Introduction
Macrophage migration inhibitory factor (MIF) was
initially named for its ability to inhibit the migration of
macrophages (1,2). However, it has since been found to
stimulate the recruitment of macrophages and other leukocytes
(3–6). MIF plays a key role in regulating
innate and adaptive immunity, autoimmunity and inflammatory
responses (7). MIF is produced by
immune and non-immune cells that are stimulated by microbial
products, complements and T cell-derived cytokines that stimulate
NF-κB signaling (7–9). Once secreted, MIF binds to chemokine
receptors, such as CXCR2 and CXCR4, activating integrin signaling
and driving the chemotaxis of monocytes and T cells (4). It also binds to the receptor CD74.
This interaction initiates a signaling cascade that activates
immune cells (10). MIF then
promotes the secretion of a broad array of proinflammatory
mediators including TNF-α, IL-1β, IL-2, IL-6, IL-8, nitric oxide,
COX-2, PGE2, chemokines and adhesion molecules via NF-κB
activation, leading to the amplification of inflammatory and immune
responses (3,7,11).
NF-κB is a key mediator in immune and inflammatory
responses. In resting cells, the NF-κB is bound by IκB, which
inhibits NF-κB signaling. A variety of stimuli, such as
lipopolysaccharide (LPS), stress and cytokines, activate IKK, the
IκB kinase, leading to its phosphorylation. This targets the
phosphorylated IκB for proteasomal degradation, and NF-κB is
released followed by translocation to the nucleus, and initiation
of the transcription of its target genes (12).
High levels of MIF are indicative of autoimmune
diseases and severe inflammatory states, such as asthma and sepsis
(13,14). Mice lacking MIF exhibit less
pulmonary inflammation and chronic colitis (13,15). Genetic alteration of human MIF
expression correlates with the severity of asthma, cystic fibrosis,
rheumatoid arthritis, inflammatory bowel disease, and ischemic
injury (13,16–19). Furthermore, MIF inhibits p53
activity, resulting in the suppression of p53-mediated apoptosis,
rendering proinflammatory responses and tumorigenesis (7,20,21). As such, overexpression of MIF has
been observed in many types of cancer (22). Accordingly, unsuccessful attempts
have been made to develop reagents that inhibit MIF such as
neutralizing antibodies and small molecule inhibitors (23,24). There is, therefore, a great need
for the development of molecules that effectively inhibit MIF,
which may be used to treat inflammatory and immune diseases as well
as cancer.
Developmental endothelial locus-1 (Del-1) is
expressed in endothelial cells and a subset of macrophages, and
regulates angiogenesis, apoptosis and cell adhesion and migration
(25–28). Del-1 binds to the leukocyte
integrin LFA-1, inhibiting the adhesion of leukocytes to vascular
endothelial cells, thereby suppressing leukocyte migration
(29). Therefore, Del-1 acts as
an endogenous anti-inflammatory molecule. Although binding of Del-1
to LFA-1 is well established, subsequent downstream signaling
events remain unclear.
Considering that MIF and Del-1 are expressed in
macrophages and are key players in the regulation of inflammation,
we aimed to investigate the involvement of Del-1 in the regulation
of MIF-1.
Materials and methods
Cell culture
RAW264.7 and HEK293T cells were obtained from the
American Type Culture Collection (Manassas, VA, USA) and were
cultivated in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% FBS and streptomycin/penicillin. THP-1 cells
were cultivated in RPMI-1640 supplemented with 10% FBS and
antibiotics. Isolation of mouse bone marrow-derived
monocytes/macrophages (BMDM) was performed as previously described
(30,31). In brief, bone marrow cells were
flushed with medium from the bones of wild-type (WT) and
Del-1-deficient (Del-1−/−) mice (provided by Professor
T. Chavakis at Dresden University, Dresden, Germany), and WT,
heterozygous (p53+/−) and homozygous (p53−/−)
mice (obtained by crossing p53 heterozygous mice, purchased from
the Jackson Laboratory, Bar Harbor, ME, USA), and then filtered on
a cell strainer. The red blood cells were lysed and the remaining
bone marrow cells were cultured in complete RPMI-1640 medium
containing murine GM-CSF (10 ng/ml) (Peprotech, Rocky Hill, NJ,
USA). After 4 days, the floating cells were removed and adherent
cells were used. The cell culture reagents were purchased from
Invitrogen Life Technologies (Carlsbad, CA, USA), unless otherwise
specified. All the animal studies were approved by the
Institutional Animal Care and Use Committees of Asan Institute and
Ewha Women’s University (Seoul, Korea).
Production of Del-1 conditioned media
using the Flp-In T-Rex system
An ~1.5 kb full-length mouse Del-1 cDNA containing
the signal sequence and an HA epitope tag was cloned into the
pcDNA5/FRT/TO plasmid (Invitrogen Life Technologies). The mDel-1
expression plasmid and pOG44 (Invitrogen Life Technologies) were
used to transfect Flp-In T-Rex293 cells, and then the cells were
cultured with 1 μg/ml doxycycline for 24 h. The conditioned media
were analyzed by immunoblotting using an anti-HA antibody (Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA).
Enzyme-linked immuno-sorbent assay
(ELISA)
RAW264.7 cells were seeded at 5×105 per
well in a PLL-coated 24-well plate, incubated at 37°C, 5%
CO2 for 12 h, and then further incubated for 24 h in the
presence of recombinant Del-1 (R&D Systems, Minneapolis, MN,
USA), LPS (E. coli 0111:B4; Sigma-Aldrich, St. Louis, MO,
USA) or both, at the concentrations of 0, 0.005, 0.05, 0.5 and 5
μg/ml. The supernatants were collected, added to a MaxiSorp 96-well
plate (Nunc A/S, Roskilde, Denmark) and incubated at 4°C for 12 h.
