1. Introduction
The development of novel technologies, such as
massively parallel DNA sequencing, has led to the identification of
several important genetic mutations in acute myeloid leukemia (AML)
(1). These include
ten-eleven-translocation oncogene family member 2 (TET2),
isocitrate dehydrogenase (IDH)1 (2) and DNA methyltransferase (Dnmt)3a
mutations (3). These mutations
are frequently found in cytogenetically normal AML (1). In particular, TET2 and IDH1/2
mutations lead to the aberrant hypermethylation signature in AML
cells (4). In addition, there are
specific alterations in the gene expression profiles, such as
homeobox (Hox)B genes, in samples with Dnmt3a
mutations compared with those without such mutations (5). These findings strongly suggest a
link between recurrent genetic alterations and aberrant epigenetic
regulation, resulting in an abnormal DNA methylation status in
myeloid malignancies. This review focuses on the current findings
of aberrant epigenetic signatures by these newly described genetic
alterations. Moreover, epigenetic aberrations resulting from
transcription factor aberrations are also described.
2. TET2 mutations
In 2009, Delhommeau et al (6) conducted a combination of molecular,
cytogenetic, comparative genomic hybridization, and single
nucleotide polymorphism analyses to identify a candidate tumor
suppressor genes common in patients with myelodysplastic syndromes
(MDS), myeloproliferative disorders and AML. They identified
inactivating mutations of the TET2 gene in approximately 15%
of patients with various myeloid malignancies, such as MDS (19%),
myeloproliferative disorders (12%), secondary AML (24%) and chronic
myelomonocytic leukemia (CMML) (22%) (6). In a recent study, Metzeler et
al (7) analyzed 427 patients
with cytogenetically normal (CN)-AML, and revealed TET2 mutations
in 23% of the patients, associated with older age (p<0.001) and
higher pre-treatment white blood cell (WBC) (p=0.04) counts
compared with wild-type TET2. A subsequent analysis of TET2
sequence and rearrangements in more than 1,000 patients by several
groups led to the establishment of the frequencies of TET2 defects
in myeloproliferative neoplasms (MPNs): approximately 12% in all
MPNs, 14% in polycythemia vera, 8% in essential thrombocythemia,
20% in primary myelofibrosis and 25% in post-MPN AML (8).
TET2 can convert 5-methylcytosine (5-mC) to
5-hydroxymethylcytosine (5-hmC) (9), which was to be an intermediate in
the demethylation of DNA. Bone marrow samples from patients with
TET2 mutations displayed uniformly low levels of 5-hmC in genomic
DNA compared to bone marrow samples from healthy controls (9). Figueroa et al (4) investigated TET2 mutant AMLs and
identified a hypermethylation phenotype, including 129
differentially methylated regions. However, the precise molecular
mechanism of TET-mediated gene regulation has not yet been
elucidated. Recently, Chen et al (10), using protein affinity
purification, demonstrated that TET2 and TET3 are associated with
O-linked β-N-acetylglucosamine (O-GlcNAc) transferase
(OGT), an enzyme that on its own catalyzes the addition of
O-GlcNAc onto serine (Ser) and threonine residues
(O-GlcNAcylation) in vivo. TET2 directly interacts
with OGT, and recruits OGT to chromatin. The authors revealed that
the OGT-mediated O-GlcNAcylation of Ser 122 of histone H2B
contributes to TET2-dependent gene transcriptional activation.
Recurrent mutations of the TET2 gene in
myeloid malignancies may disrupt these epigenetic machineries,
leading to aberrant gene regulation, and may play a role in the
pathogenesis of these diseases.
