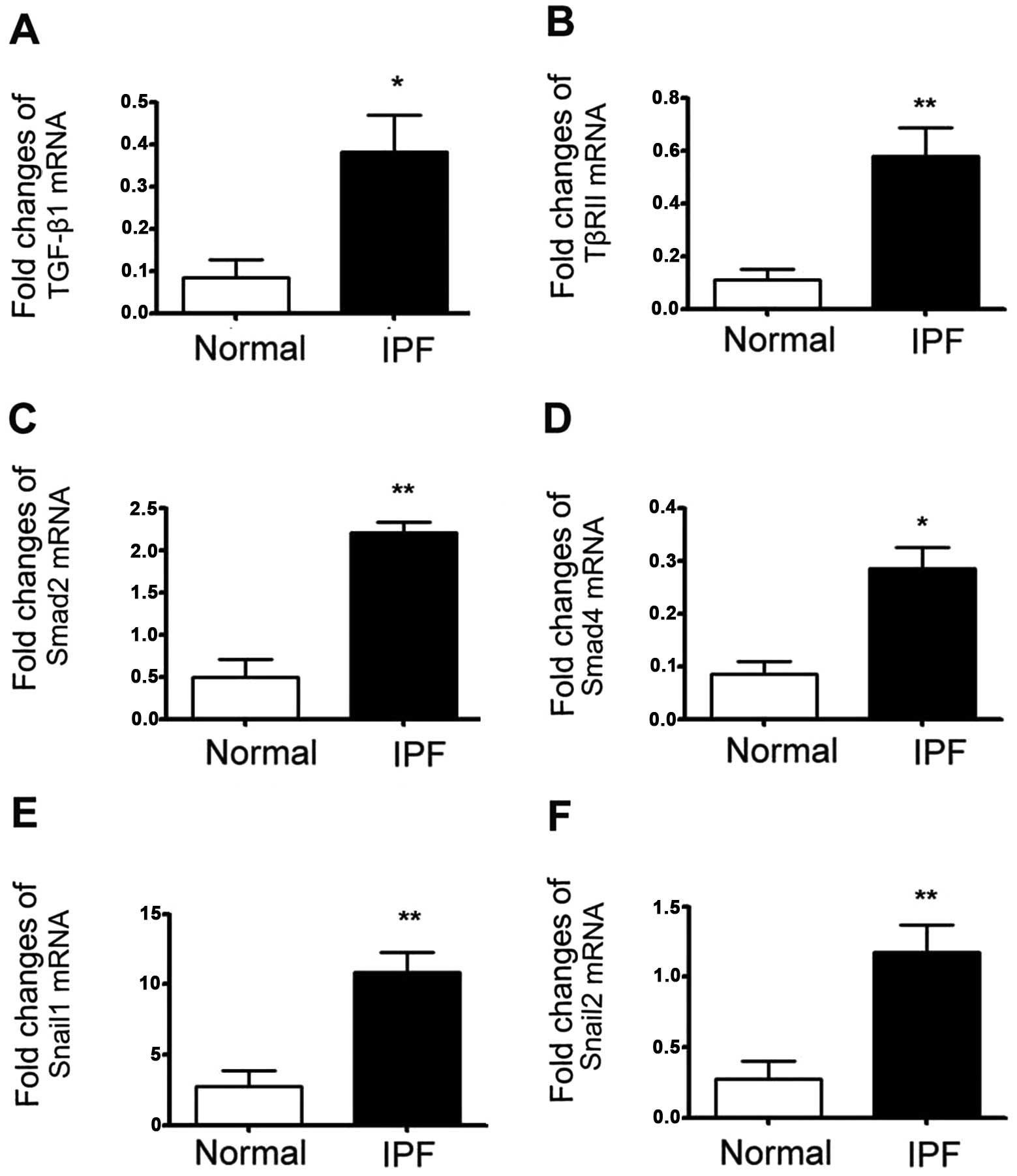
TGF-β1-dependent EMT-related mRNA
expression is increased in fibrotic lung tissues of patients with
IPF
We wished to investigate whether TGF-β1-dependent
EMT-related mRNA expression was upregulated in lungs from patients
with IPF. IPF and control lung tissues were lysed in RL buffer and
whole RNA was extracted according to the manufacturer's
instructions. Real-time RT-PCR was then performed to determine the
mRNA expression levels of TGF-β1, TβRII, Smad2, Smad4, Snail1 and
Snail2. We found that compared to the normal lung tissue, the mRNA
expression of TGF-β1, TβRII, Smad2, Smad4, Snail1 and Snail2 was
significantly increased in the fibrotic human lungs. These results
indicated that the mRNA levels are overexpressed during EMT in
fibrotic tissue (Fig. 1).