Serial dilutions of recombinant MIF protein were used as the
standard. After being washed with 0.1% PBS-Tween-20 (PBST) three
times, the plate was blocked with PBST containing 0.3% skim milk
for 2 h at room temperature. The plate was then washed, incubated
with a rabbit anti-MIF antibody (ab7207, Abcam, Cambridge, MA, USA)
at room temperature for 4 h, washed again, and incubated with an
HRP-conjugated anti-rabbit antibody (Cell Signaling Technology,
Boston, MA, USA) at room temperature for 1 h. After four washes,
the plate was incubated with the TMB Plus 2 (Kem-En-Tec
Diagnostics, Taastrup, Denmark) substrate and the reaction was
stopped with 1 N HCl. Absorbance was read at 450 nm on a SpectraMax
190 Microplate Reader (Molecular Devices, Sunnyvale, CA, USA).
Immunoblot analysis
Cells were washed with ice-cold PBS and lysed in
PRO-PREP protein extraction solution (Intron, Seongnam, Korea).
Lysates were separated on a 12% SDS-PAGE gel and transferred to
PVDF membranes. The membranes were blocked and probed with
antibodies against phospho-IκB-α (pS32/pS36; BD Pharmingen, San
Diego, CA, USA), IκB-α (C-21), p53 (DO-1) (both from Santa Cruz
Biotechnology), α/β-tubulin, or actin (both from Cell Signaling
Technology). After washing, the blots were incubated with the
appropriate HRP-conjugated secondary antibodies (Cell Signaling
Technology) and developed with the SuperSignal West Pico
Chemiluminescent Substrate kit (Thermo Fisher Scientific, Waltham,
MA, USA).
Immunocytochemistry
THP-1 cells were plated at 2×105 per well
in a PLL-coated 24-well plate. The cells were pretreated with
recombinant Del-1 for 1 h, and then stimulated with LPS for 10 min.
The plate was placed on ice for 10 min to stop reactions. The cells
were washed with ice-cold PBS, fixed with 4% paraformaldehyde for
20 min, washed, and stained with antibody against p65 (Cell
Signaling Technology) for 12 h at 4°C. The cells were washed again,
incubated with Alexa 488-conjugated secondary antibody (Invitrogen
Life Technologies) for 1 h and DAPI for 10 min at room temperature,
mounted with Fluoromount G, and observed under a confocal
microscope (LSM 710; Zeiss, Oberkochen, Germany) at a magnification
of ×200.
Luciferase assay
One day prior to transfection, 293 T cells were
plated at a density of 3×105 cells per well in a 12-well
plate. The cells were transfected with an NF-κB luciferase
construct using Effectene transfection reagent (Qiagen, Hilden,
Germany) in Opti-MEM (Invitrogen Life Technologies), according to
the manufacturer’s instructions. Twelve hours later, the cells were
treated with TNF-α at the indicated concentrations in the control
or Del-1-conditioned media. Luciferase activity was measured 24 h
post-treatment using a luciferase reporter assay system (Promega,
Madison, WI, USA) and a Luminometer (Victor X3, Perkin-Elmer,
Waltham, MA, USA), according to the manufacturer’s
instructions.
Real-time RT-PCR
Total RNA was isolated using TRIzol (Invitrogen Life
Technologies) and cDNA was synthesized using the ImProm-II reverse
transcriptase kit (Promega). The cDNA was amplified using
LightCycler 480 SYBR-Green I Master and a LightCycler 480 machine
(Roche, Mannheim, Germany). Primer sequences used were: Del-1,
forward: 5′-CTTGGTAGCAGCCTGGCTTT-3′ and reverse: 5′-GCC
TTCTGGACACTCACAGG-3′; MIF, forward: 5′-CGCACA GTACATCGCAGTG-3′ and
reverse: 5′-GGCAGCGTTCATG TCGTAAT-3′; 18S forward:
5′-CGCGGTTCTATTTTGT TGGT-3′ and 18S reverse: 5′-AGTCGGCATCGTTTA
TGGTC-3′. The following PCR cycle was used: 95°C for 15 min; 50
cycles of 30 sec at 95°C, 30 sec at 60°C and 30 sec at 72°C; and
95°C for 15 min. Melting curve analyses were performed on all PCR
products to ensure that specific PCR products were generated. Data
were analyzed using the comparative Ct method (32), and the levels of mRNA were
expressed as the relative fold change.
Statistical analysis
Data are presented as the means ± standard deviation
(SD). Data were compared using the Student’s t-test. P<0.05 was
considered to indicate statistical significance.