3. IDH1/2 mutations
Mardis et al (2), using massively parallel DNA
sequencing to obtain a very high level of coverage of a primary,
cytogenetically normal, de novo genome for AML with minimal
maturation (AML-M1) and a matched normal skin genome, identified 12
acquired mutations within the coding sequences of genes and 52
somatic point mutations in conserved or regulatory portions of the
genome. Many of them were mutations that have already been
identified, such as NRAS and nucleophosmin (NPM)1, but they found a
novel mutaton of the IDH1 gene (2). They found that the IDH1 gene
mutation was present in 15 out of the 187 AML genomes and was
strongly associated with a CN status. Boissel et al
(11) analyzed both IDH1
mutations and IDH2 mutations in a cohort of 520 adults with AML
homogeneously treated in the French Acute Leukemia French
Association (ALFA) 9801 and 9802 trials. The prevalence of IDH1 and
IDH2 mutations was 9.6 and 3.0%, respectively, mostly associated
with a CN status. Schnittger et al (12) conducted larger study. They
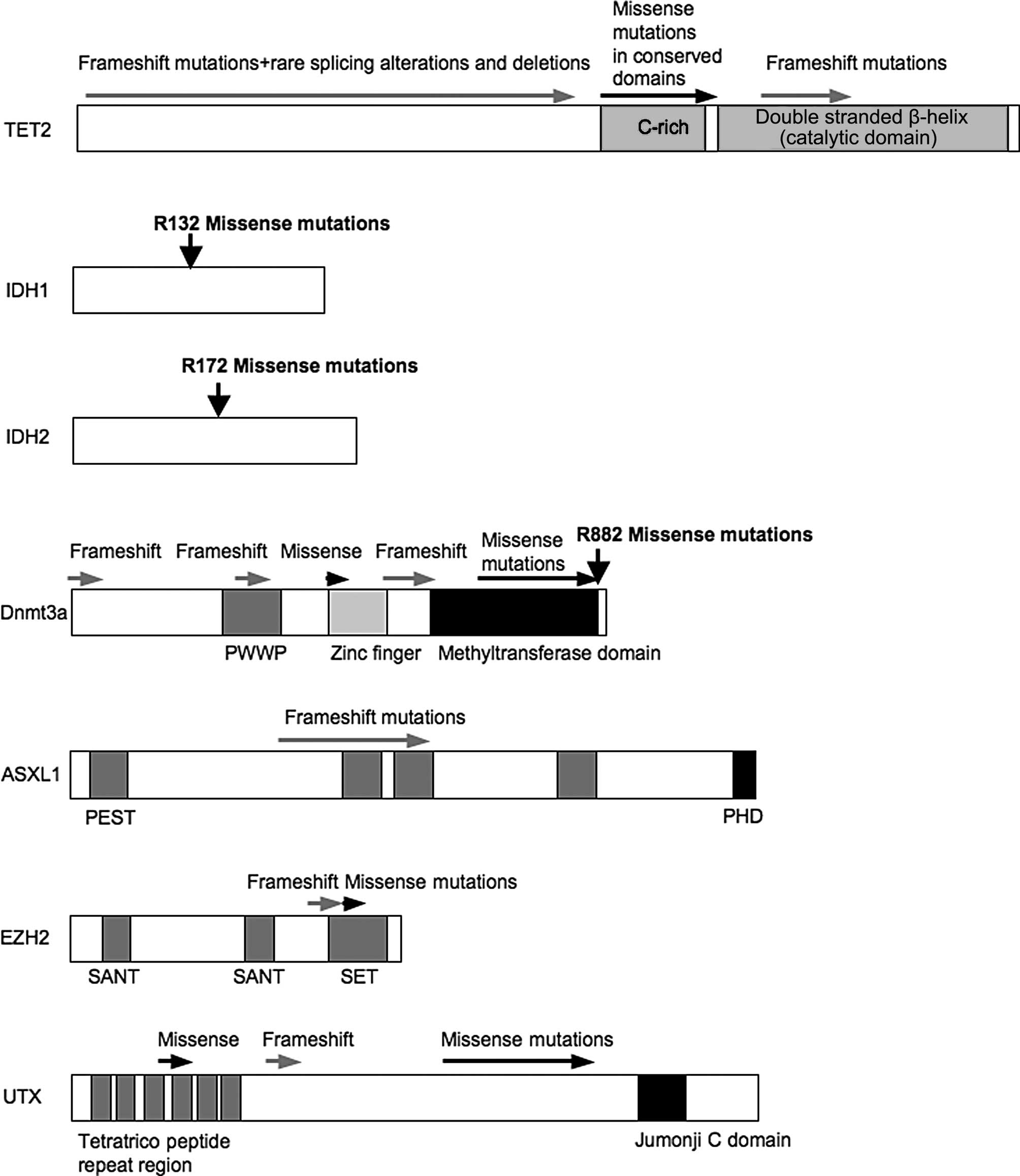
analyzed IDH1R132 mutations (Fig.
1) in 1,414 AML patients and detected IDH1 mutations in 6.6% of
1,414 AML patients with a clear prevalence in the intermediate risk
karyotype group (10.4%, p<0.001). They also detected IDH1
mutations with strong associations not only with NPM1 mutations
(p<0.001) but also with partial tandem duplications (PTDs) in
the mixed lineage leukemia (MLL) gene (p=0.020).
