Introduction
Cardiovascular disease (CVD), a leading cause of
morbidity and mortality in Western society, is caused mainly by
atherosclerosis (1).
Epidemiological studies suggest that estrogen protects women
against CVD before menopause (2),
and numerous animal studies have shown that estrogen significantly
protects against the development of atherosclerosis (3–5).
However, randomized controlled trials of postmenopausal estrogen
therapy have demonstrated mixed CVD effects (6,7).
These findings underscore the complexity of the cardiovascular
effects of estrogen, and the underlying mechanisms by which
estrogen regulates cardiovascular biology are of great value and
warrant further investigation.
Reverse cholesterol transport (RCT) is proposed to
be a primary atheroprotective property of high-density lipoprotein
(HDL) and its major protein, apolipoprotein (apo)A-1, which promote
efflux of excess cholesterol from macrophages in atherosclerotic
lesions and then transport it to the liver for degradation and
excretion (8). Cholesterol efflux
is the initial and most likely rate-limiting step in RCT and plays
a pivotal role in maintaining intracellular cholesterol levels and
preventing foam cell formation (9–11).
Recent studies have indicated that vascular smooth muscle cells
(VSMCs) are also capable of accumulating lipid and form foam-like
cells in vitro and in atherosclerotic plaques (12–14). Moreover, VSMC-derived foam cells
have been demonstrated to acquire phagocytotic activity similar to
macrophages (15) and express
cytokines or chemokines to promote intimal foam cell accumulation
(16). These studies suggest that
lipid accumulation in VSMCs contributes to atherosclerosis
development. Thus, promotion of cholesterol efflux from VSMCs may
potentially inhibit VSMC-derived foam cell formation and the
development of atherosclerosis. However, unlike macrophages, little
is known about the regulation of cholesterol efflux and cholesterol
transporters such as ATP-binding cassette transporters ABCA1 and
ABCG1 and scavenger receptor B1 (SR-B1) (8) in VSMCs.
Previous studies have focused on the
anti-proliferative and anti-migratory effects of estrogen on VSMCs
(17–19). Nevertheless, the contributions of
estrogen to cholesterol efflux and VSMC-derived foam cell formation
are relatively unexplored. In the present study, we aimed to
ascertain whether 17β-estradiol (E2) promotes cholesterol efflux
from VSMCs and inhibits VSMC-derived foam cell formation, as well
as the underlying mechanisms.
Materials and methods
Chemicals
E2, monoclonal anti-α-smooth muscle actin (F-3777),
geranylgeranyl pyrophosphate (GGPP), ICI 182,780,
1,3-bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole
dihydrochloride (MPP) and
4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a] pyrimidin-3-yl]
phenol (PHTPP) were obtained from Sigma-Aldrich (St. Louis, MO,
USA). Phenol red-free Dulbecco’s modified Eagle’s medium (DMEM) was
purchased from HyClone (Logan, UT, USA). Charcoal stripped fetal
bovine serum (FBS) was obtained from Gibco-BRL (Grand Island, NY,
USA).
22-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-23,24-bisnor-5-cholen-3β-ol
(NBD-cholesterol) and
4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene
(BODIPY® 493/503) were obtained from Invitrogen (Grand
Island, NY, USA). ApoA-1 was purchased from Calbiochem (San Diego,
CA, USA). HDL and oxidized low-density lipoprotein (ox-LDL) were
obtained from XieSheng Biotechnology (Beijing, China).
Propyl-pyrazole triol (PPT), diarylpropionitrile (DPN) and the
cholesterol assay kit (catalog no. 10007640) were obtained from
Cayman Chemical (Ann Arbor, MI, USA). Rabbit polyclonal antibodies
against ABCA1 and ABCG1 were obtained from Novus Biologicals
(Littleton, CO, USA). Rabbit polyclonal antibody against liver X
receptor (LXR)α was obtained from Abnova (Taipei, Taiwan). Rabbit
polyclonal antibody against glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) was purchased from Epitomics (Burlingame, CA,
USA). Horseradish peroxidase (HRP) goat-anti-rabbit IgG was
obtained from Abcam (Cambridge, MA, USA).
Cell culture
Eight-week-old female C57BL/6 mice were purchased
from the Experimental Animal Center of Medical School of Xi’an
Jiaotong University (Shaanxi, China). Primary mouse VSMCs were
obtained using the tissue explant method, as previously published
(20). Briefly, mice were
euthanized by CO2 asphyxiation followed by cervical
dislocation. The protocol for the present study was approved by the
Institutional Ethics Committee for Animal Experiments of Xi’an
Jiaotong University, China. The aortic segments were placed into an
ice-cold 60-mm dish containing DMEM. The media of aorta was
isolated surgically and minced into small pieces. The pieces were
then placed into 60-mm dishes and cultured in phenol red-free DMEM,
supplemented with 10% charcoal-stripped FBS containing 100 U/ml
penicillin and 100 mg/ml streptomycin at 37°C in 5% CO2.
The VSMCs used between passages 2 and 5 had a VSMC purity >95%
(determined by immunofluorescence staining for α-smooth muscle
actin). Quiescent VSMCs were obtained by incubation with serum-free
medium for 24 h prior to performing all of the experimental
procedures.
Cholesterol efflux assay
The cholesterol efflux assay was performed as
previously described with minor modifications (21). The VSMCs (4×104
cells/well) cultured in 12-well plates were treated with various
concentrations of E2 (1–100 nM) or vehicle (ethanol at a final
concentration <0.01%) for 18 h, followed by the equilibration of
NBD-cholesterol (1 μg/ml) for an additional 6 h in the presence of
E2. NBD-cholesterol-labeled cells were washed with
phosphate-buffered saline (PBS) and incubated in DMEM, DMEM
containing apoA-1 (15 μg/ml) or HDL (50 μg/ml) for 6 h. The
fluorescence-labeled cholesterol released from the cells into the
medium was measured with a multilabel counter (PerkinElmer,
Waltham, MA, USA). Cholesterol efflux was expressed as a percentage
of fluorescence in the medium relative to the total amount of
fluorescence (cells and medium). Specific efflux to apoA-1 or HDL
was calculated by subtracting the non-specific efflux to DMEM
alone.
Detection of cellular lipid droplets in
VSMCs and VSMC-derived foam cells
The VSMCs were seeded in chamber slides at a density
of 1×105 cells/chamber and pretreated with either E2
(1–100 nM) or vehicle for 2 h and then incubated with ox-LDL (50
μg/ml) for 72 h in either the presence or absence of E2. For
detecting lipid accumulation in VSMCs, BODIPY staining was
performed as previously described with minor modifications
(22). The cells were washed
twice with ice-cold PBS, followed by 4% paraformaldehyde fixation
for 1 h at room temperature. Then the cells were stained with
BODIPY® 493/503 working solution (10 μg/ml in PBS) for
30 min at room temperature and rinsed twice with PBS. The images
were captured and analyzed with NIS-Elements imaging software
(Nikon, Tokyo, Japan).
Quantitative measurement of intracellular
cholesteryl ester content
The VSMCs were plated into 6-well plates
(1×105 cells/well) and treated as described above. The
cellular lipids were extracted with hexane/isopropanol (3/2, v/v).