Results
Extracellular Del-1 protein inhibits
constitutive and LPS-induced MIF production in macrophages
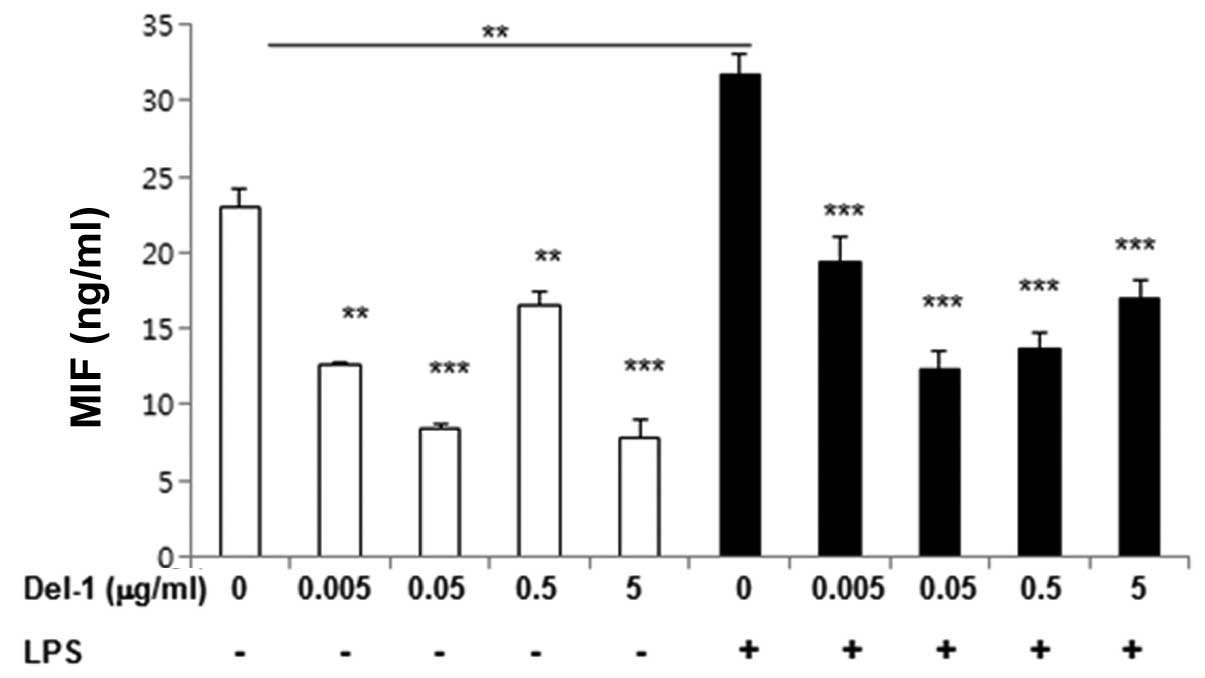
To determine whether Del-1 regulated the production
of MIF in macrophages, RAW264.7 macrophages were treated with Del-1
in the absence or presence of LPS, and secreted MIF was assessed by
ELISA. Levels of secreted MIF were reduced in the media harvested
from the cells treated with Del-1 compared to control cells, in the
LPS-treated and -untreated groups (Fig. 1). These results indicated that
Del-1 may be involved in the downregulation of MIF.
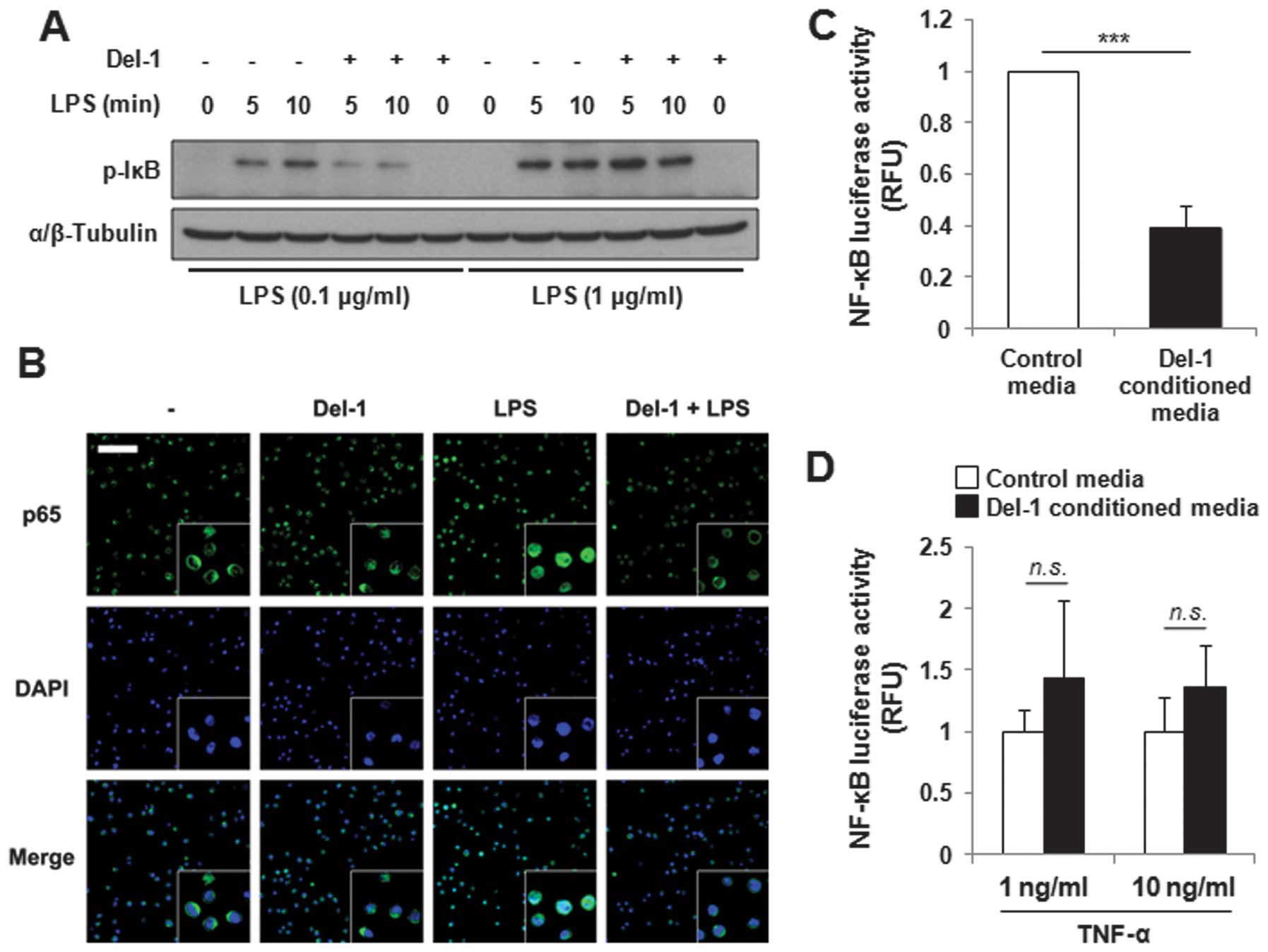
Del-1 suppresses NF-κB activation in
macrophages in a stimulation intensity-dependent manner
As MIF expression is promoted by the activation of
NF-κB (9,33), the regulation of LPS-activated
NF-κB activity by Del-1 was investigated. Levels of phospho-IκBα
and IκBα, both of which are key players in the modulation of NF-κB
activity, were examined by immunoblot analysis in RAW264.7 mouse
macrophages or THP-1 human monocytes. The cells were pretreated
with human recombinant Del-1 prior to LPS stimulation.