IDH1/2 function at a crossroads of cellular
metabolism in lipid synthesis, cellular defense against oxidative
stress, oxidative respiration and oxygen sensing signal
transduction (13). Recently, it
was demonstrated that AML patients harboring IDH1 and 2 mutations
displayed aberrant hypermethylation (4). In a recent study by Duncan et
al (14), the authors used a
gene-targeting approach by the addition of ‘knocking-in’ a single
copy of the most frequently observed IDH1 mutation, R132H, into a
human cancer cell line and profiled changes in DNA methylation at
over 27,000 CpG dinucleotides relative to wild-type parental cells.
They found that IDH1(R132H/WT) mutation induces widespread
alterations in DNA methylation, including hypermethylation of 2010
and hypomethylation of 842 CpG loci. Furthermore, they showed that
the direction of IDH1(R132H/WT)-mediated DNA methylation change was
largely dependent upon pre-existing DNA methylation levels,
resulting in the depletion of moderately methylated loci (14).
Consistent with another study (7), IDH1/2 mutations were shown to be
mutually exclusive with mutations in TET2 (p=0.009) (4). In fact, these mutations led to the
production of an abnormal metabolite in the cell,
2-hydroxyglutarate, which inhibits the hydroxylation of 5-mC by
TET2 (4). In agreement with these
results, Metzeler et al (7) also demonstrated that IDH1/2
mutations were less frequent in TET2-mutated than in TET2 wild-type
patients (p<0.001), suggesting that these mutations are mutually
exclusive.
4. Dnmt mutations
Dnmts are enzymes that catalyze the addition of
methyl groups to the cytosine residue of CpG dinucleotides.
Aberrant CpG island methylation has long been hypothesized to
contribute to the pathogenesis of cancer (15). Using massively parallel DNA
sequencing (16), Ley et
al (3) identified a somatic
mutation in Dnmt3a, sequencing from 116.4 billion base pairs of
sequence and 99.6% diploid coverage of the genome of cells from a
patient with AML with a normal karyotype. Further analysis revealed
Dnmt3a mutations in 62 of 281 AML patients (22.1%). The structure
of Dnmt3a, and the region where mutations frequently occur, is
presented in Fig. 1. Yan et
al (5) revealed that although
Dnmt3a mutations do not dramatically alter global DNA methylation
levels in AML genomes, there were alterations of specific gene DNA
methylation patterns and/or gene expression profiles, such as
HoxB genes, in samples with Dnmt3a mutations as compared
with those without such changes (5). Consistent with these data, the
Dnmt3a mutation, which frequently occurs in arginine (R)882, showed
reduced enzymatic activity in vitro (5).
5. ASXL1 mutations
The additional sex comb-like 1 (ASXL1) gene
belongs to a family of three identified members that encode
proteins regulating chromatin remodeling. The ASXL proteins contain
a C-terminal plant homeodomain (PHD) finger and belong to the
polycomb and trithorax complexes, which regulate the genetic
program of stem cells. ASXL1 is involved in the regulation of
histone methylation through the cooperation with heterochromatin
protein-1 to modulate the activity of lysine-specific demethylase 1
(17), a histone demethylase for
K4 of histone H3, K9 of histone H3 and also important for global
DNA methylation (18). Recent
studies have identified mutations of the ASXL1 gene in
various types of myeloid malignancies, including approximately 10%
of MDS (19), 10% of MPNs
(20) and 40% of CMML cases
(19). Recently, Chou et
al (21) examined ASXL1
mutations in exon 12 in 501 adults with de novo AML. ASXL1
mutations were detected in 54 patients (10.8%), 8.9% among those
with the normal karyotype and 12.9% among those with abnormal
cytogenetics. The mutation was closely associated with older age,
the male gender, isolated trisomy 8, runt-related transcription
factor 1 (RUNX1) mutation, the expression of human leukocyte
antigen-DR and CD34, but was inversely associated with t(15;17),
complex cytogenetics, internal tandem duplication of Fms-like
tyrosine kinase 3, NPM1 mutations, Wilms tumor 1 mutations and the
expression of CD33 and CD15. Patients with ASXL1 mutations had a
shorter overall survival than patients without these mutations, but
the mutation was not an independent adverse prognostic factor in
multivariate analysis (21).
However, hematopoietic fuction of ASXL1 remains unclear, since the
study of ASXL1 knockout mice revealed only the mild hematopoietic
phenotype (22).
