Introduction
Liver fibrosis is a wound-healing response to
various chronic liver injuries caused by alcoholism, persistent
viral and parasite infections, or hereditary metal overload
(1,2). Hepatic stellate cells (HSCs) play a
pivotal role in liver fibrogenesis. During liver injury, quiescent
HSCs transdifferentiate into activated HSCs, also termed
myofibroblast (MFB)-like cells. This cell type provides the main
components of the extracellular fibrotic matrix, and also produces
an array of pro-inflammatory cytokines and chemokines involved in
the development of liver fibrosis (3,4).
Thus, HSCs are considered the most important target for therapeutic
intervention to prevent the development of hepatic fibrosis
(1,5).
There is accumulating evidence that the
lipopolysaccharide (LPS)/Toll-like receptor 4 (TLR4) signaling
pathway in HSCs plays a key role in liver fibrogenesis (6,7).
Previous studies have demonstrated that the levels of the LPS
endotoxin are elevated in experimental models of hepatic fibrosis
(6) and in patients with liver
cirrhosis (8,9), and that the administration of
antibiotics that reduce the prevalence of LPS can inhibit
fibrogenesis and TLR4 expression in the liver (10). In chronic liver diseases, upon the
disruption of the intestinal barrier function, the increase in
intestinal permeability leads to the translocation of
intestine-derived bacterial products to the liver via the portal
vein (10,11). Although Kupffer cells are
considered to be the main targets of bacterial products in the
liver, there is also evidence that HSCs, and not Kupffer cells, are
the primary targets of TLR4 ligands, through which they induce a
fibrogenic phenotype (6). Both
quiescent and activated HSCs express TLR4 and are thereby highly
responsive to even low concentrations of LPS. TLR4 signaling was
identified as the molecular link between inflammation and hepatic
fibrosis (6,12,13). LPS activates the TLR4/myeloid
differentiation 2 (MD2) signaling complex through binding to serum
and cell surface proteins, including LPS-binding protein (LBP) and
CD14. This subsequently activates downstream effectors such as
nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) (14). Finally, these pathways regulate
the expression of pro-inflammatory cytokines and genes that control
cell survival and apoptosis (14).
LPS/TLR4 signaling in HSCs is essential for the
development of liver fibrosis. Mice with mutations in the genes
coding for TLR4, CD14, LBP and myeloid differentiation factor 88
show reduced liver fibrosis after bile duct ligation (BDL) or
treatment with carbon tetrachloride (6,10,15). Seki et al (6) analyzed the cell-specific molecular
mechanism underlying the role of LPS/TLR4 signaling on liver
fibrosis. They demonstrated that chimeric mice that carry
TLR4-mutant Kupffer cells and TLR4-intact HSCs develop pronounced
fibrosis, while mice that carry TLR4-intact Kupffer cells and
TLR4-mutant HSCs develop minimal fibrosis after BDL, indicating
that TLR4 is crucial for the induction of hepatic fibrosis in HSCs,
but not in Kupffer cells. LPS/TLR4 signaling in HSCs induces the
production of chemokines, which recruit Kupffer cells at HSC sites
and allow unrestricted activation of HSCs by the Kupffer
cell-derived protein known as transforming growth factor-β (TGF-β)
(5,6,13).
Thus, inhibition of LPS/TLR4 signaling in HSCs appears to be a
promising strategy for the prevention of liver fibrosis.
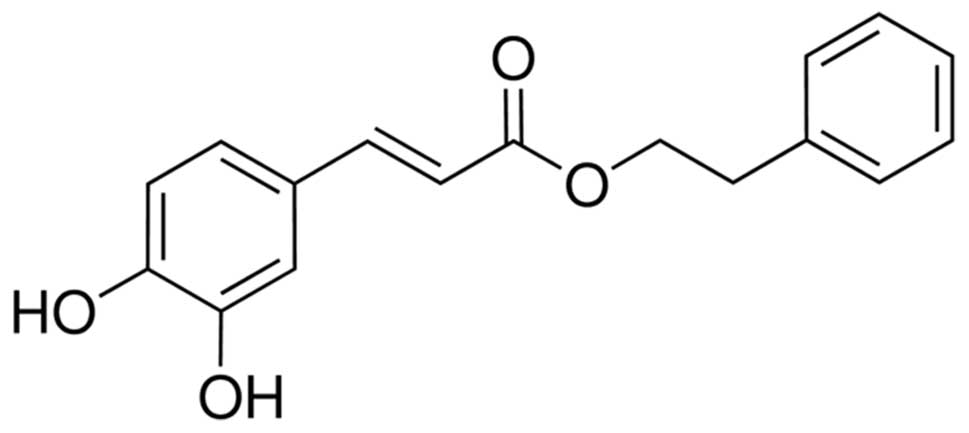
Caffeic acid phenethyl ester (CAPE) (Fig. 1) is one of the main medicinal
components of propolis, which is a naturopathic medicine collected
by honeybees from buds and exudates of conifer trees and plants
(16,17). A number of important biological
activities have been reported for CAPE, such as antioxidant
(18,19), anti-inflammatory (20) and anticancer activities (21). Previous studies have indicated
that CAPE is a potent inhibitor of NF-κB (20,22,23). For example, CAPE inhibits
Helicobacter pylori-induced NF-κB and AP-1 DNA binding
activities in a dose and time-dependent manner in gastric
epithelial cells (24). In
another study conducted on the colorectal carcinoma cell line,
HCT116, CAPE affected tumor necrosis factor-α (TNF-α)-dependent
IκBα degradation and the subsequent nuclear accumulation of NF-κB
(p65) through the direct inhibition of the inhibitory protein κB
kinase (IKK), as well as through the activation of the nuclear
factor (erythroid-derived 2)-like 2 (Nrf2) pathway (22).
Since LPS/TLR4 signaling plays a crucial role in
liver fibrosis in HSCs, the aim of this study was to investigate
the effects of CAPE on the pro-inflammatory and fibrogenic
phenotypes of LPS-stimulated HSCs.
Materials and methods
Animals and reagents
Male Wistar rats, weighing 450–500 g, were used in
the experiments, and were provided by the Laboratory Animal Center
of Kunming Medical University. The study was performed in
accordance with the principles for the care and use of laboratory
animals approved by The Research Ethics Committee of the Kunming
General Hospital of PLA (no. K2010-008).
Dulbecco’s modified Eagle’s medium (DMEM) and fetal
bovine serum (FBS) were obtained from Gibco-BRL (Los Angeles, CA,
USA). CAPE, LPS, TGF-β1, Necodenz, proteinase E and collagenase II
were purchased from Sigma-Aldrich (St. Louis, MO, USA). TRIzol,
M-MuLV reverse transcriptase, Platinum SYBR-Green qPCR
SuperMix-UDG, and enzyme-linked immunosorbent assay (ELISA) kits
for assaying rat interleukin-6 (IL-6) and monocyte chemoattractant
protein-1 (MCP-1) were provided by Invitrogen Life Technologies
(San Diego, CA, USA). ELISA kits for rat NF-κB were purchased from
Wkea Med Supplies Corp. (Changchun, China).