TGF-β1-dependent EMT- related protein
expression is upregulated in fibrotic lung tissues of patients with
IPF
We also performed western blot analysis for the
detection of the levels of proteins involved in the
TGF-β1-dependent EMT program, such as TGF-β1, TβRI, TβRII, TβRIII,
Smad2/3, p-Smad2, p-Smad3, Snail, p-AKT and α-SMA. Tissue lysates
were obtained by RIPA buffer, and primary antibodies were probed to
detect the expression of EMT-related proteins. Fig. 2A shows the representative results
of western blot analysis. Compared with the normal control tissue,
in the lung tissue obtained from patients with IPF, the levels of
TGF-β1-dependent EMT-related proteins, such as TGF-β1, TβRI, TβRII,
TβRIII, Smad2/3, p-Smad2, p-Smad3, Snail, p-AKT and α-SMA were
significantly increased (Fig.
2B).
 | Figure 2Representative results of (A) western
blot analysis for epithelial-mesenchymal transition (EMT)-related
proteins. (B) Tissue lysates were prepared and relative TGF-β1,
TβRI, TβRII, TβRIII, Smad2/3, p-Smad2, p-Smad3, Snail, p-AKT and
α-SMA protein levels were determined by western blot analysis and
normalized to β-actin or GAPDH (when the targeted protein has a
similar molecular weight with β-actin, we used GAPDH instead of the
internal control gene). Data were presented as the means ± SE.
*P<0.05, **P<0.01. Idiopathic pulmonary
fibrosis (IPF), n=5; control, n=3. |
Characterization of human lung fibroblast
cultures
In order to explore the potential MET process,
cultures of primary fibroblasts from fibrotic lung tissue were
established from sterile peripheral lung tissue biopsies. After 3–4
weeks, fibroblasts ‘crawled’ from the explants and proliferated to
form a single layer of adherent cells (Fig. 3B). The cells were then trypsinized
and passaged. At up to 8 serial passages, all cells displayed
typical spindle-shaped morphology under a phase-contrast light
microscope (Fig. 3A) and stained
positive for vimentin, collagen I (COLI), collagen III (COLIII),
fibronectin (FN), TGF-β1, TβRI, TβRII, TβRIII, Snail and Smad2/3
(Fig. 4). All subsequent
experiments were performed using subconfluent quiescent cultures of
human lung fibroblasts between passages 4 and 8 to maintain
comparability. These data showed that human lung fibroblasts were
successfully cultured and that TGF-β1-dependent EMT markers were
expressed by these cells.
