Introduction
Transmissible spongiform encephalopathies (TSEs) or
prion diseases are fatal neurodegenerative disorders of the central
nervous system (CNS) in humans [Kuru, Creutzfeldt-Jakob disease
(CJD), fatal familial insomnia (FFI) and Gerstmann Straussler
Sheinker syndrome (GSS)] and animals (bovine spongiform
encephalopathy in cattle and scrapie in sheep or goats) (1,2).
TSEs are characterized by the spongiform degeneration of the CNS,
astrogliosis and the deposition of amyloid fibrils in the brain
(1,3). In prion diseases, the normal
cellular form of the prion protein (PrPC) undergoes a
conformational conversion to the β-sheet-rich scrapie isoform
(PrPSc), which is partially resistant to protease
digestion (4,5). The conformational change into
PrPSc occurs through unknown molecular mechanisms.
One of the mechanisms of neuronal cell death in
prion diseases is apoptosis, as apoptotic neurons have been
observed in the brains of scrapie-infected sheep and patients with
CJD (6,7). The synthetic human prion protein
peptide [PrP (106-126)] maintains many characteristics of
PrPSc. These include the ability to form amyloid fibrils
and induce apoptosis in primary rat hippocampal cultures (8), primary mouse cerebellar cultures
(9), GH3 rat clonal pituitary
cells (10), as well as in mouse
retinae (11). Cortical neuron
cells treated with the PrP fragment (106-126) have been shown to
become neurotoxic, develop dysfunctional mitochondria and display
increased prion-mediated neurotoxicity associated with the
induction of Bax translocation to the mitochondria (12,13). These characteristics of the
106-126 sequence of the prion protein render it a useful, in
vitro model for the study of the pathogenesis of prion diseases
(10).
FTY720
{2-amino-2-[2-(4-n-octylphenyl)ethyl]-1,3-propanediol
hydrochloride} is synthetically derived from myriocin (ISP-1), a
metabolite isolated from the ascomycete Isaria sinclarii
(14). The pharmacokinetics of
FTY720 have been characterized extensively, and have shown clinical
efficacy in phase 3 clinical trials involving patients with
multiple sclerosis (MS) (15). It
is a prodrug that is phosphorylated by type 2 sphingosine kinase to
form FTY720-phosphate (FTY720-p) (16). In addition to its role in T-cell
sequestration, the lipophilic nature of FTY720 allows it to readily
cross the blood-brain barrier and exert a number of direct effects
on the CNS (17,18). These include the regulation of
myelination and microglial activation following injury,
proliferation and the migration of neural precursor cells toward
injury sites, as well as the potentiation of growth-factor
regulated neuronal differentiation and survival (18–22). FTY720 is capable of increasing the
production of brain-derived neurotrophic factor (BDNF), an
endogenous neuroprotectant, in neuronal cultures (17,23). Thus, some physiological effects of
FTY720 are related to its neuroprotective effects. However, to the
best of our knowledge, the effects of FTY720 on neurodegenerative
diseases, including prion-mediated neurotoxicity have not yet been
reported.
The present study focused on the effects of FTY720
on PrP (106-126)-induced apoptosis and whether FTY720 can be used
to prevent mitochondrial dysfunction in prion diseases. We
demonstrate that the treatment of neuronal cells with FTY720
inhibits PrP (106-126)-induced neurotoxicity and mitochondrial
dysfunction by blocking the phosphorylation of c-jun N-terminal
kinase (JNK). This suggests that FTY720 has therapeutic potential
in mitochondrial dysfunction-related neurodegenerative disorders,
including prion diseases.
Materials and methods
Cell culture
Human neuroblastoma cells (SH-SY5Y) were obtained
from the American Type Culture collection (ATCC; Rockville, MD,
USA). The cells were cultured in minimum essential medium (MEM)
(HyClone Laboratories, Logan, UT, USA) that contained 10% fetal
bovine serum (Invitrogen-Gibco, Grand Island, NY, USA) and
gentamycin (0.1 mg/ml), in a humidified incubator maintained at
37ºC and 5% CO2.
PrP (106-126) treatment
Synthetic PrP (106-126) (sequence,
Lys-Thr-Asn-Met-Lys-His-Met-Ala-Gly-Ala-Ala-Ala-Ala-Gly-Ala-Val-Val-Gly-Gly-Leu-Gly)
peptides were synthesized from Peptron (Seoul, Korea). The peptides
were dissolved in sterile DMSO at a concentration of 12.5 mM and
stored at −80ºC.
Lactate dehydrogenase (LDH) assay
Cytotoxicity was assessed by LDH assay in
supernatant medium, using an LDH Cytotoxicity Detection kit (Takara
Bio, Inc., Tokyo, Japan) according to the manufacturer’s
instructions. LDH activity was determined by measuring the
absorbance at 490 nm using a microplate reader (Spectra Max M2;
Molecular Devices, LLC, Sunnyvale, CA, USA).
Annexin V assay
Apoptosis was assessed by Annexin V assay in the
detached cells using an Annexin V assay kit (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) according to the
manufacturer’s instructions. The number of Annexin V-positive cells
was determined by measuring the fluorescence at excitation 488 nm
and emission 525/30 using Guava easyCyte HT System (Millipore,
Billerica, MA, USA).