Cell isolation, culture and
identification
HSCs were prepared by in situ sequential
perfusion of proteinase E/collagenase II at 37°C and
density-gradient centrifugation, as previously described (25,26). Briefly, the rats were anesthetized
with pentobarbital and the livers were perfused first with
Ca2+- and Mg2+-free solution for 10 min at
37°C, and next with 0.05% (w/v) collagenase II and 0.01% proteinase
E solution for 30 min at 37°C. The digested livers were excised,
suspended in D-Hanks solution and filtered through a sterile gauze.
Residual hepatocytes were removed by two low-speed centrifugations
(50 × g) at 4°C for 2 min. The filtered suspension was centrifuged
at 540 × g at 4°C for 5 min. The HSC-enriched fraction was obtained
by centrifugation with a triple-layered (9, 11 and 17%) Nycodenz
cushion at 1,400 × g at 4°C for 20 min. The cells in the upper
layer were washed twice and incubated, on Corstar®
6-well plastic plates, with DMEM medium containing 10 mM HEPES,
penicillin (100 U/ml), streptomycin (100 μg/ml) and 10% FBS, at
37°C in a humidified atmosphere containing 5% CO2. Cell
viability was determined by Trypan blue staining. The purity of the
isolated HSCs was analyzed by flow cytometry: all cells were
stained with propidium iodide and the HSCs were stained with the
monoclonal primary anti-desmin antibody and then with the secondary
antibody FITC-anti-IgG (Abcam, Cambridge, UK). HSCs spontaneously
underwent transdifferentiation to MFB-like HSCs on plastic plates,
and HSCs on day 7 of the primary culture were considered as
myofibroblastic cells/activated HSCs when they expressed all
features of MFBs.
Nitrite assay
HSCs from day 7 of primary culture were trypsinized
and seeded in 6-well culture plates at a density of
5×105 cells/well for 12 h. The cells were pre-treated
with various concentrations of CAPE (0.0, 5.0, 10.0, 20.0 and 40.0
μM) for 2 h, and then stimulated with LPS (100 ng/ml) for 24 h. At
the end of these treatments, nitrite, the oxidation product of
nitric oxide (NO), was quantified in conditioned medium from HSCs,
using a spectrophotometric assay as previously described (27). Absorbance values were converted to
nitrite concentrations using standard curves obtained from
spectrophotometric analysis of known concentrations of potassium
nitrite, prepared in the same culture medium.
Quantitative reverse
transcription-polymerase chain reaction (qRT-PCR)
To analyze the expression of genes coding for
pro-inflammatory mediators, HSCs were treated as described in the
nitrite assay. For the analysis of the collagen gene, HSCs from
days 3 and 7 (activated HSCs) of the primary culture were used.
HSCs were pre-treated with various concentrations of CAPE for 2 h,
then treated with 100 ng/ml LPS, 5 ng/ml TGF-β1 or the vehicle
[phosphate-buffered saline (PBS)] for 24 h. At the end of the
treatment, total RNA from the HSCs was extracted using TRIzol
reagent. The concentration and purity of total RNA was
spectrophotometrically assessed at 260 and 280 nm. The RNA was
reverse transcribed into cDNA using a M-MuLV reverse transcriptase
with RNase inhibitor. Oligonucleotide primers for quantitative PCR
were designed with the freely available Primer Premier 5.0
software, based on the mRNA sequences of MCP-1, IL-6,
inducible nitric oxide synthase (iNOS), collagen type I α1 (col1A1)
and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) genes. The
primer sequences were as follows: col1A1 forward,
5′-TGGTTTCCCTGGTGCTGATG-3′ and reverse, 5′-CTGCCAGTGAGACCCTTGG-3′;
iNOS forward, 5′-CAGGTCGGCCATTACTGTGT-3′ and reverse,
5′-CGATGCACAACTGGGTGAAC-3′; MCP-1 forward,
5′-CTGTGCTGACCCCAATAAGGA-3′ and reverse,
5′-GCTTGAGGTGGTTGGGAAAAG-3′; IL-6 forward,
5′-ATGAGAAAAGAGTTGTGCAATGG-3′ and reverse,
5′-GGAACTCCAGAAGACCAGAGC-3′; and GAPDH forward,
5′-CCCAGAACATCATCCCTGCAT-3′ and reverse,
5′-CATACTTGGCAGGTTTCTCCA-3′. PCR reactions were carried out on an
ABI 7500 Thermal Cycler (Applied Biosystems, Foster City, CA, USA).
We prepared on ice a reaction mixture containing 1 μl cDNA, 12.5 μl
Platinum SYBR-Green qPCR SuperMix-UDG and 1 μl of each primer, and
added H2O to a final volume of 25 μl. The reaction
conditions included an initial step at 95°C for 5 min, followed by
40 cycles at 95°C for 15 sec, 58°C for 10 sec and 72°C for 10 sec.
Each sample was analyzed in duplicate PCR reactions. We analyzed
the qRT-PCR data using the comparative Ct (ΔΔCT) method, as
previously described (28).
Cytokine ELISA
MCP-1 and IL-6 protein concentrations were measured
in HSC-conditioned medium using available ELISA kits, according to
the manufacturer’s instructions. Optical densities (OD) were
measured using an ELISA plate reader (Bio-Rad, Hercules, CA, USA)
at 450 nm wavelength.
Western blot analysis
The preparation of whole-cell lysates was carried
out as previously described (29). Cytosolic extracts were prepared
using the Cytoplasmic Protein Extraction kit (BioTeke, Beijing,
China) according to the instructions of the manufacturer. Western
blot analysis was performed as described in a previous study
(30). Equal amounts of protein
were resolved on a 10% SDS-PAGE gel and transferred onto a PVDF
membrane. The blot was then blocked with 5% non-fat dry milk and
probed overnight with primary antibodies, followed by incubation
with HRP-conjugated secondary antibodies. The primary rabbit
antibodies used were the following: anti-p-IκBα, anti-IκBα and
anti-GAPDH (all at 1:1,000 dilution; Cell Signaling Technology,
Inc., Danvers, MA, USA); anti-iNOS (1:200) and anti-col1A1
antibodies (1:2,000) (both from LifeSpan BioSciences Inc., Seattle,
WA, USA). Protein bands were visualized using a chemiluminescent
reagent.
NF-κB translocation assay
The cells were pre-treated with various
concentrations of CAPE for 2 h and then treated with 100 ng/ml LPS
for 30 min. At the end of the treatment, nuclear extracts were
isolated by using the Nuclear Extract kit (Active Motif, Carlsbad,
CA, USA). Briefly, cells were collected in a PBS/phosphatase
inhibitors solution and lysed in a buffer containing DTT and a
cocktail of protease inhibitors as per the manufacturer’s
recommendations. Solubilized proteins were then separated from cell
debris by centrifugation at 14,000 × g for 30 min. The protein
concentration of the nuclear fraction was measured, and the protein
content was adjusted to obtain the same concentration in all the
samples. The translocation of NF-κB was measured by the
corresponding ELISA kit following the manufacturer’s
recommendations.
Statistical analysis
All data are presented as the means ± SD.
Statistical analysis was performed using SPSS software 13.0, with a
P-value <0.05 considered to indicate a statistically significant
difference.
