Introduction
Over the past decade, a novel estrogen receptor
(ER), GPR30, now known as G protein-coupled estrogen receptor 1
(GPER1), was identified as an orphan seven-transmembrane G
protein-coupled receptor (GPCR) by multiple laboratories (1–3).
As a functional membrane receptor, GPER1 is distinct from the
classical nuclear ERs, including ERα and ERβ). GPER1 mediates rapid
non-genomic responses to estrogen stimulation which occur within
seconds or minutes, such as cAMP production, calcium mobilization
and the activation of intracellular kinases, including
mitogen-activated protein kinase (MAPK), phosphoinositide-3-kinase
(PI3K), protein kinase A (PKA) and protein kinase C (PKC) (4–6).
These responses usually cannot be explained by the classical
nuclear ERα and ERβ signaling pathways, which perform their
functions through binding to estrogen response element (ERE)
located within the regulatory region of target genes, and usually
take hours to days to exert an effect on cells (7). Estrogen, tamoxifen (a selective ER
modulator), 4-hydroxytamoxifen (OTH), ICI 172,780 (an antagonist of
ERα and ERβ) and some phytoestrogens are ligands of GPER1 (8).
Herba Epimedii (Yin Yang Huo) is a
traditional Chinese herbal medicine that is widely used in the
treatment of impotence, involuntary ejaculation, rheumatism,
osteoporosis and bone fractures in China for thousands of years
(9). Icariin (ICA) is a
prenylated flavonol glycoside isolated from plants in the genus
Epimedium, and icaritin (ICT) is a derivative hyroxylated at
the 3,7-positions of ICA. Both ICA and ICT are important bioactive
components of Herba Epimedii. They exert a number of
beneficial cellular effects, including promoting apoptosis,
stimulating angiogenesis, inducing osteogenic differentiation and
upregulating extracellular matrix synthesis (10–12). Furthermore, ICA and ICT are also
regarded to be active phytoestrogens, exerting
estrogenic/anti-estrogenic effects due to their similar chemical
structures to genistein, a well-known phytoestrogen (13). It has been reported that low
concentrations of ICT (0.1 nM to 1 μM) promote MCF-7 cell growth
(14,15), while high concentrations of ICT
(>1 μM) have been shown to inhibit breast cancer cell
proliferation, indicating a diphasic regulatory action of ICT
(16). The proliferative effects
of ICA and ICT on estrogen-dependent breast cancer cells are mainly
due to the activation of nuclear ERα- and ERβ-mediated signaling
pathways (17). However, little
is known about the interactions between GPER1 and ICA or ICT.
Epidermal growth factor receptor (EGFR) is the
prototypical member of the family of transmembrane receptor
tyrosine kinases. Increasing evidence suggests that the activation
of EGFR is involved in GPER1-mediated multiple downstream events
(18–20). Among these events, the
GPER1-mediated activation of the EGFR-MAPK pathway plays a pivotal
role in estrogen-dependent tumor cell proliferation, including
breast, endometrial, ovarian and thyroid cancer (14,21–24).
In the present study, we used an ERα- and
ERβ-negative breast cancer cell line (SKBr3) as a model to identify
the possible mechanisms that ICA and ICT promote breast cancer cell
proliferation in a non-genomic GPER1-mediated pathway.
Materials and methods
Chemicals
ICA was purchased from the Chinese National
Institute for Food and Drug Control (Beijing, China). ICT was
purchased from Tauto Biotech (Shanghai, China). 17β-estradiol (E2),
DMSO and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) were purchased from Sigma (St. Louis, MO, USA). G-1
(a GPER1 agonist), AG-1478 (an EGFR inhibitor) and PD98059 (a MAPK
inhibitor) were obtained from Cayman Chemical Co. (Ann Arbor, MI,
USA). G-15 (a GPER1 antagonist) was purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Antibodies against ERK2
or phospho-ERK1/2 were obtained from Boster Biotechnology (Wuhan,
China).
Cell culture
SKBr3 breast carcinoma cells were obtained from the
Chinese Type Culture Collection, Chinese Academy of Sciences (CAS;
Shanghai, China) and were maintained in Dulbecco’s modified Eagle’s
medium (DMEM) with 4.5 g/l glucose (high-glucose) and 0.37% sodium
bicarbonate (Gibco, Rockville, MD, USA). In routine culture, the
cells were supplemented with 10% fetal bovine serum (FBS) and 1X
antibiotic mix (1×104 U/l penicillin A and 100 mg/l of
streptomycin) and grown at 37°C in a humidified atmosphere of 95%
air/5% CO2. Thee days before the cells were treated, the
medium was replaced with phenol-red-free DMEM supplemented with 4.5
g/l glucose and 0.37% sodium bicarbonate (Gibco), supplemented with
5% charcoal-dextran stripped FBS (CDT-FBS; Gibco) to eliminate
endogenous estrogen throughout the whole experimental period.