Western blot analyses
The SH-SY5Y cells were lysed in lysis buffer (25 mM
HEPES; pH 7.4, 100 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 0.1
mM DTT and protease inhibitor mixture). Proteins were
electrophoretically resolved on a 10–15% sodium dodecyl sulfate
(SDS) gel and immunoblotting was performed as previously described
(5). Equal amounts of lysate
protein were resolved on a 10–15% SDS-polyacrylamide gel and
electrophoretically transferred onto a nitrocellulose membrane.
Immunoreactivity was detected through sequential incubation with
horseradish peroxidase-conjugated secondary antibodies and ECL
reagents. The antibodies used for immunoblotting were caspase-3,
phospho-JNK, Bax, cytochrome c (both from Cell Signaling
Technology, Inc., Beverly, MA, USA) and β-actin (Sigma-Aldrich, St.
Louis, MO, USA). Images were examined using the Fusion-FX7 imaging
system (Vilber Lourmat, Marne-la-Vallée, France).
Immunofluorescence staining
The SH-SY5Y cells cultured on glass slides were
fixed with cold acetone, blocked with 5% fetal bovine serum in TBST
and incubated with mouse phosphorylated JNK (p-JNK; Cell Signaling
Technology) and rabbit active caspase-3 antibodies (R&D
Systems, Minneapolis, MN, USA) overnight at 4ºC. After being washed
with TBST, the cells were incubated with goat anti-mouse IgG
conjugated with Alexa Fluor® 488 (green) and goat
anti-rabbit IgG conjugated with Alexa Fluor® 546 (red).
The cells were washed with TBST, mounted with fluorescence mounting
medium and observed under a fluorescence microscope (Nikon Eclipse
80i; Nikon Corp., Tokyo, Japan). Images were captured using a Nikon
digital camera, and processed with the appropriate software
(Diagnostic Instruments, Victoria Park, Australia).
Cellular fractionation
The SH-SY5Y cells were resuspended in mitochondrial
buffer (210 mm sucrose, 70 mm mannitol, 1 mm EDTA, 10 mm HEPES),
broken by a 26-gauge needle, and centrifuged at 700 × g for 10 min.
The post-nuclear supernatant was centrifuged at 10,000 × g for 30
min. The pellet was used as the mitochondrial fraction and the
supernatant was used as the cytosolic fraction. Total proteins were
obtained and subjected to western blot analysis.
Mitochondrial transmembrane potential
(MTP) assay
The changes in MTP were evaluated using a cationic
fluorescent indicator (JC-1; Molecular Probes, Eugene, OR, USA),
which aggregates in intact mitochondria (red fluorescence)
indicating high or normal MTP and low MTP when it remains in a
monomeric form in the cytoplasm (green fluorescence). The SH-SY5Y
cells were incubated in MEM containing 10 ml JC-1 at 37ºC for 15
min, washed with PBS and subsequently transferred to a clear
96-well plate. JC-1 aggregate fluorescence emission was measured at
583 nm, with an excitation wavelength of 526 nm. JC-1 monomer
fluorescence intensity was also measured with both excitation and
emission wavelengths at 525 and 530 nm, respectively using a
microplate reader (SpectraMax M2; Molecular Devices) or a Guava
easyCyte HT System. The SH-SY5Y cells were cultured on cover slips
in a 24-well plate, incubated in MEM containing 10 ml JC-1 at 37ºC
for 15 min and then washed with PBS. Finally, the cells were
mounted with DakoCytomation fluorescent mounting medium and
visualized under a fluorescence microscope.
Statistical analysis
All data are expressed as the the means ± standard
deviation (SD), and the data were compared using the Student’s
t-test, as well as ANOVA and Duncan multiple range tests with the
SAS statistical package. In the figures, mean values denoted by a
common alphabetical symbol do not differ significantly. Bars
labeled with different letters indicate significant differences
among each group of bars according to Duncan’s test
(p<0.05).