Subsequently, the cells were collected and lysates were
immunoblotted using anti-phospho-IκBα and anti-IκBα antibodies.
Notably, Del-1 decreased phospho-IκBα protein levels in monocytes
stimulated with a low concentration of LPS (0.1 μg/ml), but not in
monocytes stimulated with a high concentration of LPS (1 μg/ml)
(Fig. 2A). The total IκBα levels
were comparable between samples treated with LPS alone and LPS plus
Del-1 (data not shown). Comparable results were obtained when
conditioned media from mouse Del-1-transfected cells (known as
Del-1-conditioned media hereafter) were used to treat macrophages,
instead of recombinant Del-1 (data not shown). In addition,
recombinant Del-1 attenuated the translocation of NF-κB to the
nucleus induced by LPS (0.1 μg/ml) (Fig. 2B). To confirm these findings,
human embryonic kidney HEK293T cells were transfected with an NF-κB
reporter plasmid and stimulated with TNF-α at varying
concentrations in Del-1-conditioned media, and luciferase activity
was measured. As expected, the cells treated with the
Del-1-conditioned media showed lower luciferase activity than the
control cells, but only when a low concentration of TNF-α was
present (Fig. 2C and D). Taken
together, these data suggest that Del-1 regulates NF-κB activity by
suppressing the phosphorylation of IκBα in the presence of low
concentrations of inflammatory stimuli.
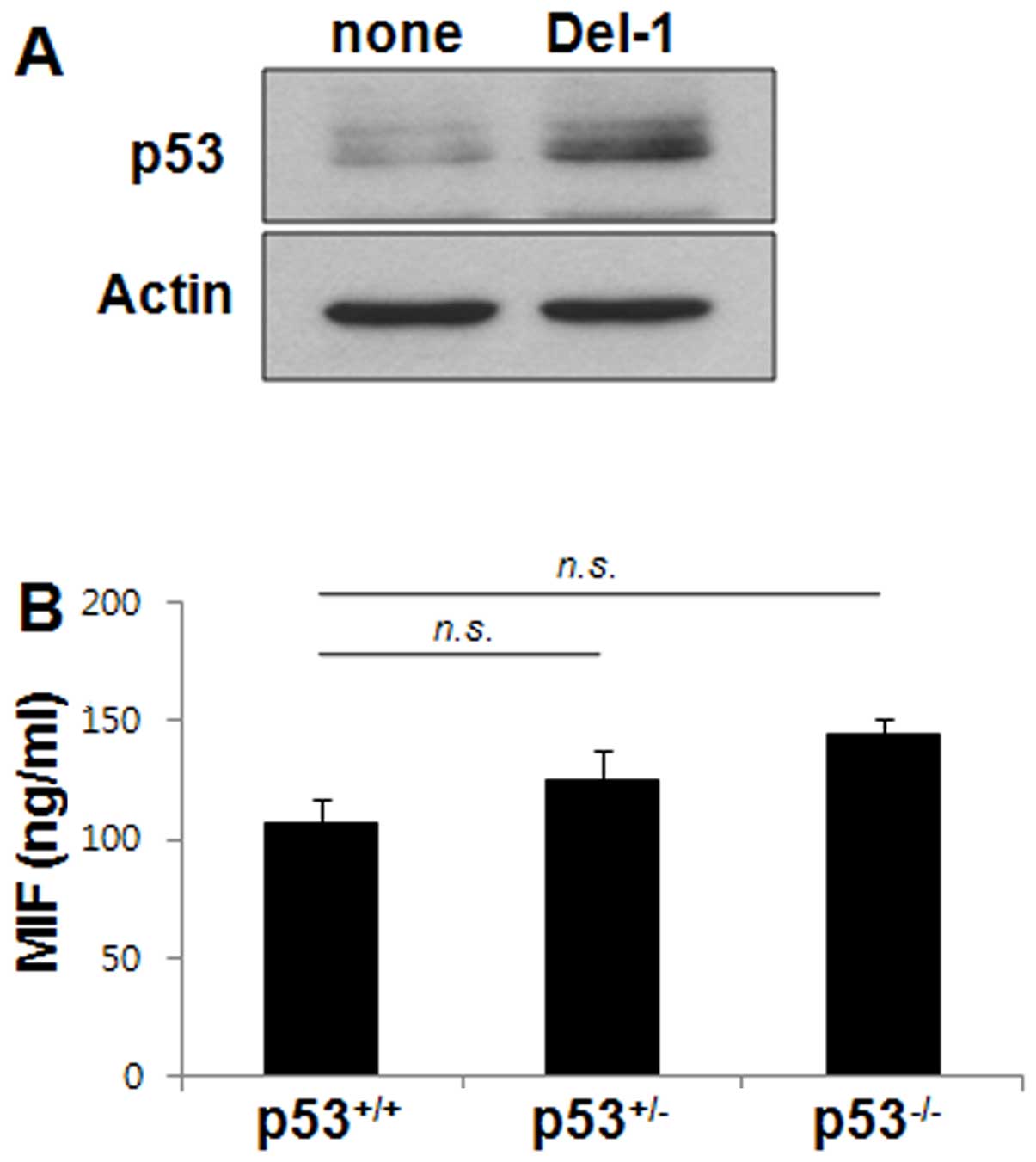
Del-1-induced p53 does not affect the
macrophage production of MIF