6. EZH2 mutations
Enhancer of zeste homolog 2 (EZH2) is located at
7q36.1 and functions as the catalytic domain of the polycomb
repressive complex 2 (PRC2), causing gene silencing by the
trimethylation of K27 of histone H3. Abnormalities of chromosome 7q
are common in myeloid malignancies (23). Recently, several studies have
identified a minimally affected region on 7q that ultimately led to
the discovery of mutations in EZH2 in several myeloid malignancies
(24–26). In myeloid neoplasms, mutations
have been described largely in poor prognosis myelodysplasia-MPNs
(10–13%), myelofibrosis (13%), and various subtypes of MDS
(24,25,27,28). EZH2 mutations are frequently
associated with uniparental disomy 7q or 7q36.1 microdeletion, but
not monosomy 7 and del(7q). EZH2 mutations in myeloid neoplasms
were spread throughout the gene and comprised of frameshift,
missense and nonsense mutations (Fig.
1), some of which were homozygous. Since the catalytic SET
domain lies at the far C-terminus, all nonsense and stop codon
mutations would be predicted to result in the loss of histone
methyltransferase activity (27).
By contrast, the mutation of a single residue (Y641) and gain of
function is observed in lymphoma (27). The fact that both activating and
inactivating mutations of EZH2 are associated with malignancy
reflects the complex role that polycomb group genes play in cell
fate decisions (29).
7. UTX mutations
Ubiquitously transcribed tetratricopeptide repeat, X
chromosome (UTX) may also play an important role in epigenetic
regulation. It is a histone H3K27 demethylase and belongs to the
polycomb group of proteins. van Haaften et al (30) demonstrated the inactivating
somatic mutations in the UTX, pointing to histone H3 lysine
methylation deregulation in multiple tumor types. They also
revealed that UTX reintroduction into cancer cells with
inactivating UTX mutations resulted in the suppression of
proliferation and marked transcriptional changes (30). Of note, in cells transfected with
wild-type UTX, SRY-related HMG-box 21 and protocadherin 19, two
genes showing significant expression changes in the transfected UTX
null lines, demonstrated a significant decrease in the
trimethylation of histone H3K27 levels upon UTX reintroduction,
suggesting that the expression changes were a direct effect of
reconstituted UTX demethylase activity (30). These data indicate that the
mutational inactivation of UTX occurs, at least in part, through
transcriptional control mechanisms.
Gelsi-Boyer et al (19) reported rare deletions of UTX and
other polycomb group protein family members in MDS. Jankowska et
al (28) recently revealed
that in a cohort of 73 patients with CMML and AML-derived CMML, two
somatic, inactivating UTX mutations were detected and four
additional missense variants that could correspond to rare
polymorphisms were identified.
8. Epigenetic aberrations result from
transcription factor aberrations – MLL rearrangement
Approximately 4–11% of patients with CN-AML present
with rearrangement of the MLL gene as the result of a PTD
within a single MLL allele (31).
MLL is a 430-kDa transcription factor and has a complex structure
that includes three AT-hook domains for DNA binding, a
methyltransferase homology domain and a SET domain (31). MLL is necessary for the
maintenance of Hox gene expression (32). The MLL SET domain can bind the
promoter of Hox genes (33). Dorrance et al (34) generated a mouse knock-in model in
which exons 5 through 11 of the murine MLL gene were targeted to
intron 4 of the endogenous Mll locus.
MllPTD/WT mice exhibit an alteration in the
boundaries of normal Hox gene expression during
embryogenesis, resulting in axial skeletal defects and increased
numbers of hematopoietic progenitor cells.
MllPTD/WT mice overexpress Hoxa7, Hoxa9 and
Hoxa10 in the spleen, bone marrow (BM) and blood. An increase in
histone H3/H4 acetylation and histone H3K4 methylation within the
Hoxa7 and Hoxa9 promoters provides an epigenetic mechanism by which
this overexpression occurs in vivo and an etiological role
for MLL-PTD gain-of-function in the pathogenesis in AML. Consistent
with this, Whitman et al (35) reported that AML patients with
MLL-PTD exhibited increased global DNA methylation vs. AML patients
with MLL-wild-type. Munoz et al (36) examined 93 adult patients with
de novo AML for incidence clinical features of MLL
rearranged AMLs. As a result, they revealed that MLL rearrangements
were detected in 13 (14%) patients. In the French-American-British
Classification, there was a significantly higher percentage of M5
sybtypes in the MLL rearranged group. MLL rearranged patients had a
lower event-free survival (EFS) (p=0.001) and a higher probability
of relapse (REL) compared to MLL wild-type patients (p=0.07).