Results
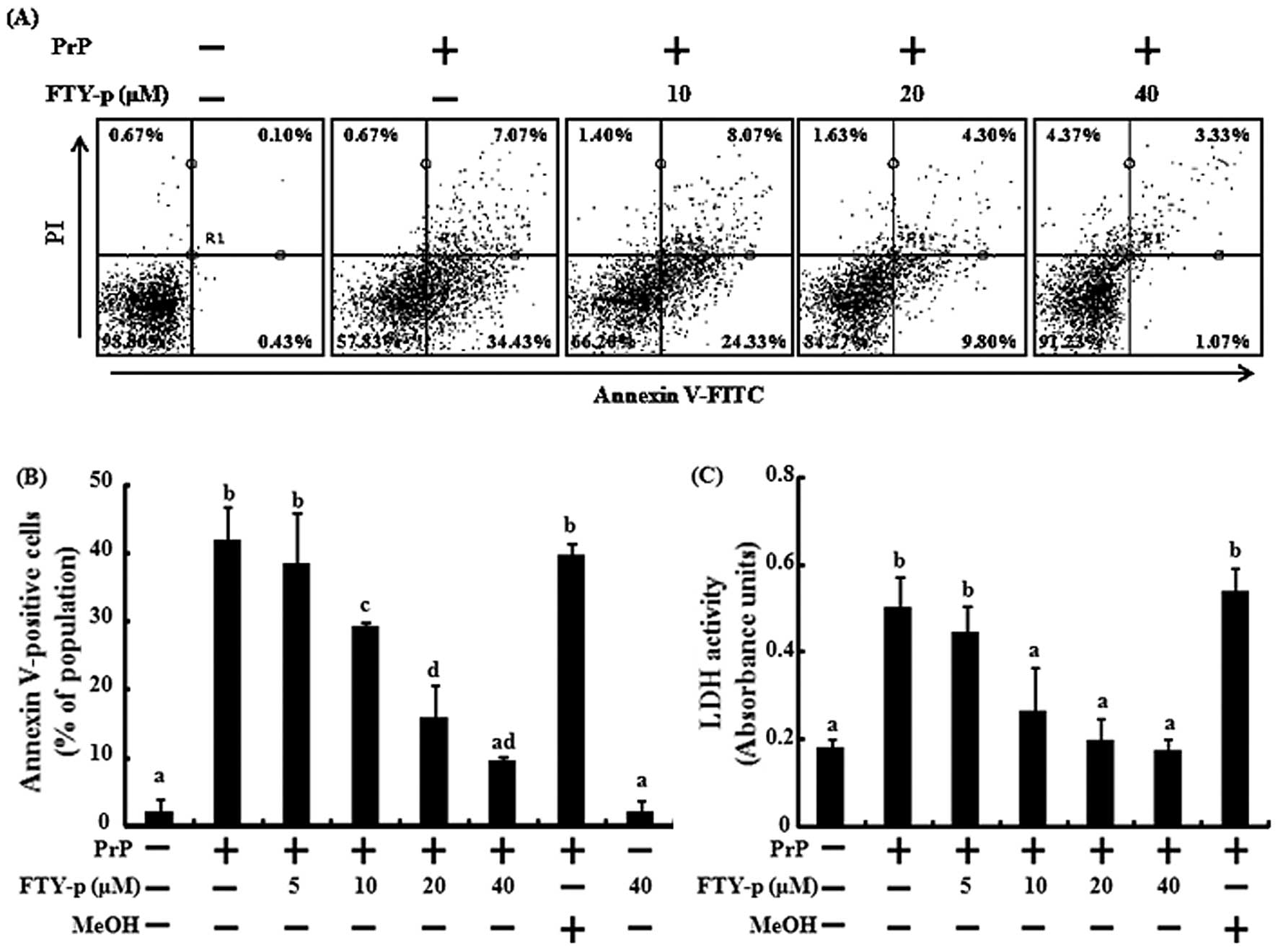
FTY720-p inhibits PrP (106-126)-induced
neuronal cell death
We used PrP (106-126) to examine PrPSc
pathogenesis through the triggering of cell death signals and
evaluated the effects of FTY720 on PrP (106-126)-induced neuronal
cell death. FTY720 is phosphorylated in vivo by sphingosine
kinase 2 to become the active drug metabolite, (S)-FTY720-p, and
only the (S)-phosphorylated form of FTY720 is capable of activating
sphingosine-1-phosphate (S1P) receptors in vitro (24,25). Therefore, we examined the effects
of FTY720-p on PrP (106-126)-induced neurotoxicity in the SH-SY5Y
cells. To examine the neuroprotective effects of FTT720-p, we
examined the effects of FTY720-p on PrP (106-126)-mediated
neurotoxicity in SH-SY5Y cells by Annexin V assay. The SH-SY5Y
cells were pre-treated with various doses of FTY720-p (5, 10, 20
and 40 μM) prior to exposure to 50 μM PrP (106-126) for 24 h. The
cells were responsive to PrP (106-126) treatment (41.5% increase in
Annexin V-positive cells), and FTY720-p had no effect on cell
viability. However, treatment with FTY720-p inhibited PrP
(106-126)-induced neuronal cell death. The effects of FTY720-p were
detected at 10 μM and were maximal at 40 μM (Fig. 1A and B). The protective effects of
FTY720-p against PrP (106-126)-mediated toxicity were further
confirmed by the determination of LDH release as a marker of
cytotoxicity. Assays of LDH activity in the cell culture
supernatants demonstrated that FTY720-p significantly inhibited PrP
(106-126)-induced cytotoxicity in SH-SY5Y cells (Fig. 1C). Consistent with these results,
immunoblot analysis of activated caspase-3 revealed that treatment
with FTY720-p markedly inhibited PrP (106-126)-induced apoptosis
(Fig. 3A and D). These results
indicated that prion-induced neuronal cell death was inhibited by
FTY720-p.