The levels of total and free cholesterol were determined by an
enzymatic, fluorometric method, using a cholesterol assay kit
according to the kit instructions (Cayman Chemical). Fluorescence
intensity was measured using excitation at 540 nm and emission at
590 nm. Cholesteryl ester content was calculated as total
cholesterol minus free cholesterol and normalized to the cellular
protein content, which was determined using a BCA protein assay kit
(Pierce, Rockford, IL, USA).
Real-time RT-PCR analysis
The VSMCs (5×105 cells) plated on 6-cm
culture dishes were incubated with E2 at various concentrations
(1–100 nM) or vehicle (ethanol) for 24 h and then rinsed twice with
PBS. Total cellular RNA was extracted by TRIzol reagent
(Invitrogen) following the manufacturer’s protocol. Reverse
transcription was carried out with 2 μg total RNA using RevertAid™
First Strand cDNA Synthesis kit (Fermentas, Burlington, CA, USA),
and the iQ SYBR-Green Supermix kit (Takara, Tokyo, Japan) was used
on an iQ5 Multicolor Real-Time PCR Detection system (Bio-Rad,
Hercules, CA, USA). Primers for the genes tested are shown in
Table I. Expression data were
normalized to GAPDH levels.
 | Table IPrimers sequences for real-time
RT-PCR. |
Table I
Primers sequences for real-time
RT-PCR.
| Gene | Forward primer
5′-3′ | Reverse primer
5′-3′ |
|---|
| ABCA1 |
CTCAGTTAAGGCTGCTGCTG |
TCAGGCGTACAGAGATCAGG |
| ABCG1 | TGTGCTGTTCG
CTGCTCTGG |
GGTAGGCTGGGATGGTGTCAAAG |
| SR-B1 |
GTTTGGTGCGCCTCTGTTTC |
CGATGCCCTTGACAGATTAG |
| LXRα |
ACGTGCAGGACCAGCTCCAA | GCAGGCGA AGGGC
AAACACT |
| GAPDH |
TCAACGGCACAGTCAAGG |
ACTCCACGACATACTCAGC |
Western blot analysis
Western blot analyses were carried out as previously
described (23). VSMC lysates
were prepared in lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl,
1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 50 mM dithiothreitol,
complete protease inhibitor cocktail). The protein concentration
was assayed using a BCA protein assay kit. An equal amount of total
proteins (30 μg) was loaded onto either 8 or 10% SDS-PAGE, and
transferred to a nitrocellulose membrane (Bio-Rad). After blocked
with 5% nonfat milk in TBST (20 mM Tris-HCl, 150 mM NaCl, pH 7.5
and 0.1% Tween-20), blots were incubated with anti-ABCA1 (1:500),
anti-ABCG1 (1:500), anti-LXRα (1:500) or anti-GAPDH (1:1,000)
antibodies overnight at 4°C. After washing with TBST, the blots
were incubated with the HRP-conjugated secondary antibody (1:5,000)
for 1 h at room temperature. Immunoreactive bands were quantified
using an enhanced chemiluminescent system of detection
(Pierce).
Transfection of siRNA
For downregulation of LXRα expression, LXRα siRNA
and negative control siRNA were synthesized by GenePharm (Shanghai,
China). The sequences for LXRα siRNA were: sense,
5′-GGCUGCAACACACAU AUGUTT-3′ and antisense, 5′-ACAUAUGUGUGUUGCAGC
CTT-3′. The sequences for negative control siRNA were: sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense, 5′-ACGUGACACGUUCG
GAGAATT-3′. One day prior to transfection, the VSMCs
(1×105/well) were seeded in 6-well plates in 2 ml DMEM
containing 10% FBS. LXRα siRNA and control siRNA were transfected
into VSMCs using Lipofectamine™ 2000 reagent (Invitrogen) at a
final concentration of 100 nM according to the manufacturer’s
protocol. Twenty-four hours post-transfection, cells were treated
with E2 (10 nM) for an additional 24 h and lysed for western blot
analysis.
Statistical analysis
All experiments were repeated at least three times.
Data are expressed as the mean ± standard error of the mean (SEM).
Intergroup differences were analyzed using one-way analysis of
variance (ANOVA) for the comparison of 3 or more groups. The
Student’s t-test was used for comparison between 2 groups. A value
of P<0.05 was considered to indicate a statistically significant
result.
Results
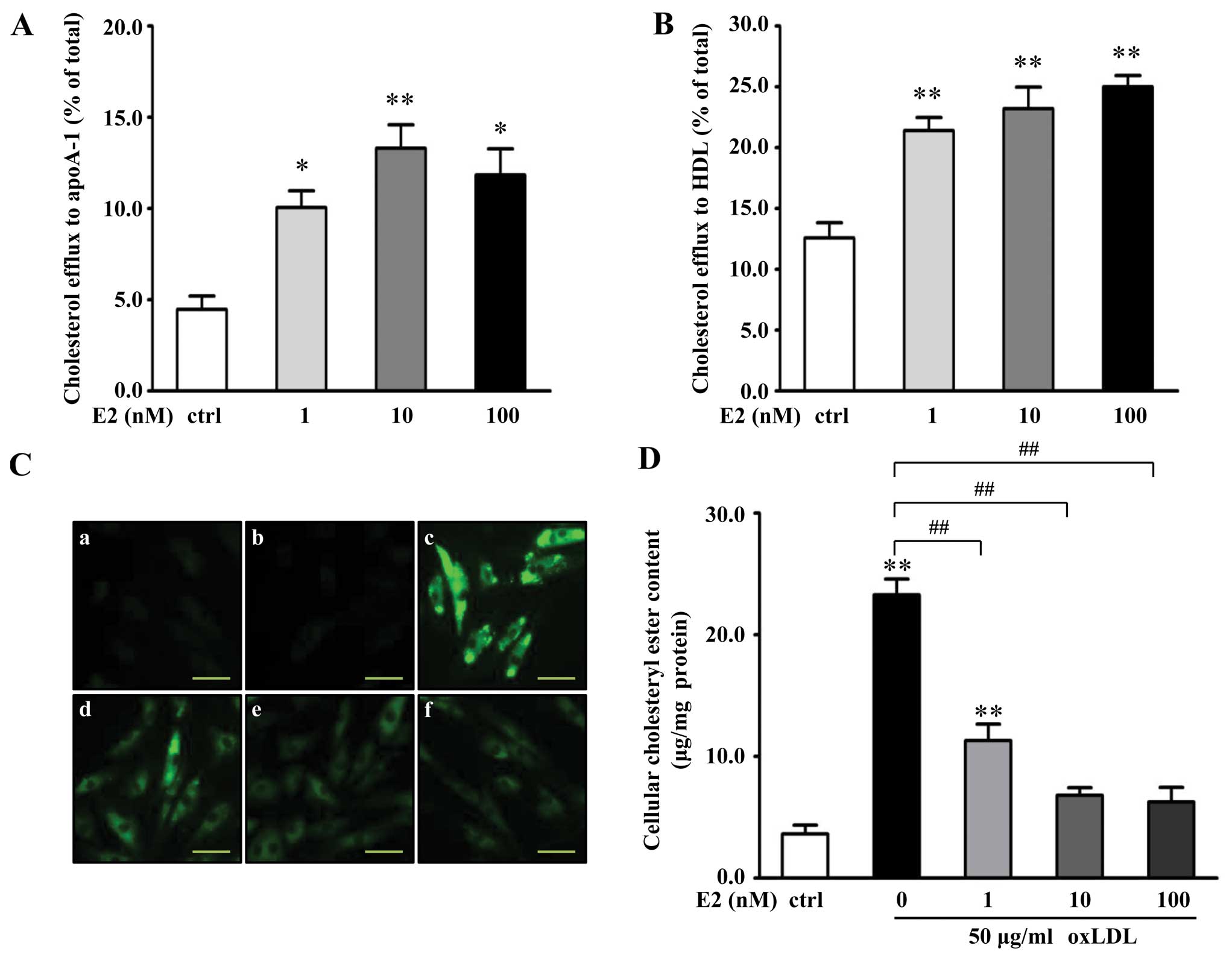
E2 promotes cholesterol efflux from VSMCs
and attenuates cholesteryl ester accumulation in VSMC-derived foam
cells
To investigate the effects of E2 on cholesterol
efflux from VSMCs, cells were pretreated with different
concentrations of E2 for 18 h, followed by incubation with
NBD-cholesterol for 6 h in the presence of E2. Cholesterol efflux
was initiated by the addition of apoA-1 (15 μg/ml) or HDL (50
μg/ml). The results (Fig. 1A and
B) revealed that cholesterol efflux to apoA-1 was significantly
increased in response to E2, and E2 dose-dependently promoted
HDL-mediated cholesterol efflux from the VSMCs. At 10 nM of E2, the
cholesterol efflux from the VSMCs to apoA-1 and HDL was increased
by 176 and 95%, respectively, compared with the controls (apoA-1 or
HDL only; P<0.01).