Results
Activation and identification of
HSCs
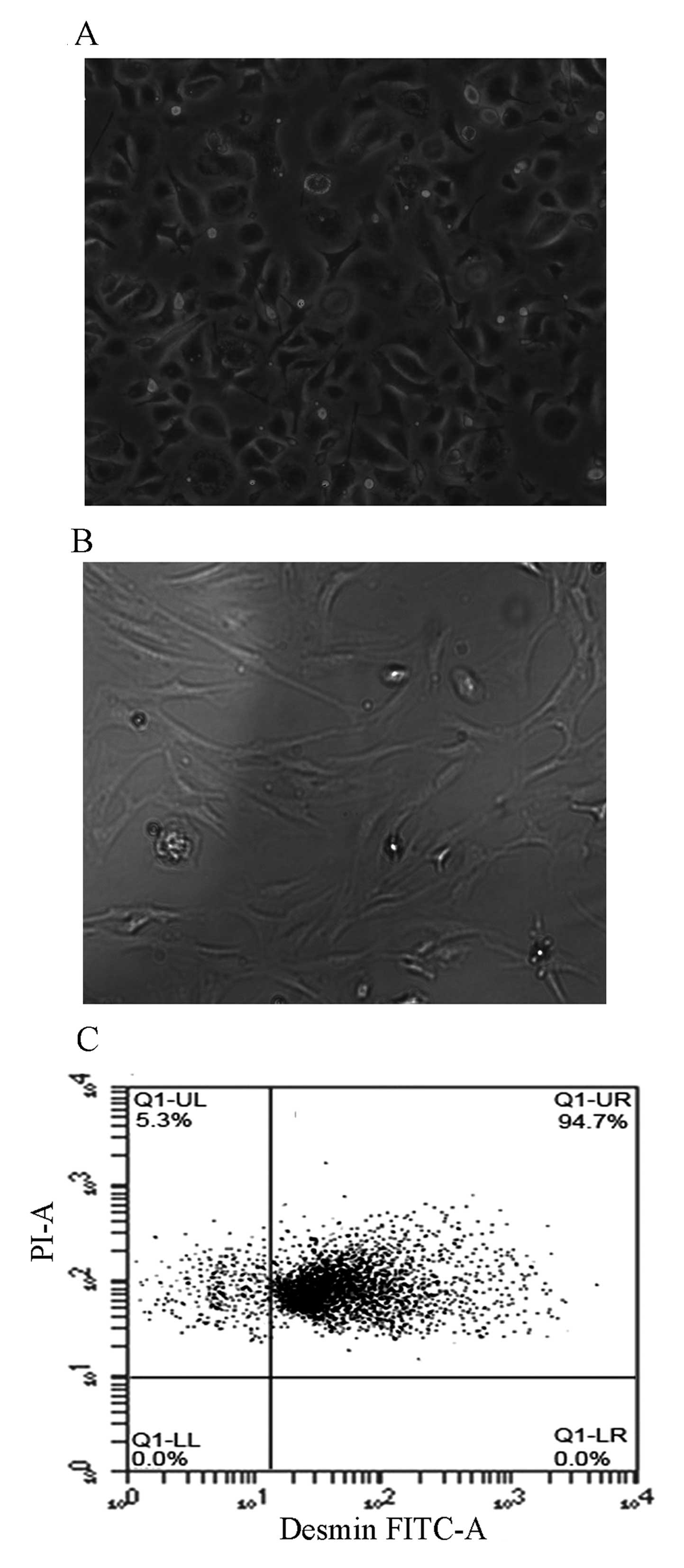
Freshly isolated HSCs from rats were cultured on
plastic plates in medium containing 10% FBS. In phase contrast
microscopic observations, the HSCs spontaneously underwent
transdifferentiation into MFB-like HSCs from day 2–3, and became
fully activated on day 7 of primary culture. Freshly isolated HSCs
were spherical. Following culture for 48 h, the cells were
spindle-shaped or spherical (Fig.
2A). On day 7 of primary culture, HSCs were fully spreading,
presenting stellate or polygonal forms (Fig. 2B). The yield rate of HSCs was
(1–6)x107/rat liver, and cell
viability was >90%, as determined by Trypan blue staining. Flow
cytometric analysis revealed that the percentage of desmin-positive
cells was 94.7% of the freshly isolated HSCs (Fig. 2C), indicating that an adequate
level of purity was achieved for the subsequent experiments.
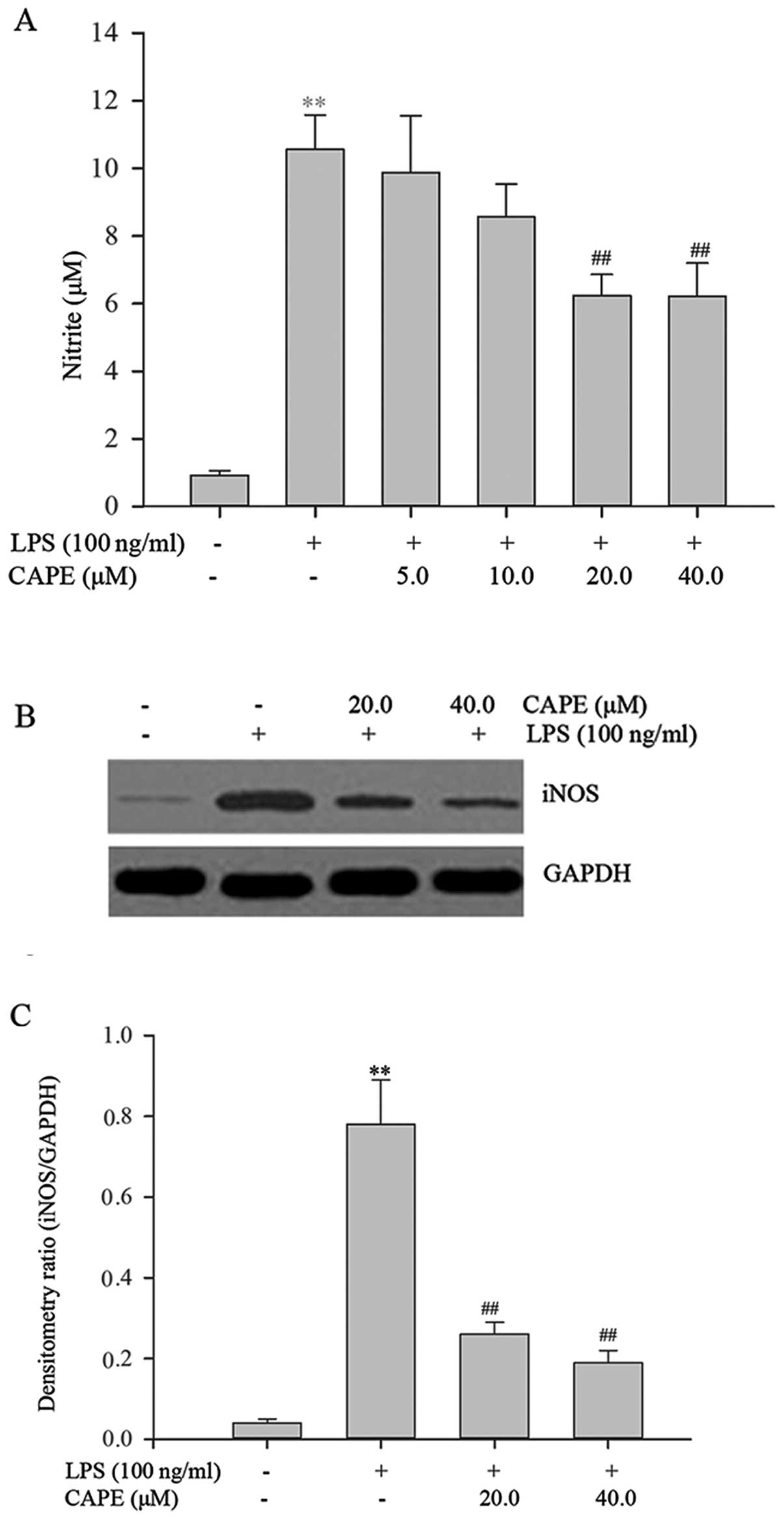
Inhibitory effects of CAPE on LPS-induced
nitrite and iNOS production in HSCs
LPS stimulation resulted in a 11.5-fold increase in
the nitrite level compared to the vehicle-treated control. CAPE
inhibited this induction in a dose-dependent manner (Fig. 3A), with 20 or 40 μM of CAPE
significantly inhibiting LPS-induced nitrite production
(P<0.01). In lactate dehydrogenase (LDH) assays, performed so as
to assess the effect of CAPE on cell toxicity, there were no
significant differences between the groups, indicating that CAPE
treatment was not toxic under the tested concentrations (data not
shown). As shown by western blot analysis, a very weak iNOS band
was detected in the vehicle-treated control (Fig. 3B). By contrast, iNOS protein
expression was markedly increased in the LPS-stimulated cells and
it was inhibited in the cells pre-treated with CAPE. Densitometric
analysis (Fig. 3C) revealed that
iNOS expression was significantly increased upon LPS treatment
compared to treatment with the vehicle, and CAPE pre-treatment
markedly and significantly reduced the LPS-induced iNOS expression
compared to LPS treatment alone (P<0.01).