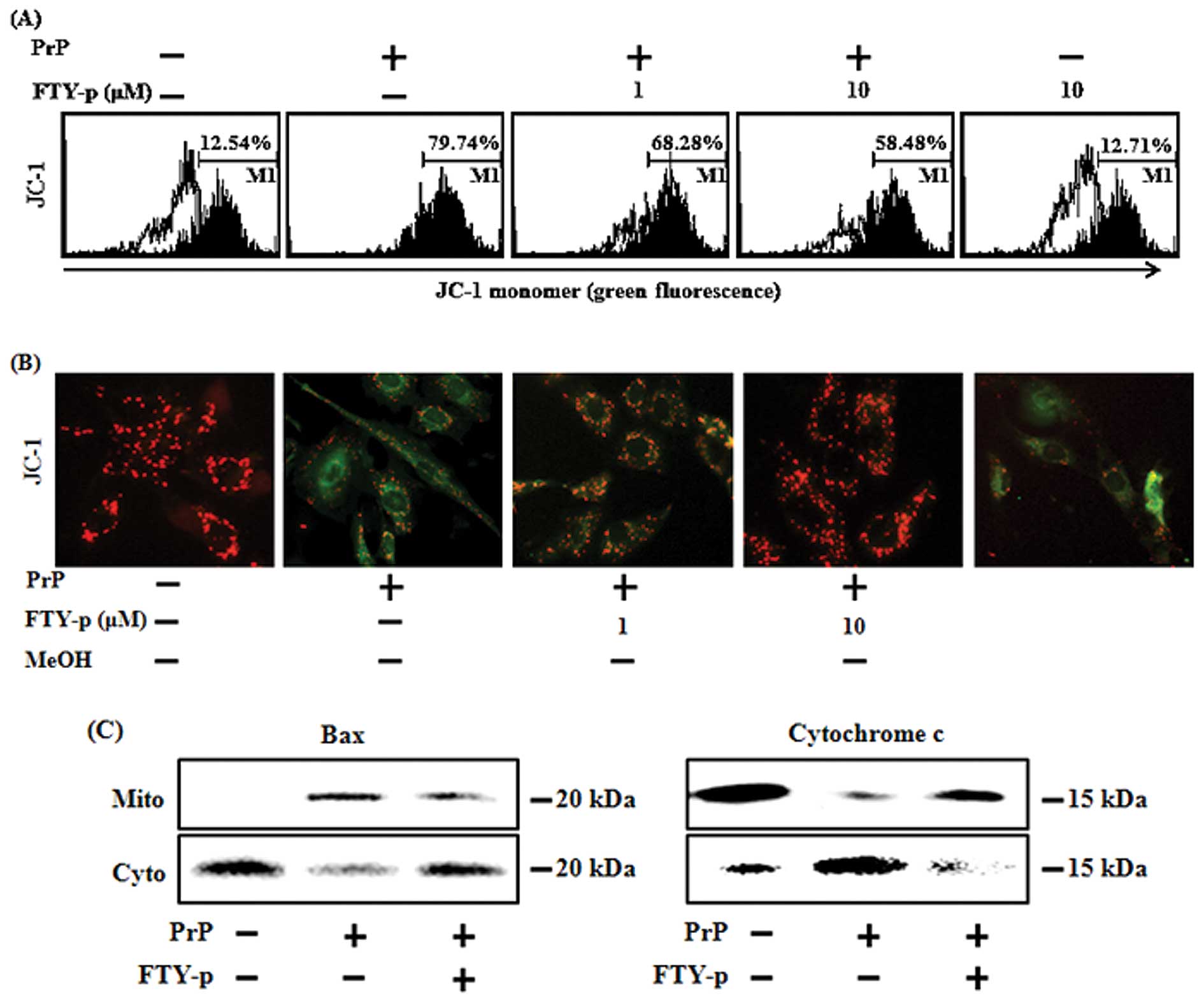
FTY720-p protects neuronal cells from PrP
(106-126)-mediated mitochondrial dysfunction
We then assessed whether the protective effects of
FTY720-p on PrP (106-126)-mediated neurotoxicity were related to
the prevention of mitochondrial dysfunction. The SH-SY5Y cells were
pre-incubated with 1 or 10 μM FTY720-p for 1 h and then exposed to
50 μM PrP (106-126). The PrP (106-126)-treated cells exhibited
increased JC-1 monomers (79.74 %), indicating low MTP values, while
treatment with FTY720-p reduced the number of PrP (106-126)-induced
JC-1 monomers (58.48%), indicating an increase in MTP values
(Fig. 2A). The fluorescence
microscopy images (Fig. 2B)
confirmed the results, depicting cells with green fluorescence
(JC-1 monomer form) following exposure to PrP (106-126), thus
indicating lower MTP values, while the control cells and FTY720-p
treated cells exhibited red fluorescence (JC-1 aggregates form),
indicating high MTP values. Given that Bax proteins act downstream
in the mitochondrial apoptotic pathway, we examined the effects of
FTY720-p on PrP (106-126)-induced Bax translocation and the release
of cytochrome c. Exposure to PrP (106-126) induced the
translocation of Bax to the mitochondria and the release of
cytochrome c release into the cytosol of SH-SY5Y cells. The
PrP (106-126)-induced Bax translocation and release of cytochrome
c were inhibited by treatment with FTY720-p (Fig. 2C). Overall these results are
consistent with the idea that FTY720-p blocks PrP (106-126)-induced
apoptosis by preventing mitochondrial dysfunction.