In addition, BODIPY staining and an enzymatic
colorimetric method were employed to assess the effect of E2 on
cholesteryl ester accumulation in VSMC-derived foam cells.
Administration of 50 μg/ml ox-LDL for 72 h significantly increased
cellular lipid droplets in the VSMCs (Fig. 1C). Treatment with E2 markedly
attenuated the ox-LDL-induced accumulation of lipid droplets in a
dose-dependent manner (Fig. 1C).
By directly measuring the intracellular cholesteryl ester content,
it was evident (Fig. 1D) that E2
dose-dependently decreased cholesteryl ester content in the VSMCs,
compared with the oxLDL-treated group. At 10 nM of E2, the
cholesteryl ester content in the VSMCs was decreased by 70%,
compared with the cells treated with ox-LDL only (P<0.01).
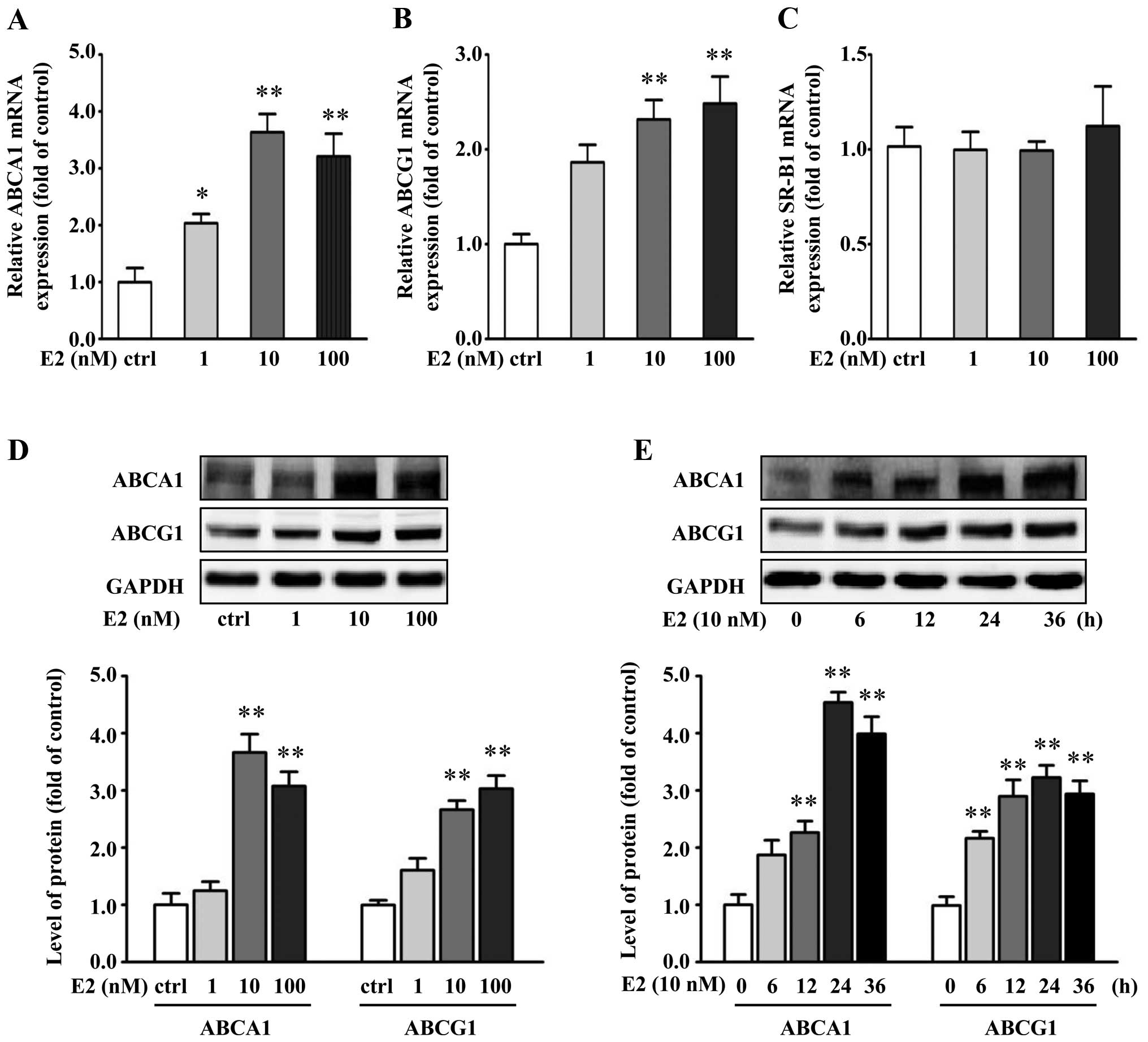
E2 increases the expression of ABCA1 and
ABCG1 in VSMCs
To study the possible mechanisms responsible for E2
action, VSMCs were treated with different concentrations of E2 for
24 h. Changes in ABCA1, ABCG1 and SR-B1 mRNA in response to E2 were
assessed by real-time RT-PCR. E2 significantly increased ABCA1 and
ABCG1 mRNA levels, whereas E2 had little effect on SR-B1 mRNA
expression in the VSMCs (Fig.