 | Figure 4Indirect immunofluorescence assay of
fibroblasts from fibrotic lung tissue. The phenotype and
epithelial-mesenchymal transition (EMT) markers of the fibroblasts
from fibrotic lung tissue was determined by indirect
immunofluorescence assay. Fibroblasts were grown on the slides and
fixed with 4% paraformaldehyde. Non-specific protein binding was
blocked by incubating the slides in PBS containing 4% bovine serum
albumin (BSA) for 1 h. The slides were then incubated overnight at
4°C with primary antibodies specific for (A) vimentin, (B) COLI,
(C) COLIII, (D) FN, (E) TGF-β1, (F) TβRI, (G) TβRII, (H) TβRIII,
(I) Snail and (J) Smad2/3. After washing, the slides were further
incubated for 1 min with either FITC- or RBITC-coupled anti-rabbit
IgG or anti-mouse IgG. After washing, the preparations were mounted
with FluorSave reagent and observed under a microscope. (K and L)
show negative control. (Bar shows 50 μm). |
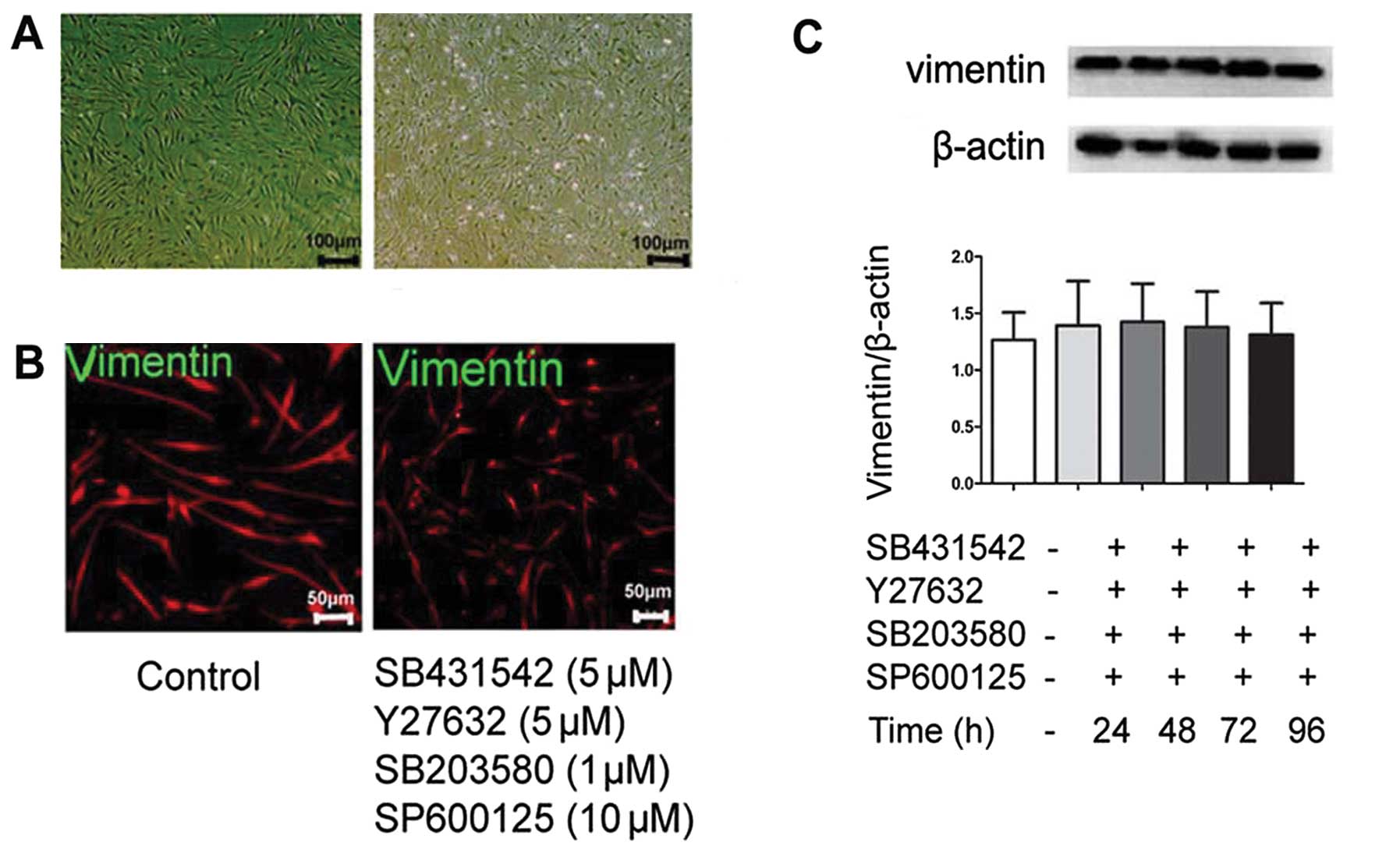
TβRI inhibitor, SB431542 does not induce
EMT reversal
We hypothesized that part of these cultured
fibroblasts from fibrotic lung tissue were derived from abnormal
alveolar epithelial cells and would undergo MET changes by possible
intervention. We first wished to examine the effect of the kinase
inhibitor, SB431542, targeting TGF-β1/TβRI activity, since TGF-β1
is the master switch of EMT (7,14,15). Subsequently, quiescent cells were
incubated with SB431542 at 5 μM or the vehicle for 24, 48, 72 and
96 h. As shown in Fig. 5A, the
addition of SB431542 at 5 μM for 96 h was insufficient to induce an
elongated morphological change of lung fibroblasts to the cubical
pattern. Furthermore, as shown by our results, SB431542 failed to
reduce the protein level of vimentin, a mesenchymal marker, as
indicated by indirect immunofluorescence and western blot analysis
(Fig. 5B and C). These data
demonstrated that inhibiting the TGF-β1 signal was insufficient to
promote the MET process.