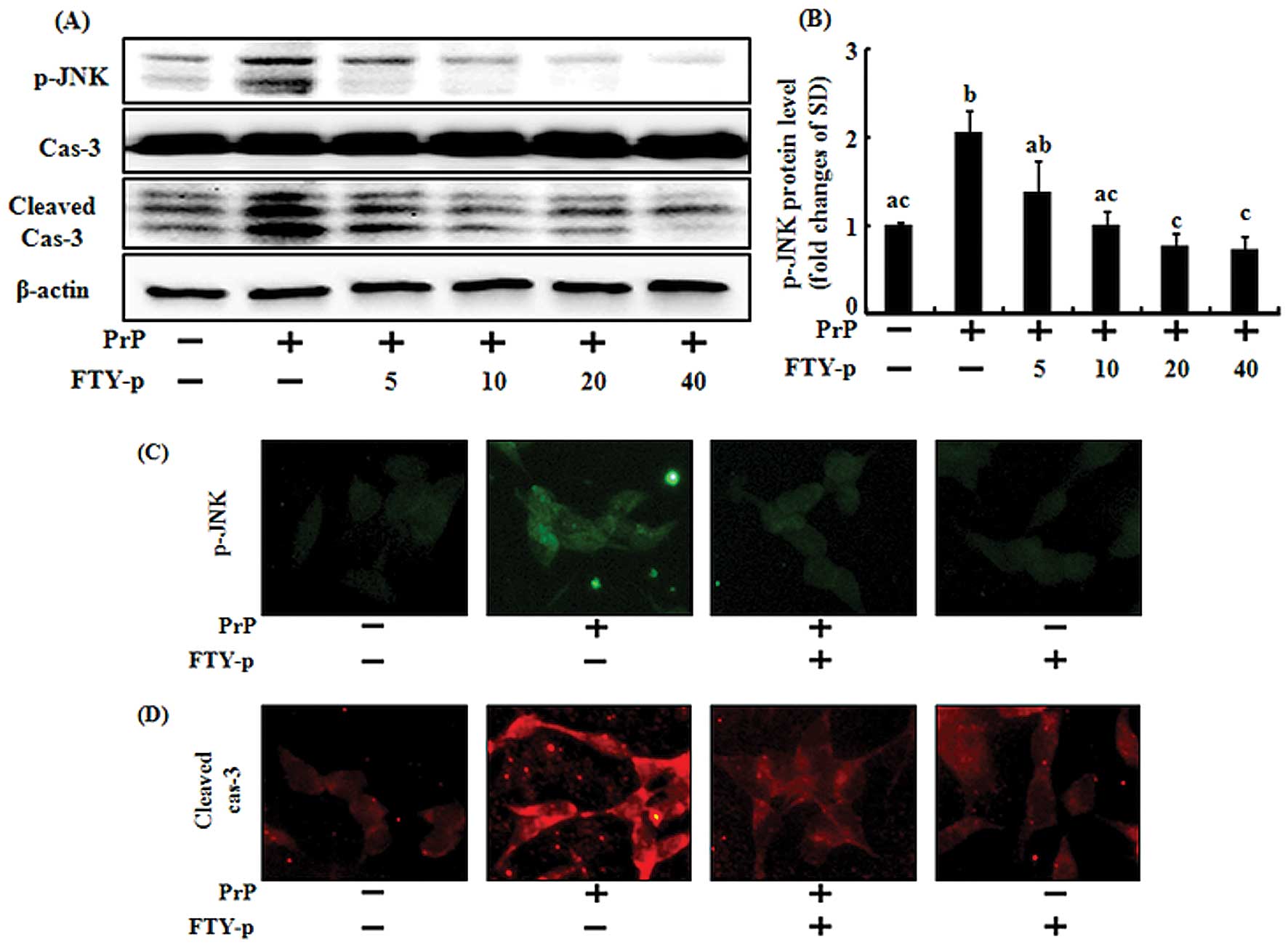
Administration of FTY720-p inhibits PrP
(106-126)-induced neurotoxicity by regulating the activation of JNK
proteins
JNK promotes the translocation of Bax to the
mitochondria and the release of mitochondrial cytochrome c,
leading to cell apoptosis (26,27). To gain insight into the molecular
mechanisms responsible for the observed biological effects of
FTY720, we examined the ability of FTY720-p to inactivate this
protein kinase. The SH-SY5Y cells were pre-incubated with various
concentrations of FTY720-p for 1 h and then exposed to PrP
(106-126) (Fig. 3). The PrP
(106-126)-treated cells displayed increased protein levels of p-JNK
(Fig. 3A). By contrast, treatment
with FTY720-p decreased p-JNK protein levels in the SH-SY5Y cells
treated with PrP (106-126) (Fig.
3A–C) in a dose-dependent manner. These results indicate that
treatment with FTY720-p prevents prion peptide-induced apoptosis by
regulating JNK activation.