MTT assay
Cell proliferation assay was determined by MTT
colorimetric assay. Briefly, the SKBr3 cells were subcultured on
the logarithmic phase supplemented with phenol-red-free
high-glucose DMEM with 5% FBS. The cells were counted and seeded in
96-well culture plates at an initial density of 3×103
cells/well and allowed to attach to the plates. After 24 h of
attachment, the culture medium was discarded, and the cells were
treated with ICA (1 nM to 10 μM), ICT (1 nM to 10 μM) or ICA (1 nM
to 10 μM) with or without G-15 (20 μM), and ICT (1 nM to 10 μM)
with or without G-15 (20 μM) in 200 μl of phenol red-free
high-glucose DMEM with 5% FBS for 48 h. The media were then removed
and replaced with 20 μl of MTT (5 mg/ml) in PBS. The plates were
incubated for 4 h at 37°C followed by the addition of 150 μl DMSO
to dissolve the purple crystals, which are products of the MTT
substrates. The absorbance was read on a microplate reader at a
wavelength of 570 nm. The proliferation rate was calculated as PR%
= absorbance of experimental group/absorbance of control group
×100%.
Semi-quantitative RT-PCR analysis
The measurement of c-fos mRNA expression was
performed by semi-quantitative RT-PCR analysis. Briefly, the SKBr3
cells in phenol red-free DMEM containing 5% FBS were cultured in
6-well plates at an initial density of 1×105
cells/plate. When the cells reached reached 70% confluency, they
were cultured in phenol red-free DMEM containing 5% FBS for 24 h.
The cells were then treated with E2 (10 nM), ICA (100 nM), or ICT
(100 nM) for 15 to 60 min with or without pre-treatment with G-15
(20 μM), AG-1478 (10 μM), or PD98059 (10 μM) for 1 h. Total
cellular RNA was extracted using TRIzol reagent (CWbiotech,
Beijing, China). First-strand cDNA was synthesized using a HiFi-
MMLV cDNA kit (from CWbiotech). PCR was then performed using
specific primer pairs. The level of 36B4 housekeeping mRNA was used
as an internal control. The primers used were as follows: c-fos
forward, 5′-AGAAAAGGAGAATCCGAA GGGAAA-3′ and reverse,
5′-ATGATGCTGGGACAGGAA GTC-3′; and 36B4 forward,
5′-CTCAACATCTCCCCCTT CTC-3′ and reverse,
5′-CAAATCCCATATCCTCGTCC-3′. The c-fos and 36B4 DNA products were
420 and 408 bp, respectively. Band intensity was quantified with
ImageJ software (National Institutes of Health). The experiments
were repeated at least 3 times.
Western blot analysis
For quantification of the ERK1/2 levels, the SKBr3
cells were serum-starved for 40 h, and were then treated with E2
(10 nM), ICA (100 nM), ICT (100 nM), or G-1 (10 nM) for 0 to 30 min
with or without pre-treatment with G-15 (20 μM) or AG-1478 (10 μM)
for 1 h. The cells were lysed in RIPA buffer and then lysates were
separated by SDS-PAGE. Total ERK and phospho-ERK were detected
using the anti-ERK2 or anti-phospho-ERK1/2 antibodies (Boster
Biotechnology). The chemiluminescent signal was revealed by the
enhanced chemiluminescence system (ECL; Pierce Biotechnology,
Rockford, IL, USA) and the protein level was detected by exposure
to x-ray film. Band intensity was quantified with ImageJ software.
Experiments were repeated at least 3 times.
Cell cycle analysis
The SKBr3 cells were seeded in 6-cm culture plates
at an initial density of 5×105 cells/plate. Twenty-four
hours after attachment, the cells were starved with phenol-red-free
high-glucose DMEM with 0.5% FBS for 3 days. The cells were then
treated with E2 (10 nM), G-1 (10 nM), ICA (100 nM), or ICT (100 nM)
with or without pre-treatment with G-15 (20 μM), PD98095 (10 μM),
or AG-1478 (10 μM). After 48 h, the cells were harvested and the
cell cycle was analyzed as previously described (25). The cell proliferation index (PI)
was calculated as PI% = (S + G2/M)/(G0/G1 + S + G2/M) ×100%.
Statistical analysis
The statistical analysis of all data was performed
using SPSS 19.0 software. Data are expressed as the means ± SD, and
the level of significance between 2 groups was assessed using a
Student’s t-test. P values <0.05 were considered to indicate
statistically significant differences.