2A–C).
To study if the increased mRNA levels of ABCA1 and
ABCG1 by E2 can lead to an increase in ABCA1 and ABCG1 protein
expression, after treatment VSMCs were determined for ABCA1 and
ABCG1 protein levels by western blot analysis. Protein levels of
ABCA1 and ABCG1 had a trend of increase similar to mRNA levels when
treated by E2 (Fig. 2D). At 10 nM
of E2, the ABCA1 and ABCG protein levels had an increase of 266 and
164%, respectively, when compared with the controls (P<0.01).
Moreover, the upregulation of ABCA1 and ABCG1 by E2 (10 nM) peaked
at 24 h, when compared with the control group (Fig. 2E).
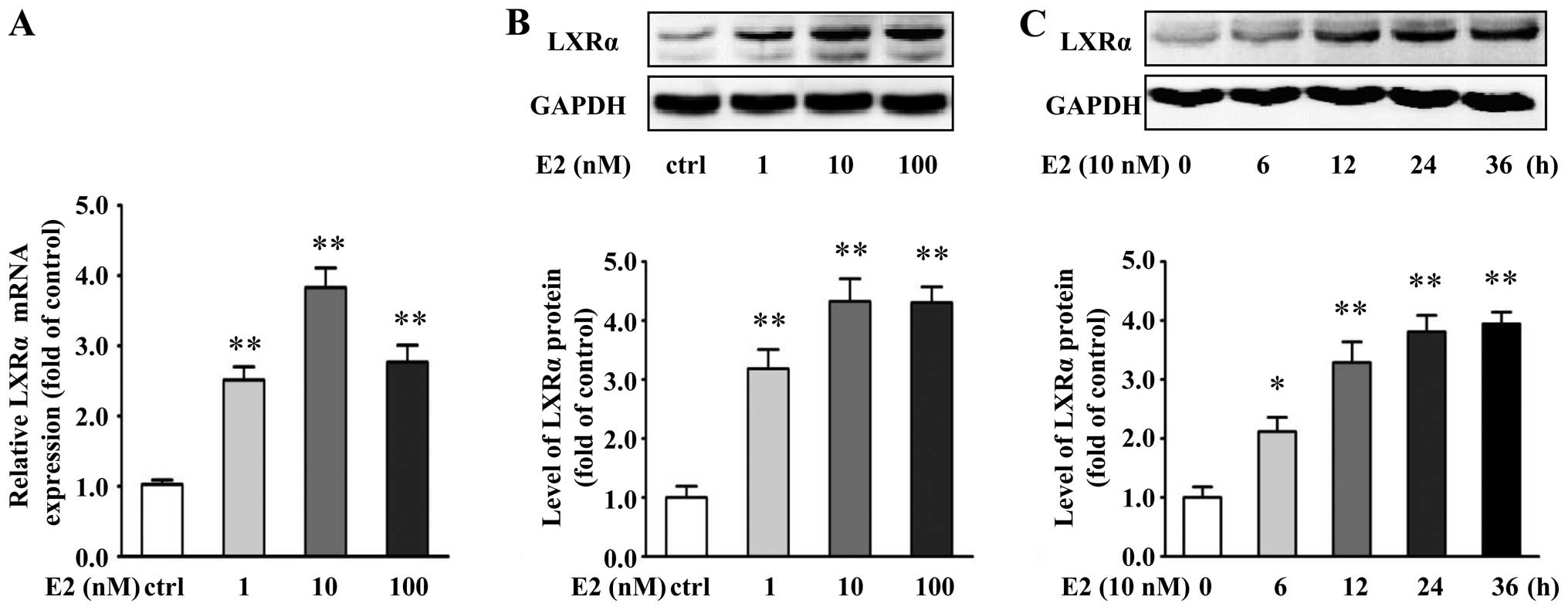
E2 induces ABCA1 and ABCG1 expression and
cholesterol efflux from VSMCs through an LXRα-dependent
pathway
To address whether LXRα is involved in the
E2-induced expression of ABCA1 and ABCG1, we determined the mRNA
and protein levels of LXRα in the E2-treated VSMCs. E2
significantly increased the mRNA and protein expression of LXRα
(Fig. 3A and B). At 10 nM of E2,
LXRα mRNA and protein levels had an increase of 282 and 333%,
respectively, compared with the controls (P<0.01). Furthermore,
E2 (10 nM) upregulated the protein levels of LXRα in a
time-dependent manner (Fig.
3C).
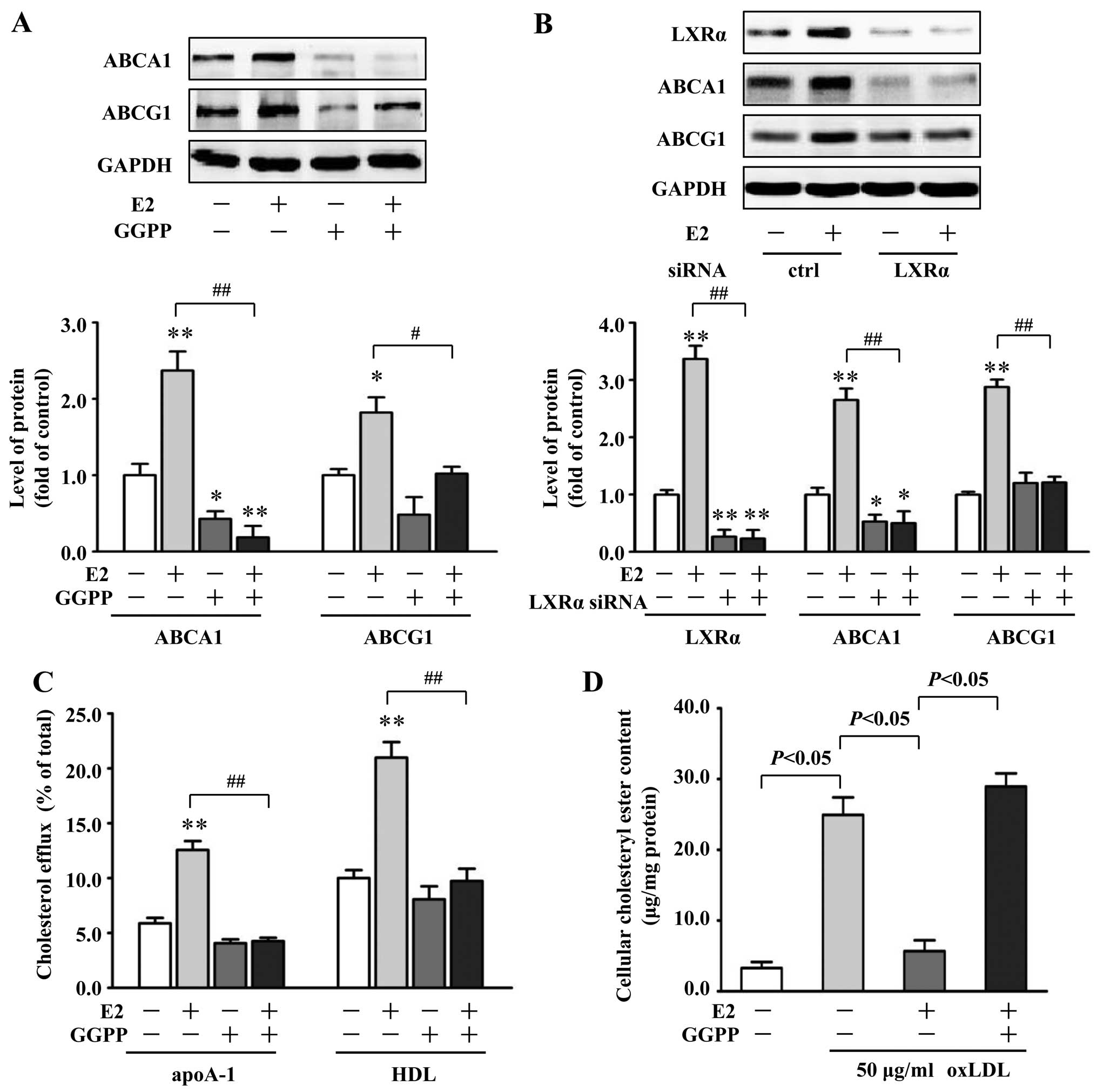
To study whether the induction of ABCA1 and ABCG1
expression by E2 is through an LXRα-dependent pathway, VSMCs were
pretreated with GGPP (20 μM), a pharmacological inhibitor of LXRα,
followed by treatment with E2 (10 nM). Compared with the VSMCs
treated with 10 nM E2 alone, coincubation with GGPP significantly
decreased ABCA1 and ABCG1 protein expression by 90% (P<0.01) and
44% (P<0.05), respectively (Fig.