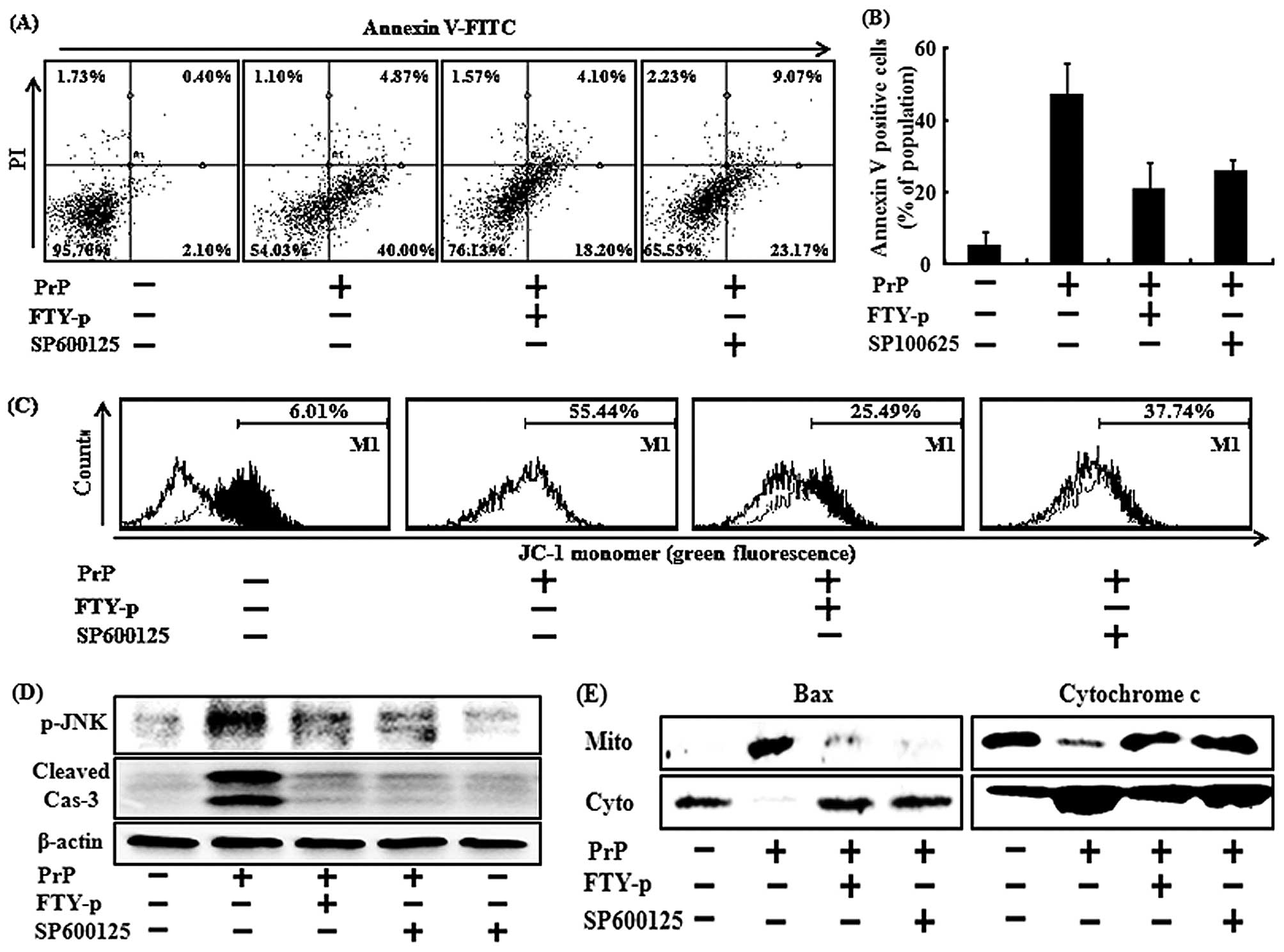
To determine whether FTY720 functions by
inactivating JNK to inhibit PrP (106-126)-induced apoptosis, the
apoptosis of the SH-SY5Y cells was induced by exposure to PrP
(106-126); the cells were either pre-treated with 10 μM of FTY720-p
or 2 μM SP600125 (a JNK inhibitor). Pre-treatment with SP600125 and
FTY720-p was sufficient to block the phosphorylation of JNK induced
by PrP (106-126) (Fig. 4D). When
SP600125 was added to the cell cultures, it prevented the increase
of Annexin V binding to membranes due to PrP (106-126) (Fig. 4A and B). The effects of SP600125
were detected at 2 μM. Treatment with 2 μM SP600125 decreased the
levels of the active form of caspase-3 to levels similar to those
observed with FTY720-p treatment (Fig. 4D).
We then we wished to determine the effects of
FTY720-induced JNK inactivation on PrP (106-126)-induced
mitochondrial dysfunction. The SH-SY5Y cells were pre-treated with
FTY720-p or SP600125 and then exposed to PrP (106-126). Treatment
with SP600125 blocked the low MTP values induced by PrP (106-126),
similar to FTY720-p treatment (Fig
4C). Treatment of the cells with SP600125 prevented the PrP
(106-126)-induced translocation of Bax and the release of
cytochrome c, as did treatment with FTY720-p, which
inhibited PrP (106-126)-mediated mitochondrial dysfunction
(Fig. 4E). These results suggest
that FTY720-p prevents PrP (106-126)-induced mitochondrial
dysfunction and apoptosis by regulating the activation of JNK.
Discussion
FTY720 has a variety of neuroprotective effects on
the CNS, protecting the CNS against MS, injury, as well as cerebral
ischemia (15,28). However, to our knowledge, the
effects of FTY720 on neurodegenerative diseases, particularly
prion-mediated diseases, have not yet been reported. In this study,
we demonstrate that FTY720 acts as a protective regulator of
neuronal cell damage induced by PrP (106-126) and that FTY720
mediates the activation of JNK. Treatment with PrP (106-126)
effectively inhibited cell survival by inducing mitochondrial
dysfunction. However, treatment with FTY720-p blocked PrP
(106-126)-mediated mitochondrial damage and apoptosis by
inactivating JNK. Similarly, the abrogation of JNK led to the
recovery of the impaired mitochondrial function and decreased cell
viability induced by PrP (106-126).