9. Evi1 overexpression
The abnormal expression of the ecotropic viral
integration site 1 (Evi1) gene, as the result of
inv(3)(q21q26)/t(3,3)(q21;q26.2) or through other unknown
mechanisms, is associated with unfavorable AML outcome (37,38). Evi1 encodes a C2H2 zinc finger
transcription factor, binding DNA in a sequence-specific manner and
functions as a repressor (39–41). Lugthart et al (42) examined the aberrant epigenetic
programming in AML patients, conducting a large-scale DNA
methylation profiling study in human Evi1 AMLs. They compared and
contrasted the abundance of cytosine methylation at 14,000 gene
promoters using the HpaII tiny fragment enrichment by
ligation-mediated PCR (HELP) assay in 26 AML patients
overexpressing Evi1 and in eight CD34+ normal bone
marrow controls. The signature contained 294 differentially
methylated genes, of which 238 (81%) were coordinately
hypermethylated. These data suggest that Evi1 contributes to
aberrant promoter DNA methylation patterning in leukemia.
10. Chromosomal translocations
From the biological standpoint, genome
AML-associated fusion proteins that result from chromosomal
translocations have been reported to aid in the establishment of
specific DNA methylation patterns in AML. For example,
promyelocytic leukemia (PML)/retinoic acid receptor (RAR)α and
RUNX1/eight-twenty one oncoprotein (ETO) have been to used recruit
both histone deacetylases and DNA methyltransferases to induce the
transcriptional repression of target genes (43).
Figueroa et al (44) recently examined the methylation
profiles of 344 patients with AML. They demonstrated that
established AML1-ETO (n=24, 7%), core binding factor β
(CBFβ)-myosin, heavy chain 11, smooth muscle (MYH11) (n=30, 9%) and
PML-RARα (n=10, 3%) leukemia entities were associated with specific
methylation profiles. In addition, they revealed that DNA
methylation profiles segregated patients with
CCAAT/enhancer-binding protein (CEBP)α aberrations from other
subtypes of leukemia. They also defined epigenetically distinct
forms of AML with NPM1 mutations. Although the molecular mechanisms
of these genetic alterations and specific methylation remain
unsolved, it is likely that the disruption of transcriptional
repressor function by chromosomal translocation may contribute to
the aberrant hypermethylation profile in AML cells.
11. PU.1 downregulation
The overall incidence of the genetic aberration of
the PU.1 gene is extremely rare in myeloid leukemia
(45–47). However, the level of PU.1
expression is critical for specifying the fate of cells and, if
perturbed, even modest decreases in PU.1 levels can lead to the
development of leukemia and lymphoma (48,49). Indeed, PU.1 is expressed at low
levels in the majority of cases of human AML (50). Similar to Evi1 (42), PU.1 interacts with Dnmt3a and
Dnmt3b (51) and suppresses its
target genes through Dnmt activity (51,52). In addition, it was recently
revealed that changes in PU.1 expression also affect several genes
(metallothionein-1, vimentin), playing a role in cellular
proliferation and differentiation, promoter DNA methylation and
histone acetylation levels in leukemia cells (52,53). Dnmt3a mutation is prevalent in
AML, and the expression of Dnmt3s is constant in AML specimens
(3). Therefore, it is possible
that Dnmt3a mutations exert their leukemogenic potential, at least
in part, through the functional impairment of PU.1 and/or Evi1
epigenetic activity as a transcriptional repressor, mainly through
the downregulation of its expression. Further studies are required
in order to fully elucidate the mechanisms involved.
12. Conclusion
The development of novel technologies has led us to
detect several important genetic mutations in AML. Along with the
development of whole genome sequencing, major genetic aberrations
have been almost completely identified. The next stage is to
determine the consequence of these molecular alterations,
particularly for these newly identified molecules. A list of
genetic aberrations with the phenotype associated with epigenetic
aberrations is summarized in Table
I.
 | Table IA list of genetic aberrations with
the phenotype associated with epigenetic aberrations. |
Table I
A list of genetic aberrations with
the phenotype associated with epigenetic aberrations.
| Genetic
aberrations | Epigenetic
aberrations | Refs. |
|---|
| TET2 mutations | TET2 can convert
5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC), which
was to be an intermediate in the demethylation of DNA. Uniformly
low levels of 5-hmC in genomic DNA compared to bone marrow samples
from healthy controls. | (9) |
| TET2 directly
interacts with O-linked β-N-acetylglucosamine
(O-GlcNAc) transferase (OGT), and recruits OGT to chromatin.