4A). Moreover, we investigated the effects of LXRα siRNA on
ABCA1 and ABCG1 expression induced by E2. LXRα siRNA reduced the
amount of LXRα protein in VSMCs by 73%, compared with the control
siRNA (P<0.01) (Fig. 4B).
Concomitantly, LXRα siRNA treatment abolished E2-induced
upregulation of ABCA1 by 81% and ABCG1 by 58% in VSMCs, compared
with the control siRNA treatment (P<0.01) (Fig. 4B). Additionally, inhibition of
LXRα activation by GGPP (20 μM) blocked the promotive effects of E2
(10 nM) on cholesterol efflux to apoA-1 and HDL (Fig. 4C) and further abrogated the
inhibitory effect of E2 on cholesteryl ester accumulation in VSMCs
(Fig. 4D). Together, these
results imply the critical role of LXRα in E2-regulated expression
of ABCA1 and ABCG1 and subsequent changes in cholesterol efflux and
cholesteryl ester accumulation in VSMCs.
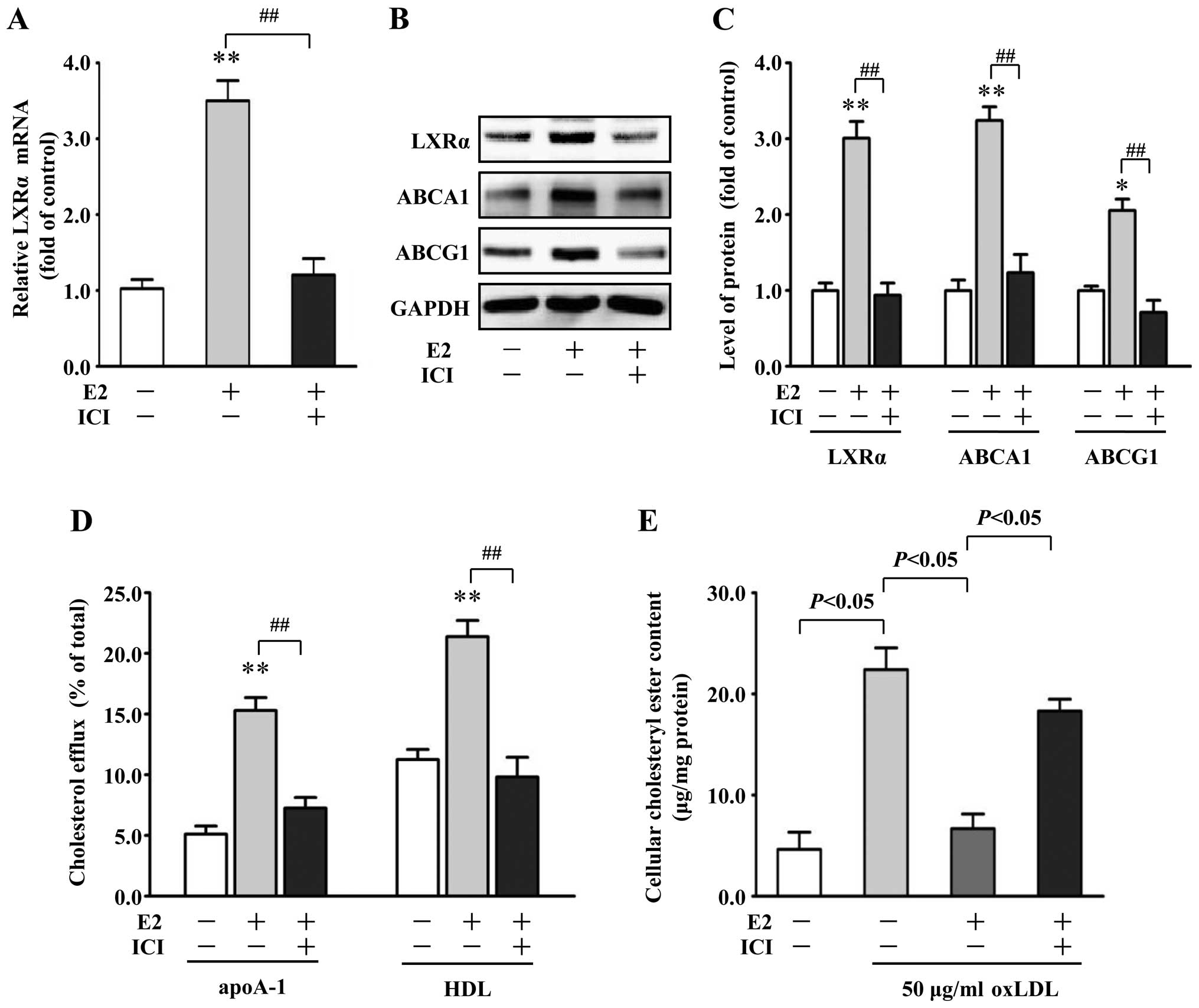
Estrogen receptor β (ERβ) mediates the
stimulatory effects of E2 on LXRα, ABCA1 and ABCG1 expression in
VSMCs
To investigate whether upregulation of LXRα by E2 is
ER-dependent, we determined LXRα mRNA expression after
pre-incubation with the nonselective ER antagonist ICI 182,780 (1
μM). ICI 182,780 effectively abolished the E2-induced increase in
LXRα mRNA expression by 71% in VSMCs, compared with the E2 alone
group (P<0.01) (Fig. 5A).