Previous studies have shown that NF-κB and p53
pathways often interact in the regulation of gene transcription
(33,34). The potential contribution of p53
to the Del-1-mediated suppression of MIF production in macrophages
was therefore examined. Del-1 enhanced the level of p53 protein in
RAW264.7 cells (Fig. 3A). MIF
protein levels were also assessed by ELISA in BMDM isolated from
p53+/+ or p53−/− or p53+/− null
for p53. The levels of MIF protein in these BMDMs were not
significantly different from one another (Fig. 3B), indicating that p53 does not
regulate MIF production in macrophages.
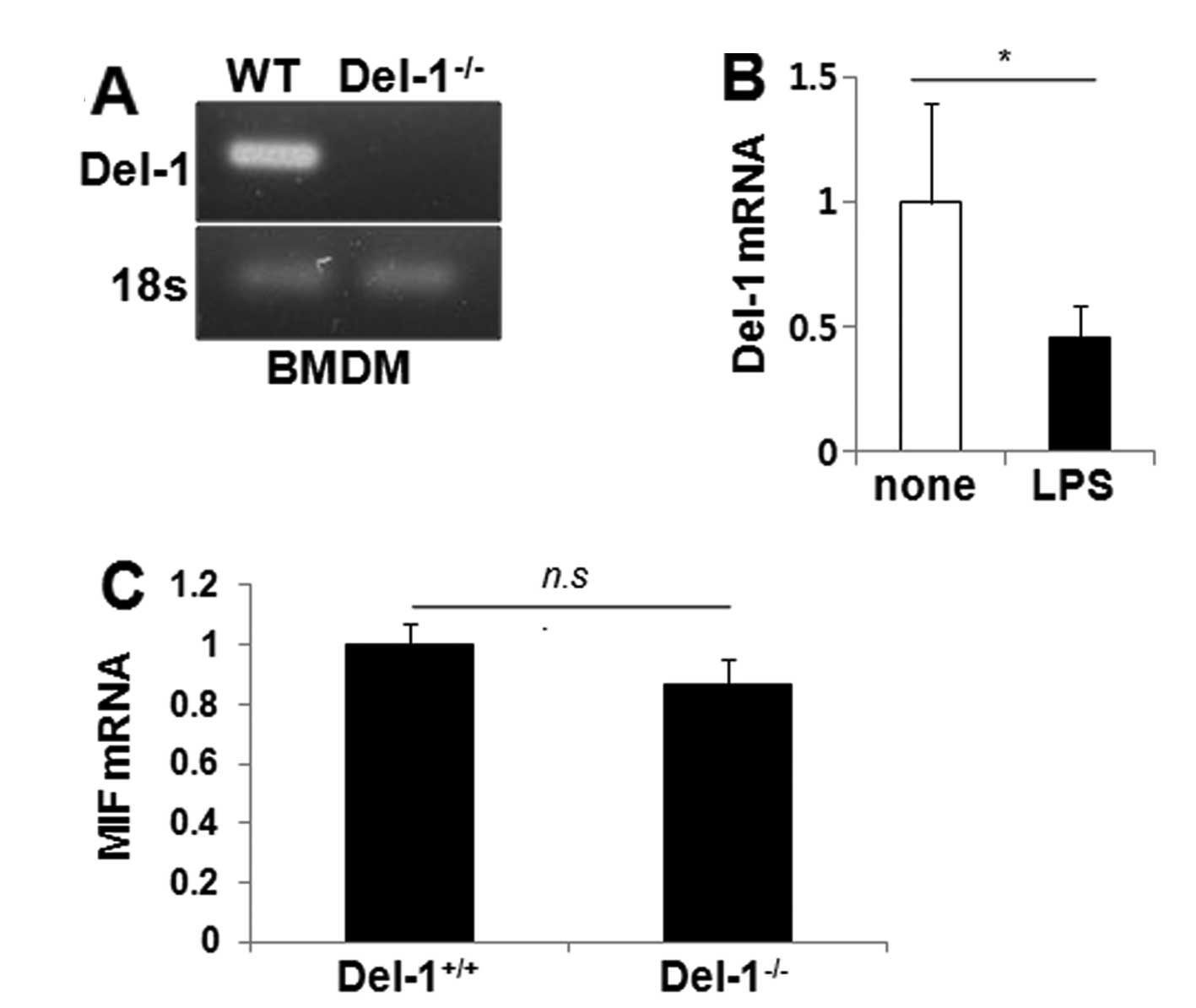
Intracellular Del-1 fails to inhibit MIF
production within macrophages
Del-1 has been reported to be expressed in a subset
of macrophage cell lines and some primary macrophages (25). To determine whether intracellular
Del-1 regulates the expression of MIF within macrophages, we first
assessed the levels of Del-1 by RT-PCR in BMDMs isolated from WT
(Del-1+/+) and Del-1−/− mice. Del-1 was
present in the WT BMDM, but not in the Del-1−/− BMDM
(Fig. 4A), and its expression
decreased following LPS stimulation in the WT BMDM (Fig. 4B), suggesting that Del-1 is
expressed in macrophages and that it could respond to inflammatory
stimuli. MIF expression in the BMDM was then assessed by
quantitative RT-PCR. No difference in the level of MIF was found
between WT and Del-1−/− BMDM (Fig. 4C). These findings suggest that
macrophage-derived Del-1 does not regulate MIF within
macrophages.