OGT-mediated O-GlcNAcylation of Ser 122 of histone H2B contributes
to TET2-dependent gene expression. | (10) |
| IDH1/2
mutations | Global
hypermethylation. | (4,14) |
| Dnmt3a
mutations | Although Dnmt3a
mutations do not dramatically alter global DNA methylation levels
in AML genomes, there were alterations of HoxB gene DNA
methylation patterns and gene expression profiles in samples with
Dnmt3a mutations as compared with those without such changes. | (5) |
| ASXL1
mutations | Unclear. The study
of ASXL1 knockout mice revealed only the mild hematopoietic
phenotype. | (22) |
| EZH2 mutations | Unclear. All
nonsense and stop codon mutations would be predicted to result in
the loss of histone methyltransferase activity. | (27) |
| UTX mutations | Inactivating
somatic mutations in UTX, pointing to histone H3 lysine methylation
deregulation in multiple tumor types. UTX reintroduction into
cancer cells with inactivating UTX mutations resulted in the
suppression of proliferation and marked transcriptional
changes. | (30) |
| MLL-PTD | From the analysis
of MLL-PTD knock-in mice, an increase in histone H3/H4 acetylation
and histone H3 lysine 4 (Lys4) methylation within the Hoxa7 and
Hoxa9 promoters was observed. | (34) |
| Evi1
overexpression | Analysis from a
large-scale DNA methylation profiling study in human EVI1 AMLs
revealed that the signature contained 294 differentially methylated
genes, of which 238 (81%) were coordinately hypermethylated. | (42) |
| AML1-ETO,
CBFβ-MYH11, PML-RARα | AML1-ETO,
CBFβ-MYH11 and PML-RARα leukemia entities are associated with
specific methylation profiles. | (44) |
| Downregulation of
PU.1 |
Metallothionein-1, vimentin
genes were upregulated, with increases in acetylated histone H3 and
H4 levels, and decreases in DNA methylation in their
promoters. | (52) |
These newly identified combinations of genetic
aberrations may lead to a refined disease classification and to the
development of rational, epigenetic or signal transduction pathway
targeted therapies.
Acknowledgements
The present study was supported in part by
Grants-in-Aid for Scientific Research (no. 23590687) from the
Ministry of Education, Science and Culture, Japan, and the Takeda
Science Foundation, a foundation from Kitasato University School of
Allied Health Sciences (Grant-in-Aid for Research Project, no.
2012-1002).
References
|
1
|
Takahashi S: Current findings for
recurring mutations in acute myeloid leukemia. J Hematol Oncol.
4:362011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mardis ER, Ding L, Dooling DJ, et al:
Recurring mutations found by sequencing an acute myeloid leukemia
genome. N Engl J Med. 361:1058–1066. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ley TJ, Ding L, Walter MJ, et al: DNMT3A
mutations in acute myeloid leukemia. N Engl J Med. 363:2424–2433.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Figueroa ME, Abdel-Wahab O, Lu C, et al:
Leukemic IDH1 and IDH2 mutations result in a hypermethylation
phenotype, disrupt TET2 function, and impair hematopoietic
differentiation. Cancer Cell. 18:553–567. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yan XJ, Xu J, Gu ZH, et al: Exome
sequencing identifies somatic mutations of DNA methyltransferase
gene DNMT3A in acute monocytic leukemia. Nat Genet. 43:309–315.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Delhommeau F, Dupont S, Della Valle V, et
al: Mutation in TET2 in myeloid cancers. N Engl J Med.
360:2289–2301. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Metzeler KH, Maharry K, Radmacher MD, et
al: TET2 mutations improve the new European LeukemiaNet risk
classification of acute myeloid leukemia: a Cancer and Leukemia
Group B study. J Clin Oncol. 29:1373–1381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Delhommeau F, Jeziorowska D, Marzac C and
Casadevall N: Molecular aspects of myeloproliferative neoplasms.
Int J Hematol. 91:165–173. 2010. View Article : Google Scholar
|
|
9
|
Ko M, Huang Y, Jankowska AM, et al:
Impaired hydroxylation of 5-methylcytosine in myeloid cancers with
mutant TET2. Nature. 468:839–843. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen Q, Chen Y, Bian C, Fujiki R and Yu X:
TET2 promotes histone O-GlcNAcylation during gene
transcription. Nature. 493:561–564. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Boissel N, Nibourel O, Renneville A, et
al: Prognostic impact of isocitrate dehydrogenase enzyme isoforms 1
and 2 mutations in acute myeloid leukemia: a study by the Acute
Leukemia French Association group. J Clin Oncol. 28:3717–3723.