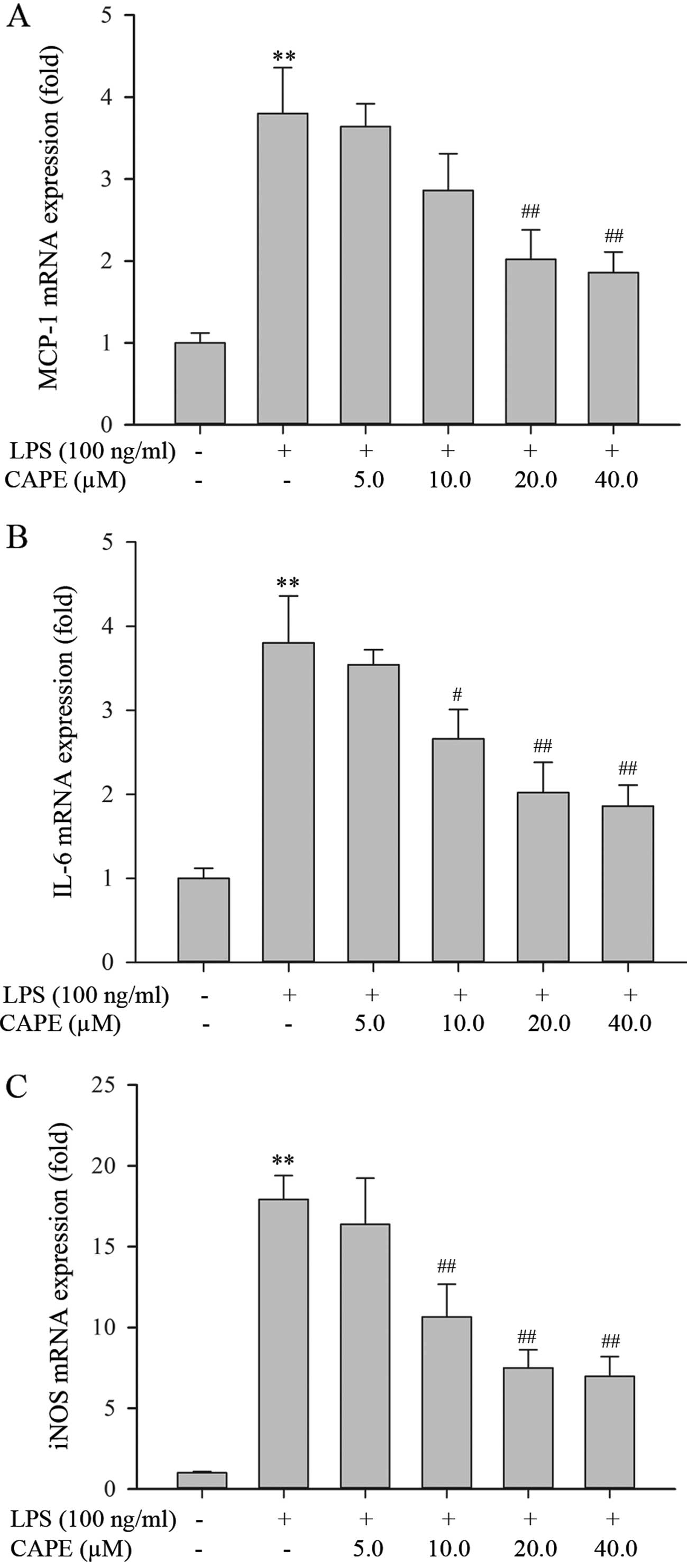
CAPE decreases the transcript levels of
pro-inflammatory mediator genes in HSCs
An earlier study suggested that LPS/TLR4 signaling
in HSCs constitutes the molecular link between inflammation and
liver fibrosis (6). It was thus
interesting to examine whether CAPE inhibits the LPS-induced
expression of genes coding for pro-inflammatory mediators. The
levels of MCP-1, IL-6 and iNOS transcripts (mRNA) were quantified
in the HSCs by qRT-PCR. Upon exposure to 100 ng/ml of LPS for 24 h,
the HSCs developed a strong pro-inflammatory phenotype, as shown by
the increased mRNA expression of MCP-1, IL-6 and iNOS (Fig. 4). CAPE pre-treatment significantly
reduced the LPS-induced mRNA expression of MCP-1, IL-6 and iNOS in
a dose-dependent manner (Fig. 4).
These data demonstrate that CAPE attenuates the pro-inflammatory
phenotype of LPS-stimulated HSCs.