Discussion
We previously demonstrated that Del-1 has an
anti-inflammatory function through its inhibition of leukocyte
adhesion (29). The present study
provides an additional mechanism by which Del-1 inhibits
inflammation by downregulating the potent proinflammatory cytokine,
MIF.
Treatment of monocytes/macrophages with recombinant
Del-1 protein attenuated IκB phosphorylation, indicating that the
decrease in MIF secretion is due to the suppression of NF-κB
signaling. However, this suppression occurred only when
monocytes/macrophages were stimulated with a low dose of LPS, but
not with a high dose of LPS. Similarly, Del-1-conditioned media
suppressed NF-κB activity only when a low dose of TNF-α was
present, suggesting that the activation of cells that act as a
first line of defense should depend on the intensity of the
stimulus. To limit unnecessary inflammatory responses caused by the
initiation of inflammation by negligible stimuli, Del-1 may be a
key player in this tolerance, by suppressing the initiation of
inflammation by trivial stimuli.
On the other hand, previous studies have suggested
that the activity of p53 and MIF is reciprocally regulated
(20,21,33). Inflammation, cell stress, and
hypoxic conditions activate hypoxia-inducible factor-1α (HIF-1α).
p53 represses HIF-1α activity and inhibits HIF-1α-dependent
transcription of multiple cytokines, including MIF (35–37). Del-1 elevated the level of p53,
which may counteract NF-κB activity (34). However, in our studies, WT BMDM
did not express decreased levels of MIF compared to BMDM with
reduced p53, suggesting that p53 activated in response to the
stimulation of macrophages with extracellular Del-1 may not play a
role in regulating MIF production in macrophages. Alternatively,
the basal level of p53 in macrophages may not be sufficiently high
to modulate MIF production.
Under inflammatory conditions, we found that Del-1
is decreased in macrophages, which is in agreement with our
previous result showing that Del-1 is decreased in vascular
endothelial cells (29). However,
the molecular mechanism underlying the inflammation-mediated
decrease in Del-1 remains to be determined. NF-κB activated by
inflammation may suppress p53 activity, which in turn decreases
Del-1 transcription (31).
Because Del-1 activates p53 and p53 decreases MIF, we initially
expected that Del-1 deficiency would increase MIF production.
However, this did not occur, suggesting that the basal level of
Del-1 in macrophages is insufficient to decrease MIF. Although we
found that Del-1 attenuates NF-κB activation, it is still premature
to conclude that Del-1 is a global inhibitor of proinflamatory
cytokine production. Further studies may clarify this issue.