2010. View Article : Google Scholar
|
|
12
|
Schnittger S, Haferlach C, Ulke M,
Alpermann T, Kern W and Haferlach T: IDH1 mutations are detected in
6.6% of 1414 AML patients and are associated with intermediate risk
karyotype and unfavorable prognosis in adults younger than 60 years
and unmutated NPM1 status. Blood. 116:5486–5496. 2010.
|
|
13
|
Reitman ZJ and Yan H: Isocitrate
dehydrogenase 1 and 2 mutations in cancer: alterations at a
crossroads of cellular metabolism. J Natl Cancer Inst. 102:932–941.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Duncan CG, Barwick BG, Jin G, et al: A
heterozygous IDH1R132H/WT mutation induces genome-wide alterations
in DNA methylation. Genome Res. 22:2339–2355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar
|
|
16
|
Wheeler DA, Srinivasan M, Egholm M, et al:
The complete genome of an individual by massively parallel DNA
sequencing. Nature. 452:872–876. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee SW, Cho YS, Na JM, et al: ASXL1
represses retinoic acid receptor-mediated transcription through
associating with HP1 and LSD1. J Biol Chem. 285:18–29. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang J, Hevi S, Kurash JK, et al: The
lysine demethylase LSD1 (KDM1) is required for maintenance of
global DNA methylation. Nat Genet. 41:125–129. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gelsi-Boyer V, Trouplin V, Adelaide J, et
al: Mutations of polycomb-associated gene ASXL1 in myelodysplastic
syndromes and chronic myelomonocytic leukaemia. Br J Haematol.
145:788–800. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Abdel-Wahab O, Manshouri T, Patel J, et
al: Genetic analysis of transforming events that convert chronic
myeloproliferative neoplasms to leukemias. Cancer Res. 70:447–452.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chou WC, Huang HH, Hou HA, et al: Distinct
clinical and biological features of de novo acute myeloid leukemia
with additional sex comb-like 1 (ASXL1) mutations. Blood.
116:4086–4094. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fisher CL, Pineault N, Brookes C, et al:
Loss-of-function additional sex combs like 1 mutations disrupt
hematopoiesis but do not cause severe myelodysplasia or leukemia.
Blood. 115:38–46. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tiu RV, Visconte V, Traina F, Schwandt A
and Maciejewski JP: Updates in cytogenetics and molecular markers
in MDS. Curr Hematol Malig Rep. 6:126–135. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nikoloski G, Langemeijer SM, Kuiper RP, et
al: Somatic mutations of the histone methyltransferase gene EZH2 in
myelodysplastic syndromes. Nat Genet. 42:665–667. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ernst T, Chase AJ, Score J, et al:
Inactivating mutations of the histone methyltransferase gene EZH2
in myeloid disorders. Nat Genet. 42:722–726. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Makishima H, Jankowska AM, Tiu RV, et al:
Novel homo- and hemizygous mutations in EZH2 in myeloid
malignancies. Leukemia. 24:1799–1804. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chase A and Cross NC: Aberrations of EZH2
in cancer. Clin Cancer Res. 17:2613–2618. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jankowska AM, Makishima H, Tiu RV, et al:
Mutational spectrum analysis of chronic myelomonocytic leukemia
includes genes associated with epigenetic regulation: UTX, EZH2,
and DNMT3A. Blood. 118:3932–3941. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sauvageau M and Sauvageau G: Polycomb
group proteins: multi-faceted regulators of somatic stem cells and
cancer. Cell Stem Cell. 7:299–313. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van Haaften G, Dalgliesh GL, Davies H, et
al: Somatic mutations of the histone H3K27 demethylase gene UTX in
human cancer. Nat Genet. 41:521–523. 2009.PubMed/NCBI
|
|
31
|
Basecke J, Whelan JT, Griesinger F and
Bertrand FE: The MLL partial tandem duplication in acute myeloid
leukaemia. Br J Haematol. 135:438–449. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu BD, Hess JL, Horning SE, Brown GA and
Korsmeyer SJ: Altered Hox expression and segmental identity in
Mll-mutant mice. Nature. 378:505–508. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Milne TA, Briggs SD, Brock HW, et al: MLL
targets SET domain methyltransferase activity to Hox gene
promoters. Mol Cell. 10:1107–1117. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dorrance AM, Liu S, Yuan W, et al: Mll
partial tandem duplication induces aberrant Hox expression in vivo
via specific epigenetic alterations. J Clin Invest. 116:2707–2716.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Whitman SP, Hackanson B, Liyanarachchi S,
et al: DNA hypermethylation and epigenetic silencing of the tumor
suppressor gene, SLC5A8, in acute myeloid leukemia with the MLL
partial tandem duplication. Blood. 112:2013–2016. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Munoz L, Nomdedeu JF, Villamor N, et al:
Acute myeloid leukemia with MLL rearrangements: clinicobiological
features, prognostic impact and value of flow cytometry in the
detection of residual leukemic cells. Leukemia. 17:76–82. 2003.