Results
GPER1 mediates the ICA or ICT-induced
proliferation of SKBr3 cells
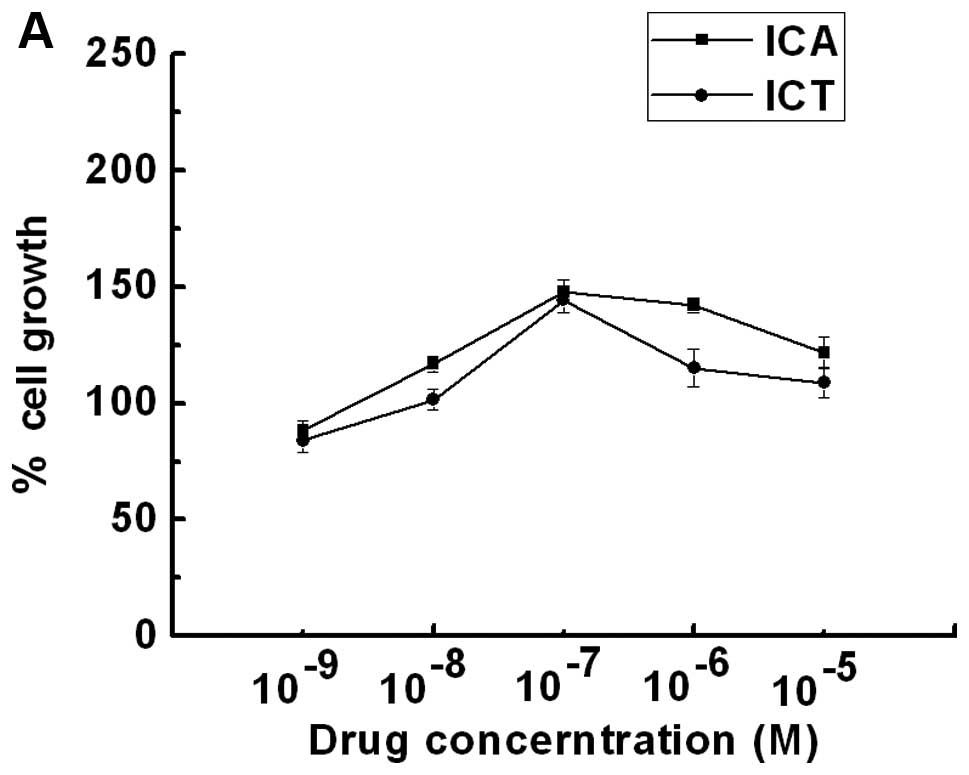
Both ICA and ICT stimulated the proliferation of the
SKBr3 ER-negative breast cancer cells in a dose-dependent manner
(Fig. 1A). The maximal cell
proliferative effects were 148 and 144% at a dose of
1×10−7 M ICA or ICT, respectively. To determine the
involvement of GPER1 in the ICA- or ICT-stimulated proliferation,
the SKBr3 cells were co-treated with G-15 (a high-affinity GPER1
antagonist, 20 μM) and ICA (100 nM) or ICT (100 nM). The
proliferation of the SKBr3 cells was completely suppressed by G-15
(Fig. 1B and C), suggesting that
the ICA- or ICT-stimulated SKBr3 cell proliferation was mediated by
the activation of GPER1.
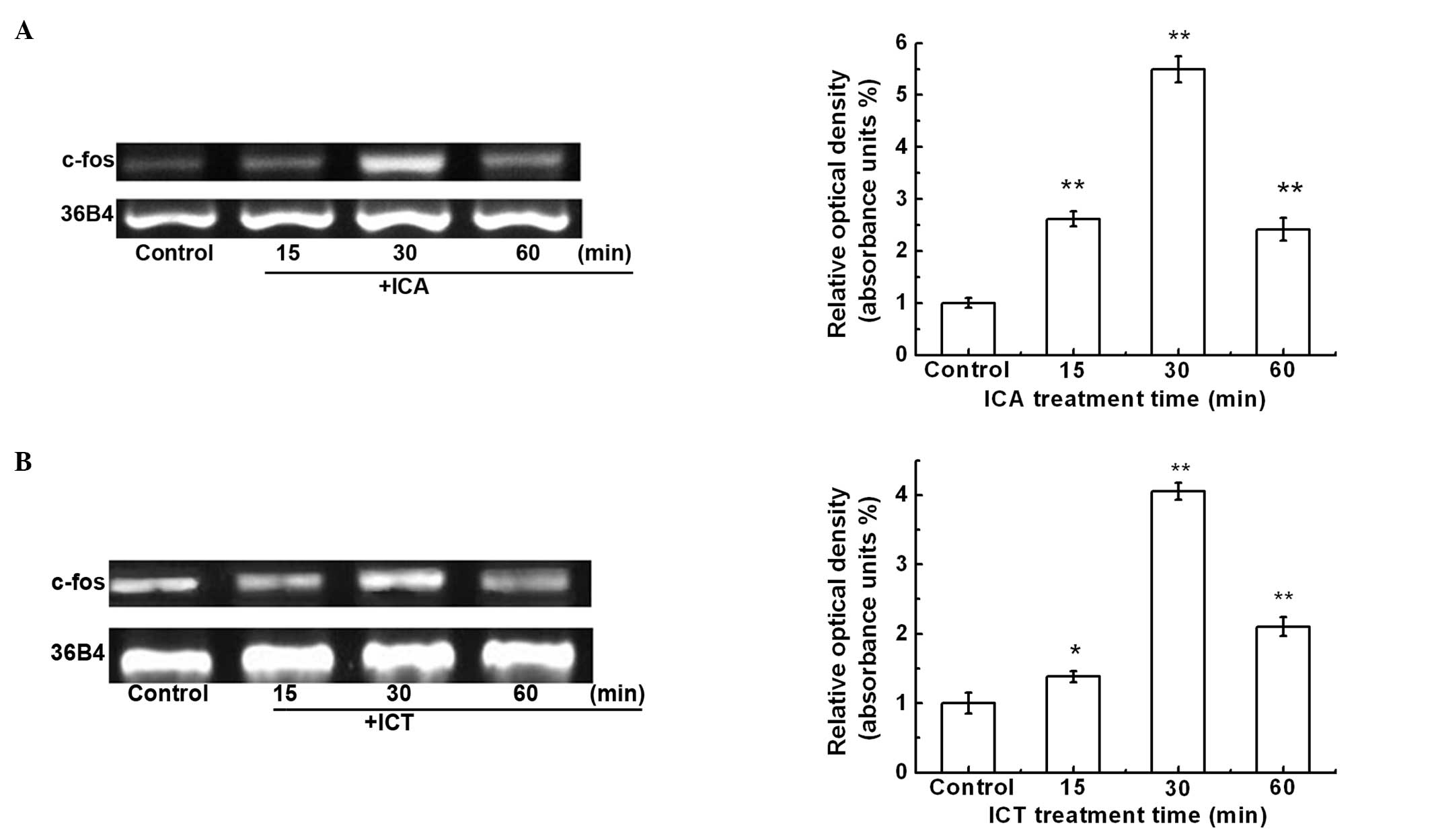
ICA and ICT stimulate c-fos mRNA
expression
The proto-gene, c-fos, is an immediate early gene
and its expression is rapidly induced by various extracellular
mitogens. In order to determine whether ICA or ICT induce c-fos
expression, the SKBr3 cells were treated with ICA (100 nM) or ICT
(100 nM) for 15, 30 and 60 min. Semi-quantitative RT-PCR analysis
was performed to measure c-fos mRNA expression (Fig. 2). The maximal c-fos mRNA
expression was detected at 30 min. c-fos mRNA expression was
increased by 5.5- and 4.1-fold in response to ICA and ICT
treatment, respectively (Fig. 2A and
B).