A combination of kinase inhibitors
targeting TβRI, ROCK, p38 MAPK and JNK does not induce the MET
process
We then aimed to determine whether a combination of
different kinase inhibitors can induce the MET potential of human
lung fibroblasts. We wished to determine the effects of 4 different
kinase inhibitors specifically targeting TβRI, ROCK, p38 MAPK and
JNK (SB431542, Y27632, SB203580 and SP60012, respectively), as
these kinases have previously been proven to play a specific role
in the EMT process (15,18–21). Firstly, to examine whether the
inhibition of the TβRI and ROCK pathways (using SB431542 and
Y27632, respectively) can induce a significant MET process, we
performed the following experiments: quiescent subconfluent cells
were treated with SB431542 and Y27632 at 5 μM for 24, 48, 72 and 96
h. The induction of the MET potential was evaluated by
morphological observation, indirect immunofluorescence and western
blot analysis for vimentin and E-cadherin expression. Although the
fibroblasts became even more elongated following treatment with the
inhibitors, they still did not assume an epithelial phenotype
(Fig. 6A). Furthermore, the
epithelial marker (E-cadherin) and mesenchymal marker (vimentin)
were not affected (Fig. 6B and
C). Taken together, these data show that the MET process of
lung fibroblasts may not be induced by the inhibition of TβRI and
ROCK.
We then used inhibitors targeting TβRI, ROCK, p38
MAPK and JNK (SB431542, Y27632, SB203580 and SP60012,
respectively), to determine whether they can induce synergistic
effects on the induction of MET. The cells were treated with 5 μM
SB431542, 5 μM Y27632, 1 μM SB203580 and 10 μM SP60012 for 24, 48,
72 and 96 h. Phase-contrast morphology showed no typical phenotypic
changes (Fig. 7A). The expression
of vimentin was not significantly affected by these inhibitors
(Fig. 7B and C). We then used
these kinase inhibitors to treat fibroblasts for an even longer
period of time (from 1 to 8 days). This effort also failed to
induce the MET process (Fig. 8).
Taken together, these results demonstrate that MET may be a
delicate process that is not easily induced in fibroblasts.
Discussion
It is increasingly being recognized that injured
epithelial cells can give rise to fibroblast-like cells and may
thus contribute to the pathogenesis of fibrosis by undergoing EMT.
In the present study, we hypothesized that part of cultured
fibroblasts from fibrotic lung tissue are derived from abnormal
epithelial cells and aimed to induce a possible MET process. We
showed that the TGF-β1-dependent EMT network (including TGF-β1,
TβRI/II/III, Smad2/3, Snail1/2 and p-AKT) was overactivated in the
lung tissues of patients with IPF. We also successfully cultured
primary human lung fibroblasts from lung explants. However, we
failed to induce MET changes in cultured fibroblasts from fibrotic
lung tissue by using kinase inhibitors targeting TβRI, RhoA, p38
MAPK and JNK, all of which have been proven to contribute to the
EMT process. Taken together, these data demonstrate that although
the EMT program exists and is activated in IPF lung tissue, the
direct induction of the MET process in fibroblasts from fibrotic
lung tissue is a more daunting job to achieve.