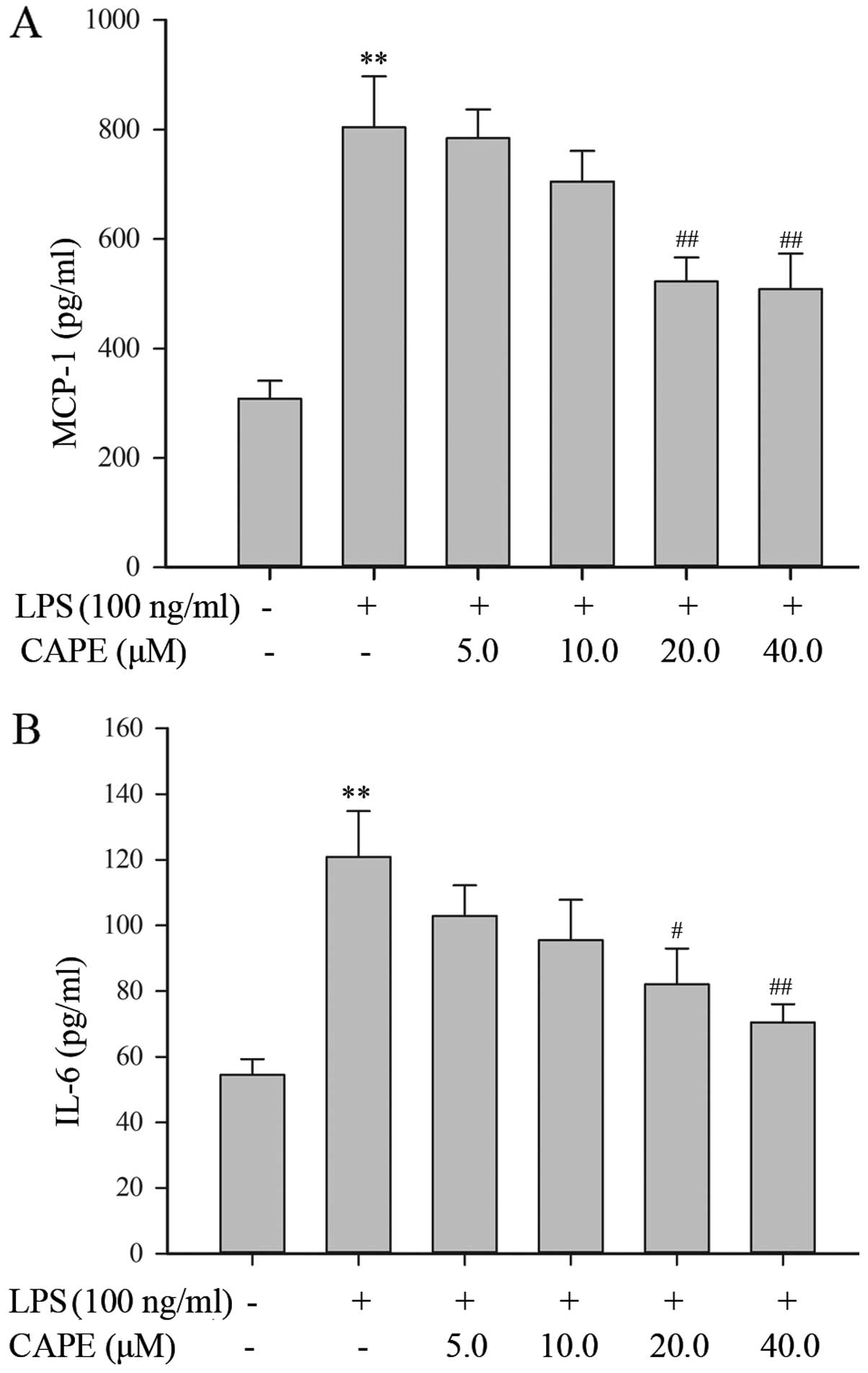
CAPE decreases pro-inflammatory cytokine
release from LPS-stimulated HSCs
In response to LPS stimulation, the HSCs secreted
considerable amounts of MCP-1 and IL-6, with a 2.6- and 2.2-fold
increase relative to the vehicle-treated control, respectively.
Following treatment with 20 or 40 μM CAPE, the production of IL-6
and MCP-1 proteins by LPS-stimulated HSCs was significantly
decreased (Fig. 5). This result
is consistent with the data obtained for mRNA expression by qRT-PCR
(Fig. 5).
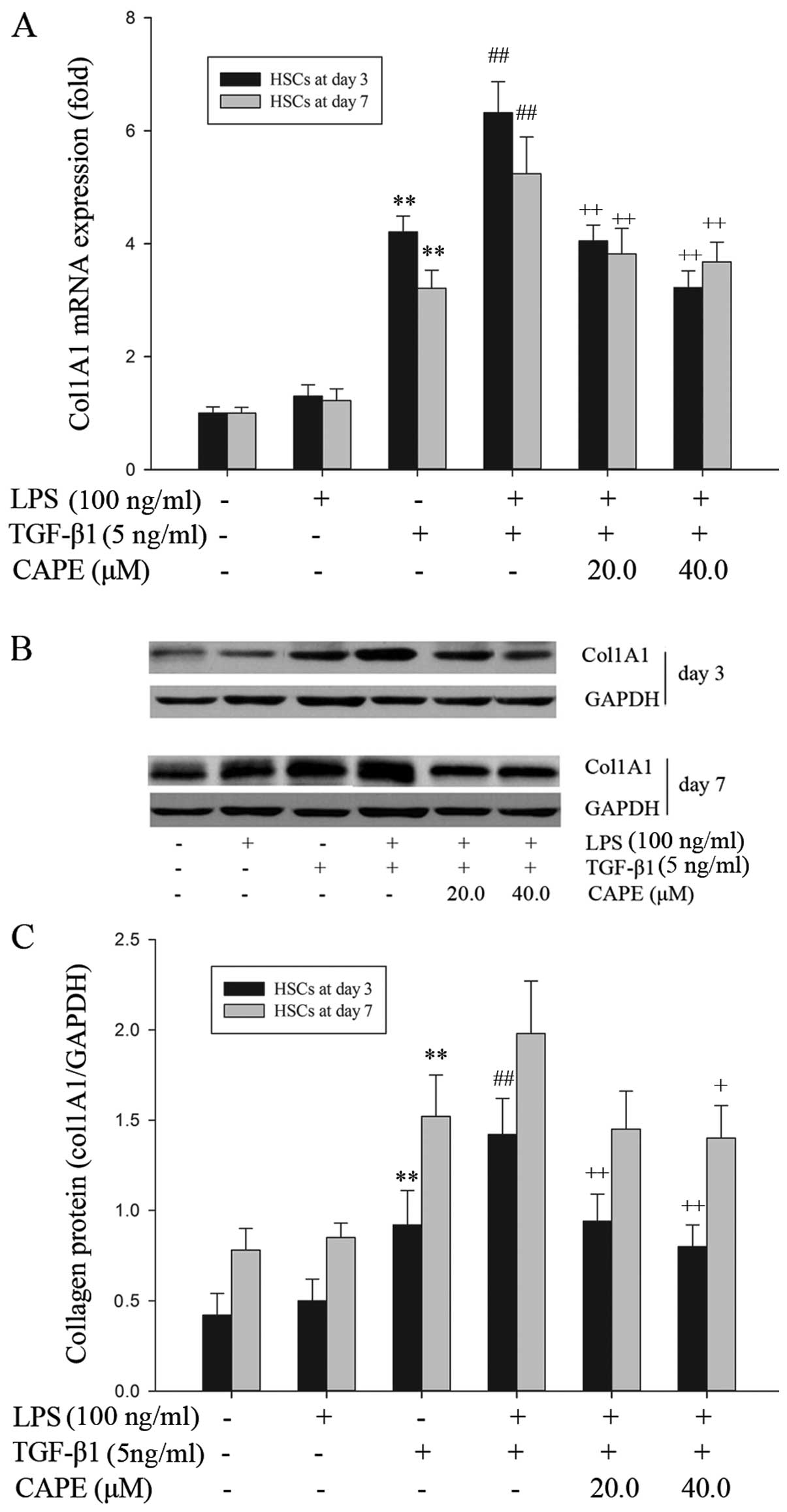
CAPE attenuates TGF-β1-induced collagen
synthesis in LPS-stimulated HSCs
Kupffer cell-derived TGF-β1 is a highly potent
profibrogenic growth factor that activates collagen synthesis in
HSCs in vivo (6,31). In the present study, we
investigated the effects of CAPE on the TGF-β1-induced fibrogenic
phenotype of LPS-stimulated HSCs showing a strong pro-inflammatory
phenotype. LPS alone did not directly induce a fibrogenic
phenotype, since collagen type 1 mRNA and protein levels were
substantially unaffected by LPS stimulation (Fig. 6). However, LPS stimulation
significantly enhanced the TGF-β1-induced mRNA expression of col1A1
in the HSCs from days 3 and 7 of primary culture, and significantly
amplified the TGF-β1-induced collagen synthesis in the HSCs from
day 3 of primary culture, as shown by qRT-PCR and western blot
analysis, respectively (Fig. 6).