FTY720, a non-selective S1P receptor agonist that
induces sustained lymphopenia and accumulates in the CNS,
represents a novel treatment modality for MS (29). In 2010, it became the first oral
drug to be approved by the Food and Drug Administration for
clinical use in the treatment of MS. In addition to its use in the
treatment of MS, FTY720 exerts pleiotropic effects on
oligodendrocytes and other neuronal cells (30). In a previous study, treatment of
rodent-derived oligodendrocyte progenitor cells with FTY720 (1 μM)
rescued them from death induced by growth factor withdrawal,
treatment with cytokines and exposure to activated microglia
conditioned medium via extracellular signal-regulated kinase 1/2
and Akt signaling (18). In
accordance with these studies, in this study, we found that FTY720
exerts neuroprotective effects against prion diseases. FTY720
hindered neuronal cell death induced by PrP (106-126) by
inactivating JNK (Figs. 1 and
3).
The association between mitochondrial function and
neurodegenerative diseases has been investigated to a certain
extent. The mitochondria are critical regulators of cell death and
a key feature of neurodegeneration (31). Mitochondrial dysfunction occurs at
an early stage and is involved in disease pathogenesis (31). The regulation of mitochondrial
homeostasis affects the progression of neurodegenerative diseases,
including Alzheimer’s and Parkinson’s diseases (32). The typical pattern of
neurotoxicity in prion diseases is due to mitochondrial damage
(13). Consistent with these
studies, in our study, FTY720 inhibited prion-mediated
mitochondrial disruption and neurotoxicity. Apoptosis was induced
in PrP (106-126)-treated cells, which decreased MTP values and
induced the translocation of Bax protein to the mitochondria
(Fig. 2). However, treatment with
FTY720-p attenuated the damaging effects induced by the prion
peptide in neuronal cells (Fig.
2).
JNK is considered a competent inducer of the release
of cytochrome c from the intermembrane space of the brain
mitochondria and of the translocation of Bax to the mitochondria,
thus initiating an essential step in mitochondrion-dependent
apoptosis (27,33). To the best of our knowledge, the
effects of FTY720 on JNK signaling in neurodegenerative disorders
have not been reported to date. In the present study, we
demonstrate that FTY720 prevents PrP (106-126)-mediated neuronal
cell mitochondrial disruption by inactivating JNK (Figs. 3 and 4).
It remains to be clarified which S1P receptors are
related to the neuroprotective effects of FTY720. Further studies
are required to determine the influence of S1P receptors on PrP
(106-126)-mediated neurotoxicity in vitro and/or in
vivo. As the Food and Drug Administration has already approved
the oral drug that readily crosses the blood-brain barrier, FTY720
is considered an attractive candidate for neuroprotection. To the
best of our knowledge the results of the current study, for the
first time, attest to its ability to promote the survival of
neuronal cells and protect them against prion-mediated neuronal
damage by protecting the mitochondria. Future studies should
include an evaluation of the therapeutic effects of FTY720 in
conjunction with prion disease in in vivo animal models, as
well as an examination of the hypothesis that prion-related
neurodegenerative diseases may be attenuated by treatment with
FTY720.
Acknowledgements
This study was supported by the National Research
Foundation of Korea Grant funded by the Korean Government
(2013R1A2A2A01009614).
References
|
1
|
Prusiner SB: Prions. Proc Natl Acad Sci
USA. 95:13363–13383. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Seo JS, Moon MH, Jeong JK, et al: SIRT1, a
histone deacetylase, regulates prion protein-induced neuronal cell
death. Neurobiol Aging. 33:1110–1120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Seo JS, Seol JW, Moon MH, Jeong JK, Lee YJ
and Park SY: Hypoxia protects neuronal cells from human prion
protein fragment-induced apoptosis. J Neurochem. 112:715–722. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Watt NT, Taylor DR, Gillott A, Thomas DA,
Perera WSS and Hooper NM: Reactive oxygen species-mediated
beta-cleavage of the prion protein in the cellular response to
oxidative stress. J Biol Chem. 280:35914–35921. 2005. View Article : Google Scholar
|
|
5
|
Jeong JK, Seo JS, Moon MH, Lee YJ, Seol JW
and Park SY: Hypoxia-inducible factor-1 α regulates prion protein
expression to protect against neuron cell damage. Neurobiol Aging.
33:1006.e1–10. 2012.
|
|
6
|
Gray F, Chrétien F, Adle-Biassette H, et
al: Neuronal apoptosis in Creutzfeldt-Jakob disease. J Neuropathol
Exp Neurol. 58:321–328. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liberski PP, Sikorska B,
Bratosiewicz-Wasik J, Gajdusek DC and Brown P: Neuronal cell death
in transmissible spongiform encephalopathies (prion diseases)
revisited: from apoptosis to autophagy. Int J Biochem Cell Biol.
36:2473–2490. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Forloni G, Angeretti N, Chiesa R, et al:
Neurotoxicity of a prion protein fragment. Nature. 362:543–546.