EMT in vivo is a complex and controversial
process, and its contribution to fibrotic disorders has not yet
been elucidated. In this study, we used fibrotic lung tissue from
patients IPF, or normal lung tissue from patients with primary
spontaneous pneumothorax, to explore the activation of the
TGF-β1-dependent EMT network in fibrotic lung tissue. Western blot
analysis and real-time RT-PCR revealed that TGF-β1 signaling
molecules involved in the EMT process, including TGF-β1, TβRI,
TβRII, TβRIII, Smad2, Smad3, Snail1 and Snail2 were upregulated and
activated in fibrotic lung tissue. This indicated that EMT in
vivo is possible and that epithelial cells may be one of the
sources of mesenchymal cells. Several parallel studies are in line
with our study. A previous study using gene array experiments
demonstrated that genes which stimulate EMT, such as TGF-β3,
lymphoid enhancer factor-1 (LEF-1) and Slug (22), were upregulated in samples from
patients with IPF. They suggested that the increased TGF-β
expression, decreased bone morphogenetic protein (BMP)-2
expression, and active BMP inhibition by gremlin created an
EMT-favoring environment in IPF lungs (22). Another study, using
immunohistochemical analysis, revealed that ATII cells assumed
mesenchymal markers, such as N-cadherin, TGF-β1 and collagen I,
indicating a possible ongoing EMT process in epithelial cells
(23). However, a previous study,
using dual-immunohistochemistry assay, demonstrated that
mesenchymal markers, such as α-SMA and vimentin were not found in
cells with epithelial markers in a bleomycin-induced pulmonary
fibrosis mouse model and patients with IPF and non-specific
interstitial pneumonia (24).
These results suggest that EMT does not occur in IPF or
bleomycin-induced pulmonary fibrosis in mice. Another possibility
is that EMT may occur in pulmonary fibrosis but the detectable
level of mesenchymal markers expressed in epithelial cells is too
low to be detected by double immunohistochemistry (24).
Although previous studies have focused on the EMT
process induced by TGF-β1 or other profibrotic cytokines (15,25–27), studies on the EMT reversal (MET)
are limited. We hypothesized that if EMT in vivo really
exists in IPF, then fibroblasts in fibrotic lung tissues may partly
originate from injured epithelial cells. We also hypothesized that
by blocking the activated signaling network in primarily cultured
fibroblasts associated with EMT, at least some fibroblasts may
abandon their mesenchymal markers and resume epithelial markers and
the EMT reversal (MET) may become possible. To this end, we used
kinase inhibitors targeting TβRI, RhoA, p38 MAPK and the JNK
pathways, all of which are involved in the EMT process. However, to
our disappointment, the treatment of fibroblasts with the kinase
inhibitors (either separately or together) failed to induce the MET
process, indicated by morphology observation, indirect
immunostaining and western blot analysis for epithelial and
mesenchymal markers (E-cadherin and vimentin). These results
indicate that the MET processs may not be the exact reversal of
EMT; the process by which cells can alter their mesenchymal
phenotype to resume an epithelial one is a much more complex
one.
To date, studies on MET in fibrotic lung fibroblasts
are limited. There are several studies, however, on MET in
epithelial cells. Epithelial cells were fist incubated with TGF-β1
or other cytokines to induce the EMT process, and subsequently,
interference methods were used to block or reverse the EMT process
(28–30). A previous study used murine renal
tubular epithelial cells to explore the possibility of the MET
process. They found that exposing cells to the TβRI inhibitor,
SB431542, combined with the ROCK inhibitor, Y27632, eliminated
detectable actin stress fibers and mesenchymal gene expression
while restoring epithelial E-cadherin and kidney-specific cadherin
(Ksp-cadherin) expression (28).
Another study used A549 and RLE-6TN (human and rat) alveolar
epithelial-like cells, demonstrating that FGF-1 plus heparin
reversed the morphological changes induced by TGF-β1 and returned
the epithelial and mesenchymal markers to the control levels
(29). However, a previous study
used primary human proximal tubule epithelial cells (RPTEC) and
immortalized (HK-2) human proximal tubule epithelial cells to show
that bone BMP-7 over a broad concentration range (0.01–100 μg/ml)
failed to attenuate TGF-β1-induced EMT in RPTEC or HK-2 cells
(30). As discussed above, we
also used fibroblasts from fibrotic lung tissue and incubated these
cells with different inhibitors of kinases involved in EMT
modulation to explore the potential of MET, but failed. Taken
together, our data, as well as data from other studies illustrate
that MET may be possible but should be induced carefully with
different treatment methods.