Upon CAPE pre-treatment, the LPS-induced upregulation of the
transcription (mRNA expression) and translation (protein synthesis)
of col1A1 was reversed in the HSCs from day 3 and 7 of primary
culture (Fig. 6). These data
suggest that CAPE attenuates the fibrogenic phenotype of
LPS-stimulated HSCs by reducing their sensitization to TGF-β1.
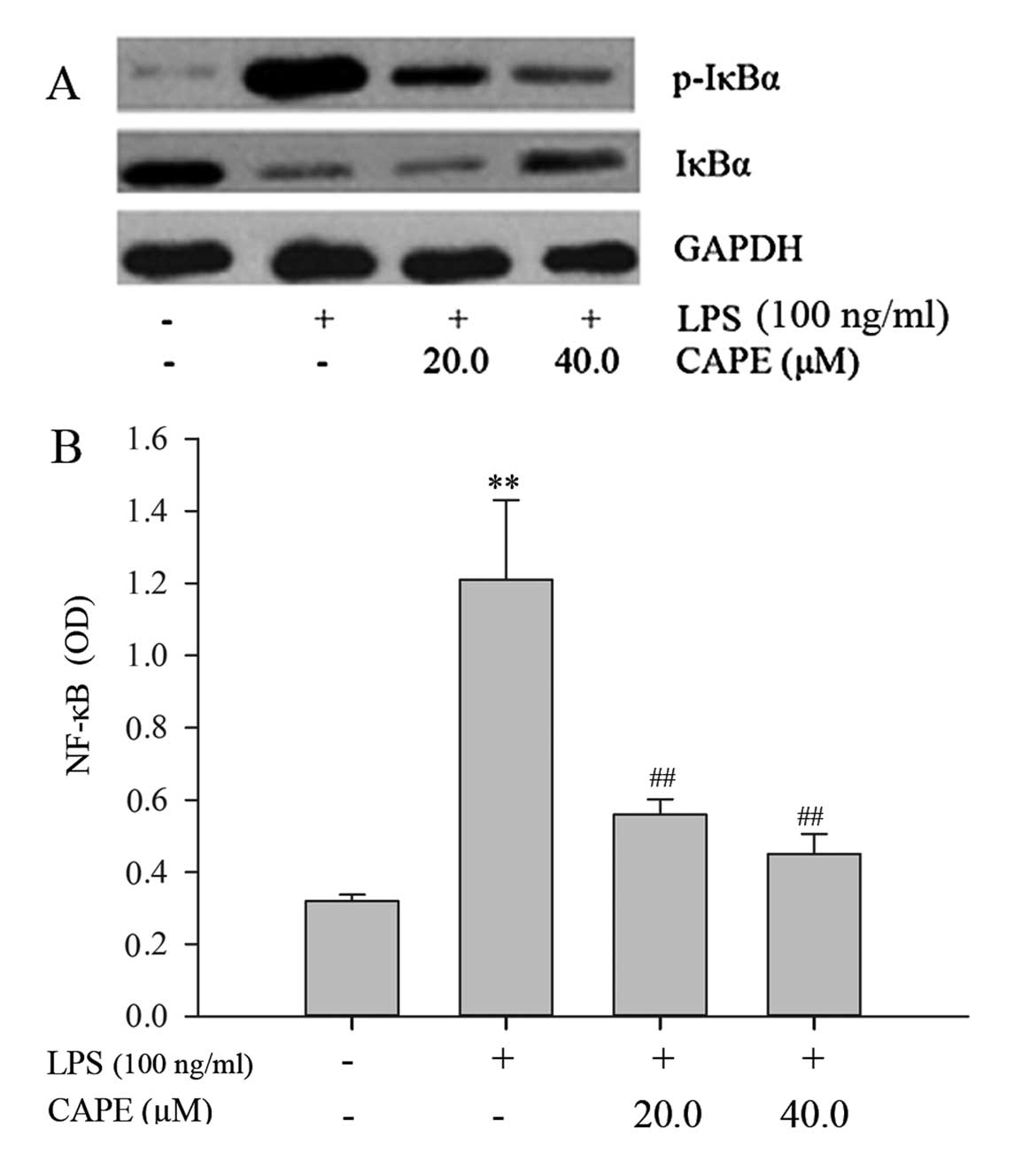
CAPE inhibits the LPS-induced
phosphorylation of IκBα and NF-κB translocation in HSCs
NF-κB is an important transcription factor,
orchestrating the production of pro-inflammatory mediators, as well
as regulating a variety of important cellular functions. To
correlate the attenuation of pro-inflammatory and fibrogenic
phenotypes by CAPE in HSCs with downstream elements of the LPS/TLR4
signal transduction pathway, we examined the phosphorylation and
degradation of IκBα by western blot analysis and the NF-κB nuclear
translocation by an ELISA assay. In response to LPS, the
phosphorylation of IκBα was markedly enhanced and IκBα was markedly
degraded. CAPE pre-treatment inhibited the phosphorylation of IκBα
and consequently prevented its degradation (Fig. 7A). LPS induced a 3.8-fold increase
in the nuclear translocation of NF-κB. Pre-treatment of the cells
with 20 and 40 μM of CAPE significantly inhibited the LPS-induced
NF-κB translocation to the nucleus by 53.7 and 62.8%, respectively
(P<0.01, compared to LPS treatment) (Fig. 7B). This result demonstrates that
the mechanism underlying the attenuation of the pro-inflammatory
and fibrogenic phenotypes of HSCs by CAPE involves the inhibition
of NF-κB signaling.
Discussion
Hepatic fibrosis is tightly linked to chronic liver
inflammation in all individuals with liver disease and in
experimental models of fibrogenesis. The molecular link between
inflammation and liver fibrosis was shown to be TLR4 signaling,
which promotes myofibroblast activation and modulates TGF-β
signaling in HSCs (6,32). Liver fibrogenesis is associated
with increased intestinal permeability (33). In hepatic sinusoids,
intestine-derived bacterial products absorbed through the
gastrointestinal wall may reach parenchymal and non-parenchymal
hepatic cells, and they are recognized by pattern recognition
receptors (PRRs) that bind to conserved microbial structures.
Typically, immune cells, such as monocytes and Kupffer cells in the
liver express basal levels of PRRs to allow an immediate and
unspecific inflammatory response to bacterial products (34,35). However, accumulating evidence
indicates that quiescent or activated HSCs also express TLR4, one
of the well-characterized PRRs, and can thereby respond to low
concentrations of LPS (6,36,37).