Moreover, in the presence of ICI 182,780, the increased protein
expression of LXRα, ABCA1 and ABCG1 by E2 (10 nM) was abrogated
(Fig. 5B and C). E2-induced
promotion of apoA-1- and HDL-mediated cholesterol efflux and
reduction of cholesteryl ester content in the VSMCs were also
effectively blocked by ICI 182,780 (Fig. 5D and E).
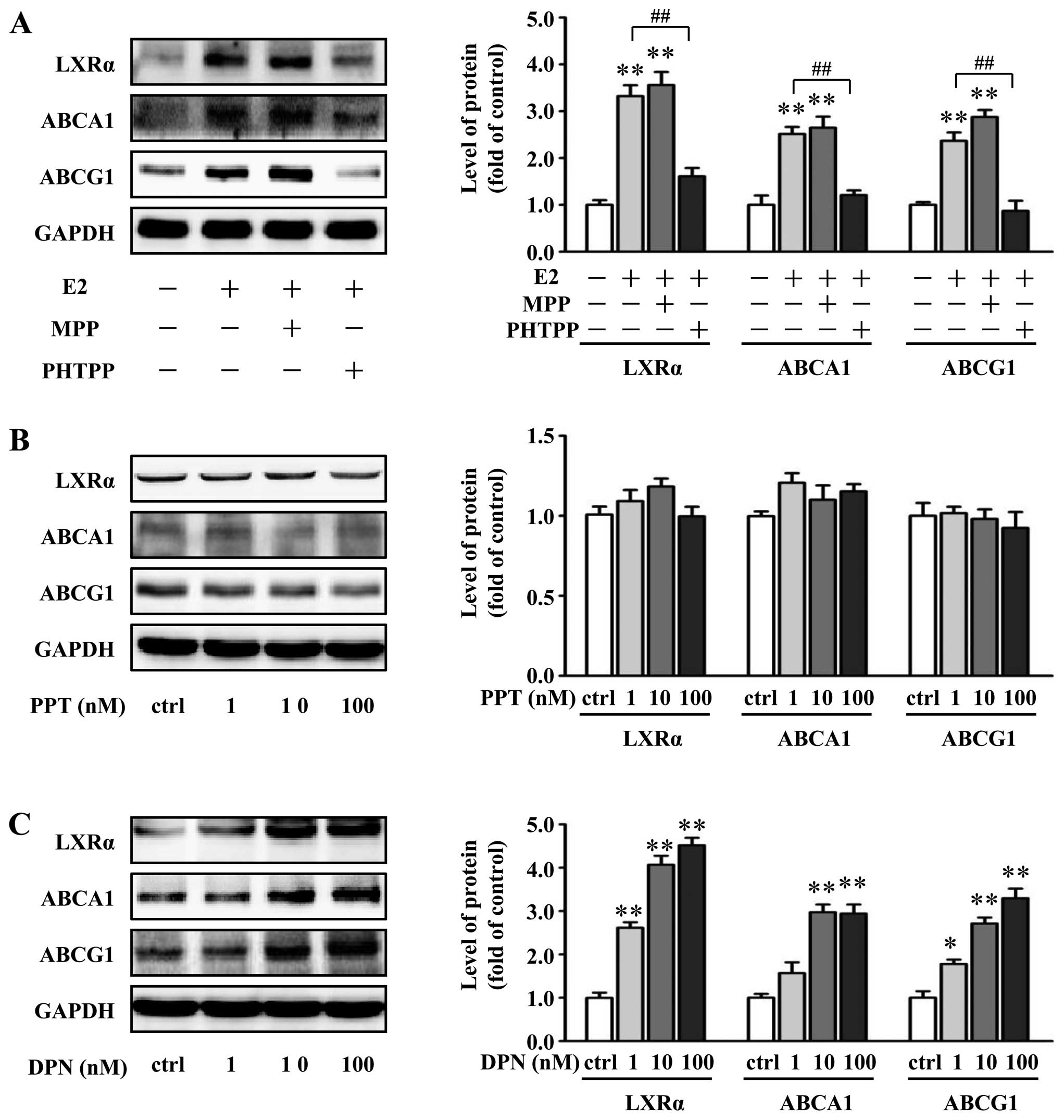
To more explicitly decipher the ER-subtype involved
in the E2-mediated upregulation of LXRα, ABCA1 and ABCG1, we
evaluated the blocking effects of a selective ERα antagonist MPP
and a selective ERβ antagonist PHTPP on the E2-induced upregulation
of LXRα, ABCA1 and ABCG1 expression. Pretreatment with PHTPP (1
μM), but not MPP (1 μM), completely inhibited the E2-induced
responses in the VSMCs (Fig. 6A).
Moreover, ERα-specific agonist PPT and ERβ-specific agonist DPN
were used to further delineate the role of specific ERs.
Importantly, DPN dose-dependently increased LXRα, ABCA1 and ABCG1
protein expression in the VSMCs, while stimulation with PPT did not
alter the protein expression of LXRα, ABCA1 and ABCG1 (Fig. 6B and C). These results indicate
that upregulation of LXRα, ABCA1 and ABCG1 by E2 is likely mediated
by ERβ.
Discussion
The formation of foam cells within the arterial wall
is thought to play a pivotal role in the development of
atherosclerotic lesions (24). In
the last decade, several studies have shown that in addition to
macrophages, VSMCs accumulate excess intracellular cholesteryl
(25,26) and give rise to a significant
number of foam cells in atherosclerotic lesions (14,27). Moreover, VSMC-derived foam cells
in vitro lose the expression of VSMC contractile markers,
transform into macrophage-like cells (15) and express monocyte chemoattractant
protein-1 (MCP-1) (16,28). These data suggest that lipid
accumulation in VSMCs may contribute to the development of
atherosclerosis. Previous research has focused on the
anti-proliferative and anti-migratory properties of estrogen on
VSMCs (29). Here, we elucidated
a novel atheroprotective effect of estrogen that E2 promotes
cholesterol efflux from VSMCs and suppresses VSMC-derived foam cell
formation.
Accumulation of cholesteryl ester stored as
cytoplasmic lipid droplets is the main characteristic of foam cells
derived from both macrophages and VSMCs (15,30). The removal of excess cholesterol
from macrophages by apoA-1 and HDL is thought to play an important
role in preventing foam cell formation (31). Unlike macrophages, little is known
regarding the regulation of cholesterol efflux and lipid
accumulation in VSMCs. Our data showed that treatment with E2 at
physiological concentrations promoted cholesterol efflux to both
apoA-1 and HDL from VSMCs and attenuated intracellular cholesteryl
ester accumulation in VSMCs. This suggests that reduced lipid
accumulation in VSMCs by E2 is, at least in part, due to an
increase in cholesterol efflux from VSMCs.
Cholesterol removal from macrophages is mediated by
ABC transporters and SR-B1. ABCA1 and ABCG1 have been demonstrated
to primarily promote cellular cholesterol efflux to apoA-1 and HDL,
respectively (8). In the present
study, we showed that E2 treatment significantly augmented both
mRNA and protein expression of ABCA1 and ABCG1 without an
alteration in SR-B1 mRNA expression in VSMCs. These data indicate
that E2 increases cholesterol efflux through the upregulation of
ABCA1 and ABCG1 in VSMCs.