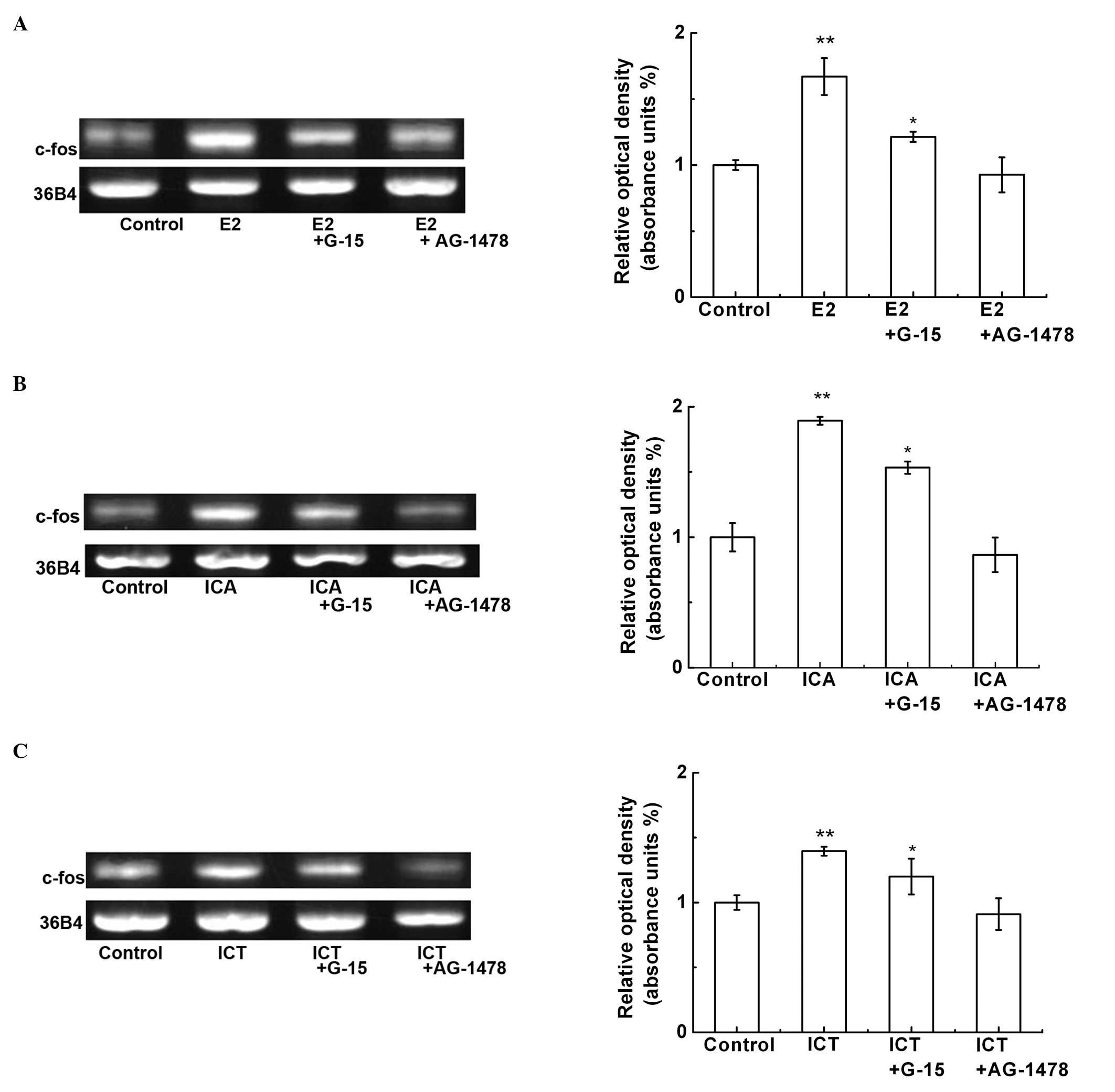
GPER1 and EGFR mediate the ICA- and
ICT-induced c-fos mRNA expression
To determine whether GPER1 and EGFR are required for
the ICA- or ICT-induced c-fos expression, the SKBr3 cells were
pre-treated with G-15 (20 μM), AG-1478 (an EGFR inhibitor, 10 μM)
for 1 h, and then treated with E2 (10 nM), ICA (100 nM) or ICT (100
nM) for 30 min (Fig. 3). G-15 and
AG-1478 markedly inhibited the E2-, ICA- or ICT-induced c-fos mRNA
expression (Fig. 3A–C). These
results indicate that ICA and ICT can mimic E2 that interacts with
GPER1 and activates downstream EFGR-mediated c-fos gene
expression.