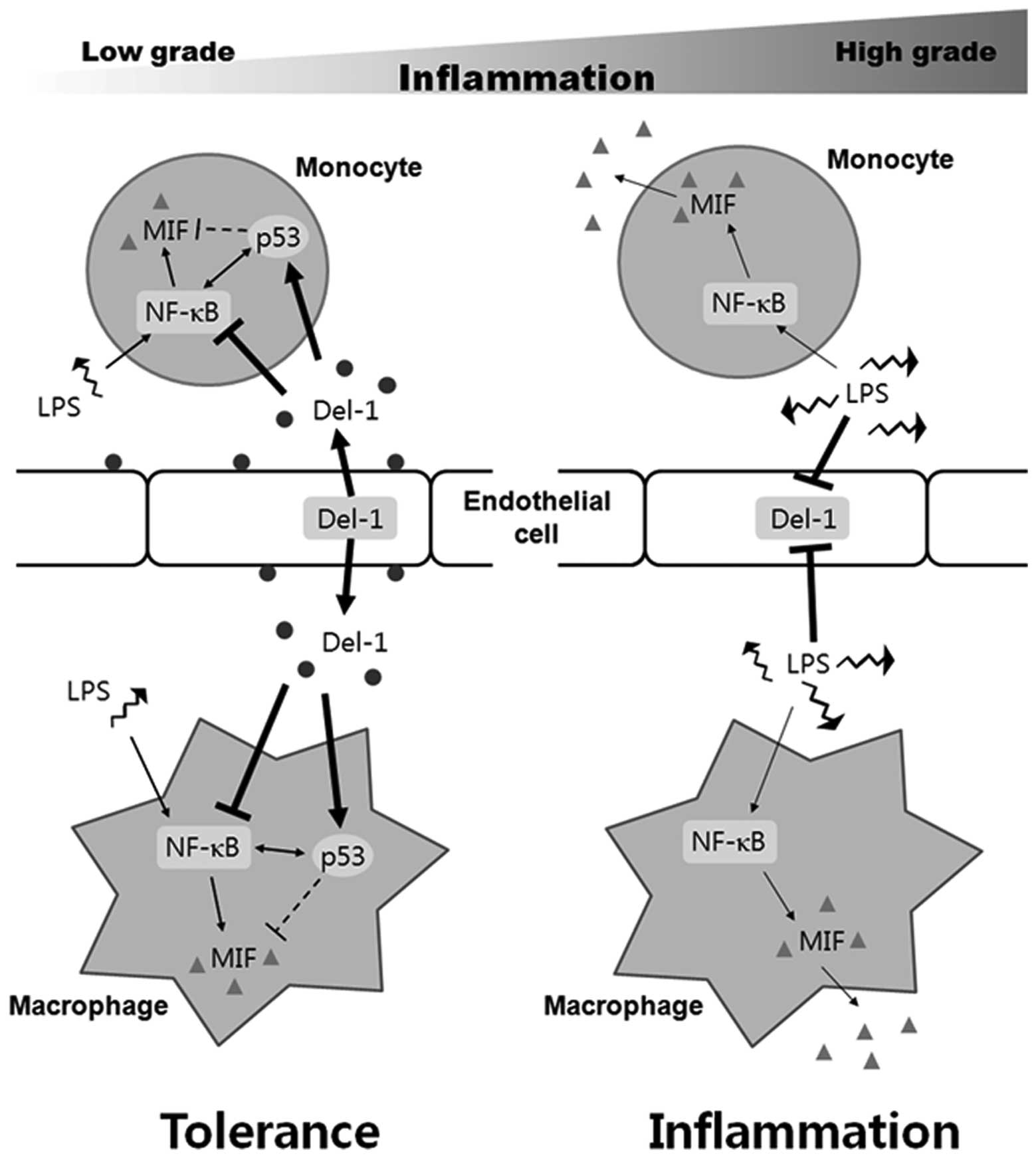
In summary, as shown in Fig. 5, once inflammation is initiated,
NF-κB suppresses p53 activity in monocytes, macrophages, and
vascular endothelial cells, all of which are first-line defense
cells, thereby repressing the anti-inflammatory molecule Del-1 and
increasing proinflammatory molecules including MIF. As a result,
beneficial inflammation ensues. High-grade inflammatory stimuli,
but not low-grade inflammatory stimuli, may inhibit Del-1
sufficiently for inflammation to be initiated. For this reason,
Del-1 may control MIF only under low-grade inflammatory conditions
or in resting cells. Taken together, these data indicate that Del-1
inhibits NF-κB-dependent proinflammatory cytokine production by
monocytes/macrophages, thereby acting as an inflammation
gatekeeper.
Acknowledgements
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and Technology
of Korea (2011-0014447). We would like to thank Dr Seok-Yong Choi
for providing a critical reading of the manuscript and general
encouragement.
References
|
1
|
Bloom BR and Bennett B: Mechanism of a
reaction in vitro associated with delayed-type hypersensitivity.
Science. 153:80–82. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
David JR: Delayed hypersensitivity in
vitro: its mediation by cell-free substances formed by lymphoid
cell-antigen interaction. Proc Natl Acad Sci USA. 56:72–77. 1966.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gregory JL, Morand EF, McKeown SJ, et al:
Macrophage migration inhibitory factor induces macrophage
recruitment via CC chemokine ligand 2. J Immunol. 177:8072–8079.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bernhagen J, Krohn R, Lue H, et al: MIF is
a noncognate ligand of CXC chemokine receptors in inflammatory and
atherogenic cell recruitment. Nat Med. 13:587–596. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yoshihisa Y, Makino T, Matsunaga K, et al:
Macrophage migration inhibitory factor is essential for eosinophil
recruitment in allergen-induced skin inflammation. J Invest
Dermatol. 131:925–931. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li J, Mo HY, Xiong G, et al: Tumor
microenvironment macrophage inhibitory factor directs the
accumulation of interleukin-17-producing tumor-infiltrating
lymphocytes and predicts favorable survival in nasopharyngeal
carcinoma patients. J Biol Chem. 287:35484–35495. 2012. View Article : Google Scholar
|
|
7
|
Calandra T and Roger T: Macrophage
migration inhibitory factor: a regulator of innate immunity. Nat
Rev Immunol. 3:791–800. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rossi AG, Haslett C, Hirani N, et al:
Human circulating eosinophils secrete macrophage migration
inhibitory factor (MIF). Potential role in asthma. J Clin Invest.
101:2869–2874. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Veillat V, Lavoie CH, Metz CN, Roger T,
Labelle Y and Akoum A: Involvement of nuclear factor-kappaB in
macrophage migration inhibitory factor gene transcription
up-regulation induced by interleukin-1 beta in ectopic endometrial
cells. Fertil Steril. 91:2148–2156. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Leng L, Metz CN, Fang Y, et al: MIF signal
transduction initiated by binding to CD74. J Exp Med.