In conclusion, although a limitation of the present
study was the small sample size used to examine the activation of
the TGF-β1-induced EMT program, our data reveal that EMT in
vivo is possible and contributes to the fibrotic process.
Although our efforts to induce the MET process in human lung
fibroblasts failed, future sutdies should focus on the MET process
directly in abnormal fibroblasts from fibrotic tissue rather than
in epithelial cells, as the MET process may not be the exact
reversal of EMT.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (no. 30971312) and the Key
Project of Beijing Municipal Education Commission Sci-Tech
Development Program (no. KZ201110025028). We would like to thank Dr
Bin You, Dr Jinbai Miao, Dr Qirui Chen, Dr Bo Tian and Dr Jin
Zhang, Department of Thoracic Surgery, Beijing Chao-Yang Hospital
for their kind supply of the lung tissue specimens from informed
patients. We also thank Professor Jun Wang, Associate Researcher
Yan Liang, Associate Researcher Xingyuan Jia, Research Assistant
Ran Miao, Research Assistant Dong Leng, Research assistant Xiaoxi
Huang, and Research Assistant Ying Wang, Beijing Key Laboratory of
Respiratory and Pulmonary Circulation Disorders, for their kind
technical assistance.
References
|
1
|
King TE Jr, Pardo A and Selman M:
Idiopathic pulmonary fibrosis. Lancet. 378:1949–1961. 2011.
View Article : Google Scholar
|
|
2
|
Selman M, Thannickal VJ, Pardo A, Zisman
DA, Martinez FJ and Lynch JP III: Idiopathic pulmonary fibrosis:
pathogenesis and therapeutic approaches. Drugs. 64:405–430. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Phan SH: Biology of fibroblasts and
myofibroblasts. Proc Am Thorac Soc. 5:334–337. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee K and Nelson CM: New insights into the
regulation of epithelial-mesenchymal transition and tissue
fibrosis. Int Rev Cell Mol Biol. 294:171–221. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Coward WR, Saini G and Jenkins G: The
pathogenesis of idiopathic pulmonary fibrosis. Ther Adv Respir Dis.
4:367–388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gharaee-Kermani M, Hu B, Phan SH and
Gyetko MR: Recent advances in molecular targets and treatment of
idiopathic pulmonary fibrosis: focus on TGFbeta signaling and the
myofibroblast. Curr Med Chem. 16:1400–1417. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chapman HA: Epithelial-mesenchymal
interactions in pulmonary fibrosis. Annu Rev Physiol. 73:413–435.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Acloque H, Adams MS, Fishwick K,
Bronner-Fraser M and Nieto MA: Epithelial-mesenchymal transitions:
the importance of changing cell state in development and disease. J
Clin Invest. 119:1438–1449. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kalluri R and Neilson EG:
Epithelial-mesenchymal transition and its implications for
fibrosis. J Clin Invest. 112:1776–1784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lan HY and Chung AC: Transforming growth
factor-beta and Smads. Contrib Nephrol. 170:75–82. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pohlers D, Brenmoehl J, Loffler I, Müller
CK, Leipner C, Schultze-Mosgau S, Stallmach A, Kinne RW and Wolf G:
TGF-beta and fibrosis in different organs - molecular pathway
imprints. Biochim Biophys Acta. 1792:746–756. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Willis BC, Liebler JM, Luby-Phelps K,
Nicholson AG, Crandall ED, du Bois RM and Borok Z: Induction of
epithelial-mesenchymal transition in alveolar epithelial cells by
transforming growth factor-beta1: potential role in idiopathic
pulmonary fibrosis. Am J Pathol. 166:1321–1332. 2005. View Article : Google Scholar
|
|
15
|
Kasai H, Allen JT, Mason RM, Kamimura T
and Zhang Z: TGF-beta1 induces human alveolar epithelial to
mesenchymal cell transition (EMT). Respir Res. 6:562005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fragiadaki M and Mason RM:
Epithelial-mesenchymal transition in renal fibrosis - evidence for
and against. Int J Exp Pathol. 92:143–150. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
American Thoracic Society; European
Respiratory Society. American Thoracic Society/European Respiratory
Society International Multidisciplinary Consensus Classification of
the Idiopathic Interstitial Pneumonias. This joint statement of the
American Thoracic Society (ATS), and the European Respiratory
Society (ERS) was adopted by the ATS board of directors, June 2001
and by the ERS Executive Committee, June 2001. Am J Respir Crit
Care Med. 165:277–304. 2002.