Induction of c-fos mRNA expression by ICA
and ICT requires ERK1/2 activation
It has been reported that GPER1/EGFR-mediated c-fos
expression occurs through the activation of the MAPK signaling
pathway (23). Thus, we
investigated whether ICA or ICT increase the phosphorylation of
ERK1/2. ICA or ICT increased ERK1/2 phosphorylation in the SKBr3
cells within 5 min (Fig. 4A and
B). The effects of ICA and ICT were mimicked by G-1 (Fig. 4C). Furthermore, G-15 and AG-1478
significantly blocked the E2-, G-1-, ICA- or ICT-induced ERK1/2
phosphorylation (Fig. 4D and E).
In addition, the MAPK inhibitor, PD98059, significantly decreased
the E2-, ICA- and the ICT-induced c-fos mRNA expression (Fig. 4F).
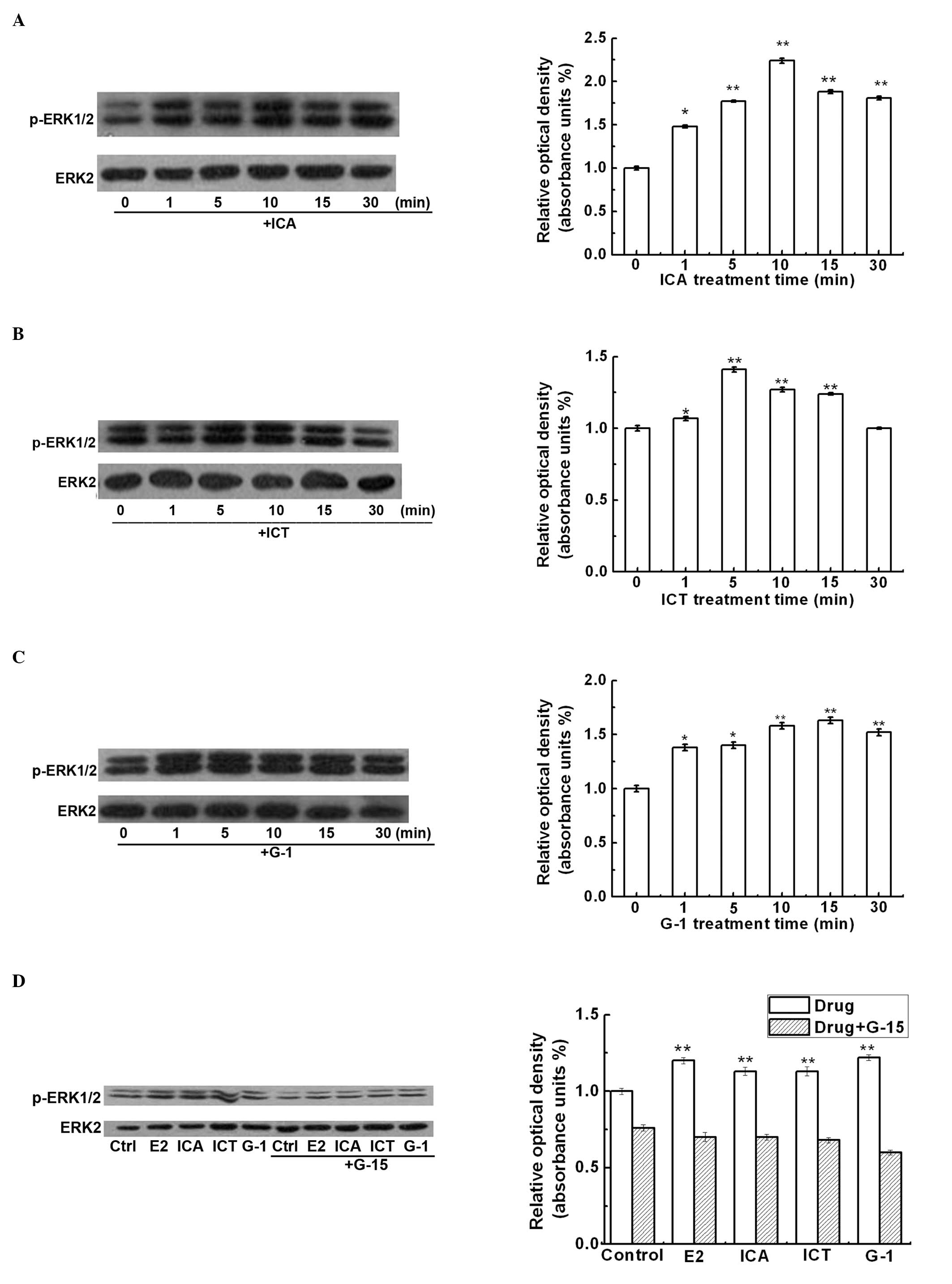 | Figure 4Icariin (ICA) or icaritin (ICT)
induces ERK1/2 phosphorylation in SKBr3 cells. SKBr3 cells were
serum-starved for 40 h before being treated for 0 to 30 min with:
(A) ICA (100 nM), (B) ICT 100 nM and (C) G-1 (10 nM). (D) SKBr3
cells were pre-treated with G-15 (20 μM) for 1 h, and then treated
with E2 (10 nM), G-1 (10 nM), ICA (100 nM) or ICT (100 nM) for 30
min. (E) SKBr3 cells were pre-treated with AG-1478 (10 μM) for 1 h,
and then treated with E2 (10 nM), G-1 (10 nM), ICA (100 nM) or ICT
(100 nM) for 30 min. Cells lysates were used for western blot
analysis for total ERK2 and phospho-ERK1/2. ERK2 protein levels
were used to normalize ERK1/2 expression. (F) SKBr3 cells were
pre-treated with PD98069 (10 μM) for 1 h, and then treated with E2
(10 nM), ICA (100 nM) or ICT (100 nM) for 30 min. RT-PCR analysis
was performed for quantification of c-fos mRNA expression. The
housekeeping gene, 36B4, was used as an internal control. The right
side panels show the means ± SD from 3 independent experiments.