Our results demonstrated that LPS induced an intense
pro-inflammatory response in HSCs, leading to a marked increase in
the nitrite level and to an upregulation in the levels of
pro-inflammatory mediators. This result is consistent with previous
studies showing that bacterial products induce a strong
pro-inflammatory phenotype of HSCs, triggering the release of
pro-inflammatory mediators and thus, contributing to tissue injury
and liver fibrosis (38). The
HSC-derived factors lead to chemoattraction of bone marrow-derived
monocytes and the accumulation of Kupffer cells in the liver
(6,39), or act in an autocrine manner to
activate HSCs (39,40).
To the best of our knowledge, the present study is
the first to demonstrate that CAPE attenuates the pro-inflammatory
and fibrogenic phenotypes of LPS-stimulated HSCs. CAPE
significantly reduced the production of the NO free radical,
nitrite, and that of the pro-inflammatory mediators, MCP-1, IL-6
and iNOS, in LPS-stimulated HSCs in vitro. Previous studies
have demonstrated that CAPE exhibits a plethora of important
biological properties, such as potent anti-inflammatory, antitumor
and antioxidant activities, and attenuates inflammation and lipid
peroxidation (23,41,42). Thus, it may be possible that CAPE
can decrease liver inflammation and fibrosis in vivo by
downregulating the secretion of chemokines from HSCs, thereby
reducing the chemoattraction of monocytes and Kupffer cells in the
liver.
As regards collagen synthesis, LPS did not directly
induce col1A1 mRNA transcription and protein expression in HSCs
in vitro. In vivo, the development of hepatic fibrosis
involves a cross-talk between HSCs and other cell types,
particularly Kupffer cells. The Kupffer cell-derived TGF-β protein
plays a crucial role in promoting HSC activation and fibrogenesis
(31,43). The results from the present study
demonstrated that LPS treatment led to an increase in
TGF-β1-induced collagen production, and CAPE decreased the
sensitivity of HSCs to TGF-β1. This effect may be mediated by the
downregulation of the TGF-β pseudoreceptor, the bone morphogenetic
protein and activin membrane bound inhibitor (BAMBI), leading to
HSC activation and sensitivity to TGF-β (6,7).
Our data demonstrated that CAPE decreased the sensitivity of HSCs
to TGF-β1, indicating that CAPE may also interfere with TGF-β
receptor signaling in LPS-stimulated HSCs.
The crucial role of LPS/TLR4 signaling in HSCs
during liver fibrogenesis has been demonstrated in previous studies
(6,13). NF-κB is a downstream element of
the LPS/TLR4 signal transduction cascade, and one of the most
ubiquitous transcription factors, regulating the expression of
genes involved in cellular proliferation, inflammatory responses
and cell adhesion (44,45). In the cytosol, NF-κB remains
inactive by forming a complex with the inhibitory protein IκBα. In
response to stimuli, IκBα kinases (IκKs) mediate IκBα
phosphorylation, the dissociation of the NF-κB/IκBα complex, and
the activation of NF-κB, which then translocates to the nucleus to
activate specific target genes (46,47). Activation of NF-κB in hepatic
cells was shown to correlate with hepatic inflammation and fibrosis
(48). Our data show that CAPE
treatment inhibits LPS-induced phosphorylation of IκBα and NF-κB
nuclear translocation in HSCs. These results indicate that the
anti-inflammatory and anti-fibrogenic effects of CAPE may associate
with the inhibition of NF-κB translocation, and result in the
downregulation of pro-inflammatory genes in LPS-stimulated
HSCs.
In conclusion, the present study demonstrates that
CAPE attenuates the LPS-induced pro-inflammatory and fibrogenic
phenotypes in rat HSCs via its effects on NF-κB signaling. This
finding provides new insight into the treatment of hepatic fibrosis
through the regulation of the TLR4 signaling pathway.
Acknowledgements
The present study was supported by the Fundamental
Research Funds (2007C250M) and the Reserve Talent Funds of Yunnan
province (2008Y003).
References
|
1
|
Friedman SL: Stellate cells: a moving
target in hepatic fibrogenesis. Hepatology. 40:1041–1043. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shaker ME, Ghani A, Shiha GE, Ibrahim TM
and Mehal WZ: Nilotinib induces apoptosis and autophagic cell death
of activated hepatic stellate cells via inhibition of histone
deacetylases. Biochim Biophys Acta. 1833:1992–2003. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Priya S and Sudhakaran PR: Cell survival,
activation and apoptosis of hepatic stellate cells: modulation by
extracellular matrix proteins. Hepatol Res. 38:1221–1232.
2008.PubMed/NCBI
|
|
4
|
Tacke F and Weiskirchen R: Update on
hepatic stellate cells: pathogenic role in liver fibrosis and novel
isolation techniques. Expert Rev Gastroenterol Hepatol. 6:67–80.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Soares JB, Pimentel-Nunes P,
Roncon-Albuquerque R and Leite-Moreira A: The role of
lipopolysaccharide/toll-like receptor 4 signaling in chronic liver
diseases. Hepatol Int. 4:659–672. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Seki E, De Minicis S, Osterreicher CH, et
al: TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med.
13:1324–1332. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yan X, Lin Z, Chen F, et al: Human BAMBI
cooperates with Smad7 to inhibit transforming growth factor-beta
signaling. J Biol Chem. 284:30097–30104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Trebicka J, Krag A, Gansweid S, et al:
Endotoxin and tumor necrosis factor-receptor levels in portal and
hepatic vein of patients with alcoholic liver cirrhosis receiving
elective transjugular intrahepatic portosystemic shunt. Eur J
Gastroenterol Hepatol. 23:1218–1225. 2011. View Article : Google Scholar
|
|
9
|
Hanck C, Rossol S, Böcker U, Tokus M and
Singer MV: Presence of plasma endotoxin is correlated with tumour
necrosis factor receptor levels and disease activity in alcoholic
cirrhosis. Alcohol Alcohol. 33:606–608. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhu Q, Zou L, Jagavelu K, et al:
Intestinal decontamination inhibits TLR4 dependent
fibronectin-mediated cross-talk between stellate cells and
endothelial cells in liver fibrosis in mice. J Hepatol. 56:893–899.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bai T, Lian LH, Wu YL, Wan Y and Nan JX:
Thymoquinone attenuates liver fibrosis via PI3K and TLR4 signaling
pathways in activated hepatic stellate cells. Int Immunopharmacol.
15:275–281. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pradere JP, Troeger JS, Dapito DH, Mencin
AA and Schwabe RF: Toll-like receptor 4 and hepatic fibrogenesis.
Semin Liver Dis. 30:232–244. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Luedde T and Trautwein C: A molecular link
between inflammation and fibrogenesis: the bacterial microflora
influences hepatic fibrosis via toll-like receptor 4-dependent
modification of transforming growth factor-beta signaling in
hepatic stellate cells. Hepatology. 47:1089–1091. 2008. View Article : Google Scholar
|
|
14
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar
|
|
15
|
Isayama F, Hines IN, Kremer M, et al: LPS
signaling enhances hepatic fibrogenesis caused by experimental
cholestasis in mice. Am J Physiol Gastrointest Liver Physiol.