1993. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brown DR, Schmidt B and Kretzschmar HA:
Role of microglia and host prion protein in neurotoxicity of a
prion protein fragment. Nature. 380:345–347. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Florio T, Thellung S, Amico C, et al:
Prion protein fragment 106-126 induces apoptotic cell death and
impairment of L-type voltage-sensitive calcium channel activity in
the GH3 cell line. J Neurosc Res. 54:341–352. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ettaiche M, Pichot R, Vincent J-P and
Chabry J: In vivo cytotoxicity of the prion protein fragment
106-126. J Biol Chem. 275:36487–36490. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jeong JK, Moon MH, Lee YJ, Seol JW and
Park SY: Autophagy induced by the class III histone deacetylase
Sirt1 prevents prion peptide neurotoxicity. Neurobiol Aging.
34:146–156. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O’Donovan CN, Tobin D and Cotter TG: Prion
protein fragment PrP-(106-126) induces apoptosis via mitochondrial
disruption in human neuronal SH-SY5Y cells. J Biol Chem.
276:43516–43523. 2001.PubMed/NCBI
|
|
14
|
Kiuchi M, Adachi K, Kohara T, et al:
Synthesis and immunosuppressive activity of 2-substituted
2-aminopropane-1,3-diols and 2-aminoethanols. J Med Chem.
43:2946–2961. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wei Y, Yemisci M, Kim HH, et al:
Fingolimod provides long-term protection in rodent models of
cerebral ischemia. Ann Neurol. 69:119–129. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kihara A and Igarashi Y: Production and
release of sphingosine 1-phosphate and the phosphorylated form of
the immunomodulator FTY720. Biochim Biophys Acta. 1781:496–502.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stessin AM, Gursel DB, Schwartz A, et al:
FTY720, sphingosine 1-phosphate receptor modulator, selectively
radioprotects hippocampal neural stem cells. Neurosci Lett.
516:253–258. 2012. View Article : Google Scholar
|
|
18
|
Miron VE, Schubart A and Antel JP: Central
nervous system-directed effects of FTY720 (fingolimod). J Neurol
Sci. 274:13–17. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harada J, Foley M, Moskowitz MA and Waeber
C: Sphingosine-1-phosphate induces proliferation and morphological
changes of neural progenitor cells. J Neurochem. 88:1026–1039.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chun J, Weiner JA, Fukushima N, et al:
Neurobiology of receptor-mediated lysophospholipid signaling. From
the first lysophospholipid receptor to roles in nervous system
function and development. Ann N Y Acad Sci. 905:110–117. 2000.
View Article : Google Scholar
|
|
21
|
Edsall LC, Pirianov GG and Spiegel S:
Involvement of sphingosine 1-phosphate in nerve growth
factor-mediated neuronal survival and differentiation. J Neurosci.
17:6952–6960. 1997.PubMed/NCBI
|
|
22
|
Jackson SJ, Giovannoni G and Baker D:
Fingolimod modulates microglial activation to augment markers of
remyelination. J Neuroinflammation. 8:762011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Deogracias R, Klein C, Matsumoto T, et al:
Expression of brain-derived neurotrophic factor is regulated by
fingolimod (FTY720) in cultured neurons. Mult Scler.
14:S2432008.
|
|
24
|
Brinkmann V, Cyster JG and Hla T: FTY720:
sphingosine 1-phosphate receptor-1 in the control of lymphocyte
egress and endothelial barrier function. Am J Transplant.
4:1019–1025. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Valentine WJ, Kiss GN, Liu J, et al:
(S)-FTY720-vinylphosphonate, an analogue of the immunosuppressive
agent FTY720, is a pan-antagonist of sphingosine 1-phosphate GPCR
signaling and inhibits autotaxin activity. Cell Signal.
22:1543–1553. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tournier C, Hess P, Yang DD, et al:
Requirement of JNK for stress-induced activation of the cytochrome
c-mediated death pathway. Science. 288:870–874. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tsuruta F, Sunayama J, Mori Y, et al: JNK
promotes Bax translocation to mitochondria through phosphorylation
of 14-3-3 proteins. EMBO J. 23:1889–1899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ehling R, Berger T and Reindl M: Multiple
sclerosis - established and novel therapeutic approaches. Cent Nerv
Syst Agents Med Chem. 10:3–15. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gonzalez-Cabrera PJ, Cahalan SM, Nguyen N,
et al: S1P(1) receptor modulation with cyclical recovery from
lymphopenia ameliorates mouse model of multiple sclerosis. Mol
Pharmacol. 81:166–174. 2012.
|
|
30
|
Kim HJ, Miron VE, Dukala D, et al:
Neurobiological effects of sphingosine 1-phosphate receptor
modulation in the cuprizone model. FASEB J. 25:1509–1518. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin MT and Beal MF: Mitochondrial
dysfunction and oxidative stress in neurodegenerative diseases.
Nature. 443:787–795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jeong JK, Moon MH, Lee YJ, Seol JW and
Park SY: Melatonin-induced autophagy protects against human prion
protein-mediated neurotoxicity. J Pineal Res. 53:138–146. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schroeter H, Boyd CS, Ahmed R, et al:
c-Jun N-terminal kinase (JNK)-mediated modulation of brain
mitochondria function: new target proteins for JNK signalling in
mitochondrion-dependent apoptosis. Biochem J. 372:359–369. 2003.
View Article : Google Scholar
|