**p<0.01 compared to the control (untreated)
group. |
ICA and ICT stimulate the proliferation
of SKBr3 cells through the GPER1-mediated modulation of the
EGFR-MAPK signaling pathway
To further confirm that ICA- or ICT-stimulated SKBr3
cell proliferation involves the GPER1-mediated activation of the
ERFR-MAPK signaling pathway, the SKBr3 cells were starved for 3
days and were then treated with E2 (10 nM), ICA (100 nM) or ICT
(100 nM) with or without pre-treatment with G-15 (20 μM), PD 98059
(10 μM) or AG-1478 (10 μM) for 2 days. The phase distribution of
the cell cycle and proliferation rate was assessed by flow
cytometry. We found that, similar to E2, ICA or ICT promoted SKBR3
cell proliferation that was blocked by pre-treatment with G-15,
AG-1478 or PD98059. These data suggest that the ICA- and
ICT-induced c-fos mRNA expression and cell proliferation of SKBr3
cells requires the activation of the EGFR-MAPK signaling pathway,
modulated by GPER1 (Table I).
 | Table IProliferation of SKBr3 cells at
various phases of the cell cycle. |
Table I
Proliferation of SKBr3 cells at
various phases of the cell cycle.
| Drug treatment | G0/G1 (%) | S (%) | G2/M (%) | PI (%) |
|---|
| Negative control | 64.1±3.8 | 2.4±0.5 | 33.6±0.6 | 35.9 |
| E2 (10 nM) | 26.5±4.2 | 35.3±0.5 | 38.3±0.9 | 73.5a |
| ICA (100 nM) | 25.9±2.7 | 61.7±0.3 | 12.3±0.8 | 74.1a |
| ICT (100 nM) | 13.8±2.6 | 50.1±0.6 | 36.1±1.1 | 86.2a |
| E2 (10 nM) + G-15 (20
μM) | 40.6±3.1 | 14.9±0.5 | 24.5±0.8 | 39.4 |
| ICA (100 nM) + G-15
(20 μM) | 42.3±1.8 | 19.8±0.2 | 18.0±1.7 | 37.7 |
| ICT (100 nM) + G-15
(20 μM) | 37.7±2.0 | 18.8±0.05 | 23.5±0.8 | 42.3 |
| E2 (10 nM) + AG-1478
(10 μM) | 34.5±5.2 | 15.1±0.6 | 20.4±0.6 | 35.5 |
| ICA (100 nM) +
AG-1478 (10 μM) | 57.4±3.2 | 20.6±0.2 | 21.9±0.6 | 42.6 |
| ICT (100 nM) +
AG-1478 (10 μM) | 66.2±4.0 | 9.5±0.2 | 24.4±0.8 | 33.8 |
| E2 (10 nM) + PD 98059
(10 μM) | 63.4±2.9 | 9.6±0.4 | 27.1±0.9 | 36.6 |
| ICA (100 nM) + PD
98059 (10 μM) | 76.0±3.5 | 13.1±0.2 | 20.9 ±0.5 | 34.0 |
| ICT (100 nM) + PD
98059 (10 μM) | 56.2±3.6 | 15.1±0.2 | 28.7±1.0 | 43.8 |
Discussion
The present study demonstrates that ICA and ICT
stimulate the proliferation of SKBr3 cells, an ERα- and
ERβ-negative breast cancer cell line, in vitro. The effects
of ICA and ICT on SKBr3 cell proliferation were mediated by rapid
non-genomic GPER1 action, which was associated with the induction
of EGFR, MAPK and c-fos expression. This ICA- and ICT-induced SKBr3
cell proliferation was inhibited by inhibitors of GPER1, EGFR and
MAPK. These results suggest that ICA and ICT possess the ability to
promote breast cancer cell growth through the activation of the
EGFR-MAPK signaling pathway, mediated by GPER1.