290:G1318–G1328. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lotfy M, Badra G, Burham W and Alenzi FQ:
Combined use of honey, bee propolis and myrrh in healing a deep,
infected wound in a patient with diabetes mellitus. Br J Biomed
Sci. 63:171–173. 2006.PubMed/NCBI
|
|
17
|
Ansorge S, Reinhold D and Lendeckel U:
Propolis and some of its constituents down-regulate DNA synthesis
and inflammatory cytokine production but induce TGF-beta1
production of human immune cells. Z Naturforsch C. 58:580–589.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wongmekiat O, Gomonchareonsiri S and
Thamprasert K: Caffeic acid phenethyl ester protects against
oxidative stress-related renal dysfunction in rats treated with
cyclosporin A. Fundam Clin Pharmacol. 25:619–626. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oktar S, Yönden Z, Aydin M, Ilhan S, Alcin
E and Ozturk OH: Protective effects of caffeic acid phenethyl ester
on iron-induced liver damage in rats. J Physiol Biochem.
65:339–344. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Toyoda T, Tsukamoto T, Takasu S, et al:
Anti-inflammatory effects of caffeic acid phenethyl ester (CAPE), a
nuclear factor-kappaB inhibitor, on Helicobacter
pylori-induced gastritis in Mongolian gerbils. Int J Cancer.
125:1786–1795. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ozturk G, Ginis Z, Akyol S, Erden G, Gurel
A and Akyol O: The anticancer mechanism of caffeic acid phenethyl
ester (CAPE): review of melanomas, lung and prostate cancers. Eur
Rev Med Pharmacol Sci. 16:2064–2068. 2012.PubMed/NCBI
|
|
22
|
Lee Y, Shin DH, Kim JH, et al: Caffeic
acid phenethyl ester-mediated Nrf2 activation and IkappaB kinase
inhibition are involved in NFkappaB inhibitory effect: structural
analysis for NFkappaB inhibition. Eur J Pharmacol. 643:21–28. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Natarajan K, Singh S, Burke TR Jr,
Grunberger D and Aggarwal BB: Caffeic acid phenethyl ester is a
potent and specific inhibitor of activation of nuclear
transcription factor NF-kappa B. Proc Natl Acad Sci USA.
93:9090–9095. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Abdel-Latif MM, Windle HJ, Homasany BS,
Sabra K and Kelleher D: Caffeic acid phenethyl ester modulates
Helicobacter pylori-induced nuclear factor-kappa B and
activator protein-1 expression in gastric epithelial cells. Br J
Pharmacol. 146:1139–1147. 2005.PubMed/NCBI
|
|
25
|
Ramm GA: Isolation and culture of rat
hepatic stellate cells. J Gastroenterol Hepatol. 13:846–851. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Weiskirchen R and Gressner AM: Isolation
and culture of hepatic stellate cells. Methods Mol Med. 117:99–113.
2005.PubMed/NCBI
|
|
27
|
Robertson DA, Hughes GA and Lyles GA:
Expression of inducible nitric oxide synthase in cultured smooth
muscle cells from rat mesenteric lymphatic vessels.
Microcirculation. 11:503–515. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cikos S, Bukovska A and Koppel J: Relative
quantification of mRNA: comparison of methods currently used for
real-time PCR data analysis. BMC Mol Biol. 8:1132007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Simone RE, Russo M, Catalano A, et al:
Lycopene inhibits NF-κB-mediated IL-8 expression and changes redox
and PPARγ signalling in cigarette smoke-stimulated macrophages.
PLoS One. 6:e196522011.
|
|
30
|
Bhoopathi P, Chetty C, Kunigal S, Vanamala
SK, Rao JS and Lakka SS: Blockade of tumor growth due to matrix
metalloproteinase-9 inhibition is mediated by sequential activation
of beta1-integrin, ERK, and NF-kappaB. J Biol Chem. 283:1545–1552.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li H, Zheng HW, Chen H, et al: Hepatitis B
virus particles preferably induce Kupffer cells to produce
TGF-beta1 over pro-inflammatory cytokines. Dig Liver Dis.
44:328–333. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schnabl B, Brandl K, Fink M, et al: A
TLR4/MD2 fusion protein inhibits LPS-induced pro-inflammatory
signaling in hepatic stellate cells. Biochem Biophys Res Commun.
375:210–214. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View
Article : Google Scholar
|
|
34
|
Frasinariu OE, Ceccarelli S, Alisi A,
Moraru E and Nobili V: Gut-liver axis and fibrosis in nonalcoholic
fatty liver disease: an input for novel therapies. Dig Liver Dis.
45:543–551. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gäbele E, Dostert K, Hofmann C, et al: DSS
induced colitis increases portal LPS levels and enhances hepatic
inflammation and fibrogenesis in experimental NASH. J Hepatol.
55:1391–1399. 2011.PubMed/NCBI
|
|
36
|
Guo J and Friedman SL: Toll-like receptor
4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis
Tissue Repair. 3:212010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Guo J, Loke J, Zheng F, et al: Functional
linkage of cirrhosis-predictive single nucleotide polymorphisms of
Toll-like receptor 4 to hepatic stellate cell responses.
Hepatology. 49:960–968. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brun P, Castagliuolo I, Pinzani M, Palu G
and Martines D: Exposure to bacterial cell wall products triggers
an inflammatory phenotype in hepatic stellate cells. Am J Physiol
Gastrointest Liver Physiol. 289:G571–G578. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Seki E, De Minicis S, Gwak GY, et al: CCR1
and CCR5 promote hepatic fibrosis in mice. J Clin Invest.
119:1858–1870. 2009.PubMed/NCBI
|
|
40
|
Seki E, de Minicis S, Inokuchi S, et al:
CCR2 promotes hepatic fibrosis in mice. Hepatology. 50:185–197.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Borrelli F, Izzo AA, Di Carlo G, et al:
Effect of a propolis extract and caffeic acid phenethyl ester on
formation of aberrant crypt foci and tumors in the rat colon.
Fitoterapia. 73(Suppl 1): S38–S43. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Juman S, Yasui N, Ikeda K, et al: Caffeic
acid phenethyl ester suppresses the production of pro-inflammatory
cytokines in hypertrophic adipocytes through
lipopolysaccharide-stimulated macrophages. Biol Pharm Bull.
35:1941–1946. 2012. View Article : Google Scholar
|
|
43
|
Kawelke N, Vasel M, Sens C, Au A, Dooley S
and Nakchbandi IA: Fibronectin protects from excessive liver
fibrosis by modulating the availability of and responsiveness of
stellate cells to active TGF-beta. PLoS One. 6:e281812011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Novotny NM, Markel TA, Crisostomo PR and
Meldrum DR: Differential IL-6 and VEGF secretion in adult and
neonatal mesenchymal stem cells: role of NFkB. Cytokine.
43:215–219. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang Y, Rangan GK, Goodwin B, Tay YC and
Harris DC: Lipopolysaccharide-induced MCP-1 gene expression in rat
tubular epithelial cells is nuclear factor-kappaB dependent. Kidney
Int. 57:2011–2022. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Karin M and Ben-Neriah Y: Phosphorylation
meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev
Immunol. 18:621–663. 2000.PubMed/NCBI
|
|
47
|
Karin M and Delhase M: The I kappa B
kinase (IKK) and NF-kappa B: key elements of proinflammatory
signalling. Semin Immunol. 12:85–98. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ribeiro PS, Cortez-Pinto H, Solá S, et al:
Hepatocyte apoptosis, expression of death receptors, and activation
of NF-kappaB in the liver of nonalcoholic and alcoholic
steatohepatitis patients. Am J Gastroenterol. 99:1708–1717. 2004.
View Article : Google Scholar
|