View Article : Google Scholar
|
|
37
|
Lugthart S, van Drunen E, van Norden Y, et
al: High EVI1 levels predict adverse outcome in acute myeloid
leukemia: prevalence of EVI1 overexpression and chromosome 3q26
abnormalities underestimated. Blood. 111:4329–4337. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Groschel S, Lugthart S, Schlenk RF, et al:
High EVI1 expression predicts outcome in younger adult patients
with acute myeloid leukemia and is associated with distinct
cytogenetic abnormalities. J Clin Oncol. 28:2101–2107
|
|
39
|
Delwel R, Funabiki T, Kreider BL,
Morishita K and Ihle JN: Four of the seven zinc fingers of the
Evi-1 myeloid-transforming gene are required for sequence-specific
binding to GA(C/T)AAGA(T/C)AAGATAA. Mol Cell Biol. 13:4291–4300.
1993.PubMed/NCBI
|
|
40
|
Matsugi T, Kreider BL, Delwel R, Cleveland
JL, Askew DS and Ihle JN: The Evi-1 zinc finger myeloid
transforming protein binds to genomic fragments containing (GATA)n
sequences. Oncogene. 11:191–198. 1995.PubMed/NCBI
|
|
41
|
Perkins AS, Fishel R, Jenkins NA and
Copeland NG: Evi-1, a murine zinc finger proto-oncogene, encodes a
sequence-specific DNA-binding protein. Mol Cell Biol. 11:2665–2674.
1991.PubMed/NCBI
|
|
42
|
Lugthart S, Figueroa ME, Bindels E, et al:
Aberrant DNA hypermethylation signature in acute myeloid leukemia
directed by EVI1. Blood. 117:234–241. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Boultwood J and Wainscoat JS: Gene
silencing by DNA methylation in haematological malignancies. Br J
Haematol. 138:3–11. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Figueroa ME, Lugthart S, Li Y, et al: DNA
methylation signatures identify biologically distinct subtypes in
acute myeloid leukemia. Cancer Cell. 17:13–27. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mueller BU, Pabst T, Osato M, et al:
Heterozygous PU.1 mutations are associated with acute myeloid
leukemia. Blood. 100:998–1007. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Vegesna V, Takeuchi S, Hofmann WK, et al:
C/EBP-beta, C/EBP-delta, PU.1, AML1 genes: mutational analysis in
381 samples of hematopoietic and solid malignancies. Leuk Res.
26:451–457. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lamandin C, Sagot C, Roumier C, et al: Are
PU.1 mutations frequent genetic events in acute myeloid leukemia
(AML)? Blood. 100:4680–4681. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
DeKoter RP and Singh H: Regulation of B
lymphocyte and macrophage development by graded expression of PU.1.
Science. 288:1439–1441. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rosenbauer F, Wagner K, Kutok JL, et al:
Acute myeloid leukemia induced by graded reduction of a
lineage-specific transcription factor, PU.1. Nat Genet. 36:624–630.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
50
|
Steidl U, Rosenbauer F, Verhaak RG, et al:
Essential role of Jun family transcription factors in PU.1
knockdown-induced leukemic stem cells. Nat Genet. 38:1269–1277.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
51
|
Suzuki M, Yamada T, Kihara-Negishi F, et
al: Site-specific DNA methylation by a complex of PU.1 and
Dnmt3a/b. Oncogene. 25:2477–2488. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Imoto A, Okada M, Okazaki T, Kitasato H,
Harigae H and Takahashi S: Metallothionein-1 isoforms and vimentin
are direct PU.1 downstream target genes in leukemia cells. J Biol
Chem. 285:10300–10309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Suzuki S, Nakano H and Takahashi S:
Epigenetic regulation of the metallothionein-1A promoter by PU.1
during differentiation of THP-1 cells. Biochem Biophys Res Commun.
433:349–353. 2013. View Article : Google Scholar : PubMed/NCBI
|