GPER1 is recognized as an important mediator for
promoting breast cancer cell proliferation through rapid
non-genomic estrogenic actions. Some phytoestrogens have been
proven to be the ligands of GPER1 (8). To the best of our knowledge, our
study demonstrates for the first time that a low concentration of
ICA and ICT (1 nM to 1 μM) induces SKBr3 cell proliferation through
the GPER1-mediated modulation of the MAPK pathway (Fig. 1). Consistent with our findings,
genistein and quercetin have been shown to stimulate MCF-7 breast
cancer cell growth through a rapid GPER1-mediated action (26). Another phytoestrogen, tectoridin,
has also been reported to induce MCF-7 cell growth through the
activation of GPRE1-mediated MAPK signaling (27).
Several in vitro studies have demonstrated
that EGFR-MAPK signaling is involved in GPER1-mediated cell
proliferation. Filardo et al demonstrated that E2 activates
ERK1/2 through the activation of EGFR by releasing heparin-bound
epidermal growth factor (EGF) in SKBr3 breast cancer cells
(21). Another study also
demonstrated that the activation of GPER1 enhances EGF production,
which triggers a rapid ERK phosphorylation and c-fos expression in
ER-negative SKBr3 and BT20 cells (23). In our study, using SKBr3 cells as
a model system, we demonstrated that the GPER1 antagonist, G-15,
inhibited ICA- and ICT-induced SKBr3 cell growth, indicating that
GPER1 mediated the ICA- and ICT-induced SKBr3 cell proliferation
(Fig. 1A and B). Importantly, we
found that the ICA- and ICT-induced c-fos mRNA expression was
markedly attenuated by the GPER1 antagonist, G-15, and the EGFR
antagonist, AG-1478, indicating that the ICA- and ICT-induced c-fos
upregulation was mediated through the activation of GPER1 and EGFR
(Figs. 2 and 3). Furthermore, we demonstrated that ICA
and ICT rapidly stimulated the phosphorylation of ERK1/2 in
serum-starved SkBr3 cells (Fig. 4A
and B), which was blocked by pre-treatment with G-15 and
AG-1478 (Fig. 4D and E).
Consistent with the above results, the MAPK inhibitor, PD98059,
blocked c-fos transcription which was induced by ICA and ICT
treatment (Fig. 4F). Considering
that the proliferation of the SKBr3 cells also required the
EGFR-dependent ERK1/2 activation, we ascertained their roles in the
proliferation of SKBr3 cells by flow cytometry. Indeed, the EGFR
antagonist, AG-1478, and the MAPK inhibitor, PD98059, abrogated the
response to ICA and ICT (Table
I). The above results demonstrated that the ICA- or
ICT-stimulated SKBr3 cell proliferation occurred through the
GPER1-mediated modulation of the ERFR-MAPK signaling pathway.
Of note, we demonstrated that both ICA and ICT
promoted SKBr3 cell proliferation through an ERα- and
ERβ-independent, non-genomic pathway, and there was no difference
in the proliferative activity between ICA and ICT. However, ICT has
been demonstrated to have stronger estrogenic activity than ICA in
MCF-7 cells through an ER-dependent pathway due to the steric
hindrance produced by glycoside moieties of ICA, which prevents
docking to the ER binding site (14,17). A possible explanation for the
equal activity of ICA and ICT to promote SKBr3 cell proliferation
is that ICA and ICT have a similar binding affinity to GPER1. The
presence of 3,7-position glycoside moieties in ICA do not hamper
binding to GPER1.
In conclusion, we demonstrate that both ICA and ICT
activate the EGFR-MAPK signaling pathway in SKBr3 cells and that
this pathway is mediated by the functional membrane, GPER1. GPER1
rapidly transduces a signal from EGFR to ERK1/2, which upregulates
c-fos expression and results in SKBr3 breast cancer cell
proliferation. The precise mechanisms responsible for the ICA or
ICT induction of c-fos expression in breast cancer cells require
further investigation. To the best of our knowledge, our findings
provide new insight into these mechanisms, demonstrating that ICA
and ICT induce the progression of ER-negative breast cancer cell
growth through the activation of GPER1.
Acknowledgements
This study was supported in part by grants from the
National Natural Science Foundation of China (Grant no. 81102890)
and the Joint Funds of the National Natural Science Foundation of
China (Grant No. U1203203)..
References
|
1
|
Owman C, Blay P, Nilsson C and Lolait SJ:
Cloning of human cDNA encoding a novel heptahelix receptor
expressed in Burkitt’s lymphoma and widely distributed in brain and
peripheral tissues. Biochem Bioph Res Co. 228:285–292.
1996.PubMed/NCBI
|
|
2
|
Kuiper GG, Enmark E, Pelto-Huikko M,
Nilsson S and Gustafsson JA: Cloning of a novel estrogen receptor
expressed in rat prostate and ovary. PNAS. 93:5925–5930. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carmeci C, Thompson DA, Ring HZ, Francke U
and Weigel RJ: Identification of a gene (GPR30) with homology to
the G-protein-coupled receptor superfamily associated with estrogen
receptor expression in breast cancer. Genomics. 45:607–617. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bouskine A, Nebout M, Bruecker-Davis F,
Benahmed M and Fenichel P: Low doses of bisphenol A promote human
seminoma cell proliferation by activating PKA and PKG via a
membrane G-protein-coupled estrogen receptor. Environ Health Persp.