LXRα, which plays a pivotal role in maintaining
macrophage cholesterol homeostasis, is the main regulator of ABCA1
and ABCG1 gene transcription (32,33). We, therefore, examined whether
LXRα is involved in the upregulation of ABCA1 and ABCG1 by E2 in
VSMCs. Notably, E2 increased LXRα expression in VSMCs. Moreover,
inhibition of LXRα activation by either GGPP or LXRα siRNA
diminished the E2-mediated ABCA1 and ABCG1 induction. GGPP also
blocked E2-induced cholesterol efflux and abrogated the inhibitory
effect of E2 on cholesteryl ester accumulation in VSMCs. These
results demonstrated the essential role of LXRα in E2-regulated
gene expression of ABCA1 and ABCG1 and promotion of cholesterol
efflux from VSMCs.
Although a number of findings regarding the gene
expression and cholesterol efflux of E2 action on human macrophages
have been previously reported, these data are controversial.
Corcoran et al (34)
reported that E2 induced a modest reduction in ABCG1 and did not
affect cholesterol efflux and ABCA1 levels in human
monocyte-derived macrophages (HMDMs). Cerda et al (35) reported that hormone therapy
reduced the mRNA levels of ABCA1, without alteration in ABCG1 and
LXRα in peripheral blood mononuclear cells (PBMCs). Kramer and Wray
(36) observed that LXRα
expression was significantly increased after estrogen withdrawal in
human macrophages. However, in a mouse study, Ribas et al
(5) reported that ABCA1 protein
expression was reduced in both bone marrow-macrophages from
ERα−/− mice and peritoneal macrophages from mice with a
myeloid-specific ERα deletion. In addition, Srivastava (37) reported that estrogen treatment
significantly increased hepatic and intestinal ABCA1 mRNA levels in
mice. Moreover, a previous study by our group also demonstrated
that E2 increased ABCA1 levels both in atherosclerotic lesions in
ApoE−/− mice and in murine Raw 264.7 macrophages
(38). Together with our present
results in VSMCs, these findings suggest species differences in the
regulation of cholesterol efflux and related gene expression
between the human and the mouse and may explain why an
atheroprotective benefit of estrogen is strongly supported by
research using animals, particularly mouse models of
atherosclerosis, but cannot be established in humans participating
in randomized controlled trials of estrogen replacement
treatment.
Estrogen exerts most of its biological action via
estrogen receptors (ERs), which are prominent members of the
nuclear receptor superfamily (39). Pre-incubation with ICI 182,780 to
antagonize ERs blocked the E2-mediated upregulation of LXRα, ABCA1
and ABCG1 expression and attenuation of cholesteryl ester
accumulation in VSMCs, indicating ER dependence. There are two
different isforms of ERs, ERα and ERβ, which are expressed in
endothelial cells, VSMCs and macrophages (29). Previous research found that ERα
may be the major mediator of the atheroprotective effects of
estrogen (40). However, recent
evidence suggests that ERα may not be involved in estrogen-mediated
protection from early lesion development (41). Moreover, Rayner et al
(42) reported that E2 via ERβ
induced an extracellular release of HSP 27 in macrophages and
prevented the development of atherosclerosis in apoE−/−
mice. In addition, Xing et al (43) showed that estrogen modulated
TNF-α-induced inflammatory responses in rat VSMCs through ERβ
activation. In the present study, treatment with specific
antagonists and agonists for ERα and ERβ clearly showed that
upregulation of LXRα, ABCA1 and ABCG1 by E2 was ERβ-dependent,
suggesting a novel mechanism for the regulation of atherosclerosis
specifically through ERβ.
In conclusion, our results indicate that in mouse
VSMCs, E2 promotes apoA-1- and HDL-mediated cholesterol efflux from
VSMCs and prevents VSMC-derived foam cell formation via
upregulation of ABCA1 and ABCG1 expression, which is mediated by
ERβ and LXRα activation. These findings provide novel insight into
the anti-atherogenic properties of estrogen and to some extent, may
explain the complexity of the cardiovascular effects of estrogen in
the human and mouse.
Acknowledgements
This study was supported by the National Science
Fund of China for Distinguished Young Scholars (NSFC 81025002 to
Z.Y.), the National Basic Research Program of China (973 program:
2012CB517804 to Z.Y.) and the National Science Foundation of China
(no. 30971219 to Y.L.).
References
|
1
|
Weber C and Noels H: Atherosclerosis:
current pathogenesis and therapeutic options. Nat Med.
17:1410–1422. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Meyer MR, Haas E and Barton M: Gender
differences of cardiovascular disease: new perspectives for
estrogen receptor signaling. Hypertension. 47:1019–1026. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hodgin JB and Maeda N: Minireview:
estrogen and mouse models of atherosclerosis. Endocrinology.
143:4495–4501. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kavanagh K, Davis MA, Zhang L, et al:
Estrogen decreases atherosclerosis in part by reducing hepatic
acyl-CoA:cholesterol acyltransferase 2 (ACAT2) in monkeys.
Arterioscler Thromb Vasc Biol. 29:1471–1477. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ribas V, Drew BG, Le JA, et al:
Myeloid-specific estrogen receptor α deficiency impairs metabolic
homeostasis and accelerates atherosclerotic lesion development.
Proc Natl Acad Sci USA. 108:16457–16462. 2011.
|
|
6
|
Hulley S, Grady D, Bush T, et al:
Randomized trial of estrogen plus progestin for secondary
prevention of coronary heart disease in postmenopausal women. Heart
and Estrogen/progestin Replacement Study (HERS) Research Group.
JAMA. 280:605–613. 1998. View Article : Google Scholar
|
|
7
|
Rossouw JE, Anderson GL, Prentice RL, et
al; Writing Group for the Women’s Health Initiative Investigators.
Risks and benefits of estrogen plus progestin in healthy
postmenopausal women: principal results from the Women’s Health
Initiative randomized controlled trial. JAMA. 288:321–333.
2002.PubMed/NCBI
|
|
8
|
Rosenson RS, Brewer HB Jr, Davidson WS, et
al: Cholesterol efflux and atheroprotection advancing the concept
of reverse cholesterol transport. Circulation. 125:1905–1919. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saeed O, Otsuka F, Polavarapu R, et al:
Pharmacological suppression of hepcidin increases macrophage
cholesterol efflux and reduces foam cell formation and
atherosclerosis. Arterioscler Thromb Vasc Biol. 32:299–307. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang X, Collins HL, Ranalletta M, et al:
Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage
reverse cholesterol transport in vivo. J Clin Invest.
117:2216–2224. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang X, Liao D, Bharadwaj U, Li M, Yao Q
and Chen C: C-reactive protein inhibits cholesterol efflux from
human macrophage-derived foam cells. Arterioscler Thromb Vasc Biol.