197:1467–1476. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Amin MA, Haas CS, Zhu K, et al: Migration
inhibitory factor up-regulates vascular cell adhesion molecule-1
and intercellular adhesion molecule-1 via Src, PI3 kinase, and
NFkappaB. Blood. 107:2252–2261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Napetschnig J and Wu H: Molecular basis of
NF-kappaB signaling. Annu Rev Biophys. 42:443–468. 2013. View Article : Google Scholar
|
|
13
|
Mizue Y, Ghani S, Leng L, et al: Role for
macrophage migration inhibitory factor in asthma. Proc Natl Acad
Sci USA. 102:14410–14415. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Calandra T, Echtenacher B, Roy DL, et al:
Protection from septic shock by neutralization of macrophage
migration inhibitory factor. Nat Med. 6:164–170. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
de Jong YP, Abadia-Molina AC, Satoskar AR,
et al: Development of chronic colitis is dependent on the cytokine
MIF. Nat Immunol. 2:1061–1066. 2001.PubMed/NCBI
|
|
16
|
Baugh JA, Chitnis S, Donnelly SC, et al: A
functional promoter polymorphism in the macrophage migration
inhibitory factor (MIF) gene associated with disease severity in
rheumatoid arthritis. Genes Immun. 3:170–176. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Plant BJ, Gallagher CG, Bucala R, et al:
Cystic fibrosis, disease severity, and a macrophage migration
inhibitory factor polymorphism. Am J Respir Crit Care Med.
172:1412–1415. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miller EJ, Li J, Leng L, et al: Macrophage
migration inhibitory factor stimulates AMP-activated protein kinase
in the ischaemic heart. Nature. 451:578–582. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nohara H, Okayama N, Inoue N, et al:
Association of the −173 G/C polymorphism of the macrophage
migration inhibitory factor gene with ulcerative colitis. J
Gastroenterol. 39:242–246. 2004.
|
|
20
|
Mitchell RA, Liao H, Chesney J, et al:
Macrophage migration inhibitory factor (MIF) sustains macrophage
proinflammatory function by inhibiting p53: regulatory role in the
innate immune response. Proc Natl Acad Sci USA. 99:345–350. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hudson JD, Shoaibi MA, Maestro R, Carnero
A, Hannon GJ and Beach DH: A proinflammatory cytokine inhibits p53
tumor suppressor activity. J Exp Med. 190:1375–1382. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cooke G, Armstrong ME and Donnelly SC:
Macrophage migration inhibitory factor (MIF), enzymatic activity
and the inflammatory response. Biofactors. 35:165–168. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stosic-Grujicic S, Stojanovic I and
Nicoletti F: MIF in autoimmunity and novel therapeutic approaches.
Autoimmun Rev. 8:244–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu L, Li Y, Sun H, et al: Current
developments of macrophage migration inhibitory factor (MIF)
inhibitors. Drug Discov Today. 18:592–600. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hanayama R, Tanaka M, Miwa K and Nagata S:
Expression of developmental endothelial locus-1 in a subset of
macrophages for engulfment of apoptotic cells. J Immunol.
172:3876–3882. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hidai C, Zupancic T, Penta K, et al:
Cloning and characterization of developmental endothelial locus-1:
an embryonic endothelial cell protein that binds the alphavbeta3
integrin receptor. Genes Dev. 12:21–33. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rezaee M, Penta K and Quertermous T: Del1
mediates VSMC adhesion, migration, and proliferation through
interaction with integrin alpha(v)beta(3). Am J Physiol Heart Circ
Physiol. 282:H1924–H1932. 2002.PubMed/NCBI
|
|
28
|
Wang Z, Kundu RK, Longaker MT, Quertermous
T and Yang GP: The angiogenic factor Del1 prevents apoptosis of
endothelial cells through integrin binding. Surgery. 151:296–305.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Choi EY, Chavakis E, Czabanka MA, et al:
Del-1, an endogenous leukocyte-endothelial adhesion inhibitor,
limits inflammatory cell recruitment. Science. 322:1101–1104. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Choi EY, Orlova VV, Fagerholm SC, et al:
Regulation of LFA-1-dependent inflammatory cell recruitment by
Cbl-b and 14-3-3 proteins. Blood. 111:3607–3614. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim H, Lee SH, Lee MN, Oh GT, Choi KC and
Choi EY: p53 regulates the transcription of the anti-inflammatory
molecule developmental endothelial locus-1 (Del-1). Oncotarget.
4:1976–1985. 2013.PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
|
|
33
|
Salminen A and Kaarniranta K: Control of
p53 and NF-kappaB signaling by WIP1 and MIF: role in cellular
senescence and organismal aging. Cell Signal. 23:747–752. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Webster GA and Perkins ND: Transcriptional
cross talk between NF-kappaB and p53. Mol Cell Biol. 19:3485–3495.
1999.PubMed/NCBI
|
|
35
|
Blagosklonny MV, An WG, Romanova LY,
Trepel J, Fojo T and Neckers L: p53 inhibits hypoxia-inducible
factor-stimulated transcription. J Biol Chem. 273:11995–11998.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Blouin CC, Pagé EL, Soucy GM and Richard
DE: Hypoxic gene activation by lipopolysaccharide in macrophages:
implication of hypoxia-inducible factor 1alpha. Blood.
103:1124–1130. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ortiz-Barahona A, Villar D, Pescador N,
Amigo J and del Peso L: Genome-wide identification of
hypoxia-inducible factor binding sites and target genes by a
probabilistic model integrating transcription-profiling data and in
silico binding site prediction. Nucleic Acids Res. 38:2332–2345.
2010. View Article : Google Scholar
|