117:1053–1058. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Filardo EJ: Epidermal growth factor
receptor (EGFR) transactivation by estrogen via the
G-protein-coupled receptor, GPR30: a novel signaling pathway with
potential significance for breast cancer. J Steroid Biochem.
80:231–238. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Revankar CM: A transmembrane intracellular
estrogen receptor mediates rapid cell signaling. Science.
307:1625–1630. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pearce ST and Jordan VC: The biological
role of estrogen receptors α and β in cancer. Crit Rev Oncol
Hematol. 50:3–22. 2004.
|
|
8
|
Prossnitz ER, Sklar LA, Oprea TI and
Arterburn JB: GPR30: a novel therapeutic target in estrogen-related
disease. Trends Pharmacol Sci. 29:116–123. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma H, He X, Yang Y, Li M, Hao D and Jia Z:
The genus Epimedium: An ethnopharmacological and phytochemical
review. J Ethnopharmacol. 134:519–541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang X, Zhu D and Lou Y: A novel
anticancer agent, icaritin, induced cell growth inhibition, G1
arrest and mitochondrial transmembrane potential drop in human
prostate carcinoma PC-3 cells. Eur J Pharmacol. 564:26–36. 2007.
View Article : Google Scholar
|
|
11
|
Chung BH, Kim JD, Kim CK, Kim JH, Won MH,
Lee HS, et al: Icariin stimulates angiogenesis by activating the
MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways in human
endothelial cells. Biochem Bioph Res Co. 376:404–408. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang L, Zhang X, Li KF, Li DX, Xiao YM,
Fan YJ, et al: Icariin promotes extracellular matrix synthesis and
gene expression of chondrocytes in vitro. Phytother Res.
26:1385–1392. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ming LG, Chen KM and Xian CJ: Functions
and action mechanisms of flavonoids genistein and icariin in
regulating bone remodeling. J Cell Physiol. 228:513–521. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang ZQ and Lou YJ:
Proliferation-stimulating effects of icaritin and desmethylicaritin
in MCF-7 cells. Eur J Pharmacol. 504:147–153. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu Y, Yan H, Hu S and Zhang J: Study on
the estrogen-like effects of Epimedium extractive. J Xi’an Jiao
tong Univ (Med Sci). 30:373–376. 2009.
|
|
16
|
Guo Y, Zhang X, Meng J and Wang ZY: An
anticancer agent icaritin induces sustained activation of the
extracellular signal-regulated kinase (ERK) pathway and inhibits
growth of breast cancer cells. Eur J Pharmacol. 658:114–122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ye HY and Lou YJ: Estrogenic effects of
two derivatives of icariin on human breast cancer MCF-7 cells.
Phytomedicine. 12:735–741. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Levin ER: Bidirectional signaling between
the estrogen receptor and the epidermal growth factor receptor. Mol
Endocrinol. 17:309–317. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Razandi M: Proximal events in signaling by
plasma membrane estrogen receptors. J Biol Chem. 278:2701–2712.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fujiwara S, Terai Y, Kawaguchi H, Takai M,
Yoo S, Tanaka Y, et al: GPR30 regulates the EGFR-Akt cascade and
predicts lower survival in patients with ovarian cancer. J Ovarian
Res. 5:352012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Filardo EJ, Quinn JA, Bland KI and
Frackelton AR Jr: Estrogen-induced activation of Erk-1 and Erk-2
requires the G protein-coupled receptor homolog, GPR30, and occurs
via trans-activation of the epidermal growth factor receptor
through release of HB-EGF. Mol Endocrinol. 14:1649–1660. 2000.
View Article : Google Scholar
|
|
22
|
Du GQ, Zhou L, Chen XY, Wan XP and He YY:
The G protein-coupled receptor GPR30 mediates the proliferative and
invasive effects induced by hydroxytamoxifen in endometrial cancer
cells. Biochem Bioph Res Co. 420:343–349. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Albanito L, Madeo A, Lappano R, Vivacqua
A, Rago V, Carpino A, et al: G protein-coupled receptor 30 (GPR30)
mediates gene expression changes and growth response to 17
beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer
cells. Cancer Res. 67:1859–1866. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vivacqua A, Bonofiglio D, Albanito L,
Madeo A, Rago V, Carpino A, et al: 17beta-estradiol, genistein, and
4-hydroxytamoxifen induce the proliferation of thyroid cancer cells
through the G protein-coupled receptor GPR30. Mol Pharmacol.
70:1414–1423. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maggiolini M: The G protein-coupled
receptor GPR30 mediates c-fos up-regulation by 17-estradiol and
phytoestrogens in breast cancer cells. J Biol Chem.
279:27008–27016. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma H, Lu Z, Sun Y, Peng T, Shuai Z, Ma Y,
et al: Selection of donor nuclei in somatic cell-mediated gene
transfer using a co-transfection method. J Reprod Develop.
53:95–104. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kang K, Lee SB, Jung SH, Cha KH, Park WD,
Sohn YC, et al: Tectoridin, a poor ligand of estrogen receptor α,
exerts its estrogenic effects via an ERK-dependent pathway. Mol
Cells. 27:351–357. 2009.PubMed/NCBI
|