|
|
18
|
Hutchison N, Hendry BM and Sharpe CC: Rho
isoforms have distinct and specific functions in the process of
epithelial to mesenchymal transition in renal proximal tubular
cells. Cell Signal. 21:1522–1531. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lv ZM, Wang Q, Wan Q, Lin JG, Hu MS, Liu
YX and Wang R: The role of the p38 MAPK signaling pathway in high
glucose-induced epithelial-mesenchymal transition of cultured human
renal tubular epithelial cells. PLoS One. 6:e228062011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kolosova I, Nethery D and Kern JA: Role of
Smad2/3 and p38 MAP kinase in TGF-β1-induced epithelial-mesenchymal
transition of pulmonary epithelial cells. J Cell Physiol.
226:1248–1254. 2011.
|
|
21
|
van der Velden JL, Guala AS, Leggett SE,
Sluimer J, Badura EC and Janssen-Heininger YM: Induction of a
mesenchymal expression program in lung epithelial cells by wingless
protein (Wnt)/β-catenin requires the presence of c-Jun N-terminal
kinase-1 (JNK1). Am J Respir Cell Mol Biol. 47:306–314.
2012.PubMed/NCBI
|
|
22
|
Selman M, Pardo A and Kaminski N:
Idiopathic pulmonary fibrosis: aberrant recapitulation of
developmental programs? PLoS Med. 5:e622008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lomas NJ, Watts KL, Akram KM, Forsyth NR
and Spiteri MA: Idiopathic pulmonary fibrosis: immunohistochemical
analysis provides fresh insights into lung tissue remodelling with
implications for novel prognostic markers. Int J Clin Exp Pathol.
5:58–71. 2012.
|
|
24
|
Yamada M, Kuwano K, Maeyama T, Hamada N,
Yoshimi M, Nakanishi Y and Kasper M: Dual-immunohistochemistry
provides little evidence for epithelial-mesenchymal transition in
pulmonary fibrosis. Histochem Cell Biol. 129:453–462. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang M, Zhang Z, Pan HY, Wang DX, Deng ZT
and Ye XL: TGF-beta1 induces human bronchial epithelial
cell-to-mesenchymal transition in vitro. Lung. 187:187–194. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim JH, Jang YS, Eom KS, Hwang YI, Kang
HR, Jang SH, Kim CH, Park YB, Lee MG, Hyun IG, Jung KS and Kim DG:
Transforming growth factor beta1 induces epithelial-to-mesenchymal
transition of A549 cells. J Korean Med Sci. 22:898–904. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fernando RI, Castillo MD, Litzinger M,
Hamilton DH and Palena C: IL-8 signaling plays a critical role in
the epithelial-mesenchymal transition of human carcinoma cells.
Cancer Res. 71:5296–5306. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Das S, Becker BN, Hoffmann FM and Mertz
JE: Complete reversal of epithelial to mesenchymal transition
requires inhibition of both ZEB expression and the Rho pathway. BMC
Cell Biol. 10:942009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ramos C, Becerril C, Montano M,
Garcia-De-Alba C, Ramirez R, Checa M, Pardo A and Selman M: FGF-1
reverts epithelial-mesenchymal transition induced by TGF-{beta}1
through MAPK/ERK kinase pathway. Am J Physiol Lung Cell Mol
Physiol. 299:L222–L231. 2010.PubMed/NCBI
|
|
30
|
Dudas PL, Argentieri RL and Farrell FX:
BMP-7 fails to attenuate TGF-beta1-induced
epithelial-to-mesenchymal transition in human proximal tubule
epithelial cells. Nephrol Dial Transplant. 24:1406–1416. 2009.
View Article : Google Scholar : PubMed/NCBI
|