28:519–526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma L, Zhong J, Zhao Z, et al: Activation
of TRPV1 reduces vascular lipid accumulation and attenuates
atherosclerosis. Cardiovasc Res. 92:504–513. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Allahverdian S, Pannu PS and Francis GA:
Contribution of monocyte-derived macrophages and smooth muscle
cells to arterial foam cell formation. Cardiovasc Res. 95:165–172.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stary HC, Chandler AB, Glagov S, et al: A
definition of initial, fatty streak, and intermediate lesions of
atherosclerosis. A report from the Committee on Vascular Lesions of
the Council on Arteriosclerosis, American Heart Association.
Arterioscler Thromb. 14:840–856. 1994. View Article : Google Scholar
|
|
15
|
Rong JX, Shapiro M, Trogan E and Fisher
EA: Transdifferentiation of mouse aortic smooth muscle cells to a
macrophage-like state after cholesterol loading. Proc Natl Acad Sci
USA. 100:13531–13536. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Higashimori M, Tatro JB, Moore KJ,
Mendelsohn ME, Galper JB and Beasley D: Role of toll-like receptor
4 in intimal foam cell accumulation in apolipoprotein E-deficient
mice. Arterioscler Thromb Vasc Biol. 31:50–57. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ueda K, Lu Q, Baur W, Aronovitz MJ and
Karas RH: Rapid estrogen receptor signaling mediates
estrogen-induced inhibition of vascular smooth muscle cell
proliferation. Arterioscler Thromb Vasc Biol. 33:1837–1843. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiang X, Zhang Y, Hou D, et al:
17beta-estradiol inhibits oleic acid-induced rat VSMC proliferation
and migration by restoring PGC-1alpha expression. Mol Cell
Endocrinol. 315:74–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang P, Xu J, Zheng S, et al:
17β-estradiol down-regulates lipopolysaccharide-induced MCP-1
production and cell migration in vascular smooth muscle cells. J
Mol Endocrinol. 45:87–97. 2010.
|
|
20
|
Metz RP, Patterson JL and Wilson E:
Vascular smooth muscle cells: isolation, culture, and
characterization. Methods Mol Biol. 843:169–176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tsai JY, Su KH, Shyue SK, et al: EGb761
ameliorates the formation of foam cells by regulating the
expression of SR-A and ABCA1: role of haem oxygenase-1. Cardiovasc
Res. 88:415–423. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Han X, Kitamoto S, Lian Q and Boisvert WA:
Interleukin-10 facilitates both cholesterol uptake and efflux in
macrophages. J Biol Chem. 284:32950–32958. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Park SH, Kim JL, Kang MK, et al: Sage weed
(Salvia plebeia) extract antagonizes foam cell formation and
promotes cholesterol efflux in murine macrophages. Int J Mol Med.
30:1105–1112. 2012.
|
|
24
|
Yuan Y, Li P and Ye J: Lipid homeostasis
and the formation of macrophage-derived foam cells in
atherosclerosis. Protein Cell. 3:173–181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rivera J, Walduck AK, Thomas SR, et al:
Accumulation of serum lipids by vascular smooth muscle cells
involves a macropinocytosis-like uptake pathway and is associated
with the downregulation of the ATP-binding cassette transporter A1.
Naunyn Schmiedebergs Arch Pharmacol. 386:1081–1093. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xue JH, Yuan Z, Wu Y, et al: High glucose
promotes intracellular lipid accumulation in vascular smooth muscle
cells by impairing cholesterol influx and efflux balance.
Cardiovasc Res. 86:141–150. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Choi HY, Rahmani M, Wong BW, et al:
ATP-binding cassette transporter A1 expression and apolipoprotein
AI binding are impaired in intima-type arterial smooth muscle
cells. Circulation. 119:3223–3231. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Klouche M, Rose-John S, Schmiedt W and
Bhakdi S: Enzymatically degraded, nonoxidized LDL induces human
vascular smooth muscle cell activation, foam cell transformation,
and proliferation. Circulation. 101:1799–1805. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nofer JR: Estrogens and atherosclerosis:
insights from animal models and cell systems. J Mol Endocrinol.
48:R13–R29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ghosh S, Zhao B, Bie J and Song J:
Macrophage cholesteryl ester mobilization and atherosclerosis.
Vascul Pharmacol. 52:1–10. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yvan-Charvet L, Wang N and Tall AR: Role
of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and
immune responses. Arterioscler Thromb Vasc Biol. 30:139–143. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao C and Dahlman-Wright K: Liver X
receptor in cholesterol metabolism. J Endocrinol. 204:233–240.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yan JQ, Tan CZ, Wu JH, et al: Neopterin
negatively regulates expression of ABCA1 and ABCG1 by the LXRα
signaling pathway in THP-1 macrophage-derived foam cells. Mol Cell
Biochem. 379:123–131. 2013.PubMed/NCBI
|
|
34
|
Corcoran MP, Lichtenstein AH, Meydani M,
Dillard A, Schaefer EJ and Lamon-Fava S: The effect of
17β-estradiol on cholesterol content in human macrophages is
influenced by the lipoprotein milieu. J Mol Endocrinol. 47:109–117.
2011.
|
|
35
|
Cerda A, Issa MH, Genvigir FD, et al:
Atorvastatin and hormone therapy influence expression of ABCA1,
APOA1 and SCARB1 in mononuclear cells from hypercholesterolemic
postmenopausal women. J Steroid Biochem Mol Biol. 138:403–409.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kramer P and Wray S: 17-Beta-estradiol
regulates expression of genes that function in macrophage
activation and cholesterol homeostasis. J Steroid Biochem Mol Biol.
81:203–216. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Srivastava RA: Estrogen-induced regulation
of the ATP-binding cassette transporter A1 (ABCA1) in mice: a
possible mechanism of atheroprotection by estrogen. Mol Cell
Biochem. 240:67–73. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liang X, He M, Chen T, et al:
17β-estradiol suppresses the macrophage foam cell formation
associated with SOCS3. Horm Metab Res. 45:423–429. 2013.
|
|
39
|
Murphy E: Estrogen signaling and
cardiovascular disease. Circ Res. 109:687–696. 2011. View Article : Google Scholar
|
|
40
|
Hodgin JB, Krege JH, Reddick RL, Korach
KS, Smithies O and Maeda N: Estrogen receptor α is a major mediator
of 17β-estradiol’s atheroprotective effects on lesion size in
Apoe−/−mice. J Clin Invest. 107:333–340. 2001.
|
|
41
|
Villablanca AC, Tenwolde A, Lee M, Huck M,
Mumenthaler S and Rutledge JC: 17beta-estradiol prevents
early-stage atherosclerosis in estrogen receptor-alpha deficient
female mice. J Cardiovasc Transl Res. 2:289–299. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rayner K, Sun J, Chen Y-X, et al: Heat
shock protein 27 protects against atherogenesis via an
estrogen-dependent mechanism: role of selective estrogen receptor
beta modulation. Arterioscler Thromb Vasc Biol. 29:1751–1756. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xing D, Feng W, Miller AP, et al: Estrogen
modulates TNF-alpha-induced inflammatory responses in rat aortic
smooth muscle cells through estrogen receptor-beta activation. Am J
Physiol Heart Circ Physiol. 292:H2607–H2612. 2007. View Article : Google Scholar : PubMed/NCBI
|